

Abstract
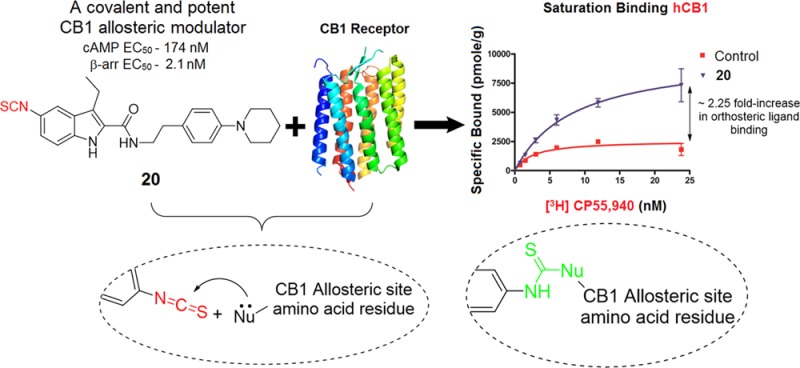
Undesirable side effects associated with orthosteric agonists/antagonists of cannabinoid 1 receptor (CB1R), a tractable target for treating several pathologies affecting humans, have greatly limited their translational potential. Recent discovery of CB1R negative allosteric modulators (NAMs) has renewed interest in CB1R by offering a potentially safer therapeutic avenue. To elucidate the CB1R allosteric binding motif and thereby facilitate rational drug discovery, we report the synthesis and biochemical characterization of first covalent ligands designed to bind irreversibly to the CB1R allosteric site. Either an electrophilic or a photoactivatable group was introduced at key positions of two classical CB1R NAMs: Org27569 (1) and PSNCBAM-1 (2). Among these, 20 (GAT100) emerged as the most potent NAM in functional assays, did not exhibit inverse agonism, and behaved as a robust positive allosteric modulator of binding of orthosteric agonist CP55,940. This novel covalent probe can serve as a useful tool for characterizing CB1R allosteric ligand-binding motifs.
Introduction
Constituents of the endocannabinoid biosignaling system include two principal cannabinoid G-protein-coupled receptors (GPCRs) 1 and 2 (CB1R and CB2R, respectively), their main endogenous cannabinergic ligands (anandamide, AEA; 2-arachidonoylglycerol, 2-AG), and enzymes responsible for endocannabinoid biosynthesis and inactivation.1−4 Expressed in various peripheral tissues, CB1R is the most abundant class-A GPCR in brain.5,6 CB1R-mediated signaling helps regulate many important physiological functions including learning, memory, and cognition, nociception, cardiovascular function, reproduction, and neuronal development. Dysregulated CB1R activity has been implicated in the pathogenesis of disease states related to these and other physiological processes such that small-molecule modulators of CB1R-mediated signaling are considered to have therapeutic potential.1,3 On the other hand, CB2R is mainly expressed in peripheral tissues, particularly immune cells7,8 as well as CNS microglia9 and has been pursued for treating pain and inflammation.10−17 In the past two decades, structurally diverse, potent, and selective CB1R orthosteric agonists have been identified with (pre)clinical efficacy in treating nausea, emesis, and multiple sclerosis and managing glaucoma, pain, and inflammatory disorders.18−20 Their salutary effects notwithstanding, CB1R orthosteric agonists have been associated with adverse events including mood alteration (euphoria, anxiety, panic), acute psychoses, and impaired cognition and motor performance, which limit their clinical utility.21 Several CB1R-selective antagonists/inverse agonists have also emerged as potential drugs for cardiometabolic diseases and nicotine- and alcohol-use disorders. Reminiscent of CB1R orthosteric agonists, however, therapeutic application of CB1R orthosteric antagonists/inverse agonists is severely restricted by the potential for unacceptable psychotropic side effects including depression, social aversion, and suicidal ideation.3,21−25
As has been demonstrated for several other class-A GPCRs, CB1R has allosteric sites spatially distinct from the orthosteric ligand-binding pocket, and allosteric modulators with CB1R selectivity vs CB2R have been identified.26−29 Engagement of CB1R by allosteric modulators is believed to induce a conformational change in the receptor that may be difficult to achieve with orthosteric ligands alone and “fine-tune” the pharmacological activity of the orthosteric ligand.30−32 Due to their generally enhanced CB1R selectivity, reduced inter-receptor promiscuity, and higher-resolution functional control of receptor information transmission, CB1R allosteric modulators are anticipated to offer several therapeutic advantages over orthosteric ligands.
