

Abstract
Objectives
We wished to evaluate the effects of an antigranulocyte-macrophage colony-stimulating factor monoclonal antibody (KB003) on forced expiratory volume in 1 s (FEV1), asthma control and asthma exacerbations in adult asthmatics inadequately controlled by long-acting bronchodilators and inhaled/oral corticosteroids.
Settings
47 ambulatory asthma care centres globally.
Primary outcome measures
Change in FEV1 at week 24.
Participants
311 were screened, 160 were randomised and 129 completed the study.
Interventions
7 intravenous infusions of either 400 mg KB003 or placebo at baseline and weeks 2, 4, 8, 12, 16 and 20.
Primary and secondary outcome measures
FEV1 at week 24, asthma control, exacerbation rates and safety in all participants as well as prespecified subgroups.
Main results
In the KB003 treated group, FEV1 at week 24 improved to 118 mL compared with 54 mL in the placebo group (p=0.224). However, FEV1 improved to 253 vs 26 mL at week 24 (p=0.02) in eosinophilic asthmatics (defined as >300 peripheral blood eosinophils/mL at baseline) and comparable improvements were seen at weeks 20 (p=0.034) and 24 (p=0.077) in patients with FEV1 reversibility ≥20% at baseline and at weeks 4 (p=0.029), 16 (p=0.018) and 20 (p=0.006) in patients with prebronchodilator FEV1 ≤50% predicted at baseline. There were no effects on asthma control or exacerbation rates. The most frequent adverse events in the KB003 group were rhinosinusitis and headache. There was no significant difference in antidrug antibody response between placebo and treated groups. There were no excess infections or changes in biomarkers known to be associated with the development of pulmonary alveolar proteinosis.
Conclusions
Higher doses and/or further asthma phenotyping may be required in future studies with KB003.
Trial registration number
NCT01603277; Results.
Keywords: THORACIC MEDICINE
Strengths and limitations of this study.
First randomised study in asthmatics with an antigranulocyte-macrophage colony-stimulating factor (GM-CSF) monoclonal antibody.
Prespecified subgroups showed a significant response in forced expiratory volume in 1 s.
Not dose ranging.
Not powered to detect differences in exacerbation rates.
GM-CSF not measured in blood or sputum to clearly identify responders prospectively.
Introduction
Asthma is a chronic disease characterised by airway inflammation, airway hyper-responsiveness and episodic bronchoconstriction. The exact etiopathogenesis of asthma is not entirely understood, and inhaled corticosteroids (ICS) are the mainstay of therapy. Some patients with asthma show little or no benefit from corticosteroids even at high doses,1 and have higher morbidity resulting in higher costs of care.2
Studies of induced sputum and airway biopsies have described two phenotypes of severe asthma, eosinophilic and neutrophilic, based on the relative number of cells present in the samples,3–5 and studies of peripheral blood of asthmatics suggest four inflammatory patterns according to eosinophil and neutrophil cut values.6 7 Development of targeted therapies for asthma and phenotype-specific clinical trials has raised interest in the eosinophilic phenotype in particular.8 9 It is generally believed that the eosinophilic phenotype has a predominantly Th2 pathogenesis, while neutrophilic asthma is associated more frequently with Th17/Th1 immune responses.10 11 Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a cytokine secreted by macrophages, T cells, mast cells, endothelial cells and fibroblasts that were initially described as haematopoietic growth factor. It is now understood that GM-CSF is a cytokine that plays a role in the activation, differentiation and survival of adaptive and innate immune cells including granulocytes, macrophages, dendritic cells and lymphocytes. GM-CSF is produced in small amounts by normal lung epithelium but in increased amounts by lung epithelial cells in asthmatics.12 Endobronchial allergen challenge in asthmatics results in increased GM-CSF immunoreactivity in lymphocytes and alveolar macrophages.13 GM-CSF levels are also higher in sputum, bronchoalveolar lavage fluid and bronchial tissue in individuals with asthma and chronic obstructive pulmonary disease.14 15 Reduced eosinophil and neutrophil apoptosis correlates with increased lung inflammatory cell numbers and severity of asthma,16 17 and GM-CSF has been shown to be an antiapoptotic factor for both these cell types.18 GM-CSF may be a key mediator in the recruitment, activation and maintenance of both these cell types in asthmatic airways15 17 19 20 because it seems to cross the boundaries between Th2 and Th17/Th1 immunity suggesting it has a role in eosinophilic and neutrophilic asthma.2 5 21 22 In addition, GM-CSF production by peripheral blood mononuclear cells from corticosteroid-resistant asthmatic individuals is insensitive to corticosteroid inhibition,23 and animal data using anti-GM-CSF antibodies support the role of GM-CSF in airway disease.24–26
KB003 is a novel, high-affinity, recombinant IgG1κ monoclonal antibody targeting GM-CSF. It neutralises GM-CSF activity by blocking its binding to GM-CSF cell surface receptors. Studies in non-human primates administered KB003 doses as high as 100 mg/kg showed no toxicology findings including lack of foamy macrophages in the lungs, which are a prodromic indicator of pulmonary alveolar proteinosis (PAP). Although circulating anti-GM-CSF antibodies have been found in otherwise healthy volunteers,27 28 the presence of such antibodies appears to be associated with the development of PAP.29 Yet, previous single-dose phase 1b studies with KB002 (the predecessor to KB003) in asthmatics and in patients with rheumatoid arthritis showed trends in improvements in forced expiratory volume in 1 s (FEV1) or Disease Activity Score using the 28 joint count, respectively, without any safety concerns.
In view of the positive trends in efficacy and an acceptable safety profile after a single dose, we speculated that multiple doses of KB003 would be beneficial in treating patients with severe asthma. As such, we conducted a randomised, double-blind, placebo-controlled trial in moderate to severe asthmatics to assess the potential benefits and safety profile resulting from neutralisation of GM-CSF with KB003 over a 24-week period.
Methods
Study population and design
The present study was a phase 2 randomised, double-blind, placebo-controlled, parallel-group, repeat-dose study over 36 weeks (including a 20-week treatment period and screening and follow-up periods) to evaluate the safety, tolerability and efficacy of KB003 in adults with asthma inadequately controlled (defined by an asthma control questionnaire (ACQ) >1.5 at baseline) despite receiving treatment with long-acting β2 agonists (LABA) and inhaled and/or oral corticosteroids. The study was approved by institutional review boards (Western Institutional Review Board on 2 July 2012—approval # 20120727, and Quorum review IRB on 18 July 2012 Quorum file # 27264). Eligible participants had physician-diagnosed asthma, a per cent predicted FEV1 between 40% and 80%, and a history of at least two asthma exacerbations in the prior year. All participants underwent a screening and run-in period of 2–4 weeks, during which baseline asthma control and adherence with study procedures and concomitant ambulatory medications were determined. During the run-in period, reversibility of airway obstruction (≥12% improvement in FEV1) with short-acting β2 agonists (SABA) was required, and baseline chest X-rays and immunoglobulin E levels were obtained. Participants who met all protocol-specified study entry criteria were randomised in a 1:1 ratio to receive 400 mg KB003 or placebo. Study drug was administered intravenously over approximately 60 min at weeks 0, 2, 4, 8, 12, 16 and 20. Subsequent follow-up visits took place at weeks 24 (primary efficacy end point) and 28. At week 32, a phone interview was conducted to collect adverse events (AEs) information. A schema of the study design is provided in figure 1.
