

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is an almost uniformly lethal disease with less than 5% survival at five years. This is largely due to metastatic disease, which is already present in the majority of patients when diagnosed. Even when the primary cancer can be removed by radical surgery, local recurrence occurs within one year in 50%-80% of cases. Therefore, it is imperative to develop new approaches for the treatment of advanced cancer and the prevention of recurrence after surgery. Tumour-targeted oncolytic viruses (TOVs) have become an attractive therapeutic agent as TOVs can kill cancer cells through multiple mechanisms of action, especially via virus-induced engagement of the immune response specifically against tumour cells. To attack tumour cells effectively, tumour-specific T cells need to overcome negative regulatory signals that suppress their activation or that induce tolerance programmes such as anergy or exhaustion in the tumour microenvironment. In this regard, the recent breakthrough in immunotherapy achieved with immune checkpoint blockade agents, such as anti-cytotoxic T-lymphocyte-associate protein 4, programmed death 1 (PD-1) or PD-L1 antibodies, has demonstrated the possibility of relieving immune suppression in PDAC. Therefore, the combination of oncolytic virotherapy and immune checkpoint blockade agents may synergistically function to enhance the antitumour response, lending the opportunity to be the future for treatment of pancreatic cancer.
Keywords: Anti-cytotoxic T-lymphocyte-associate protein 4, Anti-programmed death receptor ligand 1, Anti-programmed death receptor 1, Immunotherapy, Oncolytic viruses, Pancreatic ductal adenocarcinoma, Pancreatic cancer, Immune checkpoint blockade inhibitors, Cancer vaccine
Core tip: The poor prognosis of pancreatic cancer appeals for a novel strategy to treat this disease. Immunotherapies such as whole cell vaccines are currently being investigated in patients with pancreatic cancer. The recent breakthrough in cancer immunotherapy with immune checkpoint inhibitors has allowed us to reverse T cell anergy and enhance antitumour immunity. Tumour-targeted Oncolytic viruses in combination with checkpoint inhibitors function synergistically to increase the tumour antigen load, relieve immune suppression in the tumour bed while enhancing the activation of the adaptive immune system. Viro-immune-checkpoint therapy should thus be considered as a novel strategy to treat pancreatic cancer.
INTRODUCTION
In the past four decades, the diagnosis of pancreatic cancer has remained accompanied by a death sentence[1]. Despite major advances in the field of cancer treatment for other solid tumours such as breast, colorectal, renal and prostate[2], the all-stages-combined 5-year survival rate for a patient with pancreatic cancer remains a devastating 7%, and only 2% for those who present with distant metastases[3]. The majority (80%) of pancreatic malignancies are pancreatic ductal adenocarcinomas (PDAC), which are presented at locally advanced or metastatic disease[1,3]. Late diagnosis is attributed to several factors, mostly vague, non-specific symptoms associated with an earlier stage compared to later stages, such as jaundice, anorexia, weight loss and gastric outlet obstructions[4]. Furthermore, the lack of a reliable screening method for non-high-risk cohorts[5] impedes the possibility of early detection in the majority of the patients, as family history only contributes to 5%-10% of pancreatic cancer incidence[6]. As a result, patients are presenting at an advanced stage, diminishing the chance of curative resection and a more manageable disease[1,4]. Moreover, PDAC is an aggressive malignancy with high propensity to spread through the lymphatic system and invade distant organs[7]. The molecular tumourigenesis of PDAC is described as a stepwise progression of a preinvasive stage, pancreatic intraepithelial neoplasia, associated with the accumulation of genetic mutations[8]. One of the earliest genetic alterations in a non-malignant precursor lesion is an activating point mutation in the KRAS2 oncogene[9,10], which is present in 90% of pancreatic cancer cases. Mutational inactivation of tumour-suppressor gene CDKN2A encoding p16 (a regulator of the G1 to S phase in the cell cycle), TP53 and SMAD4 which occur in higher grade lesions are also commonly found[11]. A distinct hallmark of pancreatic cancer is its complex tumour stroma composed of a strategic array of cells. The PDAC stroma is highly heterogenous inhabiting fibroblasts, pancreatic stellate cells, immune cells, blood vessels and extracellular matrix, however very few infiltrating effector T cells[12] (Figure 1). The proliferative nature of the stromal pancreatic stellate cells, termed desmoplasia, accounts for their high turnover rate, invasiveness[12,13] and hypoxic microenvironment[14]. Consequently, the tumour stroma is not only a mechanical barrier for treatment delivery and efficacy, but also an active contributor to tumour progression[15,16].
Figure 1.
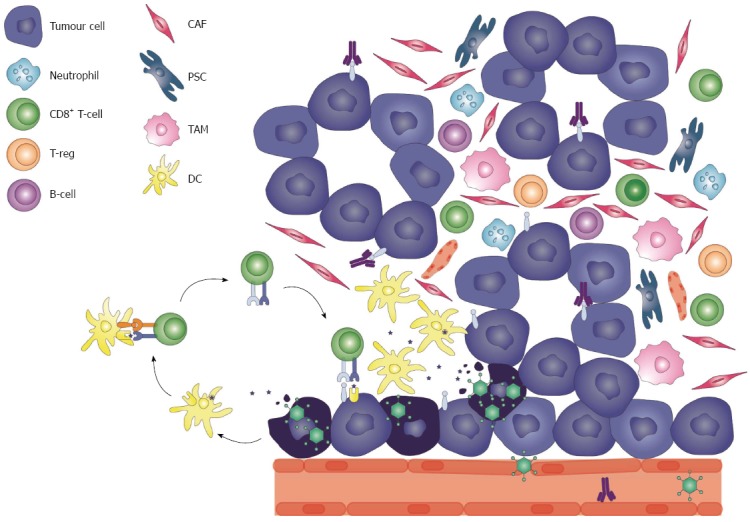
Synergistic effects of viro-immunotherapy. Systemic administration of a tumour-targeted oncolytic virus (TOV) leads to vasculature destruction allowing the invasion and infection of the tumour cells. As the TOV selectively replicates within a tumour cell, it infects neighboring cells leading to the amplification of the virotherapy and induction of the innate immune system. Direct oncolysis allows the release of tumour-associated antigens into the microenvironment, which are presented by antigen-presenting dendritic cells (DCs) to naïve T cells in the draining lymph nodes, activating the adaptive tumour-specific response. The administration of an immune checkpoint blockade antibody such as an anti-PD-L1 antibody provides an added benefit of reversing T cell anergy. This allows the antitumour response induced by the virotherapy to be enhanced and sustained. The combination of viro-immunotherapy may have significant synergistic effects in such an immunosuppressive microenvironment as seen in pancreatic ductal adenocarcinoma. CAF: Cancer-associated fibroblast; ; PSC: Pancreatic stellate cell; TAM: Tumour-associated macrophage.
This review will briefly discuss the current management of pancreatic cancer and introduce the immunotherapies in development for pancreatic cancer treatment. Lastly, we will highlight a novel emerging area of pre-clinical and clinical research, viro-immune-checkpoint blockade therapy combination strategies.
CURRENT MANAGEMENT OF PANCREATIC CANCER
Surgical resection
For the 20% of patients who present with early disease, surgical resection is the treatment of choice, and the only curative option[16,17]. Nonetheless, even after complete resection, the prognosis remains disappointing, hence the incorporation of adjuvant gemcitabine or 5-fluorouracil and/or chemoradiation into the standard of care[18]. Results from randomized controlled trials[19-21] demonstrating increased overall survival (OS) with postoperative therapy is considered to be one of the most important advances in the treatment of pancreatic cancer[16]. Similarly, neoadjuvant chemotherapy may be offered to improve surgical margins of borderline resectable tumours[22-25].
Locally advanced and metastatic disease
Gemcitabine or gemcitabine-based combination chemotherapy is the long established first line treatment for advanced pancreatic cancer, however the median survival rate is approximately 9 mo[16,26,27]. More recently, an advantage on the survival and quality of life was shown with FOLFIRINOX (folinic acid, 5-fluorouracil, irinotecan, oxaliplatin) compared to gemcitabine alone; this regimen significantly improved the OS, progression-free survival and objective response rate of patients with pancreatic cancer[28]. Similar results were also observed with nab-paclitaxel plus gemcitabine[29]. Approximately 10% of patients receiving these regimens are surviving two years, which is a rare event in advanced disease[30]. However, both regimens are associated with increased toxicities thus may only be offered to patients with good performance status[28,29]. The care of patients with poor performance status or metastatic disease remains palliative, and gemcitabine-based therapies have limited efficacy.
Targeted therapy
In the last 10 years, targeted therapy has revolutionised current cancer treatment and has paved the way for personalised medicine[31]. Due to the genetically heterogeneous nature of pancreatic cancer[32] targeted therapies such as small molecule inhibitors and monoclonal antibodies have been sought to inhibit constitutively-active cell surface signaling molecules. Nonetheless, results of phase I-III clinical trials (summarized by Seicean et al[33]) are disappointing, and the observed resistance is most likely due to the high frequency of KRAS2 mutations and upregulation of alternate signaling pathways[16,34]. To date, erlotinib, a small molecule inhibitor of the epidermal growth factor receptor inhibitor, is the only approved agent, in combination with gemcitabine, which offers a very modest but statistically significant increase in survival of two weeks[35].
