

Abstract
Background
Idiopathic pulmonary fibrosis (IPF) is a chronic interstitial lung disease characterised by progressive loss of lung function. Its clinical course is variable but ultimately fatal. There is a need for a multicentre patient registry incorporating longitudinal clinical data and biological samples to improve understanding of the natural history of IPF and contemporary practice patterns.
Methods/design
The Idiopathic Pulmonary Fibrosis–PRospective Outcomes (IPF-PRO) registry is a national IPF registry in the USA. This registry will enrol approximately 300 patients with newly diagnosed IPF over 2 years at approximately 14 tertiary pulmonary care sites. Participants will be followed for 3–5 years and will receive usual care, as defined by their physician. Clinical data from the year prior to diagnosis will be collected from medical record review on enrolment. Subsequently, data on diagnostic evaluations, pulmonary function tests, physical examinations, laboratory data and clinical events will be collected at routine clinical visits and via a call centre. Participants will complete patient-reported outcome questionnaires at enrolment and then at approximately 6-month intervals. Blood samples for cellular, genetic and transcriptomic analyses will be collected at the same intervals.
Results
The first results from the IPF-PRO registry will be presented in 2015.
Conclusions
The IPF-PRO registry will improve understanding of the natural history of IPF, its impact on patients and current practice in the diagnosis and care of patients with IPF. The registry will establish a repository of biological samples from a well-characterised patient population for future research.
Clinical trial number
Keywords: Interstitial Fibrosis, Rare lung diseases
Key messages.
Little is known about the experience of patients with IPF from the development of symptoms through diagnosis and disease progression.
The IPF-PRO registry is a national IPF patient registry in the USA. This manuscript presents the design of the registry.
Data from this registry will help to address important questions about the course of IPF, predictors of disease progression, quality of life and serum biomarkers.
Background
Idiopathic pulmonary fibrosis (IPF) is a chronic and ultimately fatal interstitial lung disease characterised by worsening lung function, dyspnoea and impaired health-related quality of life.1 Estimates of the public health burden of IPF vary widely depending on the disease definition used and the population studied, but recent estimates suggest an annual incidence in the USA of between 6.8 and 17.4 cases per 100 000 people.2 The incidence of IPF increases with age,1 and may be as high as 93.7 cases per 100 000 person-years among Medicare beneficiaries aged over 65.3 IPF carries a poor prognosis, with a median survival time of 2–3 years.1
Positive phase III trial results have been announced for nintedanib4 and pirfenidone,5 and in 2014, both these therapies were approved for the treatment of IPF by the US Food and Drug Administration. As these therapies are adopted into routine clinical practice, more information is needed on practice patterns and the characteristics of patients with IPF. To date, most research on the natural history of IPF has come from single-centre cohorts or clinical trials. As clinical trials in IPF have enrolled patients with relatively mild impairment of lung function and comparatively low mortality rates,6 the generalisability of these data to the broad population of patients with IPF may be limited. Furthermore, little information is available on the experience of patients with IPF, from the development of symptoms through formal diagnosis and disease progression. Patients are often misdiagnosed and delayed in receiving a diagnosis of IPF,7 8 with one study reporting a median delay of 2.2 years between the onset of dyspnoea and initial evaluation at a tertiary care centre.9
Once diagnosed, the clinical course of IPF is highly variable among individual patients.10–13 Clinical events such as acute exacerbations of IPF are associated with high morbidity and mortality,14–16 but their frequency and significance have not been well characterised in multicentre studies. Changes in pulmonary function have been evaluated as prognostic markers in predictive models,17–19 but these models require further validation in broader populations of patients with IPF.
Advances in understanding of the pathogenesis of IPF have fuelled increasing interest in molecular biomarkers. Genome-wide association studies have identified several variants associated with the presence of IPF,20–22 and the risk of disease progression.21 23 These studies and mechanistic discoveries have formed the rationale for several relatively small, exploratory studies of potential molecular biomarkers.24 25 To date, however, no biomarker has been demonstrated to have clinical utility in a large population of patients with IPF. A large registry incorporating longitudinally collected clinical and outcomes data with serial biological samples would help advance our understanding of biomarkers in IPF.