Figure 1.

Study schema: thick arrows denote dosing with KB003 or placebo; thin arrow denotes the primary end point assessment (forced expiratory volume in 1 s); a=week 32 visit was a phone call.
Throughout the study, medical history, concomitant medication use, physical examinations, arterial oxygen saturation (SaO2), and clinical laboratory analyses (haematology, urinalysis and chemistry, including surfactant protein D (SP-D)) were obtained. In addition, pregnancy tests for females, blood samples for pharmacokinetic (PK) and anti-KB003 antibody determinations, ECGs, and AEs were collected. To monitor asthma, lung function (spirometry and daily peak flow rates), asthma exacerbations, ACQ, asthma symptoms and rescue bronchodilator use (daily diary) were collected. The study was overseen by an independent Data Safety and Monitoring Committee, the purpose of which was to act in an advisory capacity to monitor the safety of the 160 participants enrolled in the study.
End points
The primary objective of the study was to evaluate the efficacy of KB003 on lung function in patients with asthma inadequately controlled by LABA and inhaled/oral corticosteroids, as measured by changes in prebronchodilator FEV1 at week 24. Secondary objectives of the study were to evaluate asthma exacerbation rates, peak expiratory flow rate (PEFR), ACQ scores, asthma symptoms, rescue short-acting bronchodilator use, safety and tolerability, PK and immunogenicity of KB003.
Sample size and other statistical considerations
On the basis of a previous study with anti-GM-CSF in asthmatics (unpublished), and to detect a difference between the placebo and KB003 groups of 6.7% in per cent predicted FEV1 over 24 weeks (80% statistical power, α=0.05, 2-tailed test), a sample size of 60 participants per group was needed. To account for an anticipated 15% dropout rate during the 24 weeks of the randomised treatment period, the target enrolment was 150 participants (75 participants per group). With respect to the exacerbation rate over 24 weeks, there was 80% statistical power to detect a difference of 0.5 exacerbations per participant between the placebo and KB003 treatment groups (α=0.05, 2-tailed test; 15% dropouts over 24 weeks), assuming approximately 1.0 exacerbation per participant over 24 weeks was expected as seen in two previous clinical studies with similar asthma populations.3 30
The Full Analysis Set (FAS) consisted of all randomised participants with a baseline value who received at least 1 dose of randomised treatment and had at least 1 treatment period measurement of lung function. The FAS was the primary analysis population and was used for the analysis of the primary end point. The Evaluable Set (ES) consisted of all participants included in the FAS who received at least four consecutive doses of study drug and had no major protocol deviations. The ES was used for efficacy analyses in prespecified subgroups.
For spirometry variables, the baseline value was defined as the last non-missing prebronchodilator value collected prior to the first dose of study medication. For daily variables (morning PEFR, asthma symptom score, rescue SABA use), the baseline average value was defined as the average of the last 7 days prior to randomisation (including the value collected predose at the randomisation visit (week 0). For the ACQ, the baseline value was the one obtained at the randomisation visit (week 0). Missing data were not imputed or replaced in any analyses except the jump to reference (J2R) imputation for the secondary analyses of spirometry parameters at week 24. Missing week 24 values were imputed using the average week 24 value in the placebo group and were analysed using an analysis of covariance (ANCOVA) model.
The summaries of absolute and per cent predicted FEV1 at baseline and at weeks 2, 4, 8, 12, 16, 20 and 24, and the analyses of the change from baseline in each parameter at week 24 using a linear mixed-effects model, were repeated for the following seven prespecified subgroups: (1) atopic asthma versus non-atopic asthma (atopy defined by at least 1 allergen in a panel of common allergens had a value ≥100 kUA/L; conversely, participants with values <100 kUA/L for common allergens tested were considered to have non-atopic asthma), (2) baseline blood eosinophils <0.3 vs ≥0.3 GI/L, (3) medium-dose versus high-dose ICS (determined in accordance with the Global Initiative for Asthma guidelines (http://www.ginasthma.org), (4) two asthma exacerbations versus >2 asthma exacerbations in the previous year, (5) high reversibility at study entry (<20% vs ≥20%), (6) prebronchodilator per cent predicted FEV1 at study entry (≤50% vs >50%) and (7) history of smoking versus never-smoked (smokers were defined as smoking ≤10 pack-year within the 12 months prior to screening).
All planned subgroup analyses were performed using the ES.
Efficacy analysis of absolute and per cent predicted FEV1
The primary analysis of the change from baseline in FEV1 over 24 weeks used a linear mixed-effects model with change from baseline in FEV1 as the dependent variable. The fixed terms in the model accounted for treatment, region (North America, Europe/Australia), and the baseline FEV1 value. The random effects in the model accounted for the repeated measurements within each study visit. PROC MIXED in SAS was used to analyse the data. ESTIMATE statements were used to obtain p values, and two-sided 95% CIs for the least square (LS) mean difference between KB003 and placebo at week 24. For the primary analysis, there was no data imputation. The primary efficacy analysis was performed using the FAS, and repeated using the ES.
Secondary analyses of FEV1 included analysis of the change from baseline at weeks 2, 4, 8, 12, 16 and 20 using the linear mixed-effects model as described above, and an analysis of the change from baseline at week 24 using J2R to impute missing values at week 24 using an ANCOVA model with terms for treatment, region (North America, Europe/Australia) and the baseline value. The change from baseline in FEV1 at weeks 2, 4, 8, 12, 16 and 20 were analysed in the FAS and ES using the linear mixed-effects model and the ANCOVA model with J2R imputation as described above.
Asthma exacerbations
At every study visit, participants were assessed for asthma exacerbations experienced since the last visit. Asthma exacerbations were defined by meeting one or more of the following criteria: (1) use of systemic corticosteroids (tablets, suspension or injection) for at least 3 days to treat worsening of asthma symptoms; (2) hospitalisation or emergency room, urgent care or physician visit for asthma worsening, requiring systemic corticosteroids or (3) in those participants taking oral corticosteroids (OCS) at study entry, at least a doubling of the OCS dose for at least 3 consecutive days. Courses of OCS separated by 7 days or more were treated as separate exacerbations.
The number and percentage of participants reporting a protocol-defined asthma exacerbation are presented by treatment and visit using the FAS. The protocol-defined exacerbation rates were compared using Poisson regression while accounting for the possibility of hyper-Poisson variability using PROC GENMOD in SAS, with fixed terms for treatment and region (North America, Europe/Australia). In addition, the number of participants with at least one protocol-defined exacerbation reported after the initiation of study drug was analysed using a logistic regression model with fixed terms for treatment and region (North America, Europe/Australia). Cox's proportional hazards model was used to compare the treatment groups with respect to time to first protocol-defined exacerbation using the ES. PROC PHREG in SAS was used to analyse the data. Participants who did not report a protocol-defined asthma exacerbation were treated in the model as censored observations and were censored at the date of the week 24 visit. The independent variables in the model were treatment group and region (North America, Europe/Australia). Kaplan-Meier plots were produced using PROC LIFETEST in SAS for visual assessment of the survival curves.