IMMUNOTHERAPY: A NOVEL STRATEGY FOR PANCREATIC CANCER
Discouraging response rates and resistance to current standard therapies has prompted the investigation of novel strategies for the treatment of pancreatic cancer. Immunotherapy is an attractive therapeutic option as it has the greatest promise to eradicate tumours with minimal toxicities to healthy tissues and furthermore, to prevent recurrence via the induction of long-term memory[36]. It has recently become more evident that the immune system plays contrasting roles in both tumour elimination and tumour progression, a phenomenon called immunosurveillance[37]. Therefore, “avoiding immune destruction” and “tumour-promoting inflammation” have been recently included as hallmark biological capabilities of tumours by Hanahan and Weinberg in their famous review, adding to already well-defined neoplastic characteristics such as sustaining proliferative signaling, evading growth suppressors and activating invasion and metastasis[38]. Transformed cells limit their presentation of neo-antigens and paralyze infiltrating immune cell functions, leading to the development of clinically-apparent tumours[39]. Therefore, main goal of immunotherapy is to breach the immune suppression induced by the tumour microenvironment (TME) while boosting or revoking effector functions[36]. The TME of PDAC, which promotes tumour growth and impedes successful delivery of traditional cytotoxic therapies, may paradoxically be a perfect candidate for manipulation by immunotherapy. Lastly, the greatest advantage of immunotherapy is its potential to induce both innate and adaptive immune responses, which are not always activated by traditional cytotoxic or targeted therapies.
THERAPEUTIC VACCINES
Several preparations of antitumour vaccines are currently being investigated in clinical trials. The objective of therapeutic vaccination is to amplify pre-existing immune responses or to prompt de novo responses[40]. The clinical potential of various types of vaccines (peptide-based[41,42], dendritic cell[43,44], whole tumour cell[45,46], and recombinant viral or bacterial vector-based vaccines[47,48]) has been demonstrated in early phase clinical trials with some promising immunological and clinical responses in patients with pancreatic cancer, however, the phase III TeloVac trial showed that adding GV1001 peptide vaccination to chemotherapy did not improve OS[49]. Emerging evidence over the last two decades has demonstrated that prime-boost protocols integrating different vectors result in effective immune responses with protective capability in animal models and human clinical trials[50,51]. Pre-clinical experiments and early phase clinical trials have indicated that the sequential use of recombinant vaccinia and avipox or fowlpox vectors expressing tumour-associated antigens (TAAs) such as carcinoembryonic antigen (CEA) and mucin (MUC)-1 and co-stimulatory molecules in a heterologous prime-boost regimen resulted in a more potent T cell response than either vaccine alone[52] and demonstrated encouraging clinical responses[53]. However, the heterologous prime-boost regimen has yet to show efficacy in a phase III trial for human pancreatic cancer[54]. The failure of previous cancer vaccine trials can be attributed to several factors including inappropriate choice of tumour antigen (only a few TAAs were selected), use of an unoptimised antigen delivery vector or vaccination schedule, or selection of the wrong patient group (late stage advanced disease). In the last decade, the most popular vaccine strategy has been based on immunisation with genetically modified tumour cells. We will describe the two most clinically advanced vaccines for treatment of pancreatic cancer below.
Algenpantucel-L
One of the most clinically advanced vaccines for advanced pancreatic cancer is a preparation of whole human allogeneic pancreatic cell lines engineered to express α-galactosyl (α-gal) transferase, which synthesizes α-gal epitopes on the surface of glycolipids and glycoproteins. Although human cells do not express α-gal epitopes[55], humans have existing anti-α-gal antibodies, which activate complement and antibody-dependent cell-mediated lysis upon binding to antigen[56]. This causes a hyperacute rejection of the allografts and a generation of an immune response against the tumour[56]. In a phase II trial of algenpantucel-L with adjuvant chemotherapy with gemcitabine and 5-FU-based chemoradiation after R0/R1 resection, the 1-year disease-free survival and OS were reached by 62% and 86% of patients, respectively[57]. The results of this trial were compared with the RTOG-9704 trial whereby patients received the same chemoradiation regimen[58]. Minor adverse effects associated with the vaccine were pain and induration at the site of injection, which resolved within a week[57]. Two multi-institutional phase III trials are currently being conducted and the results are highly anticipated (clinicaltrials.gov: NCT01072981, NCT01836432).
Granulocyte-macrophage colony-stimulating factor vaccine
The granulocyte-macrophage colony-stimulating factor (GM-CSF) vaccine (G-VAX) is another promising whole cell vaccine that induces a potent anti-tumour immune response. GM-CSF is a widely used immunomodulator because it is a potent monocyte, leukocyte and eosinophil attractant. GM-CSF-expressing irradiated tumour cells were shown to induce a long-lasting immune response involving both CD4+ and CD8+ T cells in a B16 melanoma model[59]. G-VAX was shown to be safe, efficacious and induce very minimal toxicities in patients with PDAC in phase I and II trials[45,46,60]. Furthermore, G-VAX induced mesothelin- (a TAA in pancreatic cancer) specific interferon (IFN)-γ-producing CD8+ T cells, which also provided evidence of in vivo cross-priming by antigen-presenting cells (APCs) in G-VAX-vaccinated patients[46,61,62]. Jaffe et al[45] observed a delayed-type hypersensitivity response in three patients, all of which remained disease-free at least 25 mo post-diagnosis. Phase II trial results by Lutz et al[60] revealed 1-year disease-free and 1-year survival rates of 67% and 85%, respectively, which correlated with the induction of a PDAC-associated antigen mesothelin-specific CD8+ T cells in human leukocyte antigen (HLA)-A1+ and A2+ patients[60], when combined with chemoradiation. This suggests that PDAC, which is considered a highly “non-immunogenic” solid tumour, can be converted to an immunogenic tumour[63]. Lutz et al[63] demonstrated post-G-VAX T cell infiltration and aggregate formation, which resulted in the upregulation of the PD-1/PD-L1 pathway, suggesting that vaccine-primed PDAC patients may respond better to other immunotherapies[63]. G-VAX with low-dose cyclophosphamide and CRS-207, a live-attenuated Listeria monocytogenes-expressing mesothelin has shown safety and increased survival in metastatic pancreatic cancer, and longer OS was accompanied by enhanced mesothelin-specific CD8+ T cell responses, regardless of the treatment arm[64]. A phase IIB trial for testing G-VAX + CRS-207 compared to chemotherapy or CR-207 alone in previously treated metastatic PDAC is currently recruiting patients (clinicaltrials.gov: NCT02004262). Nonetheless, studies have also demonstrated that GM-CSF-producing vaccines can play conflicting roles in the antitumour response[65]. Serafini et al[66] demonstrated that a high-dose GM-CSF-producing B78H1 cell line resulted in the production of GM-CSF-induced Gr1+/CD11b+ myeloid-derived suppressor cells (MDSCs) that impaired antigen-specific T cell responses. CD14+HLA-DR-/lo MDSCs were also detected in the peripheral blood of GM-CSF-vaccinated metastatic melanoma patients and were absent in healthy donors or patients receiving non-GM-CSF-based vaccines[67]. The suppressive activity of the identified MDSCs involved the production of transforming growth factor (TGF)-β[67]. A study demonstrated that chemotherapy-derived inflammatory responses accelerated the formation of MDSCs in the PDAC microenvironment, and was associated with poorer prognosis[68]. Moreover, GM-CSF production induced by oncogenic KRAS has also been shown to promote neoplastic precursors of PDAC[69]. Therefore, these findings demand caution when administrating GM-CSF in patients with PDAC.
ANTI-IMMUNE CHECKPOINT AGENTS
Immune checkpoint blockade
Immune checkpoints are crucial for maintaining self-tolerance under normal physiological conditions and protecting healthy tissues during an immunological response[70]. They consist of co-stimulatory and co-inhibitory receptors that regulate T cell activation. Tumours have evolved to exploit immune checkpoints to avoid recognition by the immune system and sustain immunosuppression. T cells are the main therapeutic targets in cancer immunotherapy, because of the major role they play in adaptive immunity. Moreover, activated CD8+ T cells directly lyse antigen-expressing cells[70]. As a result, antibodies targeting T cell co-stimulator and co-inhibitory receptors “wake up” the immune system, unleashing an antitumour response, first demonstrated in animal models[71,72]. Today, the clinical activity of these agents has been tested in various cancer types[73-78] and exciting results have shined a new light on the field of cancer immunotherapy. Most importantly, the ability of these anti-immune checkpoint receptor agents to relieve immunosuppression or boost a pre-existing immune response provides further rational for combining immunotherapeutic strategies.
Anti-cytotoxic T-lymphocyte-associate protein 4 inhibitors
Frequently credited as one of the most important developments for cancer immunotherapy is the anti-cytotoxic T-lymphocyte-associate protein (CTLA)-4 humanized antibody, ipilimumab (Yervoy®; Bristol-Myers Squibb, New York City, NY, United States), which was approved by United States’ Food and Drug Administration in 2011 for the treatment of metastatic melanoma[73,79]. CTLA-4 is a co-negative regulator on T cells that binds with high affinity to the B7 family of accessory molecules on APCs during the T cell priming phase, thus competes with the CD28 co-stimulatory molecule on T cell[80-82]. Its two major roles are the downmodulation of helper T cell activity, and the enhancement of the regulator T cell (Treg) suppressive function[70]. Loss of CTLA-4 leads to massive lymphoproliferation, autoimmunity and death in mice[83]. Consequently, CTLA-4 may be blocked in order to relieve immunosuppression and un-restrain pre-existing T cell responses. CTLA-4 efficacy was questioned at first, because of its non-specificity to tumour cells, however, it was demonstrated that partial inhibition with a monoclonal antibody could achieve a therapeutic window[71]. Nonetheless, as these immunomodulatory mechanisms exist to attenuate chronic autoimmune inflammation, certain toxicities were expected. CTLA-4 blockade compromised tolerance to normal tissue antigens, causing immune-related adverse events such as colitis and hypophysitis, which were mostly resolved with corticosteroid therapy[73]. Ipilimumab as a single agent (3.0 mg/kg/dose) was tested in advanced pancreatic patients however was shown to be ineffective[84]. In a phase I study, gemcitabine and escalating doses of i.v. tremelimumab (6, 10 or 15 mg/kg) were administered to patients with metastatic pancreatic cancer, demonstrating a safe and tolerable profile, where 2/19 patients receiving 15 mg/kg achieved partial responses[85]. Results from a randomized controlled trial comparing ipilimumab as a single agent (10 mg/kg/dose) and ipilimumab + G-VAX favored the second arm (OS = 3.6 mo vs 5.7 mo, 1-year survival 7% vs 27%, respectively) and an increase in mesothelin-specific T cells and enhancement of the T cell repertoire were observed[86], further implying the benefit of combining immunotherapeutic strategies. These patients also experienced declines in CA 19-9 levels[86].