IPF has been shown to have a detrimental effect on health-related quality of life, with dyspnoea and cough being the primary drivers of this impairment.26 27 A limited body of literature supports the use of specific patient-reported outcome measures in IPF,28–30 yet few studies have characterised the impact of disease progression on activities of daily living, functional status and overall well-being. Thus, there remains a need to explore the validity and clinical utility of patient-reported outcome measures in prospective studies in patients with IPF.
The Idiopathic Pulmonary Fibrosis–PRospective Outcomes (IPF-PRO) registry is a national IPF registry in the USA. It is funded by Boehringer Ingelheim and managed as a collaborative project by the Duke Clinical Research Institute (DCRI), Boehringer Ingelheim and clinical site investigators. The objectives of the IPF-PRO registry are as follows: (1) to describe patients’ interactions with the healthcare system, including diagnostic procedures, in the 12 months prior to formal diagnosis of IPF; (2) to describe the course of IPF, including physiological changes, interactions with the healthcare system and death; (3) to characterise patient-reported outcomes in patients with IPF using self-administered questionnaires; and (4) to obtain biological samples at multiple time points from a well-characterised patient population for future research. In this manuscript, we describe the design of the IPF-PRO registry and how the data collected will expand our understanding of the natural history of IPF, current practice patterns and the impact of IPF on patients.
Methods
Study design
The IPF-PRO registry is a multicentre, observational registry of patients with newly diagnosed IPF. The registry will target 300 patients for enrolment over 2 years at approximately 14 sites in the USA. Participants will be followed for a minimum of 3 years and up to 5 years. The protocol will be approved by the relevant Institutional Review Boards (IRBs) and/or local Independent Ethics Committees prior to patient enrolment at each site. The registry is registered on clinicaltrials.gov (NCT01915511).
Participants
To be eligible to participate in the registry, patients must be aged ≥40 years with a diagnosis of IPF confirmed at a tertiary pulmonary care centre according to current international guidelines within 3 months of the registry enrolment date. At patient enrolment, each site investigator will complete an investigator assessment form recording the diagnostic category most supported by the cumulative clinical data (definite IPF, probable IPF or possible IPF according to American Thoracic Society/European Respiratory Society/Japanese Respiratory Society/Latin American Thoracic Association definitions based on High-resolution computed tomography patterns and surgical lung biopsy results1). Patients must be able to read and write in English and provide written informed consent.
Exclusion criteria include malignancy, other than skin cancer, within the past 5 years; being on a lung transplant waiting list or participating in a randomised clinical trial at the time of enrolment. Patients enrolled in a non-interventional registry are eligible for inclusion, and patients may enter a randomised clinical trial after enrolment in the IPF-PRO registry.
Physicians at the enrolling sites will be instructed to approach every eligible patient receiving care at their clinic about participation in the registry and to document reasons for non-participation. As IPF-PRO is a non-interventional registry, enrolled patients will receive usual care for IPF as defined by the treating physician.
Data collection
Retrospective data collection
Clinical data from the 12 months prior to diagnosis will be collected from patient records at the time of enrolment and will include demographics; medical history; symptoms; comorbidities; vital signs; diagnostic procedures; pulmonary function tests; laboratory tests; 6 min walk test; concomitant medications including oxygen requirements; participation in pulmonary rehabilitation programmes and other interactions with the healthcare system, including hospitalisations and referrals.
Prospective data collection by sites
Enrolling sites will abstract data from the medical record at 6-month intervals throughout the study. Information will be collected on vital status; diagnostic evaluations; pulmonary function tests; 6-minute walk test; high resolution CT results; physical examinations; laboratory data; clinical events; comorbidities; participation in a pulmonary rehabilitation programme; healthcare utilisation, including changes to oxygen requirements, hospitalisations and outpatient visits; inclusion on a lung transplant list or having received a lung transplant and enrolment in a clinical trial. At each 6-month interval, site investigators will indicate whether the diagnosis has changed since the last point of data entry, and, if so, will enter details regarding the new diagnosis.