Exemplars of well-studied, structurally distinct CB1R-selective allosteric ligands are shown in Figure 1. These include 5-chloro-3-ethyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (1, Org27569)26 and 1-(4-chlorophenyl)-3-(3-(6-(pyrrolidin-1-yl)pyridine-2-yl)phenyl)urea (2, PSNCBAM-1),27 two CB1R allosteric modulators that emerged from initial structure–activity relationship (SAR) studies on high-throughput screening (HTS) leads. Although 1 and 2 exhibit several characteristic properties of allosteric modulators, they elicit markedly divergent effects on the affinity and efficacy of the standard cannabinoid-receptor orthosteric ligand 2-[(1R,2R,5R)-5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol (CP55,940). Compounds 1 and 2 behave as both a positive allosteric modulator (PAM) of CP55,940 binding affinity and a negative allosteric modulator (NAM) of CP55,940 signaling efficacy and potency. Additionally, endogenous CB1R allosteric modulators have been identified and characterized. The nonclassical eicosanoid (5S,6R,7E,9E,11Z,13E,15S)-5,6,15-trihydroxyicosa-7,9,11,13-tetraenoic acid (3, lipoxin A4), whose traditional biological target is the formyl peptide receptor FPR1, was also shown to function as CB1R PAM of orthosteric ligand binding and adenylyl cyclase activity.28 The endogenous steroid 1-((3S,8S,9S,10R,13S,14S,17R)-3-hydroxy-10,12,13-trimethyl-2,3,4,7,8,9,10,11,12,13,14,15,16,17-tetradecahydro-1H-cyclopenta[a]phenanthren-17-yl)ethan-1-one (4, pregnenolone) acts as a CB1R NAM functionally (CB1R-mediated ERK1/2 phosphorylation) without any effect on orthosteric agonist binding affinity.29 The dopamine transport inhibitor RTI-37133 and the PPAR-α agonist fenofibrate have also been suggested to act at a CB1R allosteric site.34 Very recently, we have shown that cannabidiol, the nonpsychoactive constituent of Cannabis sativa, exhibits negative allosteric modulation at CB1R.35 Additionally, ligands displaying positive allosteric modulation of orthosteric ligand’s binding and function have been reported recently.36,37 These collective findings substantiate the existence and functional significance of allosteric sites on CB1R whose pharmacological modulation of endogenous/orthosteric ligand activity could be exploited for therapeutic ends.
Figure 1.

Representative CB1R allosteric modulators reported in the literature.
Although it has been a decade since the first CB1 NAM (1) was reported, no new CB1 NAM with improved potency/efficacy has been identified that has been studied in vivo. To date, very limited in vivo studies with 1 and 2 have been reported where these NAMs have shown moderate efficacy.27,38−40 Another major limitation associated with these two compounds is that they exhibit CB1R inverse agonism in addition to having NAM activity.26,41,42 For establishing therapeutic utility of CB1 NAMs, there is a need for developing potent and efficacious CB1R NAMs that lack inverse agonism so as to avoid related side effects.
To inform rational drug design aimed at therapeutic CB1R allosteric modulation, it is critical to expand our currently limited knowledge of the structural properties and functional influence of the receptor’s allosteric ligand-binding site(s). Although candidate atomic-level interactions involved in GPCR ligand recognition and functionally productive engagement can be extrapolated from ligand–receptor cocrystals, a CB1R crystal structure has remained elusive, and its inherently static nature precludes direct observation of structure–function correlates of CB1R (allosteric) ligands. Although homology modeling and mutation studies have allowed some characterization of the properties of CB1R’s allosteric ligand-binding domain, these approaches per se cannot afford direct experimental observation of the molecular nature of ligand–CB1R interaction and its consequences for cell signaling, since even conservative, single amino-acid mutations may alter inadvertently receptor conformation and function.41−44 We have incorporated an alternative approach for interrogating directly the structure–function correlates of ligand binding to druggable protein targets (enzymes, GPCRs) in their functional state and under physiological conditions.45−49 Globally, this experimental paradigm, termed ligand-assisted protein structure (LAPS), integrates information from point mutations, molecular modeling, and peptide-level tandem mass spectrometry studies on ligand–receptor complexes to identify amino acid residues within (or in the immediate vicinity of) the ligand-binding domain critical to ligand engagement and activity.49 Pharmacologically active ligands of diverse chemical classes purpose-designed to carry reactive groups as high-affinity, site-directed covalent probes are key elements foundational to the LAPS experimental paradigm. Various reactive groups, both electrophilic (e.g., isothiocyanate, benzophenone, etc.) and photoactivatable (trifluoromethyl diazirine, aliphatic/aromatic azides, etc.) type, can be incorporated at key positions into a noncovalent parent ligand to render the parent ligand capable of reacting in a chemically defined manner with a distinct amino acid specie.50−52 For this purpose, and in recognition of the importance of cysteine residues to protein structure and function, we have successfully exploited the spontaneous, preferential reactivity at physiological pH between isothiocyanate (NCS)-functionalized electrophilic ligands and target-protein cysteine nucleophiles.46,53−56 The isothiocyanate functionality exhibits pronounced reactivity toward amine nucleophiles and sulfhydryl groups of cysteine with poor ability to react with other nucleophiles such as alcohol or water.
