

Abstract
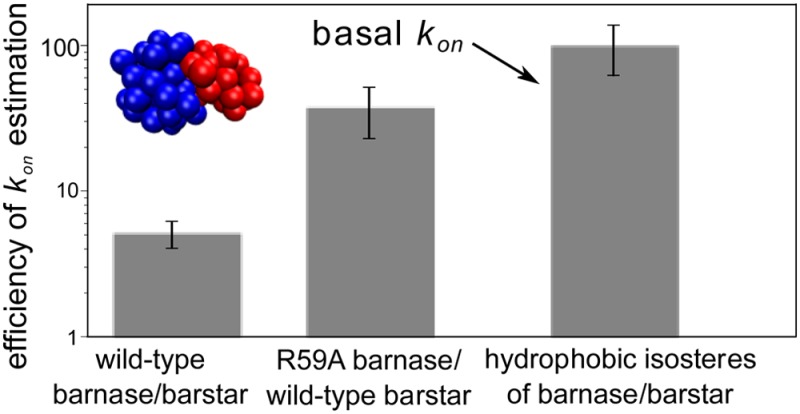
An essential baseline for determining the extent to which electrostatic interactions enhance the kinetics of protein–protein association is the “basal” kon, which is the rate constant for association in the absence of electrostatic interactions. However, since such association events are beyond the milliseconds time scale, it has not been practical to compute the basal kon by directly simulating the association with flexible models. Here, we computed the basal kon for barnase and barstar, two of the most rapidly associating proteins, using highly efficient, flexible molecular simulations. These simulations involved (a) pseudoatomic protein models that reproduce the molecular shapes, electrostatic, and diffusion properties of all-atom models, and (b) application of the weighted ensemble path sampling strategy, which enhanced the efficiency of generating association events by >130-fold. We also examined the extent to which the computed basal kon is affected by inclusion of intermolecular hydrodynamic interactions in the simulations.
Introduction
Of fundamental interest to biology is the extent to which electrostatic interactions enhance the rate of protein–protein association. An essential baseline for determining the magnitude of these rate enhancements is the “basal” kon, which is the rate constant for association in the absence of electrostatic interactions.1 In principle, the basal kon should be measured in the same solvent environment using the hydrophobic isosteres—that is, hypothetical mutants with molecular shapes that are identical to those of the wild-type proteins, but are entirely uncharged. However, due to the difficulty of engineering hydrophobic isosteres, experimental studies have instead estimated the basal kon by measuring the kon for the wild-type proteins at various salt concentrations and extrapolating to the limit of infinite salt concentration where electrostatic interactions would be completely screened.1
An alternative approach is to construct the exact hydrophobic isosteres in silico by setting all partial charges of the wild-type proteins to zero and directly computing the basal kon by simulating the association of the hydrophobic isosteres. Ideally, such simulations would involve the use of flexible molecular models in order to capture conformational changes during the association process. However, since the weak associations of completely hydrophobic proteins are beyond the milliseconds time scale,1−7 it has only been feasible to directly compute the basal kon using rigid, models with atomically detailed simulations.2 Theoretical estimates of the basal kon have also been made using spherical models with orientational constraints3−6 and applications of transition-rate theory to rigid, atomistic models.7
Here, for the first time, we directly computed the basal kon for a protein–protein association process using flexible models with molecular simulations. We focused on barnase and barstar, which are among the most rapidly associating proteins by virtue of their complementary electrostatic surfaces.2 Our simulations employed flexible, pseudoatomic protein models of barnase and barstar that were designed by Frembgen-Kesner and Elcock8 to retain the molecular shapes, electrostatic potentials, and diffusion properties of the corresponding atomistic models at the experimental ionic strength (50 mM).9 The same authors have demonstrated that the use of these models with standard “brute force” simulations can reproduce the experimental kon values of both the wild-type and mutant protein pairs. However, they were unable to carry out such simulations to obtain a statistically robust estimate of the kon for the hydrophobic isosteres (i.e., the basal kon) due to the large computational cost.8
A critical feature of our study is the application of the weighted ensemble (WE) strategy11 to enhance the sampling of rare events, e.g. the slow association of completely hydrophobic, uncharged proteins. Although the WE strategy has been previously applied to protein binding processes using Brownian dynamics (BD) simulations,11,12 these studies were carried out without the inclusion of HIs) between, and within, the diffusing proteins. In the absence of explicit solvent, it has been demonstrated that the translational and rotational diffusion coefficients of flexible protein models are drastically underestimated unless intramolecular HIs are included in the simulations.13 In addition, the neglect of intermolecular HIs in previous BD studies of protein binding processes2,5,14 is likely to have contributed to their consistent overestimation of association rate constants.8 Importantly, our simulations were validated by computing the kon values for both wild-type barnase and its R59A mutant, which associates more slowly than wild-type barnase with barstar,9 and comparing the computed values to experiment.