Data collection by central call centre
Participants will be contacted by interviewers from the DCRI centralised call centre at 6-month intervals to confirm vital status and any hospitalisations or unscheduled healthcare encounters since their last visit to the enrolling centre. These interviews will be conducted for all participants, including those who fail to attend clinical appointments and cannot be reached at the enrolling centre.
Patient-reported outcomes
The following self-administered questionnaires will be completed by patients at baseline and approximately every 6 months thereafter, at the closest regularly scheduled clinic visit: the Short Form Health Survey (SF-12),28 31 32 the EuroQoL (EQ-5D),33 the St George's Respiratory Questionnaire28 34 35 and the cough domains of the Cough and Sputum Assessment Questionnaire.36 37
Biological sampling
Whole blood from registry participants will be collected at baseline for future DNA analysis and centralised storage of plasma, serum and RNA samples. The registry protocol does not include a priori hypotheses or analysis plans for the biological samples; it is anticipated that these analyses will be driven by proposals from study investigators. The IPF-PRO registry informed consent is written in a manner that allows for future biological research using collected samples. Specific research studies proposing to use those samples as part of a separate analysis will proceed through IRB review as appropriate. Only de-identified patient information will be provided for use in future biomarker or genetic studies.
Administrative structure
The registry will be administered by Committees with representation from DCRI, Boehringer Ingelheim and site investigators (figure 1 and table 1). Proposals from site or external investigators for analyses of registry data, including research based on stored biological samples, will be assessed with regard to scientific value, feasibility and appropriateness of analytic methodology.
Figure 1.
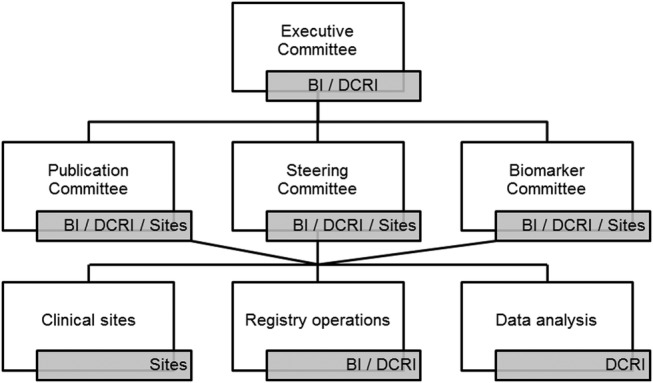
Registry organisational chart. The Idiopathic Pulmonary Fibrosis–PRospective Outcomes (IPF-PRO) registry will be administered by Committees with representation from Duke Clinical Research Institute (DCRI), Boehringer Ingelheim (BI) and site investigators (sites).
Table 1.
Registry organisation: committee roles and responsibilities
| Committee | Key roles and responsibilities |
|---|---|
| Executive Committee |
|
| Steering Committee |
|
| Publications Committee |
|
| Biomarker Committee |
|
Data analysis
A detailed analysis plan will be approved by the Executive Committee before every data analysis begins. We expect new research questions to be triggered by information that becomes available during the course of the registry. Standard approaches to the analysis of observational data will be used.38 39 All data analyses will be performed by the DCRI.
Discussion
As a national US registry for IPF, the IPF-PRO registry provides the opportunity to characterise the diagnosis and clinical management of IPF, markers and predictors of disease progression, and patient-reported and clinical outcomes over time. An inclusive population of patients recruited across multiple tertiary care centres will support generalisability of the findings to the broader population of patients with IPF. Retrospective review will enable comprehensive description of disease course and clinical practice patterns from the onset of symptoms to diagnosis. Enrolment of only newly diagnosed patients with IPF provides a consistent starting point from which longitudinal assessments can begin. Over a follow-up period of up to 5 years, the registry will track the evolution of clinical practice, including changes in referral and treatment patterns. Biological samples from patients who have been well characterised at discrete time points will provide opportunities to explore biomarkers, genetic and transcriptomic variants associated with disease progression. We anticipate that this detailed information will help to address important questions about the progression, management and outcomes of IPF.