The successful design and utilization of covalent affinity probes in LAPS and other experimental applications to help characterize experimentally the orthosteric ligand-binding domains of CB1R and other cannabinoid-system protein therapeutic targets60−62,47,57,58 prompted the current work aimed at generating a focused library of electrophilic and photoaffinity probes carrying covalently reacting groups and targeted to the CB1R allosteric site(s). The design approach was based on the rational derivatization of two well-studied CB1R allosteric ligands, 1 and 2 (Figure 1), guided by the existing SAR data. Our results document the successful extension of the application of orthosteric CB1R covalently reactive probes to the receptor’s allosteric site(s) and constitute the first identification and functional profiling of a novel, covalent, allosteric CB1R affinity probe.
Chemistry
From precedent structure–activity relationship studies conducted by us40 and others59−64 on the two CB1R allosteric modulators 1 and 2, we identified two sites on the molecule, the C3 and C5 positions of 1, and the C-4 position of 2, important to the overall allosteric activity of these compounds. A focused library of seven analogues with electrophilic (isothiocyanate) or photoaffinity (azide or benzophenone) warheads placed at the terminal carbon of the C3 side chain and at the C5 position on parent molecule 1, and at the C-4 positon of 2 were synthesized, generating the novel analogues 19, 20, 25, 26, 33, 34, and 36 containing covalently reacting groups (Schemes 1–5).
Scheme 1. Synthesis of Piperidinyl Phenethylamine 8.

Reagents and conditions: (a) piperidine, K2CO3, anhyd NMP, 135 °C, 12 h; (b) CH3NO2, NH4OAc, reflux, 2 h; (c) (i) NaBH4, MeOH, 5 °C to rt, 2 h; (ii) NiCl2·6H2O, NaBH4, THF:MeOH (95:5), 0 °C, 3 h.
Scheme 5. Synthesis of Diarylureas 33, 34, and 36.

Reagent and conditions: (a) neat, rt, 1 h; (b) Ba(OH)2, Pd[P(Ph)3]4, DME:H2O (5:2), 150 °C, M.W., 15 min; (c) Raney-nickel, H2, MeOH, rt, 3 h; (d) triphosgene, Et3N, toluene, 70 °C, 3 h; (e) Et3N, DCM, 0 °C, 6 h; (f) (i) TPP, reflux, 4 h, benzene; (ii) CS2, 40 °C, 12 h; (g) triphosgene, Et3N, toluene, 70 °C, 3 h; (h) THF, 0 °C to rt, 6 h.
The novel indole-2-carboxamide analogues (19, 20, 25, and 26) of 1 were constructed as shown in Schemes 3 and 4. The C5-substituted indole rings were synthesized with an efficient method that utilizes Fisher cyclization on a mixture of azo and hydrazone of the corresponding diazonium salts (Scheme 2). The final carboxamide derivatives were synthesized using carbodiimide based amidation of the substituted indole-2-carboxylic acids with piperidinyl phenethylamine synthesized as per Scheme 1.
Scheme 3. Synthesis of 3-Ethylindole-2-carboxamides 19 and 20.

Reagents and conditions: (a) dioxane:H2O (10:1), KOH, reflux, 2 h, acidic workup; (b) EDCI, HOBt, DIPEA, NMP, rt, overnight; (c) NiCl2·6H2O, NaBH4, THF:CH3OH (13:1), −5 °C, 1 h; (d) t-BuONO, TMSN3, THF, rt, 3 h, (e) di(2-pyridyl)thionocarbonate, CH2Cl2, rt, 15 min.
Scheme 4. Synthesis of 5-Chloroindole-2-carboxamides 25 and 26.