Methods
The protein model and energy function
The wild-type and mutant pairs of barnase and barstar were represented using flexible, pseudoatomic models developed by Frembgen-Kesner and Elcock.8 Full details of these models are provided in ref (5). Briefly, the generation of these models began with all-atom models of the wild-type proteins, which were based on the crystal structure of the barnase–barstar complex (PDB code: 1BRS);15 the same models were used for both the unbound and bound states. Approximately one pseudoatom was then used to represent every three amino acid residues (33 pseudoatoms for the 110 residues of barnase and 27 pseudoatoms for the 89 residues of barstar). For the wild-type proteins and R59A mutant barnase, the effective charge method16 was used to derive effective charges for the pseudoatomic models such that the electrostatic potentials of the corresponding all-atom models were reproduced. Electrostatic potentials were obtained by numerically solving the nonlinear Poisson–Boltzmann equation under experimental conditions (pH 8, 25 °C, and ionic strength of 50 mM).9 Pseudoatoms were then positioned and sized to replicate the electron density envelope of the all-atom model. To generate models of the exact hydrophobic isosteres of barnase and barstar, we started with the pseudoatomic models of the wild-type proteins and set all effective charges to zero.
The energy function consisted of a single intramolecular term involving flexible, harmonic bonds between the pseudoatoms and intermolecular terms for electrostatic and nonelectrostatic interactions. To maintain the molecular shapes of the proteins, three bonds per pseudoatom were formed on average. All intermolecular electrostatic interactions between pseudoatoms were calculated using the Debye–Hückel equation; intramolecular electrostatic interactions were omitted. Nonelectrostatic interactions were calculated using a very weak Go̅-type potential energy function with a shallow well depth (ε = 0.1 kcal/mol). Thus, native contacts were only slightly rewarded by a weakly attractive Lennard-Jones-like potential and nonnative contacts were penalized by a purely repulsive potential.17,18 The well-depth was kept at a minimal value in order to avoid implicitly double counting the attractive electrostatic interactions, which are assumed to be a primary driving force for the formation of the barnase-barstar complex.9 Two pseudoatoms were considered to form a native contact if any non-hydrogen atoms of the residues in the all-atom model were within 5.5 Å of each other in the crystal structure of the native complex, yielding a total of 34 intermolecular native contacts.
Weighted Ensemble (WE) Simulations
All simulations were carried out using the WE path sampling strategy,11 as implemented in the WESTPA software package (https://westpa.github.io/westpa).10 In this strategy, a large number of simulations, or trajectory “walkers”, are started in parallel from the initial state and iteratively evaluated at fixed time intervals τ for resampling in which walkers are either replicated or combined to maintain a similar number of walkers per bin along a progress coordinate toward the target state. Rigorous management of the statistical weights associated with each walker ensures that no bias is introduced into the dynamics.
In this study, the WE strategy was applied using steady-state simulations within the framework of the Northrup–Allison–McCammon (NAM) method.19 This framework involves the definition of two concentric spherical surfaces with radii b and q that correspond to center-to-center separation distances for barnase and barstar. The inner sphere, or b surface, represents the initial, unbound state, and the outer sphere, or q surface, is an absorbing surface that is positioned at a much larger separation distance (q ≫ b) to avoid wasting computational effort sampling the indefinite drifting apart of the proteins. Each WE simulation was started from 24 configurations of the unbound state in which barnase and barstar were randomly oriented at a center-to-center separation distance of b. A walker was continued until the pair of proteins either exceeded a separation distance q or satisfied the criterion for the target state for successful association, i.e. reaching a threshold value, Qrxn, in the fraction of native intermolecular contacts, Q, that reproduces the experimental kon for the wild-type proteins. Consistent with previous brute force simulations,8b and q were set to 100 and 500 Å, respectively. Upon reaching the q surface, a walker was “recycled” by starting a new walker from the unbound state with the same statistical weight thereby maintaining a steady state and enforcing a constant effective protein concentration (3.2 μM). Upon reaching a particular Qrxn value, a walker was effectively recycled after completing the WE simulation by removing the walker and its replicas prior to calculating the kon.