Anti-PD-1 and anti-PD-L1 inhibitors
A second immune checkpoint is in the peripheral tissue at a site of inflammation, whereby the activation of a T cell is limited by the programmed death receptor (PD)-1[70]. Thus, CTLA-4 regulates T cell “priming” or activation whereas PD-1 regulates T cell effector function within tissues and tumours[70]. PD-1 is a cell surface coinhibitory receptor expressed on activated T cells, natural killer (NK) cells, monocytes and B cells. Its expression is associated with impaired or exhausted T cells[87], and is more broadly expressed than CTLA-4[70]. Its two known ligands, PD-L1 (B7-H1) and PD-L2 (B7-DC)[88,89], are commonly overexpressed on tumour cells, while PD-1 itself is highly expressed on tumour-infiltrating lymphocytes (TILs) in many cancers[88,90]. Expression of PD-L1 was first shown to inhibit antitumour T cell mediated responses in animal models[72,88,91], thus determining its expression pattern in solid tumours may be an important biomarker for assessing a patient’s suitability for anti-PD-1/PD-L1 therapy[70]. Engagement of PD-1 with its ligand leads to negative regulation of lymphocyte activation[92]. The antibodies currently in clinical development target both PD-1 and PD-L1. Several studies have investigated the clinical significance of PD-1 and its ligand in pancreatic cancer[93-97]. PD-L1 and PD-1 expression was higher in pancreatic cancer tissues compared to normal pancreatic tissues, and was associated with poorer tumour differentiation and immunosuppression. PD-L1 expression is inversely correlated with TILs which is associated with poorer prognosis[94,96]. Nivolumab, an anti-PD-1 monoclonal antibody, has shown promise in patients with solid tumours such as non-small cell lung cancer, prostate cancer, renal cell carcinoma, colorectal cancer and melanoma[77] however has not shown induce objective responses in pancreatic cancer[98]. An explanation for the failure is most likely due to the poor immunogenicity of PDAC, whereby stromal components such as pancreatic stellate cells inhibit tumour-infiltrating CD4+ and CD8+ T cell access to the primary tumour[99-101]. Consequently, attempts at improving tumour lymphocyte-infiltration and TME trafficking consist of priming the immune response with G-VAX[99]. As described above, Lutz et al[63] demonstrated G-VAX as a neoadjuvant therapy for resectable PDACs induced tertiary lymphoid aggregates and infiltration of PD-L1+ cells within these aggregates. G-VAX + PD-1/PD-L1 blockade however may be more clinically significant as anti-PD-1/PD-L1 agents are notably less toxic than ipilimumab, most likely due to the specificity of PD-L1 in the peripheral tissue[77,98,102]. Soares et al[99] demonstrated upregulation of PD-L1 expression in both human and murine PDAC tissues when IFN-γ-producing CD8+ T cells infiltrate the TME post-G-VAX administration, a process of adaptive resistance. This provides rationale for the use of anti-PD-1 agents in G-VAX-primed patients with PDAC.
Predicting responses to anti-immune checkpoint agents
As demonstrated by Soares et al[99], determining the mechanism of immune resistance is important for determining the efficacy of immunotherapies. The upregulation of PD-L1 on solid tumour cells has been explained by one of two mechanisms: innate or adaptive immune resistance[70]. Innate immune resistance is a result of constitutive oncogenic signaling and is independent of the TME[103]. The latter describes an upregulation of PD-L1/2 in response to inflammatory signals (e.g., IFN-γ) produced by an active antitumour immune response[104]. The difference between these two mechanisms may be the underlying differentiation between responders and non-responders, accounting for the heterogeneity of the expression levels of PD-1 ligands[70]. A study in pre-clinical sarcoma models identified tumour-specific neo antigens as a major class of T cell rejection antigens following anti-CTLA-4 and/or anti-PD-1 therapy, demonstrating tumour-specific mutant antigens are not only targets of immune checkpoint blockade agents but are also reactivated following treatment[105]. Although the presence of TILs and an “immune-active” microenvironment has been shown to be predictive of responses to immune checkpoint blockade clinically[106,107], the identification of predictive biomarkers in immunotherapy has been a challenge due to highly dynamic changes in the TME[108]. Nonetheless, patients with a pre-existing immune-active TME (e.g., IDO expression and Treg infiltration), and increased expression of PD-L1 on tumour and stromal cells induced by adaptive immune resistance, will most likely be the most receptive to anti-PD-1 blockade[108,109]. The PDAC TME is a major factor in determining responses to treatment because of its complex, desmoplastic and highly immunosuppressive nature. An anti-CD40 treatment on monocytes within the TME in a KPC mouse model of pancreatic cancer demonstrated increased survival when combined with gemcitabine and nab-paclitaxel[110]. Both CD4+ and CD8+ T cells and APCs were activated in the lymph nodes and at the tumour site and Treg cells were significantly reduced, demonstrating even the most immunosuppressive TME can be altered to respond to immune checkpoint blockade[110]. It has been proposed that immune-cell exclusion may be the reason why certain immunotherapies fail; Feig et al[111] showed that anti-CTLA-4 and anti-PD-L1 treatment resistance can be reversed when fibroblast activation protein (FAP) positive cancer-associated fibroblasts (CAFs) are depleted from the TME in the KPC model[111,112]. Similarly, Beatty et al[75] observed tumour regression in the KPC murine model was mediated by CD40-activated macrophages.
ONCOLYTIC VIROTHERAPY
Viruses are a strategic tool for targeting tumours for two reasons: viruses activate similar pathways as tumours, and viral infections activate both the innate and adaptive immune responses[113]. Moreover, tumours create a niche of innate and adaptive immune suppression, which not only protect the tumour from the host immune system, but also limits its ability to respond to viral infection[39]. The therapeutic potential of viruses was first observed in early case reports of tumour regression following a naturally occurring viral infection[114]. Tumour-targeted oncolytic viruses (TOVs) are viruses that selectively infect, replicate in, and lyse tumour cells, sparing healthy, normal tissues. TOVs can be inherently tumour-selective, i.e., are naturally nonpathogenic to humans and sensitive to antiviral signaling[115] or depend on oncogenic signaling pathways such as constitutively-activated Ras[116,117]. TOVs can also be genetically engineered to be tumour-selective, which often involve deletions of genes required for replication in normal tissues[116] such as thymidine kinase and vaccinia growth factor (VGF) for vaccinia virus (VV)[118] or thymidine kinase UL23 gene in herpes simplex virus (HSV)-1[119]. More recently, gene silencing by RNA interference technology to achieve tumour selectivity has also been utilized[117]. A second strategy involves the insertion of a tumour-specific promoter[120,121] that drives the expression of a gene necessary for viral replication in order to restrict its replication in tumour cells[117]. TOVs can also be designed to express cell surface receptors unique to tumour cells[122,123], which allow specific tropism[117]. TOVs can thus be engineered to increase safety, efficacy and tissue tropism, including being armed with immunomodulatory transgenes[116]. The advantages of TOVs are their specificity, very modest toxicity, low probability for resistance and most importantly, their induction of an inflammatory cascade and engagement of the adaptive immune system[116]. Unlike the pharmacokinetics of traditional drugs, the therapeutic dose of TOVs increases with time as the virus replicates and spreads to neighboring cells[116], and if armed with a therapeutic gene, each viral progeny will also carry the transgene, enhancing the therapeutic effect[124]. Although TOVs directly lyse infected malignant cells, causing acute tumour debulking, it is the ability of the virus to spread and potentiate an inflammatory response that allows the destruction of a tumour, distinguishing TOVs from vaccines or immune adjuvants[39,125]. TOVs target multiple cellular pathways, therefore the risk of tumour resistance development is low[116]. TOVs have also been shown to act synergistically with conventional chemotherapy and radiation[126-128].
The first TOV to undergo a clinical trial was ONYX-015 (dl1520), an adenovirus deficient in the E1B gene[129]. The gene product E1B-55 kDa protein was originally thought to sequester p53, inactivating it and as a result, allowing replication in a cell[130]. O’Shea et al[131] later determined that it is differential viral RNA export between normal and cancer cells which accounts for ONYX-015’s tumour-selectivity. The first approved oncolytic virus to date is H101 (Oncorine; Shanghair Sunway Biotech, Shanghai, China), which was approved in combination with chemotherapy in China in 2005 for the treatment of head and neck cancers[129].
ONCOLYTIC VIROTHERAPY FOR PANCREATIC CANCER
Adenovirus
ONYX-015 was the first TOV used in a clinical trial for pancreatic cancer. ONYX-015 was administered intratumourally under endoscopic ultrasound-guidance into patients with locally advanced adenocarcinoma of the pancreas or metastatic disease in phase I/II trials[132]. The treatment was well-tolerated in most patients, however no objective responses were seen with ONYX-015 as a single agent and only 2/21 patients experienced mild responses when combined with gemcitabine[132]. A second adenovirus vector carries a deletion in the E1A gene[133]. E1A normally binds to the retinoblastoma protein, forcing cells to prematurely enter the S phase of the cell cycle. Since most pancreatic cancers harbor a mutation in CDKN2A[134], the E1A protein is unnecessary for entry of the TOV into cancer cells. Furthermore a double-deleted (E1A and E1B19) adenovirus demonstrated increase potency and selectivity in pancreatic cancer models[135,136]. This demonstrates that TOVs can be genetically engineered to increase selectivity and efficacy while maintaining their potency. Adenovirus selectivity has also been improved by engineering tumour-specific promoters such as a human CEA promoter[137] or by substituting the adenovirus serotype 5 fiber knob with the fiber knob from serotype 3[138]. The potency of TOVs can also be improved further by engineering them with therapeutic genes that stimulate the immune system and/or improve direct oncolysis. Adenovirus ZD55-IL-24 expressing IL-24 locally in pancreatic tumours in immune competent mice inhibited tumour growth and induced a stronger T cell response compared to its backbone virus, as measured by IL-6 and IFN-γ levels[139].