Other secondary end points
The following secondary end points were collected and analysed in the study: (1) PEFR using the highest recorded value of three acceptable efforts, the average morning PEFR (defined as the highest value of 3 acceptable efforts) at each visit being calculated for each participant over the prior week; (2) asthma symptoms score using the average of the responses to four daytime asthma symptom questions in the daily diary, the average daily symptom score (defined as the average of the responses to 4 questions in the daily dairy) at each visit being calculated for each participant over the prior week; (3) nocturnal awakenings using the responses to the question ‘Did you wake up with asthma symptoms?’ in the daily diary, the total number of nights with a nocturnal awakening at each visit being calculated for each participant over the prior week; (4) rescue short-acting bronchodilator (SABA and/or short-acting antimuscarinic agent (SAMA)) use using the daily number of puffs captured in the daily diary, the average daily number of puffs of rescue short-acting bronchodilator (SABA and/or SAMA) at each visit being calculated for each participant over the prior week and (5) ACQ score31 using the first five items related to asthma symptoms calculated for each participant at each visit, the ACQ5 score being calculated for each participant at each visit. For each of these secondary end points, the change from baseline at weeks 2, 4, 8, 12, 16, 20 and 24 was analysed using a linear mixed-effects model. The fixed terms in the model accounted for treatment, region (North America, Europe/Australia) and the baseline values. The random effects in the model accounted for the repeated measurements within each study participant. p Values for the LS mean difference between KB003 and placebo by visit were calculated along with two-sided 95% CIs.
Pharmacokinetics analysis
A KB003 PK model was developed using data from an earlier study (study KB003-04) and a phase 1 study conducted in healthy adult volunteers (study KB003-01). In study KB003-04, blood for serum KB003 assay was collected from a subset of participants prior to dose administration at weeks 0, 4, 8, 12, 16 and 20; at approximately 1 h after the end of infusion at weeks 0 and 20; and during a scheduled clinic visit at weeks 24 and 28. In study KB003-01, a single intravenous infusion of 1, 3, or 10 mg KB003 per kg of body weight was administered over 1 h. The PK of KB003 was described adequately by a two-compartment linear model with first-order elimination. Body weight did not influence the PK of KB003. The individual post hoc estimates from the population PK model were used to derive the individual exposure metrics (area under the concentration-time curve (AUC), maximum observed concentration (Cmax) and half-life (T1/2)) of KB003.
Immunogenicity analysis
A validated electrochemiluminescence assay method was used for sample testing. The determination of antidrug antibodies consisted of three sequential steps: (1) screening, (2) confirmation and (3) titer determination. A predose/postdose ratio was evaluated for the combined placebo and KB003 groups. This ratio established what could be considered a significant increase in postdose signal response from the corresponding prefirst dose sample (emergent immune response). In order to compare predose and postdose samples analysed on different plates, the screening signal response was normalised against the pooled negative control signal response specific to the plate where the sample was analysed. Using this calculated normalised signal response, each participant in the placebo and KB003 groups was analysed for a predose/postdose ratio for all postdose time points. A 95% upper limit was established using all the predose/postdose ratio data. Any sample with a predose/postdose ratio above the 95% upper limit was considered a meaningful increase in signal response. Using the calculated predose/postdose ratio, samples and participants were identified that corresponded to an increase in signal response from baseline.
For AEs with >5% frequency, Fisher exact tests were applied to compare groups, without any multiplicity adjustment for the significance level.
Results
In total, 311 asthmatics were screened across 47 clinical sites between July 2012 and June 2013, of whom 160 were enrolled in the study. Of these, 128 participants completed the study: 111 completed all study visits including the week 32 follow-up visit and 107 received all seven doses of either study drug or placebo. Subject dispositions are summarised in figure 2 and table 1. All participants who were randomised in the study received at least one dose. Participants were randomly assigned to study treatment in accordance with the randomisation schedule. The randomisation scheme was stratified according to the use of chronic OCs (yes, no) and region (North America, Australia, Europe). The protocol included an expectation that between 20% and 40% of participants would be on treatment with both chronic oral and ICS in the study; however, there were actually fewer participants than anticipated in this category (approximately 10%). Therefore, the use of chronic OCs (yes, no) strata was not included in analyses as a factor in models or as a subgroup variable. In addition, due to the small number (n=13) of participants randomised in Australia, participants randomised in Australia were combined with participants randomised in Europe, where region is indicated as a factor in analysis models. Participants were randomised in a ratio of 1:1 (400 mg KB003: placebo) at the randomisation visit, the day of first infusion (week 0).
Figure 2.

Participant disposition during the trial (see text for details).
Table 1.
Disposition of participants
| KB003 (n=78) |
Placebo (n=82) |
|
|---|---|---|
| Full Analysis Set * | 74 (94.9) | 76 (92.7) |
| Safety Set† | 78 (100.0) | 82 (100.0) |
| Evaluable Set‡ | 64 (82.1) | 65 (79.3) |
| Received all 7 doses of study drug | 56 (71.8) | 51 (62.2) |
| Completed all study visits including week 32 visit | 57 (73.1) | 54 (65.9) |
| Discontinued the study early | 14 (17.9) | 18 (22.0) |
| Primary reason for early study discontinuation | ||
| Adverse event | 1 (7.1) | 2 (11.1) |
| Non-compliance/lost to follow-up | 2 (14.3) | 1 (5.6) |
| Pregnancy | 0 | 0 |
| Protocol deviation | 6 (42.9) | 10 (55.6) |
| Consent withdrawal | 5 (35.7) | 4 (22.2) |
| Death | 0 | 0 |
| Investigator withdrew participation from study | 0 | 1 (5.6) |
Percentages for primary reason for early study discontinuation are based on the number of discontinued participants in each treatment group. All other percentages are based on the number of randomised participants in each treatment group.
*The Full Analysis Set consisted of all randomised participants with a baseline value who received at least 1 dose of study drug and had at least 1 treatment period measurement.
†The Safety Set consisted of all randomised participants who received at least 1 dose of study drug.
‡The Evaluable Set consisted of all participants included in the Full Analysis Set who received at least 4 consecutive doses of study drug and had no major protocol deviations.
Demographics and baseline characteristics for the FAS are summarised in table 2. The demographic and key baseline characteristics across the analysis sets and across treatment groups both within and across the analysis sets were comparable. All participants were treated with LABA/ICS, and 10% of all participants also received OCS. The doses of ICS/LABA, exacerbation rates as well as eosinophils and doses of ICS/LABA were comparable between groups. Comorbidities were not collected and exacerbation rates as well as eosinophils were not different between groups at baseline.
Table 2.