For each barnase-barstar pair, five independent WE simulations were performed with different initial random seeds for BD propagation. In each simulation, the configurational space of the protein pairs was divided into 760 bins along a progress coordinate that was intended to capture the slowest protein motions of the association process. We used a progress coordinate that consisted of three zones: (a) a “far” zone involving the distance between barnase and barstar, (b) an “intermediate” zone involving the RMS deviation of barstar from its bound-state position following alignment of barnase, and (c) a “near” zone involving the same RMS deviation metric as in part b and the fraction of native contacts between barnase and barstar. Simulations were evaluated for resampling at fixed time intervals τ (or iterations) of 2 ns to maintain 24 walkers per bin. Each simulation was carried out for 1000 iterations, or a molecular time of 2 μs (defined as Nτ where N is the number of iterations).
Propagation of Dynamics
The dynamics of our WE simulations were propagated using the UIOWA-BD software,13,20 which is the same BD engine that was used for the brute force simulations by Frembgen-Kesner and Elcock.8 Consistent with these simulations, our WE simulations were performed at a constant temperature of 25 °C using a standard BD algorithm with the inclusion of hydrodynamic interactions (HIs) via calculation of the diffusion tensor using the equations of Rotne and Prager21 and Yamakawa;22 the same values were used for the hydrodynamic radii of the pseudoatoms to reproduce the translational diffusion coefficients of the corresponding all-atom protein models by the hydrodynamics program HYDROPRO;23 and a time step of 0.25 ps was used throughout the simulations.
Calculation of kon values
For each barnase-barstar pair, the kon value was computed from each of five independent WE simulations using conformations that were sampled every 20 ps once a steady state was achieved (Figure S1, Supporting Information). These values were then averaged. All WE simulations were sufficiently long to yield relative percent uncertainties in the average kon of <20% (Figure S2, Supporting Information). Uncertainties in the average kon values were represented by calculating 95% confidence intervals. The kon from each WE simulation was calculated using the NAM method according to the following equation:19
where k(b) and k(q) are the diffusion rate constants for achieving separation distances of b and q, respectively, and β is the probability of successful collisions, i.e. that a simulation starting from the unbound state with a separation distance of b (100 Å) reaches the bound state before drifting apart to a separation distance of q (500 Å). Assuming that the motions of the binding partners are isotropic, k(b) and k(q) are given by the Smoluchowski result; k(r) = 4Dr, where D is the relative translational diffusion coefficient of the two proteins (i.e., the sum of their corresponding diffusion coefficients). As done for the brute force simulations by Frembgen-Kesner and Elcock,8 we used the estimate from HYDROPRO23 for D (2.672 × 102 Å2ps–1). The β value was calculated using the following equation:
where fSSbound is the steady-state flux into the bound state and fSS is the steady-state flux into the q surface. As evident in the above equations, the influence of HIs is considered in our calculation of the probability of successful collisions (β), but only approximately on the diffusion of the two proteins by using the sum of their diffusion coefficients (D).24
Calculation of WE Efficiency
For each barnase–barstar pair, we determined the efficiency of a single WE simulation relative to brute force simulation in computing the kon for each of five independent WE simulations; these efficiencies were then averaged and uncertainties in the efficiencies were determined by calculating the 95% confidence intervals. The efficiency of each WE simulation was calculated using the following equation:
where tBF and tWE are the wall-clock times required by brute force simulation and the WE simulation, respectively, to generate the same number of independent (uncorrelated) association events using the same computing resource (i.e., 256 CPU cores of 2.3 GHz AMD Interlagos processors). Association events were considered independent if, within the period between the event and one correlation time before the event, their corresponding trajectories did not share a common simulation segment. The correlation time was determined by monitoring autocorrelation of the flux into the bound state as a function of the lag time and identifying the first lag time that results in zero autocorrelation (within a 95% confidence interval; see Figure S2, Supporting Information). Since it was not practical to directly obtain tBF for all of the barnase-barstar pairs (i.e., the hydrophobic isosteres), we estimated tBF in a consistent manner for each pair using the following equation:
where MBF is the number of trajectories in a brute force simulation to generate the same number of independent association events observed in a WE simulation–given that the brute force trajectories are terminated when the proteins either associate or reach a separation distance of q according to the NAM method; 0.02 days/trajectory/core is the average wall-clock time that would be required to complete a single brute force trajectory before the proteins reach a separation distance of q; and β (as defined above) is the probability calculated by WE for a single brute force trajectory to generate a successful association event before dissociating to a separation distance of q.