Extant research on the natural history of IPF is limited and comes primarily from single-centre observational studies and clinical trial populations. Data from these trials, most of which recruited patients with IPF with mild or moderate impairment of lung function and without certain comorbidities, suggest that the decline in forced vital capacity in patients with IPF is approximately 150–200 mL/year.4 10 40–43 Acute exacerbations of IPF are an important feature of the clinical course of IPF in some patients, and are associated with high rates of hospitalisation and mortality.14–16 44 The IPF-PRO registry will help characterise the progression of IPF, including the burden of acute exacerbations and other clinical events, in a diverse patient population.
Patient-reported outcomes are integral to the study of progressive, incurable diseases like IPF, and comprehensive understanding of the effect of practice patterns on patients’ quality of life and well-being provides the foundation for optimal patient-centred care.45 In a recent review of five studies, patients with IPF identified timely diagnosis, expectations of treatment, access to tertiary care centres/centres of excellence and health-related quality of life as particularly relevant and important.26 Patient-reported outcome measures collected in the IPF-PRO registry may help to improve collaborative decision-making between patients and providers that effectively incorporates patient values and priorities.
The IPF-PRO registry will enrol only patients with a new diagnosis of IPF from tertiary care centres, rather than a community-based study population. This inclusion criterion was chosen to reflect current practice in the diagnosis of IPF, which, according to international guidelines, should involve multidisciplinary discussion by experts in the diagnosis of interstitial lung disease.1 Differences between patients who choose to participate and those who do not may limit the generalisability of the results; however, attempts will be made to characterise the population of patients who are eligible but do not enrol in the registry.
The IPF-PRO registry design has several unique strengths. Recently, a multidisciplinary group of investigators highlighted the need for a biorepository linked to clinical data to inform future translational research.46 The IPF-PRO registry will directly respond to this need by prospectively describing the clinical course of patients from the time of diagnosis with IPF through subsequent clinical events and will include both patients on FDA-approved therapies and patients on investigational therapies. Linkage of these longitudinal clinical data to serial biological specimens will provide a key resource to support future translational research. Further, the call centre used in the IPF-PRO registry will help to overcome the issue of missing data that has beset many of the clinical trials conducted in patients with IPF by enabling comprehensive and uniform capture of outcomes data over the entire study period. A similar call centre has provided systematic data collection for similar studies with an interview completion rate of over 95%.
Findings from the IPF-PRO registry will complement those from single-centre studies and from other regional and international IPF registries, including the state-wide Daniel and Joan Beren Pennsylvania IPF (PA-IPF) Registry,47 the Australian IPF registry,48 the German INSIGHTS-IPF registry,49 the PROOF registry in Belgium and Luxembourg50, the Canadian CARE-PF registry51 and the European IPF Network registry,52 53 which has established a large biobank of biological samples. The IPF-PRO registry will build on this work in three major ways. First, the collection of serial biosamples will support examination of trends in biomarkers of disease progression in a well-phenotyped population over time, which to our knowledge has not yet been explored in multicentre US-based IPF populations. Second, this registry will collect detailed information from real-world IPF populations on patient-reported outcomes such as symptom impact and quality of life, which may be significantly reduced in IPF. Further, collection of longitudinal patient-reported outcome measurements will allow analysis of changes in these metrics from the first months of a new diagnosis through disease progression, which to date has not been fully explored. Third, the IPF-PRO registry will collect detailed information about newly approved IPF therapies including patterns of usage. By targeting a large population of newly diagnosed patients with IPF from tertiary care centres across the US, collecting detailed longitudinal clinical and outcomes data and serial biological specimens, this registry will enable clinical and translational studies aimed at addressing knowledge gaps in this population.
Conclusions
The IPF-PRO registry is a national, longitudinal registry that will provide detailed insight into the progression of IPF, in addition to a robust repository of well-phenotyped biological samples from a diverse population of patients with IPF. The data collected in this registry will expand our understanding of the natural history of IPF, current practice patterns and the impact of the disease on patient-reported and clinical outcomes over time.
Acknowledgments
Medical writing assistance, supported financially by Boehringer Ingelheim Pharmaceuticals, Inc, was provided by Clare Ryles and Wendy Morris of Fleishman-Hillard Group, Ltd during the preparation of this manuscript.
Footnotes
Contributors: All authors contributed to the design of the IPF-PRO registry and to the writing of this manuscript. EO is responsible for the overall content as guarantor.