Demographics and baseline characteristics (Safety Set and Full Analysis Set)
| Safety Set |
Full Analysis Set |
|||
|---|---|---|---|---|
| KB003 (n=78) |
Placebo (n=82) |
KB003 (n=74) |
Placebo (n=76) |
|
| Demographics | ||||
| Age, years | ||||
| Mean (SD) | 52.9 (11.95) | 53.1 (10.30) | 52.9 (11.84) | 53.3 (10.38) |
| Median | 53.5 | 54.0 | 53.5 | 54.0 |
| Range | 22–73 | 19–75 | 22–73 | 19–75 |
| Gender, n (%) | ||||
| Female | 44 (56.4) | 48 (58.5) | 44 (59.5) | 46 (60.5) |
| Male | 30 (43.6) | 34 (41.5) | 30 (40.5) | 30 (39.5) |
| Race, n (%) | ||||
| Asian | 2 (2.6) | 3 (3.7) | 2 (2.7) | 2 (2.6) |
| Black or African American | 7 (9.0) | 9 (11.0) | 6 (8.1) | 8 (10.5) |
| White | 69 (88.5) | 69 (84.1) | 66 (89.2) | 65 (85.5) |
| Other | 0 | 1 (1.2) | 0 | 1 (1.3) |
| Ethnicity, n (%) | ||||
| Hispanic or Latino | 4 (5.1) | 7 (8.5) | 4 (5.4) | 6 (7.9) |
| Not Hispanic or Latino | 74 (94.9) | 75 (91.5) | 70 (94.6) | 70 (92.1) |
| Baseline characteristics | ||||
| Height, cm | ||||
| Mean (SD) | 168.37 (9.986) | 167.31 (8.477) | 167.97 (9.891) | 166.93 (8.405) |
| Median | 167.80 | 167.57 | 167.0 | 167.25 |
| Range | 148.0–190.5 | 152.0–185.4 | 148.0–188.0 | 152.0–185.4 |
| Weight, kg | ||||
| Mean (SD) | 83.05 (17.452) | 82.26 (16.951) | 82.44 (17.551) | 82.49 (17.006) |
| Median | 81.35 | 82.44 | 79.55 | 82.70 |
| Range | 42.6–120.0 | 48.0–123.0 | 42.6–120.0 | 48.0–123.0 |
| BMI, kg/m2 | ||||
| Mean (SD) | 29.340 (6.0031) | 29.365 (5.6555) | 29.259 (6.0292) | 29.592 (5.7388) |
| Median | 29.209 | 29.440 | 29.008 | 29.560 |
| Range | 13.46–46.99 | 17.99–42.79 | 13.46–46.99 | 17.99–42.79 |
| Percentage predicted FEV1 | ||||
| Mean (SD) | 56.636 (10.6441) | 56.483 (10.8880) | 56.440 (10.6499) | 57.638 (9.5821) |
| Median | 56.760 | 55.815 | 56.185 | 56.690 |
| Range | 34.59–77.82 | 25.09–77.45 | 34.59–77.82 | 39.81–77.45 |
| FEV1, L | ||||
| Mean (SD) | 1.774 (0.5536) | 1.713 (0.4678) | 1.752 (0.5425) | 1.736 (0.4595) |
| Median | 1.760 | 1.630 | 1.700 | 1.650 |
| Range | 0.97–3.03 | 0.69–3.10 | 0.97–3.03 | 1.09–3.10 |
| ACQ5 score | ||||
| Mean (SD) | 2.86 (0.705) | 2.87 (0.763) | 2.84 (0.711) | 2.89 (0.786) |
| Median | 2.86 | 2.86 | 2.86 | 2.86 |
| Range | 1.6–4.3 | 1.3–4.7 | 1.6–4.3 | 1.3–4.7 |
Percentages are based on the number of randomised participants in the Full Analysis Set or Safety Set in each treatment group. Baseline values are defined as the last non-missing values collected prior to first dose of study drug.
ACQ, Asthma Control Questionnaire; BMI, body mass index; FEV1, forced expiratory volume in 1 s.
Change from baseline by visit in absolute and per cent predicted prebronchodilator FEV1 in KB003 and placebo groups in the FAS and ES over 24 weeks are presented in figure 3A, B, respectively. At week 24, the primary end point, improvement in mean FEV1 in the KB003 group was 118 mL compared with 54 mL in the placebo group (p=0.224).
Figure 3.
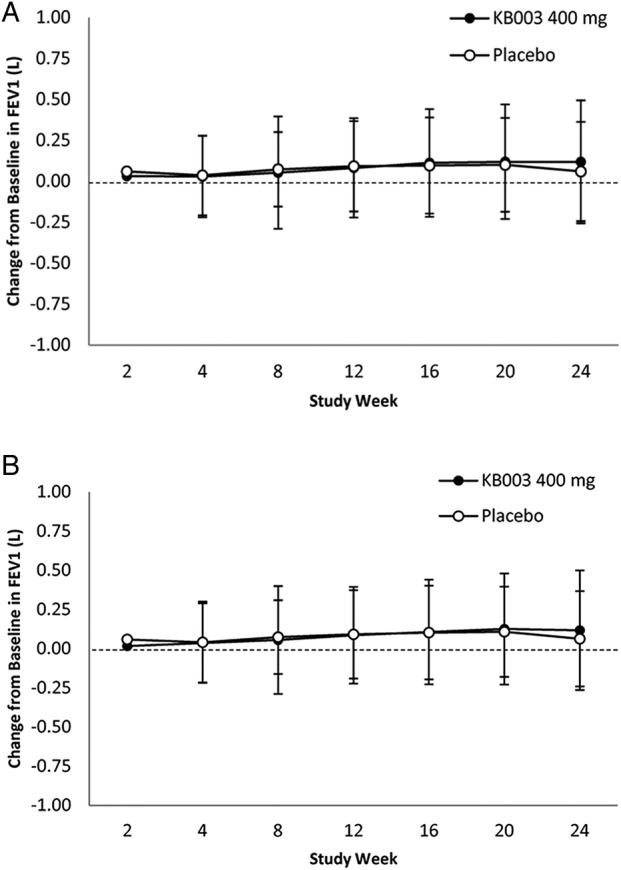
(A) Mean change±SD in forced expiratory volume in 1 s (FEV1). At baseline: KB003 n=74; placebo n=76. Full Analysis Set (all participants who received at least 1 dose); (B) only participants who receive 4 doses of KB003 or placebo. At baseline: KB003 n=64; placebo n=65. Close circles=KB003 recipients.
There were no differences in mean cumulative asthma exacerbations by visit over 24 weeks in the KB003 and placebo groups in the FAS and ES; data for the FAS are presented in figure 4. There were no differences with placebo in asthma exacerbation rates over 24 weeks (KB003=0.398 vs placebo=0.349). In addition, no drug effect was observed on PEFR, asthma symptoms, nocturnal awakenings, rescue SABA use, ACQ scores or peripheral blood eosinophilia in the study population.
Figure 4.
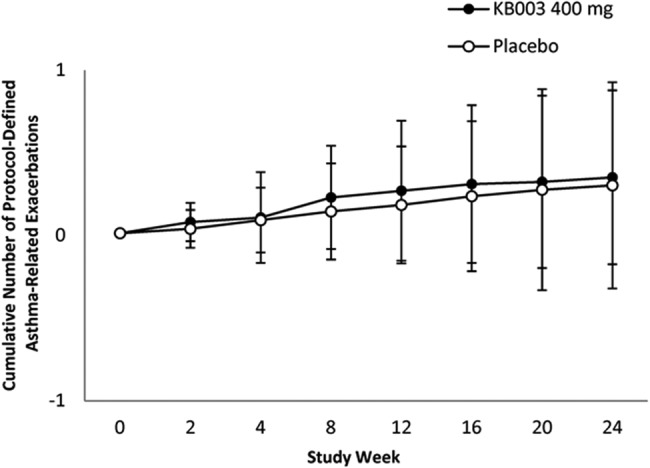
Cumulative number of exacerbations in participants who received at least one dose of KB003 or placebo. Close circles=KB003 recipients (see text for details) (FEV1, forced expiratory volume in 1 s; LS, least square).