Results and Discussion
Our general strategy for computing kon values from our simulations was to first identify a criterion for successful association that reproduces the experimental kon for wild-type barnase and barstar. Next, we validated the simulations by using this criterion to calculate the kon for R59A mutant barnase and wild-type barstar, which associates 9-fold more slowly than the wild-type proteins,9 and comparing the calculated kon to the experimental value. Finally, we used this criterion to estimate the basal kon, i.e., the kon for the hydrophobic isosteres in which all effective charges of the wild-type proteins are set to zero. Following the brute force simulations by Frembgen-Kesner and Elcock,8 our criterion for successful association was to reach a threshold value, Qrxn, in the fraction of native intermolecular contacts, Q; dynamics were propagated using the same BD engine with the inclusion of intramolecular HIs to achieve realistic diffusive properties of the individual proteins; and kon values were calculated according to the NAM method (see Methods).19
Validation of the Simulation Strategy
Figure 1 shows the computed kon as a function of Qrxn for all five independent WE simulations of each barnase–barstar pair. The experimental kon for wild-type barnase and barstar (2.86 × 108 M–1 s–1)9 was reproduced when using Qrxn values of 0.27 and 0.56 for simulations with and without intermolecular HIs, respectively. These values differ slightly from those determined by Frembgen-Kesner and Elcock using brute force simulations and the same protein models (0.32 and 0.47, respectively)8 due to more frequent monitoring of the reaction criterion (every 20 ps instead of 100 ps); thus, our WE simulations are less likely to have missed conformations that satisfy the reaction criterion. Importantly, using the Qrxn values that we have identified, the computed kon values for R59A barnase and wild-type barstar are in excellent agreement with experiment, regardless of whether or not intermolecular HIs were included (Figure 2; see also Table S1, Supporting Information). The reproduction of experimental kon values for both wild-type and mutant pairs of barnase and barstar is consistent with results from brute force simulations,8 providing validation of our WE simulation protocol. Relative to the basal kon, our computed kon values for wild-type barnase and barstar are 53- and 103-fold faster with and without intermolecular HIs, respectively. These rate enhancements are solely due to the electrostatic interactions between the wild-type proteins given the omission of intramolecular electrostatic interactions in our simulations.
Figure 1.
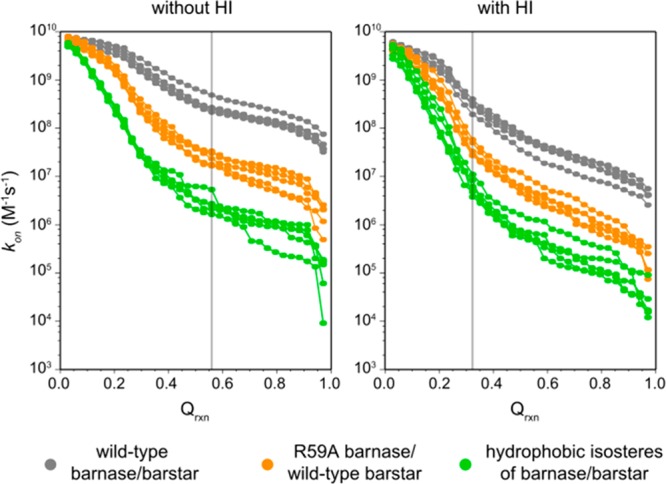
Computed kon values for each barnase-barstar pair from each of five independent WE simulations as a function of the fraction of intermolecular native contacts Qrxn. Results from simulations without and with the inclusion of intermolecular HI are shown in the left and right panels, respectively. The vertical gray line in each panel indicates the Qrxn value that reproduces the experimental kon for the wild-type pair for simulations without and with HI (0.56 and 0.27, respectively) and was used for calculating kon values for the mutant pairs in that panel.