HSV
Two oncolytic HSV-1 vectors are currently in clinical trials for the treatment of pancreatic cancer. HF10 is a non-engineered, naturally occurring oncolytic HSV that demonstrated regression in 1/6 of the patients treated[140,141]. OncoVex GM-CSF is a ∆34.5 and ICP47-deleted mutant expressing GM-CSF, whereby the deletions allow for tumour-selective replication and inhibition of protein-kinase R activation, respectively[142]. Phase I/II trials in various solid tumours demonstrated OncoVex GM-CSF to be well-tolerated at high and repeated doses[143,144]. A phase I clinical trial with OncoVex GM-CSF in patients with unresectable pancreatic cancer is underway.
Poxviruses
The most widely studied poxvirus is VV, which is highly immunogenic and produces a strong cytotoxic T cell response[145] and circulating neutralizing antibodies which can be detected decades later[146]. For its crucial role in the eradication of smallpox, much has been learned about its potential role in immunotherapy today. The Lister strain of vaccinia remarkably showed no replication degradation even under the hypoxic conditions of PDAC[147]. A second Lister strain, thymidine kinase-deleted replicating VV armed with IL-10 demonstrated superior and long-lasting antitumour immunity in both a subcutaneous pancreatic cancer model and a Kras-p53 mutant-transgenic pancreatic cancer model after systemic delivery compared to its unarmed backbone virus[148]. Myxoma virus, a rabbit-specific poxvirus combined with gemcitabine resulted in 100% long-term survival in Pan02-engrafted immunocompetent intraperitoneal dissemination models of pancreatic cancer[149]. The only poxvirus to be tested in clinical trials is a non-replicative VV that expresses the pancreatic TAAs CEA and MUC-2[150]. The vaccine also includes a triad of costimulatory molecules, B7.1 (CD80), ICAM-1 (intra-cellular adhesion molecule-1) and LFA-3 (leukocyte function-associated antigen-3) (TRICOM) (PANVAC-VF)[150]. GM-CSF was also used as an adjuvant following each vaccination of PANVAC-VF. Phase I trials demonstrated antigen-specific antitumour responses in 62.5% of patients enrolled and antibody responses against VV was observed in all ten patients, which was associated with an increase in survival (15.1 mo vs 3.9 mo)[48]. A phase III clinical trial for the treatment of metastatic pancreatic cancer after failing treatment with gemcitabine, however, was terminated after failing to reach its primary efficacy endpoint[151].
Other pre-clinical TOVs for pancreatic cancer therapy
Parvovirus, measles virus and reovirus have also demonstrated pre-clinical activity in pancreatic cancer models. Parvoviruses particularly demonstrated enhanced IL-2-activated NK responses against PDAC cells[152,153]. An armed measles virus (MV), MV-purine nucleoside phosphorylase (PNP)-anti-prostate stem cell antigen, that expresses the prodrug convertase PNP, which then activates the prodrug fludarabine, was shown to enhance the oncolytic efficacy of the virus in gemcitabine-resistant PDAC cells[154]. Reovirus is another promising TOV for pancreatic cancer therapy, particularly because its selectivity depends on the cellular activity of Ras, which is constitutively active in pancreatic cancer[155]. Reolysin® (Oncolytics Biotech Inc., Calgary, AB, Canada) a reovirus administered intraportally resulted in decreased metastatic tumour volumes in the liver of immunocompetent animal models[156,157]. A phase II study of Reolysin® in combination with gemcitabine in patients with advanced PDAC has been completed (clinicaltrials.gov: NCT00998322). A two-armed randomized phase II study of carboplatin and paclitaxel plus Reolysin® vs carboplatin and paclitaxel alone in recurrent or metastatic pancreatic cancer is currently being conducted by the United States National Cancer Institute (NCI-8601/OSU-10045).
RATIONALIZING VIRO-IMMUNE-CHECKPOINT COMBINATION THERAPY
A understanding how antitumour immunity is regulated allows us to recognize barriers against effective immunotherapy delivery and furthermore, allow for the development of rational combination therapies aiming targeting these mechanisms[108,158,159]. This approach allows therapies to work synergistically and also has the potential to benefit a broader patient population[108]. Tumours have evolved to avoid immune recognition and/or destruction at every stage in the antitumour response, therefore targeting more than one immune resistance mechanism will enhance antitumour immunity.
An important immunological barrier in cancer immunotherapy is the tolerance towards self-antigens. Tumours downregulate their antigenicity through various mechanisms in response to selective pressure by the immune system, a process called “immunoediting”[37]. Therefore, in order to raise an effective antitumour response, the immunological tolerance must be broken to allow tumour antigen-specific cytotoxic T cell responses[158]. This can be achieved by increasing the tumour load and/or enhance antigen presentation[108]. TOVs can initiate selective infection and replication in the tumour bed, exposing TAA, disrupting the immunotolerance employed by the tumour while re-engaging adaptive immune effector responses[39]. Combining an agent that can cause disruption to the tumour bed i.e., an oncolytic virus, with a novel antitumour immunomodulating agent such as anti-PD-1/PD-L1 antibodies can maximize immune-stimulating and immune-recruiting inflammatory responses[39]. Specifically, TOV lysis induces the release of tumour antigens into the microenvironment, which are then cross-presented to T cells in the draining lymph nodes by APCs[159] (Figure 1). This allows T cell infiltration to the tumour bed. Next, T cell dysfunction must be reversed[108,158]. Immune checkpoint inhibitors alleviate immunosuppression, allowing the elimination of the tumour by the adaptive immune system[70]. TOVs in combination with immune checkpoint inhibitors can therefore potentiate and activate the immune system synergistically, ultimately creating a pro-inflammatory environment. Pre-existing TILs are strong prognostic predictors in cancer[106]. This is extremely relevant for tumours with poor immune-cell infiltration, such as pancreatic cancer, which would depend on TOV-infection mediated lymphocyte infiltration for an enhanced response to immune checkpoint blockade. Zamarin et al[160] demonstrated constrained replication of an intratumoural-injected Newcastle disease virus in a B16 melanoma model. Lymphocytic infiltrates, however, were detected in both TOV-injected and non-TOV-injected tumours, and rendered the tumours sensitive to CTLA-4 blockade. The antitumour activity was dependent on CD8+ T cells, NK cells and type I and II IFNs[160]. Ipilimumab with or without talimogene laherparapvec, is in early clinical testing in patients with unresected melanoma (clinicaltrials.org: NCT01740297). Interestingly, an MV engineered to express CTLA-4 or PD-L1 antibodies delayed tumour progression and prolonged median OS in B16 melanoma models[161]. Finally, TOVs have demonstrated a tolerable toxicity profile, whereby flu-like symptoms are the most common adverse events, and in fact, most of the side effects seen so far in the combination regiment are related to the immune checkpoint blockade inhibitor[162]. Dias et al[163] suggested an oncolytic adenovirus expressing CTLA-4 locally might reduce systemic side effects normally induced with anti-CTLA-4 antibodies alone.
OVERCOMING OBSTACLES IN ONCOLYTIC VIRUS DELIVERY
The main issue with virotherapy is systemic delivery for targeting metastatic cancer cells. Intravenous administration is more practical, especially for treatment of a tumour in a hard-to-reach location such as the pancreas, and with the majority of patients presenting with advanced or metastatic disease. However, nonimmune human serum and existing anti-TOV antibodies may neutralize the TOV in the bloodstream. Furthermore, non-specific hepatic and splenic sequestration of the TOV and ineffective extravasation into the tumours are important issues[164]. Currently, studies in pre-clinical models aim to overcome these obstacles. These include chemical modification of viral coat proteins by conjugation of biocompatible polymers e.g. polyethylene glycosylation[165,166], using mesenchymal stem cell carrier systems to deliver the TOV to the tumour bed[167-169], and increasing vessel permeabilization[170,171].
In PDAC, however, the biggest hurdle may not be the host immune system, but the TME. The TME has played a significant role in not only acting as a physical barrier to deliver treatments, but it also in the development of resistance to conventional drugs. The TME remains a problem for successful TOV treatment. The TOV must be able to spread in the hypoxic and densely stromal-rich TME in order to attract enough attention to induce antitumour immunity[172]. Breaching the stromal barrier in PDAC is needed for TOVs to access the cancer cells[173]. Paradoxically, a recent study by Ilkow et al[174] demonstrated that the cross-talk between CAFs and cancer cells actually lead to increased permissibility of TOV-based therapeutics. Tumour cells producing TGF-α reprogrammed CAFs, dampening levels of anti-viral transcripts. This allowed the cells to be more sensitive to VV, vesicular stomatitis virus and maraba MG1 TOVs. The reprogrammed CAFs produced fibroblast growth factor (FGF)-2 which suppressed levels of retinoic acid-inducible gene I and increased the susceptibility of the tumour cells to virus[175]. This study also demonstrated that an FGF2-expressing TOV has improved therapeutic efficacy by sensitizing the tumour cells to virotherapy and is particularly relevant to pancreatic cancers, where CAFs are a major component of the tumour stroma[175]. It is important to note that not only the patient’s existing immune system may impede successful TOV therapy, but that the enhanced antitumour response by combinatory approaches (e.g., the inclusion of immune-checkpoint inhibitors) may also impede successful TOV infection, spread and engagement of the immune system. This stresses the importance of determining strategic combinations, dosing and timing schedules in future studies.