Funding: The IPF-PRO registry is funded by Boehringer Ingelheim Pharmaceuticals, Inc and coordinated by the Duke Clinical Research Institute.
Competing interests: VG, SG and CSC are employees of Boehringer Ingelheim Pharmaceuticals, Inc. DCRI has received payment from Boehringer Ingelheim Pharmaceuticals, Inc for coordination of the IPF-PRO registry.
Ethics approval: The protocol was approved by the relevant Institutional Review Boards and/or local Independent Ethics Committees prior to patient enrolment at each site.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: No additional data are available.
References
- 1.Raghu G, Collard HR, Egan JJ et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788–824. doi:10.1164/rccm.2009-040GL [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nalysnyk L, Cid-Ruzafa J, Rotella P et al. Incidence and prevalence of idiopathic pulmonary fibrosis: review of the literature. Eur Respir Rev 2012;21:355–61. doi:10.1183/09059180.00002512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raghu G, Chen SY, Yeh WS et al. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: incidence, prevalence, and survival, 2001–11. Lancet Respir Med 2014;2:566–72. doi:10.1016/S2213-2600(14)70101-8 [DOI] [PubMed] [Google Scholar]
- 4.Richeldi L, du Bois RM, Raghu G et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2071–82. doi:10.1056/NEJMoa1402584 [DOI] [PubMed] [Google Scholar]
- 5.King TE Jr, Bradford WZ, Castro-Bernardini S et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2083–92. doi:10.1056/NEJMoa1402582 [DOI] [PubMed] [Google Scholar]
- 6.King TE Jr, Albera C, Bradford WZ et al. All-cause mortality rate in patients with idiopathic pulmonary fibrosis. Implications for the design and execution of clinical trials. Am J Respir Crit Care Med 2014;189:825–31. doi:10.1164/rccm.201311-1951OC [DOI] [PubMed] [Google Scholar]
- 7.Collard HR, Tino G, Noble PW et al. Patient experiences with pulmonary fibrosis. Respir Med 2007;101:1350–4. doi:10.1016/j.rmed.2006.10.002 [DOI] [PubMed] [Google Scholar]
- 8.Schoenheit G, Becattelli I, Cohen AH. Living with idiopathic pulmonary fibrosis: an in-depth qualitative survey of European patients. Chron Respir Dis 2011;8:225–31. doi:10.1177/1479972311416382 [DOI] [PubMed] [Google Scholar]
- 9.Lamas DJ, Kawut SM, Bagiella E et al. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med 2011;184:842–7. doi:10.1164/rccm.201104-0668OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;183:431–40. doi:10.1164/rccm.201006-0894CI [DOI] [PubMed] [Google Scholar]
- 11.Martinez FJ, Safrin S, Weycker D et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med 2005;142:963–7. doi:10.7326/0003-4819-142-12_Part_1-200506210-00005 [DOI] [PubMed] [Google Scholar]
- 12.Mura M, Porretta MA, Bargagli E et al. Predicting survival in newly diagnosed idiopathic pulmonary fibrosis: a 3-year prospective study. Eur Respir J 2012;40:101–9. doi:10.1183/09031936.00106011 [DOI] [PubMed] [Google Scholar]
- 13.Nathan SD, Shlobin OA, Weir N et al. Long-term course and prognosis of idiopathic pulmonary fibrosis in the new millennium. Chest 2011;140:221–9. doi:10.1378/chest.10-2572 [DOI] [PubMed] [Google Scholar]
- 14.Collard HR, Moore BB, Flaherty KR et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2007;176:636–43. doi:10.1164/rccm.200703-463PP [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collard HR, Yow E, Richeldi L et al. Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir Res 2013;14:73 doi:10.1186/1465-9921-14-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song JW, Hong SB, Lim CM et al. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011;37:356–63. doi:10.1183/09031936.00159709 [DOI] [PubMed] [Google Scholar]
- 17.Dimmock AEF, Chinchilli VM, Criner GJ et al. The GAP Index predicts mortality in a statewide cohort of patients with idiopathic pulmonary fibrosis [abstract]. Am J Respir Crit Care Med 2013;187:A6086. [Google Scholar]