Figure 2.
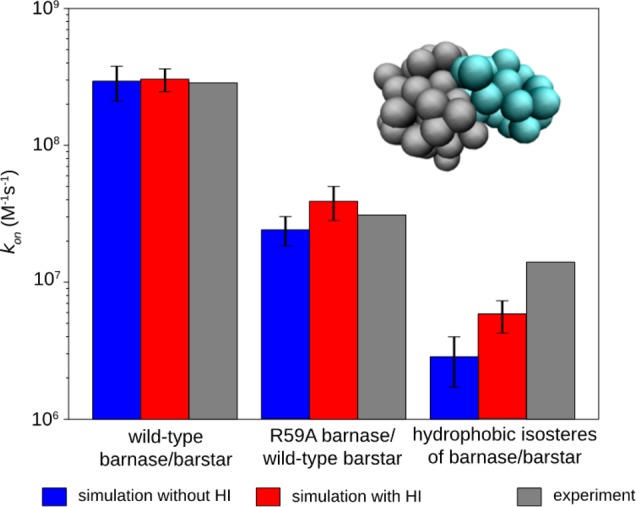
Comparison of computed and experimental9kon values on a log scale. Reported values from simulation (with and without intermolecular HI) are averages from five independent WE simulations with the error bars representing 95% confidence intervals. The pseudoatomic protein model of barnase (gray) and barstar (cyan) is shown in the upper right corner.
Estimation of the Basal kon
The basal kon computed from our simulations with and without intermolecular HIs are (2.85 ± 1.30) × 106 M–1 s–1 and (5.79 ± 0.17) × 106 M–1 s–1, respectively. At the effective protein concentration maintained in our simulations (3.2 μM), these rate constants correspond to time scales beyond tens of milliseconds. Our computed basal kon values are similar to those using less computationally intensive strategies; in particular, the use of spherical models with orientational constraints3−6 has provided estimates in the range of 105–106 M–1 s–1 and the use of rigid, atomistic models in either the application of transition-rate theory7 or direct BD simulation of protein–protein association2 has yielded estimates of ∼1 × 106 M–1 s–1. The similarity of our estimates to these previous estimates suggests that flexible models may not be essential for obtaining realistic estimates of kon values for proteins such as barnase and barstar that do not undergo significant conformational changes upon binding (the Cα RMS deviation between the crystal structures of the unbound25,26 and bound15 conformations is only 0.5 Å for both barnase and barstar). However, it has not been possible to directly estimate the basal kon with uncertainties of <100% using standard BD simulations with rigid, atomistic models since the association events were much slower in the absence of electrostatic forces.2 On the other hand, our WE simulations with flexible molecular models enable significantly more precise calculations of the kon (uncertainties of 22–46%) and could therefore be used for even more complicated binding processes, including ones that involve large conformational changes. Notably, our computed kon values are significantly lower than that obtained by experiment from extrapolation to infinite salt concentration (1.4 × 107 M–1 s–1),1 suggesting that the favorable electrostatic interactions between the proteins are not completely eliminated at high salt concentrations.
Effect of Intermolecular HIs on the Kinetics of Association
Although the inclusion of intermolecular HIs has no effect on the ability of the simulation model to reproduce the effects of mutation on the kon, for a fixed value of Qrxn, the inclusion of intermolecular HIs significantly slows down the rate of association for all three pairs of the barnase-barstar system (Figures 2 and 3). Surprisingly, the extent to which kon decreases is essentially the same for wild-type and R59A mutant pairs (e.g., by ∼5-fold at Qrxn = 0.27). In contrast, the impact of intermolecular HIs in the brute force simulations by Frembgen-Kesner and Elcock was more pronounced for slower associating mutants of barnase such as R59A in which the electrostatic interactions with barstar are diminished.8 On the basis of these results, it was predicted that the impact would be the most pronounced for the hydrophobic isosteres of barnase and barstar. However, the enhanced sampling provided by the WE strategy reveals no statistical difference between the impact of the intermolecular HIs on the kon for the wild-type and R59A mutant pairs. For the hydrophobic isosteres, our results are inconclusive. Although it was possible to obtain statistically robust estimates of the basal kon—which was the primary goal of this work–our simulations did not reach the level of precision in the ratio of the kon values with and without intermolecular HIs that would be required to determine the effect of HIs on the association kinetics relative to the wild-type pair (note the large confidence intervals in Figure 3). For future studies of this effect, significantly greater sampling using a larger number of simulations and/or longer simulations would be required to achieve a sufficient level of precision in the computed kon values, particularly in the absence of intermolecular HIs.