CONCLUSION
The poor prognosis of pancreatic cancer due in part to the limited efficacy of conventional and targeted therapies, appeals for a novel strategy to treat this disease. It has become very clear that the immune system has the greatest potential to selectively destroy tumours, and when it is strategically induced, a durable benefit can be achieved. Past and present studies have defined means for tumour escape from immune surveillance and have developed immunotherapies to counteract these mechanisms. However, with the various escape strategies leading to low immunogenicity and highly immunosuppressive tumour beds, a successful control of tumour growth by immunotherapy does not come without various obstacles and challenges. Future steps include the development of immune-monitoring strategies for the identification of biomarkers, to establishment guidelines to assess clinical end points of immunotherapy and finally to evaluate combination therapeutic strategies to maximize clinical benefit[176]. The ability of TOVs to stimulate inflammation, deliver genes and immunomodulatory agents as well as reduce tumour burden by direct cell lysis, allows them to be important therapeutic vectors for a highly immunosuppressed tumour such as PDAC. Immune checkpoint blockade agents can then reverse T cell anergy and further boost OV-induced responses. As this combinatory approach may exist as a double-edged sword, it is crucial to determine appropriate timing, dosing and sequence schedules of each agent.
Footnotes
Supported by The United Kingdom Charity Pancreatic Cancer Research Fund, Pancreatic Cancer Research United Kingdom; and Ministry of Sciences and Technology of China, No. 2013DFG32080.
Conflict-of-interest statement: The above-mentioned authors of this manuscript hereby declare that they do not have any conflict-of-interest related to the work submitted herein.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 30, 2015
First decision: September 29, 2015
Article in press: December 14, 2015
P- Reviewer: Aglietta M, Kleeff J, Liu XE, Tsuchikawa T, Zhang ZM S- Editor: Gong ZM L- Editor: A E- Editor: Ma S
References
- 1.Sener SF, Fremgen A, Menck HR, Winchester DP. Pancreatic cancer: a report of treatment and survival trends for 100,313 patients diagnosed from 1985-1995, using the National Cancer Database. J Am Coll Surg. 1999;189:1–7. doi: 10.1016/s1072-7515(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 2.Gralow J, Ozols RF, Bajorin DF, Cheson BD, Sandler HM, Winer EP, Bonner J, Demetri GD, Curran W, Ganz PA, et al. Clinical cancer advances 2007: major research advances in cancer treatment, prevention, and screening--a report from the American Society of Clinical Oncology. J Clin Oncol. 2008;26:313–325. doi: 10.1200/JCO.2007.15.4088. [DOI] [PubMed] [Google Scholar]
- 3.Atlanta: American Cancer Society; 2015. Cancer Facts & Figures 2015; pp. 1–56. [Google Scholar]
- 4.Stathis A, Moore MJ. Advanced pancreatic carcinoma: current treatment and future challenges. Nat Rev Clin Oncol. 2010;7:163–172. doi: 10.1038/nrclinonc.2009.236. [DOI] [PubMed] [Google Scholar]
- 5.Canto MI, Harinck F, Hruban RH, Offerhaus GJ, Poley JW, Kamel I, Nio Y, Schulick RS, Bassi C, Kluijt I, et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut. 2013;62:339–347. doi: 10.1136/gutjnl-2012-303108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi C, Hruban RH, Klein AP. Familial pancreatic cancer. Arch Pathol Lab Med. 2009;133:365–374. doi: 10.5858/133.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–1249. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- 8.Ottenhof NA, Milne AN, Morsink FH, Drillenburg P, Ten Kate FJ, Maitra A, Offerhaus GJ. Pancreatic intraepithelial neoplasia and pancreatic tumorigenesis: of mice and men. Arch Pathol Lab Med. 2009;133:375–381. doi: 10.5858/133.3.375. [DOI] [PubMed] [Google Scholar]
- 9.Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997;57:2140–2143. [PubMed] [Google Scholar]
- 10.Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, Hruban RH, Maitra A, Kinzler K, Vogelstein B, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142:730–733.e9. doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feldmann G, Beaty R, Hruban RH, Maitra A. Molecular genetics of pancreatic intraepithelial neoplasia. J Hepatobiliary Pancreat Surg. 2007;14:224–232. doi: 10.1007/s00534-006-1166-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18:4266–4276. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chu GC, Kimmelman AC, Hezel AF, DePinho RA. Stromal biology of pancreatic cancer. J Cell Biochem. 2007;101:887–907. doi: 10.1002/jcb.21209. [DOI] [PubMed] [Google Scholar]
- 14.Erkan M, Reiser-Erkan C, Michalski CW, Deucker S, Sauliunaite D, Streit S, Esposito I, Friess H, Kleeff J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia. 2009;11:497–508. doi: 10.1593/neo.81618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6:1186–1197. doi: 10.1158/1535-7163.MCT-06-0686. [DOI] [PubMed] [Google Scholar]
- 16.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 17.Shaib Y, Davila J, Naumann C, El-Serag H. The impact of curative intent surgery on the survival of pancreatic cancer patients: a U.S. Population-based study. Am J Gastroenterol. 2007;102:1377–1382. doi: 10.1111/j.1572-0241.2007.01202.x. [DOI] [PubMed] [Google Scholar]
- 18.Rossi ML, Rehman AA, Gondi CS. Therapeutic options for the management of pancreatic cancer. World J Gastroenterol. 2014;20:11142–11159. doi: 10.3748/wjg.v20.i32.11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalser MH, Ellenberg SS. Pancreatic cancer. Adjuvant combined radiation and chemotherapy following curative resection. Arch Surg. 1985;120:899–903. doi: 10.1001/archsurg.1985.01390320023003. [DOI] [PubMed] [Google Scholar]
- 20.Neoptolemos JP, Stocken DD, Friess H, Bassi C, Dunn JA, Hickey H, Beger H, Fernandez-Cruz L, Dervenis C, Lacaine F, et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N Engl J Med. 2004;350:1200–1210. doi: 10.1056/NEJMoa032295. [DOI] [PubMed] [Google Scholar]
- 21.Oettle H, Neuhaus P, Hochhaus A, Hartmann JT, Gellert K, Ridwelski K, Niedergethmann M, Zülke C, Fahlke J, Arning MB, et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: the CONKO-001 randomized trial. JAMA. 2013;310:1473–1481. doi: 10.1001/jama.2013.279201. [DOI] [PubMed] [Google Scholar]
- 22.Gillen S, Schuster T, Meyer Zum Büschenfelde C, Friess H, Kleeff J. Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010;7:e1000267. doi: 10.1371/journal.pmed.1000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lemmens VE, Bosscha K, van der Schelling G, Brenninkmeijer S, Coebergh JW, de Hingh IH. Improving outcome for patients with pancreatic cancer through centralization. Br J Surg. 2011;98:1455–1462. doi: 10.1002/bjs.7581. [DOI] [PubMed] [Google Scholar]
- 24.Takahashi H, Ohigashi H, Gotoh K, Marubashi S, Yamada T, Murata M, Ioka T, Uehara H, Yano M, Ishikawa O. Preoperative gemcitabine-based chemoradiation therapy for resectable and borderline resectable pancreatic cancer. Ann Surg. 2013;258:1040–1050. doi: 10.1097/SLA.0b013e31829b3ce4. [DOI] [PubMed] [Google Scholar]
- 25.Nanda RH, El-Rayes B, Maithel SK, Landry J. Neoadjuvant modified FOLFIRINOX and chemoradiation therapy for locally advanced pancreatic cancer improves resectability. J Surg Oncol. 2015;111:1028–1034. doi: 10.1002/jso.23921. [DOI] [PubMed] [Google Scholar]
- 26.Burris HA, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 27.Heinemann V, Boeck S, Hinke A, Labianca R, Louvet C. Meta-analysis of randomized trials: evaluation of benefit from gemcitabine-based combination chemotherapy applied in advanced pancreatic cancer. BMC Cancer. 2008;8:82. doi: 10.1186/1471-2407-8-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardière C, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 29.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:2140–2141. doi: 10.1056/NEJMc1412266. [DOI] [PubMed] [Google Scholar]
- 31.Gerber DE. Targeted therapies: a new generation of cancer treatments. Am Fam Physician. 2008;77:311–319. [PubMed] [Google Scholar]
- 32.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seicean A, Petrusel L, Seicean R. New targeted therapies in pancreatic cancer. World J Gastroenterol. 2015;21:6127–6145. doi: 10.3748/wjg.v21.i20.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chong CR, Jänne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19:1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 36.Finn OJ. Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann Oncol. 2012;23 Suppl 8:viii6–viii9. doi: 10.1093/annonc/mds256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 38.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 39.Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14:559–567. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 40.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–489. doi: 10.1038/nature10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gjertsen MK, Buanes T, Rosseland AR, Bakka A, Gladhaug I, Søreide O, Eriksen JA, Møller M, Baksaas I, Lothe RA, et al. Intradermal ras peptide vaccination with granulocyte-macrophage colony-stimulating factor as adjuvant: Clinical and immunological responses in patients with pancreatic adenocarcinoma. Int J Cancer. 2001;92:441–450. doi: 10.1002/ijc.1205. [DOI] [PubMed] [Google Scholar]