Examination of predefined subgroups
Eosinophilic asthma: In participants with baseline blood eosinophils ≥0.3 GI/L, there was a significant FEV1 improvement in the KB003 group at week 24 (figure 5A): LS mean: KB003, 0.253 L; placebo, 0.026 L (95%, 2-sided CI 0.038 to 0.414), p=0.020.
Figure 5.
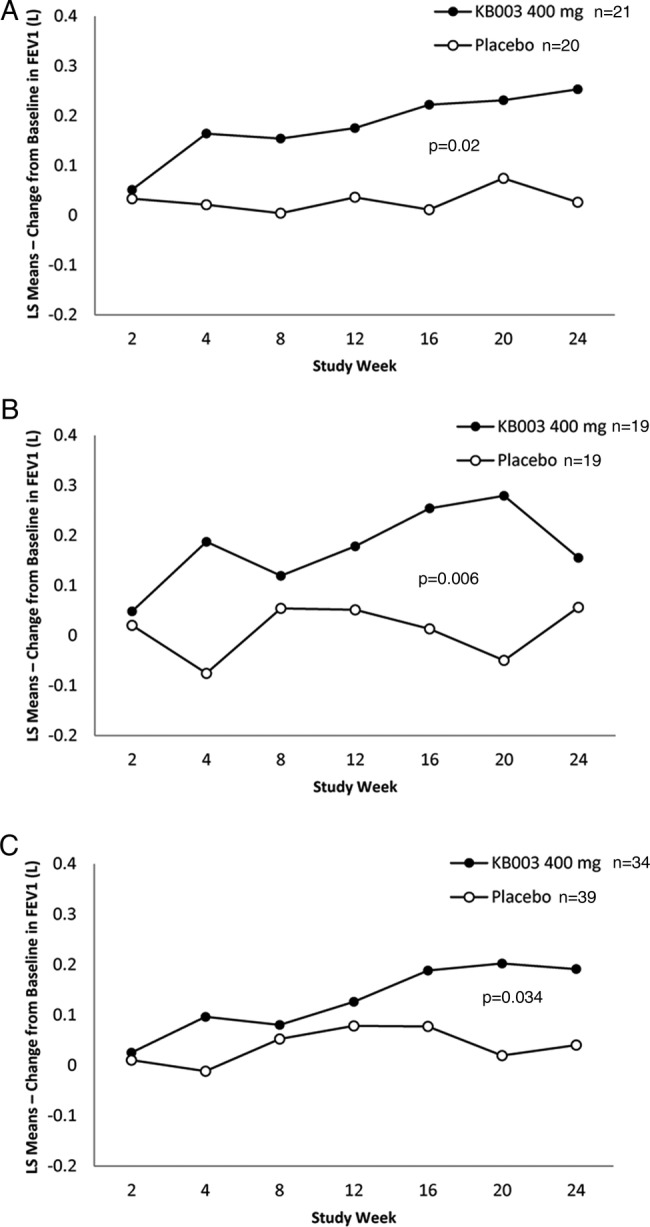
Changes from baseline in primary end point in three of the predetermined subgroups (see text for details). (A) Eosinophilic asthmatics at baseline; (B) severe airflow obstruction at randomisation and (C) augmented postbronchodilator reversibility (>20% improvement in FEV1) at baseline. Close circles=KB003 recipients (FEV1, forced expiratory volume in 1 s; LS, least square).
Prebronchodilator FEV1 ≤50%: In participants with prebronchodilator FEV1 ≤50% at study entry, there were significant FEV1 improvements in the KB003 group at week 4 (figure 5B): LS mean: KB003, 0.187; placebo, −0.076 (95%, 2-sided CI 0.029 to 0.498), p=0.029, at week 16: LS mean: KB003, 0.254; placebo, 0.013 (95%, 2-sided CI 0.044 to 0.438), p=0.018, and at week 20: LS mean: KB003, 0.279; placebo, −0.050 (95%, 2-sided CI 0.100 to 0.559), p=0.006.
Highly reversible FEV1: In participants with ≥20% reversibility at study entry, there was a significant FEV1 improvement in the KB003 group at week 20 (figure 5C): LS mean: KB003, 0.202; placebo, 0.019 (95%, 2-sided CI 0.015 to 0.353), p=0.034 and a trend towards FEV1 improvement at week 24: LS mean: KB003, 0.191; placebo, 0.040 (95%, 2-sided CI −0.01 to 0.320), p=0.077.
We found 10 participants (8 who received KB003) in whom the characteristics of eosinophilia, a low FEV1 at baseline, coupled with a history of ≥2 exacerbations in the previous year, were associated with significant improvements in FEV1 (table 3) in six of eight participants, which were not accompanied by reductions of ACQ.
Table 3.
FEV1 changes in participants with eosinophilia, airways reversibility at baseline and history of >2 asthma exacerbations in the previous year
| Subject ID | Study drug | BMI (kg/m2) (Gender) | Baseline |
W24/ET |
||||
|---|---|---|---|---|---|---|---|---|
| FEV1 % predicted (%) | FEV1 (L) | ACQ5 Score | FEV1 % predicted (%) | FEV1 (L) | ACQ5 Score | |||
| 121 001 | KB003 | 40.55 (F) | 58.62 | 1.53 | 1.0 | 79.69 | 2.08 | 3.4 |
| 150 002 | KB003 | 31.52 (F) | 53.26 | 1.39 | 4.0 | 74.33 | 1.94 | 2.2 |
| 401 002 | KB003 | 29.55 (F) | 43.13 | 1.13 | 4.0 | 70.61 | 1.85 | 4.2 |
| 505 001 | KB003 | 33.95 (M) | 50.92 | 1.66 | 3.2 | 48.47 | 1.58 | 2.4 |
| 508 005 | KB003 | 30.83 (M) | 46.58 | 2.11 | 2.6 | 64.02 | 2.90 | 0.8 |
| 606 002 | KB003 | 23.14 (F) | 52.04 | 1.53 | 1.2 | 63.61 | 1.87 | 1.8 |
| 149 003 (ET after W8) | KB003 | 29.41 (F) | 39.78 | 1.09 | 3.8 | 47 | 1.29 | 1.6 |
| 602 004 (ET after W8 | KB003 | 28.31 (F) | 59.69 | 1.94 | 3.0 | 44.31 | 1.44 | 3.0 |
| 130 001 | Placebo | 28.11 (F) | 47.46 | 1.31 | 2.0 | 36.59 | 1.01 | 2.4 |
| 505 003 | Placebo | 33.96 (F) | 38.97 | 1.06 | 2.6 | 45.59 | 1.24 | 1.6 |
ACQ, Asthma Control Questionnaire; BMI, body mass index; ET, early termination; FEV1, forced expiratory volume in 1 s; W, week.
Pharmacokinetics and immunogenicity
The individual post hoc estimates from the PK model for KB003 parameters were as follows: AUC, 12 488 μg h/mL (range 7486–18 244); median T1/2, 706 h (range 530–883); Cmax, 69 942 ng/mL (range 64 010–78 938) and Cmax at steady state (Cmax-ss), 78 074 ng/mL (range 67 837–90 988). Using a calculated predose/postdose ratio analysis, 7 of 77 participants in the KB003 group developed antibodies in response to KB003 compared with 4 of 77 participants in the placebo group.