Figure 3.
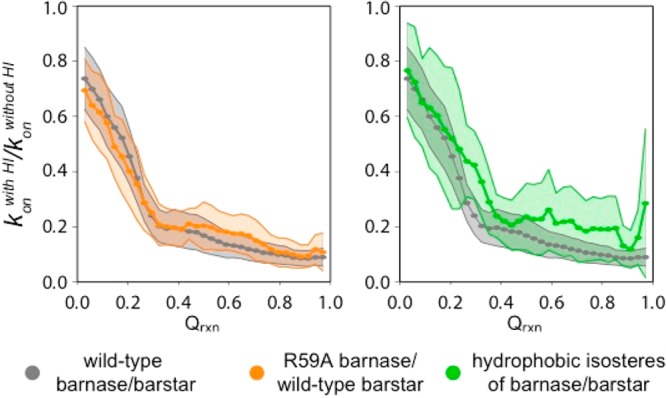
Ratio of association rate constants konwith HI/kon computed from simulations with and without intermolecular HIs as a function of the fraction of intermolecular contacts Qrxn. The shaded regions represent 95% confidence intervals for averages (filled circles) from five independent WE simulations.
Efficiency of WE Simulation
Finally, it would not have been practical to obtain converged estimates of the basal kon without the use of the WE strategy. In addition, a highly scalable, parallel implementation of this strategy was essential since it would have otherwise required >2 years to carry out the simulations using a serial implementation. To determine the efficiency of parallelized WE vs brute force simulation in estimating the kon, we compared the wall-clock time that would be required of WE vs brute force simulation (both using the NAM framework) to generate the same number of independent (uncorrelated) association events using the same computing resource (256 CPU cores of 2.3 GHz AMD Interlagos processors). Figure 4 shows the efficiencies of WE simulations relative to brute force simulations for each barnase-barstar pair (see also Table S2, Supporting Information). For the wild-type pair, a WE simulation was 6-fold more efficient than brute force simulation with the inclusion of intermolecular HI. This efficiency increased to 46-fold for the R59A mutant pair and ultimately 131-fold for the hydrophobic isosteres. In the latter case, brute force simulation using the same flexible protein models would be highly impractical, requiring 386 days in wall-clock time to generate the same number of association events (>1000) as a single WE simulation, which required only 3 days. The greater efficiency of WE observed for the slower processes (i.e., increasing with the barrier height) is consistent with previous WE studies of other rare events.12,27−29
Figure 4.
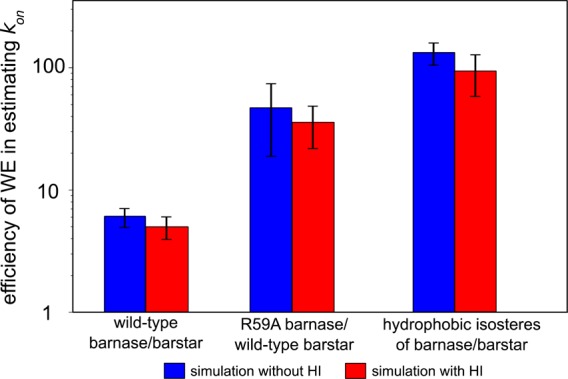
Average efficiencies of WE relative to brute force simulation in computing the kon. Full details for estimating efficiencies are described in Methods. Uncertainties represent 95% confidence intervals for averages from five independent WE simulations.