Reagents and conditions: (a) (i) ethanolamine, EtOH, reflux, 14 h; (ii) KOH, dioxane:H2O (4:1), reflux, overnight; (b) Boc-anhydride, THF, aq NaHCO3 soln, 0 °C (3 h) to rt (24 h); (c) EDCI, HOBT, DIPEA, NMP, rt, overnight; (d) TFA:CH2Cl2 (1:10), rt, 3 h; (e) K2CO3, CuSO4, CH3OH:H2O (20:1), TfN3 in CH2Cl2, rt, 18 h; (f) di(2-pyridyl)thionocarbonate, CH2Cl2, rt, 15 min.
Scheme 2. Synthesis of Substituted Indole-2-carboxylates 15a, 15b, and 15c.

Reagent and conditions: (a) NaNO2, HCl, 0 °C, 1 h; (b) NaOEt, EtOH, 12 h, reflux; (c) CH3COONa, EtOH, 0 °C, 3 h; (d) 20% H2SO4, EtOH, reflux, 24 h.
N-Arylation of commercially available 4-fluorobenzaldehyde (5) with piperidine gave 4-piperidinylbenzaldehyde (6).65 To access the substituted nitrostyrene 7, we employed the Henry reaction on 6 in the presence of ammonium acetate in nitromethane as a solvent. Direct conversion of 7 to the desired amine 8 using LiAlH4, according to a previously published protocol, required 48 h, and the product was isolated in low yield.66 Alternatively, a route involving first the reduction of the double bond on 7 with NaBH4 followed by reduction of nitro group with in situ-generated nickel borohydride gave 8 in high yield (Scheme 1).
Scheme 2 describes the synthesis of the key substituted-indole esters. The alkylation of ethyl acetoacetate using (substituted) alkyl halides (11) in the presence of sodium ethoxide gave β-ketoesters (12) as the first step. Condensation of 12 with freshly prepared diazonium salts 10 (obtained from substituted anilines 9) in the presence of sodium acetate gave a mixture of azos (13) and hydrazones (14) as per the Japp–Klingeman reaction. These intermediates (13, 14) were isolated as a mixture by passing the reaction crude product over a small silica gel column, and in the case of 12c the reaction yielded only the azo compound 13c.67 This was followed by Fisher cyclization in 20% ethanolic sulfuric acid to give the chloro indole ester (15a), the nitro indole ester (15b), and the phthalimido indole ester (15c) in 57–71% yields.
The azido (19) and isothiocyanate (20) analogues of 1 were constructed as depicted in Scheme 3. Base-catalyzed hydrolysis of the nitro indole ester (15b) gave acid (16b) in high yield. Coupling of 16b with 8 yielded the nitro indole-2-carboxamide (17). Similarly, 15a was used to synthesize 1. The nitro functionality of 17 was efficiently reduced to the amino indole-2-carboxamide (18) using in situ-generated nickel borohydride. Treatment of 18 with a mixture of tert-butyl nitrite and azido trimethylsilane yielded 5-azido-3-ethyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (19). Compound 18 was also served as a precursor to synthesize the corresponding isothiocyanate analogue (20) in the presence of di-2-pyridyl thionocarbonate (DPT) at room temperature.
Phthalimido indole ester (15c) was treated with ethanolamine to give a lactam intermediate which was hydrolyzed using KOH to give the acid (21) (Scheme 4). The amino group in 21 was protected using Boc-anhydride to provide 22 which upon coupling with 8 in the presence of EDCI afforded 23. TFA-mediated deprotection of the NH-Boc group on 23 led to the amino analogue (24). The azido analogue (25) was then obtained by treating 24 with in situ-generated trifluoromethanesulfonyl azide. Direct conversion of 24 into the corresponding isothiocyanate analogue (26) was carried out at room temperature using DPT.
Analogues of 2 (33, 34, and 36) were synthesized as shown in Scheme 5. Treatment of 2,6-dibromopyridine (27) with neat pyrrolidine (28; excess) gave pyrrolidinyl bromopyridine (30) in quantitative yield. Microwave-accelerated Suzuki coupling of 29 with m-nitrophenylboronic acid in the presence of catalytic Pd(PPh3)4 afforded intermediate 30 which was further reduced to amine (31) in the presence of Raney-nickel under a hydrogen atmosphere. The isocyanate intermediate was synthesized in situ by treating 31 with triphosgene in the presence of triethylamine and was then reacted with 4-azidoaniline (32) in the presence of triethylamine to afford the desired product 33. It was then converted to the isothiocyanate analogue (34) by treating 33 with triphenylphosphine followed by exposure to CS2 (Staudinger/Aza-Wittig reaction).68 Commercially available benzophenone aniline (35) upon treatment with triphosgene in the presence of triethylamine gave corresponding isocyanate which was further reacted with 31 to yield the desired benzophenone-containing probe (36).