- 18.du Bois RM, Weycker D, Albera C et al. Ascertainment of individual risk of mortality for patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;184:459–66. doi:10.1164/rccm.201011-1790OC [DOI] [PubMed] [Google Scholar]
- 19.Ley B, Ryerson CJ, Vittinghoff E et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012;156:684–91. doi:10.7326/0003-4819-156-10-201205150-00004 [DOI] [PubMed] [Google Scholar]
- 20.Fingerlin TE, Murphy E, Zhang W et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet 2013;45:613–20. doi:10.1038/ng.2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noth I, Zhang Y, Ma SF et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med 2013;1:309–17. doi:10.1016/S2213-2600(13)70045-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seibold MA, Wise AL, Speer MC et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med 2011;364:1503–12. doi:10.1056/NEJMoa1013660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peljto AL, Zhang Y, Fingerlin TE et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA 2013;309:2232–9. doi:10.1001/jama.2013.5827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ley B, Brown KK, Collard HR. Molecular biomarkers in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 2014;307:L681–91. doi:10.1152/ajplung.00014.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vij R, Noth I. Peripheral blood biomarkers in idiopathic pulmonary fibrosis. Transl Res 2012;159:218–27. doi:10.1016/j.trsl.2012.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belkin A, Swigris JJ. Patient expectations and experiences in idiopathic pulmonary fibrosis: implications of patient surveys for improved care. Expert Rev Respir Med 2014;8:173–8. doi:10.1586/17476348.2014.880056 [DOI] [PubMed] [Google Scholar]
- 27.Swigris JJ, Stewart AL, Gould MK et al. Patients’ perspectives on how idiopathic pulmonary fibrosis affects the quality of their lives. Health Qual Life Outcomes 2005;3:61 doi:10.1186/1477-7525-3-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swigris JJ, Brown KK, Behr J et al. The SF-36 and SGRQ: validity and first look at minimum important differences in IPF. Respir Med 2010;104:296–304. doi:10.1016/j.rmed.2009.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swigris JJ, Fairclough D. Patient-reported outcomes in idiopathic pulmonary fibrosis research. Chest 2012;142:291–7. doi:10.1378/chest.11-2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swigris JJ, Esser D, Conoscenti CS et al. The psychometric properties of the St. George's Respiratory Questionnaire (SGRQ) in patients with idiopathic pulmonary fibrosis: a literature review. Health Qual Life Outcomes 2014;12:124 doi:10.1186/s12955-014-0124-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tomioka H, Imanaka K, Hashimoto K et al. Health-related quality of life in patients with idiopathic pulmonary fibrosis—cross-sectional and longitudinal study. Intern Med 2007;46:1533–42. doi:10.2169/internalmedicine.46.6218 [DOI] [PubMed] [Google Scholar]
- 32.Ware J Jr, Kosinski M, Keller SD. A 12-Item Short-Form Health Survey: construction of scales and preliminary tests of reliability and validity. Med Care 1996;34:220–33. doi:10.1097/00005650-199603000-00003 [DOI] [PubMed] [Google Scholar]
- 33.Rabin R, de Charro F. EQ-5D: a measure of health status from the EuroQol Group. Ann Med 2001;33:337–43. doi:10.3109/07853890109002087 [DOI] [PubMed] [Google Scholar]
- 34.Barr JT, Schumacher GE, Freeman S et al. American translation, modification, and validation of the St. George's Respiratory Questionnaire. Clin Ther 2000;22:1121–45. doi:10.1016/S0149-2918(00)80089-2 [DOI] [PubMed] [Google Scholar]
- 35.Jones PW. St. George's Respiratory Questionnaire: MCID. COPD 2005;2:75–9. doi:10.1081/COPD-200050513 [DOI] [PubMed] [Google Scholar]
- 36.Crawford B, Monz B, Hohlfeld J et al. Development and validation of a cough and sputum assessment questionnaire. Respir Med 2008;102:1545–55. doi:10.1016/j.rmed.2008.06.009 [DOI] [PubMed] [Google Scholar]