Conclusions
In conclusion, we have directly computed the basal kon for a protein–protein association process for the first time using flexible models with molecular simulations. In particular, we computed the basal kon for the barnase-barstar system using highly efficient WE simulations. Our computed basal kon is significantly lower than that obtained by experiment from extrapolation to infinite salt concentration, suggesting that the electrostatic interactions are not completely eliminated at high salt concentrations. This result underscores the importance of directly computing the basal kon using the true hydrophobic isosteres of the proteins under regular salt concentrations—a goal that can only be achieved by molecular simulation. Relative to our basal kon, the electrostatic interactions of the wild-type proteins enhance the rate of association by >130-fold. As demonstrated by Frembgen-Kesner and Elcock using brute force simulations,8 the inclusion of intermolecular HIs significantly decreases the computed kon values for both wild-type and mutant pairs. However, the extensive sampling provided by our WE simulations has revealed that the extent by which the kon is reduced is the same for both the wild-type and R59A mutant pairs. For the hydrophobic isosteres, the relative extent to which the kon was affected by the intermolecular HIs was inconclusive due to insufficient precision in the ratio of the kon with and without intermolecular HI. Finally, our results demonstrate that WE simulations are orders of magnitude more efficient than brute force simulation in providing converged estimates of rate constants for the slow associations of proteins in the complete absence of electrostatic interactions. The computation of such rate constants is otherwise impractical when using flexible protein models—even when these models are coarse-grained. Given its high efficiency, the simulation strategy used in this study would be useful for even more complicated systems, including those that undergo large conformational changes upon binding.
Acknowledgments
We thank Adrian Elcock for valuable discussions and making the UIOWA-BD software available. We also thank Alex DeGrave and Adam Pratt for critical reading of the manuscript. Financial support was provided by NSF CAREER MCB-0846216, NIH 1R01GM115805-01, and a DAAD graduate research grant. Computational resources were provided by NSF CNS-1229064 and the University of Pittsburgh’s Center for Simulation and Modeling.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.5b10747.
Figures S1 (average calculated kon values as a function of time) and S2 (representative autocorrelation of the flux into the bound state) and Tables S1 (average calculated kon values) and S2 (average efficiencies of weighted ensemble vs brute force) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Schreiber G.; Haran G.; Zhou H. X. Fundamental aspects of protein-protein association kinetcs. Chem. Rev. 2009, 109, 839–860. 10.1021/cr800373w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabdoulline R. R.; Wade R. C. Simulation of diffusional association of barnase and barstar. Biophys. J. 1997, 72, 1917–1929. 10.1016/S0006-3495(97)78838-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northrup S. H.; Erickson H. P. Kinetics of protein-protein association explained by Brownian dynamics computer simulation. Proc. Natl. Acad. Sci. U. S. A. 1992, 89, 3338–3342. 10.1073/pnas.89.8.3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. X. Enhancement of protein-protein association rate by interaction potential: accuracy of prediction based on local Boltzmann factor. Biophys. J. 1997, 73, 2441–2445. 10.1016/S0006-3495(97)78272-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C. J.; Kimura S. R.; Delisi C.; Vajda S. Kinetics of desolvation-mediated protein-protein binding. Biophys. J. 2000, 78, 1094–1105. 10.1016/S0006-3495(00)76668-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosshauer M.; Baker D. Realistic protein-protein association rates from a simple diffusional model neglecting long-range interactions, free energy barriers, and landscape ruggedness. Protein Sci. 2004, 13, 1660–1669. 10.1110/ps.03517304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsallaq R.; Zhou H. X. Prediction of protein-protein association rates from a transition-state theory. Structure 2007, 15, 215–224. 10.1016/j.str.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Frembgen-Kesner T.; Elcock A. H. Absolute protein-protein association rate constants from flexible coarse-grained Brownian dynamics simulations: The role of intermolecular hydrodynamic interactions in barnase-barstar association. Biophys. J. 2010, 99, L75–L77. 10.1016/j.bpj.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber G.; Fersht A. R. Rapid, electrostatically assisted association of proteins. Nat. Struct. Biol. 1996, 3, 427–431. 