Safety profile
Safety evaluations included all randomised participant who received at least one dose of randomised treatment. All AE variables were categorised and summarised using relative frequencies of the least 5% in any of the groups. Fisher exact tests were applied to compare groups, without any multiplicity adjustment for the significance level. AEs were coded using MedDRA V.16.1 and are summarised in table 4, and serious AEs (SAEs) are summarised in table 5. Infusion-related reactions were mild to moderate, affecting four participants in the KB003 group and two in the placebo group. All events were either self-limiting or were treated with medication and resolved without sequelae. Three participants discontinued study drug and withdrew from the study due to AEs: one participant in the KB003 group after hospitalisation for a suicide attempt, one on placebo because of hospitalisation for chronic septic arthritis, and a third, also on placebo, who was withdrawn from the study after receiving two doses of study drug due to non-serious infusion-related reactions. One participant in the KB003 group experienced a decreased absolute neutrophil count (ANC) below 1.5×103/μL at week 16 (1.42×103/μL), which returned to 1.92×103/μL (higher than baseline, 1.66×103/μL) by week 20 after seven doses. The second participant, in the placebo group, was found to have a decreased ANC below 1.5×103/μL at week 20 (1.23×103/μL) after receiving the last of the seven doses directed by the study protocol.
Table 4.
Summary of adverse events
| System organ class preferred term | KB003 (n=78) |
Placebo (n=82) |
Total (n=160) |
|---|---|---|---|
| Participants reporting at least 1 infusion-related reaction | 4 | 2 | 6 |
| All infusion-related reactions reported | 6 | 10* | 16 |
| General disorders and administration site conditions | 0 | 5 | 5 |
| Fatigue | 0 | 2 | 2 |
| Influenza-like illness | 0 | 2 | 2 |
| Infusion site pain | 0 | 1 | 1 |
| Gastrointestinal disorders | 1 | 2 | 3 |
| Diarrhoea | 0 | 0 | 0 |
| Nausea | 1 | 0 | 1 |
| Tongue pruritus | 0 | 2 | 2 |
| Investigations | 1 | 0 | 1 |
| Body temperature increased | 1 | 0 | 1 |
| Nervous system disorder | 3 | 0 | 3 |
| Dizziness | 1 | 0 | 1 |
| Headache | 2 | 0 | 2 |
| Psychiatric disorders | 0 | 1 | 1 |
| Anxiety | 0 | 1 | 1 |
| Skin and subcutaneous tissue disorder | 1 | 2 | 3 |
| Rash | 1 | 2 | 3 |
Events are tabulated by each incidence; a reaction may have occurred multiple times in a single participant.
Source: Listing 16.2.7.3 (appendix 16.2).
*Nine of the 10 events reported for the placebo group occurred in a single participant.
Table 5.
Summary of all serious adverse events
| System organ class preferred term | KB003 (n=78) | Placebo (n=82) | Total (n=160) |
|---|---|---|---|
| Participants reporting at least 1 serious adverse event | 5 | 2 | 7 |
| All serious adverse events reported* | 6 | 4 | 10 |
| Cardiac disorders | 1 | 0 | 1 |
| Acute myocardial infarction | 1 | 0 | 1 |
| Gastrointestinal disorders | 0 | 1 | 1 |
| Diarrhoea | 0 | 1 | 1 |
| General disorders and administrative conditions | 0 | 1 | 1 |
| Thrombosis in device | 0 | 1 | 1 |
| Immune system disorders | 0 | 1 | 1 |
| Anaphylactic reactions | 0 | 1 | 1 |
| Infections and Infestations | 3 | 1 | 4 |
| Appendicitis | 1 | 0 | 1 |
| Arthritis bacterial | 0 | 1 | 1 |
| Pneumonia | 2 | 0 | 2 |
| Psychiatric disorders | 1 | 0 | 1 |
| Suicide attempt | 1 | 0 | 1 |
| Respiratory, thoracic and mediastinal disorders | 1 | 0 | 1 |
| Hypoxia* | 1 | 0 | 1 |
Source: Listing 16.2.7.5 (appendix 16.2).
*Hypoxia was reported as a separate event concurrent with one of the pneumonia cases.
There was no difference between the KB003 and the placebo groups in SP-D, which has been described as a biomarker associated with the development of PAP.32 33
Discussion
In this 24-week study conducted in 160 adults with moderate to severe uncontrolled asthma, KB003 did not provoke improvement in prebronchodilator FEV1 in the overall study population. We wished to explore FEV1 as primary end point for three reasons: (1) the size of the study would allow for statistical power, (2) the evidence collected on a previous phase 1b study in asthmatics (unpublished) in which trends were seen in FEV1 improvements (120 mL) within 28 days and (3) because other biologics which reduce asthma exacerbations also improve FEV1 and improve asthma control.9 34 Indeed, in this present study, we found significant FEV1 improvements in participants with peripheral blood eosinophils >300 cells/mL, high FEV1 reversibility (≥20% improvement following SABA use) or prebronchodilator FEV1 ≤50% at baseline. Given that the FEV1 response to biologics can be variable depending on the patient population35–37 and, possibly, on the biological mechanism of action, the three prespecified phenotypic characteristics of peripheral blood eosinophilia, low baseline FEV1, and high acute reversibility postbronchodilators in a population with a history of frequent asthma exacerbations are markers of poorly controlled asthma. All participants in this study were receiving LABA and medium or high doses of ICS (some including OCS), but the changes in FEV1 were independent of the dose of corticosteroids used by participants (data not shown). This suggests that the statistically significant FEV1 improvements seen are unlikely related to undertreatment with corticosteroids.
Although the FEV1 improvements were consistent over time, no improvements in asthma control or reduction in exacerbations were observed possibly due to the low exacerbation rate and the reduced small sample size used for these secondary analyses.
The reasons why 400 mg KB003 administered once monthly did not yield benefits in non-eosinophilic asthma are less clear and remain speculative. It is possible that lower concentrations of GM-CSF are needed for an antiapoptotic effect on eosinophils than on neutrophils: 100 pg/mL vs 100 ng/mL, respectively.18 38 Thus, in eosinophilic asthma where the eosinophil but not neutrophil numbers are high, the levels of GM-CSF in the lungs and airways may be very low (pg/mL) but still effective on eosinophils, and these low GM-CSF levels might have been neutralised at 400 mg dosing of KB003. Conversely, in neutrophilic asthma, where antiapoptosis of neutrophils may be important, the GM-CSF levels in lungs/airways required for such an effect may have to be higher (ng/mL). Higher amounts of KB003 may therefore be needed to neutralise these higher levels of GM-CSF in neutrophilic asthma. In addition, the duration of exposure of inflammatory cell precursors in asthmatics to GM-CSF may determine their differentiation into different effector cells.39 The duration of neutralisation of GM-CSF needed for effects on neutrophils versus eosinophils may be different and may require different concentrations of injected antibody. If neutrophil generation or activation requires only a short exposure to GM-CSF, then neutralisation by KB003 will need to be maintained constantly and may require higher or more frequent dosing than the regimen we used in our study. Finally, it is possible that the priming and activation of eosinophils and neutrophils in asthma may occur not only via the GM-CSF pathway, or that it occurs in different ‘compartments’ of the body (eg, submucosa vs airway lumen vs circulation). For example, it has been reported that baseline airway inflammation in intrinsic and extrinsic asthma is characterised by eosinophilic inflammation and the presence of the Th1 cytokine interferon γ, and that GM-CSF treatment allows eosinophils to respond to lower concentrations of eotaxin via integrin activation and induction of PKCβII-mediated L-plastin phosphorylation.40 Given this, one of the limitations of the present study is that we did not measure levels of GM-CSF in blood or the lung compartment.