- 42.Ramanathan RK, Lee KM, McKolanis J, Hitbold E, Schraut W, Moser AJ, Warnick E, Whiteside T, Osborne J, Kim H, et al. Phase I study of a MUC1 vaccine composed of different doses of MUC1 peptide with SB-AS2 adjuvant in resected and locally advanced pancreatic cancer. Cancer Immunol Immunother. 2005;54:254–264. doi: 10.1007/s00262-004-0581-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kimura Y, Tsukada J, Tomoda T, Takahashi H, Imai K, Shimamura K, Sunamura M, Yonemitsu Y, Shimodaira S, Koido S, et al. Clinical and immunologic evaluation of dendritic cell-based immunotherapy in combination with gemcitabine and/or S-1 in patients with advanced pancreatic carcinoma. Pancreas. 2012;41:195–205. doi: 10.1097/MPA.0b013e31822398c6. [DOI] [PubMed] [Google Scholar]
- 44.Bauer C, Dauer M, Saraj S, Schnurr M, Bauernfeind F, Sterzik A, Junkmann J, Jakl V, Kiefl R, Oduncu F, et al. Dendritic cell-based vaccination of patients with advanced pancreatic carcinoma: results of a pilot study. Cancer Immunol Immunother. 2011;60:1097–1107. doi: 10.1007/s00262-011-1023-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jaffee EM, Hruban RH, Biedrzycki B, Laheru D, Schepers K, Sauter PR, Goemann M, Coleman J, Grochow L, Donehower RC, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol. 2001;19:145–156. doi: 10.1200/JCO.2001.19.1.145. [DOI] [PubMed] [Google Scholar]
- 46.Laheru D, Lutz E, Burke J, Biedrzycki B, Solt S, Onners B, Tartakovsky I, Nemunaitis J, Le D, Sugar E, et al. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: a pilot study of safety, feasibility, and immune activation. Clin Cancer Res. 2008;14:1455–1463. doi: 10.1158/1078-0432.CCR-07-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marshall JL. Novel vaccines for the treatment of gastrointestinal cancers. Oncology (Williston Park) 2005;19:1557–1565; discussion 1566, 1568 passim. [PubMed] [Google Scholar]
- 48.Kaufman HL, Kim-Schulze S, Manson K, DeRaffele G, Mitcham J, Seo KS, Kim DW, Marshall J. Poxvirus-based vaccine therapy for patients with advanced pancreatic cancer. J Transl Med. 2007;5:60. doi: 10.1186/1479-5876-5-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Middleton G, Silcocks P, Cox T, Valle J, Wadsley J, Propper D, Coxon F, Ross P, Madhusudan S, Roques T, et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): an open-label, randomised, phase 3 trial. Lancet Oncol. 2014;15:829–840. doi: 10.1016/S1470-2045(14)70236-0. [DOI] [PubMed] [Google Scholar]
- 50.Draper SJ, Heeney JL. Viruses as vaccine vectors for infectious diseases and cancer. Nat Rev Microbiol. 2010;8:62–73. doi: 10.1038/nrmicro2240. [DOI] [PubMed] [Google Scholar]
- 51.Hill AV, Reyes-Sandoval A, O’Hara G, Ewer K, Lawrie A, Goodman A, Nicosia A, Folgori A, Colloca S, Cortese R, et al. Prime-boost vectored malaria vaccines: progress and prospects. Hum Vaccin. 2010;6:78–83. doi: 10.4161/hv.6.1.10116. [DOI] [PubMed] [Google Scholar]
- 52.Hodge JW, McLaughlin JP, Kantor JA, Schlom J. Diversified prime and boost protocols using recombinant vaccinia virus and recombinant non-replicating avian pox virus to enhance T-cell immunity and antitumor responses. Vaccine. 1997;15:759–768. doi: 10.1016/s0264-410x(96)00238-1. [DOI] [PubMed] [Google Scholar]
- 53.Marshall JL, Gulley JL, Arlen PM, Beetham PK, Tsang KY, Slack R, Hodge JW, Doren S, Grosenbach DW, Hwang J, et al. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J Clin Oncol. 2005;23:720–731. doi: 10.1200/JCO.2005.10.206. [DOI] [PubMed] [Google Scholar]
- 54.Harrop R, John J, Carroll MW. Recombinant viral vectors: cancer vaccines. Adv Drug Deliv Rev. 2006;58:931–947. doi: 10.1016/j.addr.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 55.Galili U, Clark MR, Shohet SB, Buehler J, Macher BA. Evolutionary relationship between the natural anti-Gal antibody and the Gal alpha 1----3Gal epitope in primates. Proc Natl Acad Sci USA. 1987;84:1369–1373. doi: 10.1073/pnas.84.5.1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rossi GR, Mautino MR, Unfer RC, Seregina TM, Vahanian N, Link CJ. Effective treatment of preexisting melanoma with whole cell vaccines expressing alpha(1,3)-galactosyl epitopes. Cancer Res. 2005;65:10555–10561. doi: 10.1158/0008-5472.CAN-05-0627. [DOI] [PubMed] [Google Scholar]
- 57.Hardacre JM, Mulcahy M, Small W, Talamonti M, Obel J, Krishnamurthi S, Rocha-Lima CS, Safran H, Lenz HJ, Chiorean EG. Addition of algenpantucel-L immunotherapy to standard adjuvant therapy for pancreatic cancer: a phase 2 study. J Gastrointest Surg. 2013;17:94–100; discussion p. 100-101. doi: 10.1007/s11605-012-2064-6. [DOI] [PubMed] [Google Scholar]
- 58.Regine WF, Winter KA, Abrams RA, Safran H, Hoffman JP, Konski A, Benson AB, Macdonald JS, Kudrimoti MR, Fromm ML, et al. Fluorouracil vs gemcitabine chemotherapy before and after fluorouracil-based chemoradiation following resection of pancreatic adenocarcinoma: a randomized controlled trial. JAMA. 2008;299:1019–1026. doi: 10.1001/jama.299.9.1019. [DOI] [PubMed] [Google Scholar]
- 59.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, Jackson V, Hamada H, Pardoll D, Mulligan RC. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci USA. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lutz E, Yeo CJ, Lillemoe KD, Biedrzycki B, Kobrin B, Herman J, Sugar E, Piantadosi S, Cameron JL, Solt S, et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A Phase II trial of safety, efficacy, and immune activation. Ann Surg. 2011;253:328–335. doi: 10.1097/SLA.0b013e3181fd271c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thomas AM, Santarsiero LM, Lutz ER, Armstrong TD, Chen YC, Huang LQ, Laheru DA, Goggins M, Hruban RH, Jaffee EM. Mesothelin-specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exp Med. 2004;200:297–306. doi: 10.1084/jem.20031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schueneman AJ, Sugar EA, Uram J, Bigelow E, Herman JM, Edil BH, Jaffee EM, Zheng L, Laheru DA. Low total lymphocyte count is associated with poor survival in patients with resected pancreatic adenocarcinoma receiving a GM-CSF secreting pancreatic tumor vaccine. Ann Surg Oncol. 2013;20 Suppl 3:S725–S730. doi: 10.1245/s10434-013-3262-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, Solt S, Dorman A, Wamwea A, Yager A, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res. 2014;2:616–631. doi: 10.1158/2326-6066.CIR-14-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Le DT, Wang-Gillam A, Picozzi V, Greten TF, Crocenzi T, Springett G, Morse M, Zeh H, Cohen D, Fine RL, et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J Clin Oncol. 2015;33:1325–1333. doi: 10.1200/JCO.2014.57.4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parmiani G, Castelli C, Pilla L, Santinami M, Colombo MP, Rivoltini L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann Oncol. 2007;18:226–232. doi: 10.1093/annonc/mdl158. [DOI] [PubMed] [Google Scholar]
- 66.Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004;64:6337–6343. doi: 10.1158/0008-5472.CAN-04-0757. [DOI] [PubMed] [Google Scholar]
- 67.Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, Castelli C, Mariani L, Parmiani G, Rivoltini L. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol. 2007;25:2546–2553. doi: 10.1200/JCO.2006.08.5829. [DOI] [PubMed] [Google Scholar]
- 68.Takeuchi S, Baghdadi M, Tsuchikawa T, Wada H, Nakamura T, Abe H, Nakanishi S, Usui Y, Higuchi K, Takahashi M, et al. Chemotherapy-Derived Inflammatory Responses Accelerate the Formation of Immunosuppressive Myeloid Cells in the Tissue Microenvironment of Human Pancreatic Cancer. Cancer Res. 2015;75:2629–2640. doi: 10.1158/0008-5472.CAN-14-2921. [DOI] [PubMed] [Google Scholar]
- 69.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–847. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 72.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, Walker J, Gonzalez I, Meeuwsen T, Fox BA, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013;73:7189–7198. doi: 10.1158/0008-5472.CAN-12-4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Silver Spring: U.S. Food and Drug Administration; 2011. FDA approves new treatment for a type of late-stage skin cancer; pp. 1–56. [Google Scholar]
- 80.Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell. 1992;71:1065–1068. doi: 10.1016/s0092-8674(05)80055-8. [DOI] [PubMed] [Google Scholar]
- 81.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 82.Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174:561–569. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 84.Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I, Rosenberg SA. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33:828–833. doi: 10.1097/CJI.0b013e3181eec14c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aglietta M, Barone C, Sawyer MB, Moore MJ, Miller WH, Bagalà C, Colombi F, Cagnazzo C, Gioeni L, Wang E, et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann Oncol. 2014;25:1750–1755. doi: 10.1093/annonc/mdu205. [DOI] [PubMed] [Google Scholar]
- 86.Le DT, Lutz E, Uram JN, Sugar EA, Onners B, Solt S, Zheng L, Diaz LA, Donehower RC, Jaffee EM, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother. 2013;36:382–389. doi: 10.1097/CJI.0b013e31829fb7a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 89.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 90.Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8:467–477. doi: 10.1038/nri2326. [DOI] [PubMed] [Google Scholar]