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- Zwier M. C.; Adelman J. L.; Kaus J. W.; Pratt A. J.; Wong K. F.; Rego N. B.; Suarez E.; Lettieri S.; Wang D. W.; Grabe M.; et al. WESTPA: An interoperable, highly scalable software package for weighted ensemble simulation and analysis. J. Chem. Theory Comput. 2015, 11, 800–809. 10.1021/ct5010615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber G. A.; Kim S. Weighted-Ensemble Brownian dynamics simulations of protein association reactions. Biophys. J. 1996, 70, 97–110. 10.1016/S0006-3495(96)79552-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojnuckarin A.; Livesay D. R.; Subramaniam S. Bimolecular reaction simulation using Weighted Ensemble Brownian dynamics and the University of Houston Brownian Dynamics program. Biophys. J. 2000, 79, 686–693. 10.1016/S0006-3495(00)76327-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frembgen-Kesner T.; Elcock A. H. Striking effects of hydrodynamic interactions on the simulated diffusion and folding of proteins. J. Chem. Theory Comput. 2009, 5, 242–256. 10.1021/ct800499p. [DOI] [PubMed] [Google Scholar]
- Gabdoulline R. R.; Wade R. C. Protein-protein association: investigation of factors influencing association rates by brownian dynamics simulations. J. Mol. Biol. 2001, 306, 1139–1155. 10.1006/jmbi.2000.4404. [DOI] [PubMed] [Google Scholar]
- Buckle A. M.; Schreiber G.; Fersht A. R. Protein-protein recognition: crystal structural analysis of a barnase-barstar complex at 2.0-A resolution. Biochemistry 1994, 33, 8878–8889. 10.1021/bi00196a004. [DOI] [PubMed] [Google Scholar]
- Gabdoulline R. R.; Wade R. C. Effective charges for macromolecules in solvent. J. Phys. Chem. 1996, 100, 3868–3878. 10.1021/jp953109f. [DOI] [Google Scholar]
- Go N. Theoretical studies of protein folding. Annu. Rev. Biophys. Bioeng. 1983, 12, 183–210. 10.1146/annurev.bb.12.060183.001151. [DOI] [PubMed] [Google Scholar]
- Takada S. Go̅-ing for the prediction of protein folding mechanisms. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 11698–11700. 10.1073/pnas.96.21.11698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northrup S. H.; Allison S. A.; McCammon J. A. Brownian dynamics simulation of diffusion-influenced bimolecular reactions. J. Chem. Phys. 1984, 80, 1517–1524. 10.1063/1.446900. [DOI] [Google Scholar]
- Elcock A. H. Molecular simulations of cotranslational protein folding: Fragment stabilities, folding cooperativity, and trapping in the ribosome. PLoS Comput. Biol. 2006, 2, e98. 10.1371/journal.pcbi.0020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotne J.; Prager S. Variational treatment of hydrodynamic interaction in polymers. J. Chem. Phys. 1969, 50, 4831–4837. 10.1063/1.1670977. [DOI] [Google Scholar]
- Yamakawa H. Transport properties of polymer chains in dilute solution - hydrodynamic interaction. J. Chem. Phys. 1970, 53, 436–443. 10.1063/1.1673799. [DOI] [Google Scholar]
- Garcia de la Torre J.; Huertas M. L.; Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 2000, 78, 719–730. 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlugosz M.; Antosiewicz J. M.; Zielinski P.; Trylska J. Contributions of far-field hydrodynamic interactions to the kinetics of electrostatically driven molecular association. J. Phys. Chem. B 2012, 116, 5437–5447. 10.1021/jp301265y. [DOI] [PubMed] [Google Scholar]
- Martin C.; Richard V.; Salem M.; Hartley R.; Mauguen Y. Refinement and structural analysis of barnase at 1.5 Å resolution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999, 55, 386–398. 10.1107/S0907444998010865. [DOI] [PubMed] [Google Scholar]
- Ratnaparkhi G. S.; Ramachandran S.; Udgaonkar J. B.; Varadarajan R. Discrepancies between the NMR and X-ray structures of uncomplexed barstar: analysis suggests that packing densities of protein structures determined by NMR are unreliable. Biochemistry 1998, 37, 6958–6966. 10.1021/bi972857n. [DOI] [PubMed] [Google Scholar]
- Zhang B. W.; Jasnow D.; Zuckerman D. M. Efficient and verified simulation of a path ensemble for conformational change in a united-residue model of calmodulin. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 18043–18048. 10.1073/pnas.0706349104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adelman J. L.; Grabe M. Simulating rare events using a weighted ensemble-based string method. J. Chem. Phys. 2013, 138, 044105. 10.1063/1.4773892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwier M. C.; Kaus J. W.; Chong L. T. Efficient explicit-solvent molecular dynamics simulations of molecular association kinetics: Methane-methane, Na+/Cl-, methane/benzene, and K+/18-crown-6 ether. J. Chem. Theory Comput. 2011, 7, 1189–1197. 10.1021/ct100626x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.