Nasopharyngitis, upper respiratory tract infection and headache were the only AEs that occurred with an overall incidence rate of 5% or greater. Among these, nasopharyngitis was the only event that occurred in more participants in the KB003 group than in the placebo group (6.4% vs 4.9%, respectively). All infusion-related reactions were either self-limiting or were treated with medication and resolved without sequelae. Ten SAEs were reported during the study, none of which were considered related to the study drug (table 5). All doses were generally well tolerated, with no safety signals of concern.
Importantly, there was no evidence of granulocytopenia (ANC <1000), monocytopenia, severe infusion reactions or pattern of AEs. There were no laboratory changes suggestive of serious or unusual infections. As part of the PAP pharmacovigilance programme for this study, SP-D, lactate dehydrogenase, oxygen saturation, chest X-rays and ≥20% decrease from baseline in FEV1 in the absence of acute bronchoconstriction were monitored for each participant. No observations indicative of signs or symptoms of PAP occurred during the treatment period or during the 3-month safety follow-up period. Indeed, spontaneously occurring and therapy-induced neutralising anti-GM-CSF antibodies have been reported both in healthy adults and in diseased populations without significant safety concerns noted. Low titres of anti-GM-CSF autoantibodies have been reported in the sera of 10–30% of healthy adults,27 and anti-GM-CSF has also been reported to be the dominant anticytokine activity in human intravenous immunoglobulin preparations from healthy human volunteers.41
Finally, using a predose/postdose ratio analysis, 7 of 77 participants in the KB003 group had an apparent confirmed emergent immune response to KB003 compared with 4 of 77 participants in the placebo group. The average serum exposure to KB003 derived from the post hoc estimates of the population PK model was comparable in patients with asthma (study KB003–04) and healthy subjects (study KB003-01), and showed an approximately linear increase in Cmax and AUC with increasing dose. The T1/2 of KB003 ranged from 639 to 808 h across the dose range in both studies.
In summary, the primary objective of the study, which was improvement in lung function with KB003 administration in patients with asthma inadequately controlled by corticosteroids, was not met in the overall population. Analyses of FEV1 in prespecified groups of participants treated with KB003 compared with placebo showed improvements over 24 weeks at a number of time points in patients with eosinophilic asthma (defined as those having blood eosinophil counts ≥0.3 GI/L at baseline), in participants who demonstrated high (≥20%) FEV1 reversibility at baseline, and in participants with a baseline per cent predicted FEV1 ≤50%, but not in other phenotypes. Further studies are required to select a dose of KB003, and a candidate asthma phenotype, for evaluating the role of GM-CSF in severe asthma or other airway conditions.
Footnotes
Contributors: JAL, GY, NAM and BS developed the study. CKO, MC, PK, DS and NAM conducted and supervised the study. BS analysed the data. NAM wrote the manuscript approved by all coauthors. Kalobios, Chiltern and study sites did the study design, execution and collection of data. Analysis and interpretation of data was done by Chiltern, Kalobios and external advisors. Kalobios decided to publish the data.
Funding: Kalobios Pharmaceuticals.
Competing interests: NAM, GY and CKO were Kalobios employees during the study conduct.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: No additional data are available.
References
- 1.Moore WC, Meyers DA, Wenzel SE et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med 2010;181:315–23. 10.1164/rccm.200906-0896OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akinbami LJ, Sullivan SD, Campbell JD et al. Asthma outcomes: healthcare utilization and costs. J Allergy Clin Immunol 2012;129(3 Suppl):S49–64. 10.1016/j.jaci.2011.12.984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wenzel SE, Schwartz LB, Langmack EL et al. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med 1999;160:1001–8. 10.1164/ajrccm.160.3.9812110 [DOI] [PubMed] [Google Scholar]
- 4.Gibson PG, Simpson JL, Saltos N. Heterogeneity of airway inflammation in persistent asthma: evidence of neutrophilic inflammation and increased sputum interleukin-8. Chest 2001;119:1329–36. 10.1378/chest.119.5.1329 [DOI] [PubMed] [Google Scholar]
- 5.Douwes J, Gibson P, Pekkanen J et al. Non-eosinophilic asthma: importance and possible mechanisms. Thorax 2002;57:643–8. 10.1136/thorax.57.7.643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nadif R, Siroux V, Oryszczyn MP et al. Heterogeneity of asthma according to blood inflammatory patterns. Thorax 2009;64:374–80. 10.1136/thx.2008.103069 [DOI] [PubMed] [Google Scholar]
- 7.Baines KJ, Simpson JL, Wood LG et al. Systemic upregulation of neutrophil α-defensins and serine proteases in neutrophilic asthma. Thorax 2011;66:942–7. 10.1136/thx.2010.157719 [DOI] [PubMed] [Google Scholar]
- 8.Molfino NA, Gossage D, Kolbeck R et al. Molecular and clinical rationale for therapeutic targeting of interleukin-5 and its receptor. Clin Exp Allergy 2012;42:712–37. 10.1111/j.1365-2222.2011.03854.x [DOI] [PubMed] [Google Scholar]
- 9.Castro M, Wenzel SE, Bleecker ER et al. Benralizumab, an anti-interleukin 5 receptor α monoclonal antibody, versus placebo for uncontrolled eosinophilic asthma: a phase 2b randomized dose-ranging study. Lancet Respir Med 2014;2:879–90. 10.1016/S2213-2600(14)70201-2 [DOI] [PubMed] [Google Scholar]
- 10.Newcomb DC, Peebles RS Jr. Th17-mediated inflammation in asthma. Curr Opin Immunol 2013;25:755–60. 10.1016/j.coi.2013.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wakashin H, Hirose K, Iwamoto I et al. Role of IL-23-Th17 cell axis in allergic airway inflammation. Int Arch Allergy Immunol 2009;149(Suppl 1):108–12. 10.1159/000211382 [DOI] [PubMed] [Google Scholar]
- 12.Soloperto M, Mattoso VL, Fasoli A et al. A bronchial epithelial cell-derived factor in asthma that promotes eosinophil activation and survival as GM-CSF. Am J Physiol 1991;260(6 Pt 1):L530–8. [DOI] [PubMed] [Google Scholar]