- 37.Gries KS, Esser D, Wiklund I. Content validity of CASA-Q cough domains and UCSD-SOBQ for use in patients with idiopathic pulmonary fibrosis. Glob J Health Sci 2013;5:131–41. doi:10.5539/gjhs.v5n6p131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Austin PC. An introduction to propensity score methods for reducing the effects of confounding in observational studies. Multivariate Behav Res 2011;46:399–424. doi:10.1080/00273171.2011.568786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kleinbaum D, Kupper LL, Nizam A et al. Applied regression analysis and other multivariable methods. 5th edn Boston, MA: Cengage Learning, 2013. [Google Scholar]
- 40.King TE Jr, Brown KK, Raghu G et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011;184:92–9. doi:10.1164/rccm.201011-1874OC [DOI] [PubMed] [Google Scholar]
- 41.Martinez FJ, de Andrade JA, Anstrom KJ et al. Randomized trial of acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2014;370:2093–101. doi:10.1056/NEJMoa1401739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raghu G, Million-Rousseau R, Morganti A et al. Macitentan for the treatment of idiopathic pulmonary fibrosis: the randomised controlled MUSIC trial. Eur Respir J 2013;42:1622–32. doi:10.1183/09031936.00104612 [DOI] [PubMed] [Google Scholar]
- 43.Richeldi L, Costabel U, Selman M et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med 2011;365:1079–87. doi:10.1056/NEJMoa1103690 [DOI] [PubMed] [Google Scholar]
- 44.Daniels CE, Yi ES, Ryu JH. Autopsy findings in 42 consecutive patients with idiopathic pulmonary fibrosis. Eur Respir J 2008;32:170–4. doi:10.1183/09031936.00176307 [DOI] [PubMed] [Google Scholar]
- 45.Institute of Medicine. Crossing the quality chasm: a new health system for the 21st century. Washington DC: National Academy Press, 2001. http://www.iom.edu/~/media/Files/Report%20Files/2001/Crossing-the-Quality-Chasm/Quality%20Chasm%202001%20%20report%20brief.pdf (accessed 11 Apr 2014). [PubMed] [Google Scholar]
- 46.White ES, Brown KK, Collard HR et al. Open-access biorepository for idiopathic pulmonary fibrosis. The way forward. Ann Am Thorac Soc 2014;11:1171–5. doi:10.1513/AnnalsATS.201406-289OI [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindell KO, Black TR, Hoffmann K et al. The Pennsylvania Idiopathic Pulmonary Fibrosis (PA-IPF) state registry [abstract]. Am J Respir Crit Care Med 2010;A2964 doi:10.1164/ajrccm-conference.2010.181.1_MeetingAbstracts.A2964 [Google Scholar]
- 48.Moodley Y, Goh N, Glaspole I et al. Australian Idiopathic Pulmonary Fibrosis Registry: vital lessons from a national prospective collaborative project. Respirology 2014;19:1088–91. doi:10.1111/resp.12358 [DOI] [PubMed] [Google Scholar]
- 49.Behr J, Hoeper MM, Kreuter M et al. Investigating significant health trends in idiopathic pulmonary fibrosis (INSIGHTS-IPF): rationale, aims and design of a nationwide prospective registry. BMJ Open Resp Res 2014;1:e000010 doi:10.1136/bmjresp-2013-000010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wuyts W, Dahlqvist C, Schlesser M et al. PROOF-registry: a prospective observational registry to describe the disease course and outcomes of idiopathic pulmonary fibrosis patients in a real-world clinical setting [abstract]. Eur Respir J 2014;44(Suppl 58):P3785. [Google Scholar]
- 51.Ryerson CJ, Tan B, Fell CD et al. The CAnadian REgistry for Pulmonary Fibroiss (CARE-PF): design and rationale of a national pulmonary fibrosis registry. Can Respir J. Published Online First: 3 Nov 2015. doi: 17206 [pii] [PubMed] [Google Scholar]
- 52.Guenther A, European IPF Network. The European IPF Network: towards better care for a dreadful disease. Eur Respir J 2011;37:747–8. doi:10.1183/09031936.00012111 [DOI] [PubMed] [Google Scholar]
- 53.European IPF Registry (eurIPFreg). http://www.pulmonary-fibrosis.net/ (accessed 11 Apr 2014).