- 91.Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, Rosenberg SA. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Geng L, Huang D, Liu J, Qian Y, Deng J, Li D, Hu Z, Zhang J, Jiang G, Zheng S. B7-H1 up-regulated expression in human pancreatic carcinoma tissue associates with tumor progression. J Cancer Res Clin Oncol. 2008;134:1021–1027. doi: 10.1007/s00432-008-0364-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang L, Ma Q, Chen X, Guo K, Li J, Zhang M. Clinical significance of B7-H1 and B7-1 expressions in pancreatic carcinoma. World J Surg. 2010;34:1059–1065. doi: 10.1007/s00268-010-0448-x. [DOI] [PubMed] [Google Scholar]
- 95.Loos M, Giese NA, Kleeff J, Giese T, Gaida MM, Bergmann F, Laschinger M, W Büchler M, Friess H. Clinical significance and regulation of the costimulatory molecule B7-H1 in pancreatic cancer. Cancer Lett. 2008;268:98–109. doi: 10.1016/j.canlet.2008.03.056. [DOI] [PubMed] [Google Scholar]
- 96.Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, Nakamura S, Enomoto K, Yagita H, Azuma M, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13:2151–2157. doi: 10.1158/1078-0432.CCR-06-2746. [DOI] [PubMed] [Google Scholar]
- 97.Chen Y, Sun J, Zhao H, Zhu D, Zhi Q, Song S, Zhang L, He S, Kuang Y, Zhang Z, et al. The coexpression and clinical significance of costimulatory molecules B7-H1, B7-H3, and B7-H4 in human pancreatic cancer. Onco Targets Ther. 2014;7:1465–1472. doi: 10.2147/OTT.S66809. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 98.Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller TL, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, Wamwea A, Bigelow E, Lutz E, Liu L, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother. 2015;38:1–11. doi: 10.1097/CJI.0000000000000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fukunaga A, Miyamoto M, Cho Y, Murakami S, Kawarada Y, Oshikiri T, Kato K, Kurokawa T, Suzuoki M, Nakakubo Y, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 2004;28:e26–e31. doi: 10.1097/00006676-200401000-00023. [DOI] [PubMed] [Google Scholar]
- 101.Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, Marshall JF, Chin-Aleong J, Chelala C, Gribben JG, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145:1121–1132. doi: 10.1053/j.gastro.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gangadhar TC, Vonderheide RH. Mitigating the toxic effects of anticancer immunotherapy. Nat Rev Clin Oncol. 2014;11:91–99. doi: 10.1038/nrclinonc.2013.245. [DOI] [PubMed] [Google Scholar]
- 103.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 104.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, Chen S, Klein AP, Pardoll DM, Topalian SL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, Alaparthy S, Berman D, Jure-Kunkel M, Siemers NO, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61:1019–1031. doi: 10.1007/s00262-011-1172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, Guida M, Hyams DM, Gómez H, Bastholt L, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med. 2011;9:204. doi: 10.1186/1479-5876-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zamarin D, Postow MA. Immune checkpoint modulation: rational design of combination strategies. Pharmacol Ther. 2015;150:23–32. doi: 10.1016/j.pharmthera.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 109.Ribas A, Tumeh PC. The future of cancer therapy: selecting patients likely to respond to PD1/L1 blockade. Clin Cancer Res. 2014;20:4982–4984. doi: 10.1158/1078-0432.CCR-14-0933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Byrne KT, Vonderheide RH, Jaffee EM, Armstrong TD. Special Conference on Tumor Immunology and Immunotherapy: A New Chapter. Cancer Immunol Res. 2015:Epub ahead of print. doi: 10.1158/2326-6066.CIR-15-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110:20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fearon DT. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol Res. 2014;2:187–193. doi: 10.1158/2326-6066.CIR-14-0002. [DOI] [PubMed] [Google Scholar]
- 113.Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther. 2011;19:1008–1016. doi: 10.1038/mt.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15:651–659. doi: 10.1038/sj.mt.6300108. [DOI] [PubMed] [Google Scholar]
- 115.Naik S, Russell SJ. Engineering oncolytic viruses to exploit tumor specific defects in innate immune signaling pathways. Expert Opin Biol Ther. 2009;9:1163–1176. doi: 10.1517/14712590903170653. [DOI] [PubMed] [Google Scholar]
- 116.Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. 2014;2:295–300. doi: 10.1158/2326-6066.CIR-14-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wong HH, Lemoine NR, Wang Y. Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles. Viruses. 2010;2:78–106. doi: 10.3390/v2010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Thorne SH, Hwang TH, O’Gorman WE, Bartlett DL, Sei S, Kanji F, Brown C, Werier J, Cho JH, Lee DE, et al. Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963. J Clin Invest. 2007;117:3350–3358. doi: 10.1172/JCI32727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252:854–856. doi: 10.1126/science.1851332. [DOI] [PubMed] [Google Scholar]
- 120.Pan W, Bodempudi V, Esfandyari T, Farassati F. Utilizing ras signaling pathway to direct selective replication of herpes simplex virus-1. PLoS One. 2009;4:e6514. doi: 10.1371/journal.pone.0006514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Doloff JC, Waxman DJ, Jounaidi Y. Human telomerase reverse transcriptase promoter-driven oncolytic adenovirus with E1B-19 kDa and E1B-55 kDa gene deletions. Hum Gene Ther. 2008;19:1383–1400. doi: 10.1089/hum.2008.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nishimoto T, Yoshida K, Miura Y, Kobayashi A, Hara H, Ohnami S, Kurisu K, Yoshida T, Aoki K. Oncolytic virus therapy for pancreatic cancer using the adenovirus library displaying random peptides on the fiber knob. Gene Ther. 2009;16:669–680. doi: 10.1038/gt.2009.1. [DOI] [PubMed] [Google Scholar]
- 123.Conner J, Braidwood L, Brown SM. A strategy for systemic delivery of the oncolytic herpes virus HSV1716: redirected tropism by antibody-binding sites incorporated on the virion surface as a glycoprotein D fusion protein. Gene Ther. 2008;15:1579–1592. doi: 10.1038/gt.2008.121. [DOI] [PubMed] [Google Scholar]
- 124.Kaur B, Cripe TP, Chiocca EA. “Buy one get one free”: armed viruses for the treatment of cancer cells and their microenvironment. Curr Gene Ther. 2009;9:341–355. doi: 10.2174/156652309789753329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sobol PT, Boudreau JE, Stephenson K, Wan Y, Lichty BD, Mossman KL. Adaptive antiviral immunity is a determinant of the therapeutic success of oncolytic virotherapy. Mol Ther. 2011;19:335–344. doi: 10.1038/mt.2010.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dai MH, Zamarin D, Gao SP, Chou TC, Gonzalez L, Lin SF, Fong Y. Synergistic action of oncolytic herpes simplex virus and radiotherapy in pancreatic cancer cell lines. Br J Surg. 2010;97:1385–1394. doi: 10.1002/bjs.7124. [DOI] [PubMed] [Google Scholar]
- 127.Pandha HS, Heinemann L, Simpson GR, Melcher A, Prestwich R, Errington F, Coffey M, Harrington KJ, Morgan R. Synergistic effects of oncolytic reovirus and cisplatin chemotherapy in murine malignant melanoma. Clin Cancer Res. 2009;15:6158–6166. doi: 10.1158/1078-0432.CCR-09-0796. [DOI] [PubMed] [Google Scholar]
- 128.Nishizaki M, Meyn RE, Levy LB, Atkinson EN, White RA, Roth JA, Ji L. Synergistic inhibition of human lung cancer cell growth by adenovirus-mediated wild-type p53 gene transfer in combination with docetaxel and radiation therapeutics in vitro and in vivo. Clin Cancer Res. 2001;7:2887–2897. [PubMed] [Google Scholar]
- 129.Yu W, Fang H. Clinical trials with oncolytic adenovirus in China. Curr Cancer Drug Targets. 2007;7:141–148. doi: 10.2174/156800907780058817. [DOI] [PubMed] [Google Scholar]
- 130.Green NK, Seymour LW. Adenoviral vectors: systemic delivery and tumor targeting. Cancer Gene Ther. 2002;9:1036–1042. doi: 10.1038/sj.cgt.7700541. [DOI] [PubMed] [Google Scholar]
- 131.O'Shea CC, Johnson L, Bagus B, Choi S, Nicholas C, Shen A, Boyle L, Pandey K, Soria C, Kunich J, et al. Late viral RNA export, rather than p53 inactivation, determines ONYX-015 tumor selectivity. Cancer Cell. 2004;6:611–623. doi: 10.1016/j.ccr.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 132.Hecht JR, Bedford R, Abbruzzese JL, Lahoti S, Reid TR, Soetikno RM, Kirn DH, Freeman SM. A phase I/II trial of intratumoral endoscopic ultrasound injection of ONYX-015 with intravenous gemcitabine in unresectable pancreatic carcinoma. Clin Cancer Res. 2003;9:555–561. [PubMed] [Google Scholar]
- 133.Heise C, Hermiston T, Johnson L, Brooks G, Sampson-Johannes A, Williams A, Hawkins L, Kirn D. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumoral efficacy. Nat Med. 2000;6:1134–1139. doi: 10.1038/80474. [DOI] [PubMed] [Google Scholar]
- 134.Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, Moskaluk CA, Hahn SA, Schwarte-Waldhoff I, Schmiegel W, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57:3126–3130. [PubMed] [Google Scholar]
- 135.Oberg D, Yanover E, Adam V, Sweeney K, Costas C, Lemoine NR, Halldén G. Improved potency and selectivity of an oncolytic E1ACR2 and E1B19K deleted adenoviral mutant in prostate and pancreatic cancers. Clin Cancer Res. 2010;16:541–553. doi: 10.1158/1078-0432.CCR-09-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cherubini G, Kallin C, Mozetic A, Hammaren-Busch K, Müller H, Lemoine NR, Halldén G. The oncolytic adenovirus AdΔΔ enhances selective cancer cell killing in combination with DNA-damaging drugs in pancreatic cancer models. Gene Ther. 2011;18:1157–1165. doi: 10.1038/gt.2011.141. [DOI] [PubMed] [Google Scholar]