- 13.Broide DH, Firestein GS. Endobronchial allergen challenge in asthma. Demonstration of cellular source of granulocyte macrophage colony-stimulating factor by in situ hybridization. J Clin Invest 1991;88:1048–53. 10.1172/JCI115366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mattoli S, Marini M, Fasoli A. Expression of the potent inflammatory cytokines, GM-CSF, IL6, and IL8, in bronchial epithelial cells of asthmatic patients. Chest 1992;101(3 Suppl):27S–9S. 10.1378/chest.101.3_Supplement.27S [DOI] [PubMed] [Google Scholar]
- 15.Saha S, Doe C, Mistry V et al. Granulocyte-macrophage colony-stimulating factor expression in induced sputum and bronchial mucosa in asthma and COPD. Thorax 2009;64:671–6. 10.1136/thx.2008.108290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duncan CJ, Lawrie A, Blaylock MG et al. Reduced eosinophil apoptosis in induced sputum correlates with asthma severity. Eur Respir J 2003;22:484–90. 10.1183/09031936.03.00109803a [DOI] [PubMed] [Google Scholar]
- 17.Laan M, Prause O, Miyamoto M et al. A role of GM-CSF in the accumulation of neutrophils in the airways caused by IL-17 and TNF-alpha. Eur Respir J 2003;21:387–93. 10.1183/09031936.03.00303503 [DOI] [PubMed] [Google Scholar]
- 18.Esnault S, Malter JS. Granulocyte macrophage-colony-stimulating factor mRNA is stabilized in airway eosinophils and peripheral blood eosinophils activated by TNF-alpha plus fibronectin. J Immunol 2001;166:4658–63. 10.4049/jimmunol.166.7.4658 [DOI] [PubMed] [Google Scholar]
- 19.Woolley KL, Adelroth E, Woolley MJ et al. Granulocyte-macrophage colony-stimulating factor, eosinophils and eosinophil cationic protein in subjects with and without mild, stable, atopic asthma. Eur Respir J 1994;7:1576–84. 10.1183/09031936.94.07091576 [DOI] [PubMed] [Google Scholar]
- 20.Hamilton JA. GM-CSF in inflammation and autoimmunity. Trends Immunol 2002;23:403–8. 10.1016/S1471-4906(02)02260-3 [DOI] [PubMed] [Google Scholar]
- 21.Kotsimbos AT, Humbert M, Minshall E et al. Upregulation of alpha GM-CSF-receptor in nonatopic asthma but not in atopic asthma. J Allergy Clin Immunol 1997;99:666–72. 10.1016/S0091-6749(97)70029-0 [DOI] [PubMed] [Google Scholar]
- 22.Simpson JL, Grissell TV, Douwes J et al. Innate immune activation in neutrophilic asthma and bronchiectasis. Thorax 2007;62:211–18. 10.1136/thx.2006.061358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larsson S, Löfdahl CG, Linden M. IL-2 and IL-4 counteract budesonide inhibition of GM-CSF and IL-10, but not of IL-8, IL-12 or TNF-alpha production by human mononuclear blood cells. Br J Pharmacol 1999;127:980–6. 10.1038/sj.bjp.0702631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohta K, Yamashita N, Tajima M et al. Diesel exhaust particulate induces airway hyperresponsiveness in a murine model: essential role of GM-CSF. J Allergy Clin Immunol 1999;104:1024–30. 10.1016/S0091-6749(99)70084-9 [DOI] [PubMed] [Google Scholar]
- 25.Yamashita N, Tashimo H, Ishida H et al. Attenuation of airway hyperresponsiveness in a murine asthma model by neutralization of granulocyte-macrophage colony-stimulating factor (GM-CSF). Cell Immunol 2002;219:92–7. 10.1016/S0008-8749(02)00565-8 [DOI] [PubMed] [Google Scholar]
- 26.Vlahos R, Bozinovski S, Chan SP et al. Neutralizing granulocyte/macrophage colony-stimulating factor inhibits cigarette smoke-induced lung inflammation. Am J Respir Crit Care Med 2010;182:34–40. 10.1164/rccm.200912-1794OC [DOI] [PubMed] [Google Scholar]
- 27.Revoltella RP, Laricchia-Robbio L, Moscato S et al. Natural and therapy-induced anti-GM-CSF and anti-G-CSF antibodies in human serum. Leuk Lymphoma 1997;26(Suppl 1):29–34. 10.3109/10428199709058597 [DOI] [PubMed] [Google Scholar]
- 28.Uchida K, Nakata K, Suzuki T et al. Granulocyte/macrophage-colony-stimulating factor autoantibodies and myeloid cell immune functions in healthy subjects. Blood 2009;113:2547–56. 10.1182/blood-2008-05-155689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kitamura T, Tanaka N, Watanabe J et al. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med 1999;190:875–80. 10.1084/jem.190.6.875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nair P, Pizzichini MM, Kjarsgaard M et al. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med 2009;360:985–93. 10.1056/NEJMoa0805435 [DOI] [PubMed] [Google Scholar]
- 31.Juniper EF, O'Byrne PM, Guyatt GH et al. Development and validation of a questionnaire to measure asthma control. Eur Respir J 1999;14:902–7. 10.1034/j.1399-3003.1999.14d29.x [DOI] [PubMed] [Google Scholar]
- 32.Honda Y, Kuroki Y, Matsuura E et al. Pulmonary surfactant protein D in sera and bronchoalveolar lavage fluids. Am J Respir Crit Care Med 1995;152(6 Pt 1):1860–6. 10.1164/ajrccm.152.6.8520747 [DOI] [PubMed] [Google Scholar]
- 33.Brasch F, Birzele J, Ochs M et al. Surfactant proteins in pulmonary alveolar proteinosis in adults. Eur Respir J 2004;24:426–35. 10.1183/09031936.04.00076403 [DOI] [PubMed] [Google Scholar]
- 34.Ortega HG, Liu MC, Pavord ID et al. Mepoluzimab treatment in patients with severe eosinophilic asthma. New Engl J Med 2014;371:1198–207. 10.1056/NEJMoa1403290 [DOI] [PubMed] [Google Scholar]
- 35.Wenzel SE, Barnes PJ, Bleecker ER et al. A randomized, double-blind, placebo-controlled study of tumor necrosis factor-alpha blockade in severe persistent asthma. Am J Respir Crit Care Med 2009;179:549–58. 10.1164/rccm.200809-1512OC [DOI] [PubMed] [Google Scholar]
- 36.Corren J, Busse W, Meltzer EO et al. A randomized, controlled, phase 2 study of AMG 317, an IL-4Ralpha antagonist, in patients with asthma. Am J Respir Crit Care Med 2010;181:788–96. 10.1164/rccm.200909-1448OC [DOI] [PubMed] [Google Scholar]
- 37.Castro M, Mathur S, Hargreave F et al. Reslizumab for poorly controlled, eosinophilic asthma: a randomized, placebo-controlled study. Am J Respir Crit Care Med 2011;184:1125–32. 10.1164/rccm.201103-0396OC [DOI] [PubMed] [Google Scholar]
- 38.Kobayashi T, Takaku Y, Kikuchi I et al. Eosinophils do not enhance the trans-basement-membrane migration of neutrophils. Int Arch Allergy Immunol 2007;143(Suppl 1):38–43. 10.1159/000101403 [DOI] [PubMed] [Google Scholar]
- 39.Hodge JM, Kirkland MA, Aitken CJ et al. Osteoclastic potential of human CFU-GM: biphasic effect of GM-CSF. J Bone Miner Res 2004;19:190–9. 10.1359/JBMR.0301232 [DOI] [PubMed] [Google Scholar]
- 40.Pazdrak K, Young TW, Straub C et al. Priming of eosinophils by GM-CSF is mediated by protein kinase CbetaII-phosphorylated L-plastin. J Immunol 2011;186:6485–96. 10.4049/jimmunol.1001868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Svenson M, Hansen MB, Ross C et al. Antibody to granulocyte-macrophage colony-stimulating factor is a dominant anti-cytokine activity in human IgG preparations. Blood 1998;91:2054–61. [PubMed] [Google Scholar]