- 137.Xu C, Sun Y, Wang Y, Yan Y, Shi Z, Chen L, Lin H, Lü S, Zhu M, Su C, et al. CEA promoter-regulated oncolytic adenovirus-mediated Hsp70 expression in immune gene therapy for pancreatic cancer. Cancer Lett. 2012;319:154–163. doi: 10.1016/j.canlet.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 138.Chu QD, Sun G, Pope M, Luraguiz N, Curiel DT, Kim R, Li BD, Mathis JM. Virotherapy using a novel chimeric oncolytic adenovirus prolongs survival in a human pancreatic cancer xenograft model. Surgery. 2012;152:441–448. doi: 10.1016/j.surg.2012.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.He B, Huang X, Liu X, Xu B. Cancer targeting gene-viro-therapy for pancreatic cancer using oncolytic adenovirus ZD55-IL-24 in immune-competent mice. Mol Biol Rep. 2013;40:5397–5405. doi: 10.1007/s11033-013-2638-8. [DOI] [PubMed] [Google Scholar]
- 140.Nawa A, Luo C, Zhang L, Ushjima Y, Ishida D, Kamakura M, Fujimoto Y, Goshima F, Kikkawa F, Nishiyama Y. Non-engineered, naturally oncolytic herpes simplex virus HSV1 HF-10: applications for cancer gene therapy. Curr Gene Ther. 2008;8:208–221. doi: 10.2174/156652308784746422. [DOI] [PubMed] [Google Scholar]
- 141.Nakao A, Kasuya H, Sahin TT, Nomura N, Kanzaki A, Misawa M, Shirota T, Yamada S, Fujii T, Sugimoto H, et al. A phase I dose-escalation clinical trial of intraoperative direct intratumoral injection of HF10 oncolytic virus in non-resectable patients with advanced pancreatic cancer. Cancer Gene Ther. 2011;18:167–175. doi: 10.1038/cgt.2010.65. [DOI] [PubMed] [Google Scholar]
- 142.Campadelli-Fiume G, De Giovanni C, Gatta V, Nanni P, Lollini PL, Menotti L. Rethinking herpes simplex virus: the way to oncolytic agents. Rev Med Virol. 2011;21:213–226. doi: 10.1002/rmv.691. [DOI] [PubMed] [Google Scholar]
- 143.Hu JC, Coffin RS, Davis CJ, Graham NJ, Groves N, Guest PJ, Harrington KJ, James ND, Love CA, McNeish I, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12:6737–6747. doi: 10.1158/1078-0432.CCR-06-0759. [DOI] [PubMed] [Google Scholar]
- 144.Harrington KJ, Hingorani M, Tanay MA, Hickey J, Bhide SA, Clarke PM, Renouf LC, Thway K, Sibtain A, McNeish IA, et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin Cancer Res. 2010;16:4005–4015. doi: 10.1158/1078-0432.CCR-10-0196. [DOI] [PubMed] [Google Scholar]
- 145.Miller JD, van der Most RG, Akondy RS, Glidewell JT, Albott S, Masopust D, Murali-Krishna K, Mahar PL, Edupuganti S, Lalor S, et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity. 2008;28:710–722. doi: 10.1016/j.immuni.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 146.Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9:64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- 147.Hiley CT, Yuan M, Lemoine NR, Wang Y. Lister strain vaccinia virus, a potential therapeutic vector targeting hypoxic tumours. Gene Ther. 2010;17:281–287. doi: 10.1038/gt.2009.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chard LS, Maniati E, Wang P, Zhang Z, Gao D, Wang J, Cao F, Ahmed J, El Khouri M, Hughes J, et al. A vaccinia virus armed with interleukin-10 is a promising therapeutic agent for treatment of murine pancreatic cancer. Clin Cancer Res. 2015;21:405–416. doi: 10.1158/1078-0432.CCR-14-0464. [DOI] [PubMed] [Google Scholar]
- 149.Wennier ST, Liu J, Li S, Rahman MM, Mona M, McFadden G. Myxoma virus sensitizes cancer cells to gemcitabine and is an effective oncolytic virotherapeutic in models of disseminated pancreatic cancer. Mol Ther. 2012;20:759–768. doi: 10.1038/mt.2011.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Petrulio CA, Kaufman HL. Development of the PANVAC-VF vaccine for pancreatic cancer. Expert Rev Vaccines. 2006;5:9–19. doi: 10.1586/14760584.5.1.9. [DOI] [PubMed] [Google Scholar]
- 151.New York: Newswire PR; 2015. Therion Reports Results of Phase 3 PANVAC-VF Trial and Announces Plans for Company Sale. [Google Scholar]
- 152.Bhat R, Dempe S, Dinsart C, Rommelaere J. Enhancement of NK cell antitumor responses using an oncolytic parvovirus. Int J Cancer. 2011;128:908–919. doi: 10.1002/ijc.25415. [DOI] [PubMed] [Google Scholar]
- 153.Dempe S, Lavie M, Struyf S, Bhat R, Verbeke H, Paschek S, Berghmans N, Geibig R, Rommelaere J, Van Damme J, et al. Antitumoral activity of parvovirus-mediated IL-2 and MCP-3/CCL7 delivery into human pancreatic cancer: implication of leucocyte recruitment. Cancer Immunol Immunother. 2012;61:2113–2123. doi: 10.1007/s00262-012-1279-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bossow S, Grossardt C, Temme A, Leber MF, Sawall S, Rieber EP, Cattaneo R, von Kalle C, Ungerechts G. Armed and targeted measles virus for chemovirotherapy of pancreatic cancer. Cancer Gene Ther. 2011;18:598–608. doi: 10.1038/cgt.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17:3351–3362. doi: 10.1093/emboj/17.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Himeno Y, Etoh T, Matsumoto T, Ohta M, Nishizono A, Kitano S. Efficacy of oncolytic reovirus against liver metastasis from pancreatic cancer in immunocompetent models. Int J Oncol. 2005;27:901–906. [PubMed] [Google Scholar]
- 157.Etoh T, Himeno Y, Matsumoto T, Aramaki M, Kawano K, Nishizono A, Kitano S. Oncolytic viral therapy for human pancreatic cancer cells by reovirus. Clin Cancer Res. 2003;9:1218–1223. [PubMed] [Google Scholar]
- 158.Escors D. Tumour immunogenicity, antigen presentation and immunological barriers in cancer immunotherapy. New J Sci. 2014;2014:pii 734515. doi: 10.1155/2014/734515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Moehler MH, Zeidler M, Wilsberg V, Cornelis JJ, Woelfel T, Rommelaere J, Galle PR, Heike M. Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation, and cross-presentation by dendritic cells. Hum Gene Ther. 2005;16:996–1005. doi: 10.1089/hum.2005.16.996. [DOI] [PubMed] [Google Scholar]
- 160.Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6:226ra32. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Engeland CE, Grossardt C, Veinalde R, Bossow S, Lutz D, Kaufmann JK, Shevchenko I, Umansky V, Nettelbeck DM, Weichert W, et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Ther. 2014;22:1949–1959. doi: 10.1038/mt.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dolgin E. Oncolytic viruses get a boost with first FDA-approval recommendation. Nat Rev Drug Discov. 2015;14:369–371. doi: 10.1038/nrd4643. [DOI] [PubMed] [Google Scholar]
- 163.Dias JD, Hemminki O, Diaconu I, Hirvinen M, Bonetti A, Guse K, Escutenaire S, Kanerva A, Pesonen S, Löskog A, et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012;19:988–998. doi: 10.1038/gt.2011.176. [DOI] [PubMed] [Google Scholar]
- 164.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Croyle MA, Callahan SM, Auricchio A, Schumer G, Linse KD, Wilson JM, Brunner LJ, Kobinger GP. PEGylation of a vesicular stomatitis virus G pseudotyped lentivirus vector prevents inactivation in serum. J Virol. 2004;78:912–921. doi: 10.1128/JVI.78.2.912-921.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Eto Y, Yoshioka Y, Mukai Y, Okada N, Nakagawa S. Development of PEGylated adenovirus vector with targeting ligand. Int J Pharm. 2008;354:3–8. doi: 10.1016/j.ijpharm.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 167.Moniri MR, Dai LJ, Warnock GL. The challenge of pancreatic cancer therapy and novel treatment strategy using engineered mesenchymal stem cells. Cancer Gene Ther. 2014;21:12–23. doi: 10.1038/cgt.2013.83. [DOI] [PubMed] [Google Scholar]
- 168.García-Castro J, Alemany R, Cascalló M, Martínez-Quintanilla J, Arriero Mdel M, Lassaletta A, Madero L, Ramírez M. Treatment of metastatic neuroblastoma with systemic oncolytic virotherapy delivered by autologous mesenchymal stem cells: an exploratory study. Cancer Gene Ther. 2010;17:476–483. doi: 10.1038/cgt.2010.4. [DOI] [PubMed] [Google Scholar]
- 169.Ling X, Marini F, Konopleva M, Schober W, Shi Y, Burks J, Clise-Dwyer K, Wang RY, Zhang W, Yuan X, et al. Mesenchymal Stem Cells Overexpressing IFN-β Inhibit Breast Cancer Growth and Metastases through Stat3 Signaling in a Syngeneic Tumor Model. Cancer Microenviron. 2010;3:83–95. doi: 10.1007/s12307-010-0041-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kottke T, Galivo F, Wongthida P, Diaz RM, Thompson J, Jevremovic D, Barber GN, Hall G, Chester J, Selby P, et al. Treg depletion-enhanced IL-2 treatment facilitates therapy of established tumors using systemically delivered oncolytic virus. Mol Ther. 2008;16:1217–1226. doi: 10.1038/mt.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tseng JC, Granot T, DiGiacomo V, Levin B, Meruelo D. Enhanced specific delivery and targeting of oncolytic Sindbis viral vectors by modulating vascular leakiness in tumor. Cancer Gene Ther. 2010;17:244–255. doi: 10.1038/cgt.2009.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wennier S, Li S, McFadden G. Oncolytic virotherapy for pancreatic cancer. Expert Rev Mol Med. 2011;13:e18. doi: 10.1017/S1462399411001876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ady JW, Heffner J, Klein E, Fong Y. Oncolytic viral therapy for pancreatic cancer: current research and future directions. Oncolytic Virotherapy. 2014;3:35–46. doi: 10.2147/OV.S53858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ilkow CS, Marguerie M, Batenchuk C, Mayer J, Ben Neriah D, Cousineau S, Falls T, Jennings VA, Boileau M, Bellamy D, et al. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat Med. 2015;21:530–536. doi: 10.1038/nm.3848. [DOI] [PubMed] [Google Scholar]
- 175.Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer. 2011;11:805–812. doi: 10.1038/nrc3153. [DOI] [PMC free article] [PubMed] [Google Scholar]