Results and Discussion
Following the discovery of 1, substantial SAR studies around it revealed that the indole-2-carboxamide scaffold is a promising template through which CB1R allosteric modulators with improved affinity, efficacy, potency, and pharmacokinetics could be generated. These studies identified several key pharmacophoric features within this structural class that influence their binding and functional properties.59,61−63 For example, the indole ring is more important for ligand affinity with the allosteric site than for its ability to modulate ligand binding at the orthosteric site (allosteric cooperativity). Alkyl chain length at the C3 position and the substitutions on the C5 position of the indole ring significantly impact allosteric affinity as well as cooperativity at CB1R.62,63 Replacing the amide linkage with ester functionality or modulating ethylene linker length between the amide bond and the phenyl ring drastically reduces the allosteric cooperativity toward binding.60,62,64 Replacing the piperidinyl group with a dimethylamino group significantly increases the allosteric cooperativity, and groups such as methyl, methylamino, nitro, and chloro but not fluoro, pyrrolidinyl, or 4-methylpiperazinyl were somewhat tolerated.62,64 Along similar lines, SAR reported around 2 from us40 and others61 has enabled identification of critical positions (especially the C-4 position) on this molecule that affects CB1R orthosteric ligand binding and downstream signaling. The commonality of the key phamacophoric features of 1 and 2, their SAR trends, and their unique and paradoxical pharmacological profile at CB1R strongly suggest that both may be acting through the same allosteric site on CB1R.
Functional Characterization
A focused library of analogues bearing reactive warheads placed at the terminal carbon of the C3 side chain and at C5 positions on parent molecule 1 (analogues 19, 20, 25, and 26) and at the C4 positon of 2 (analogues 33, 34, and 36) were biochemically evaluated in a series of assays. As these analogues were expected to potentially exhibit the “affinity vs efficacy paradox” similar to the parent compounds,26,41 and as a CB1 NAM having functional potency but no effect on orthosteric ligand binding has been identified,29 we chose to first characterize these newly synthesized analogues in two key functional assays. We characterized both the parent compounds and their analogues in CHO-K1 cells stably expressing human CB1R (hCB1R) by using the PathHunter β-arrestin and HitHunter cAMP cell-based functional assays. The PathHunter technology indexes the ability of a test agent to affect the recruitment and binding of the pleiotropic scaffold protein β-arrestin following kinase phosphorylation of agonist-bound CB1R, a process that uncouples the phosphorylated CB1R from its cognate G protein (routinely, Gi), subsequently targeting the receptor for internalization and enabling the recruitment of signal transducers by the internalizing CB1R−β-arrestin complex. The HitHunter assay indexes the ability of a test agent to modulate forskolin-stimulated cellular adenylyl cyclase activity (i.e., cellular cAMP formation). Notably, β-arrestin-mediated signaling is independent of both G proteins and classical second messengers, whereas the HitHunter cAMP assay reflects signal transduction dependent upon G-proteins and cAMP as second messenger.41,69,70
The parent compounds as well as their covalent analogues inhibited cellular CB1R-dependent β-arrestin recruitment and cAMP accumulation with nanomolar potencies (Figures 2 and 3 and Tables 1 and 2). The negative allosteric modulatory activity of both 1 and 2 in the β-arrestin and cAMP assays is congruent with previous observations (Table 1 and 2).41 Covalent analogues of 1 exhibited a 14- to 83-fold greater potency, and slightly greater efficacy, in inhibiting β-arrestin recruitment as compared to their effect on cellular cAMP accumulation (Figure 2, Table 1). Similarly, covalent analogues of 2 also exhibited greater potency in the β-arrestin vs cAMP assay, but the magnitude of the difference (3- to 19-fold) was not as great as that displayed by the covalent analogues of 1 (Figure 3, Table 2). Among these probes, 20 was the most potent inhibitor of β-arrestin recruitment (EC50 = 2 nM) and exhibited appreciable activity in the cAMP assay (EC50 = 174 nM) (Table 1). Compound 20 was more potent and efficacious than the parent compound, 1, in both β-arrestin and cAMP assays (Figure 2, Table 1) and exhibited the highest functional selectivity (83-fold) for β-arrestin vs cAMP. When the azide group was attached to the terminal carbon of the ethyl chain at the C3 position (25), the ability of the analogue to inhibit forskolin-stimulated cAMP accumulation (EC50 = 1120 nM), as well as its ability to inhibit β-arrestin recruitment (EC50 = 64 nM), was significantly compromised. Interestingly, placing the isothiocyanate group at the terminal carbon of the alkyl chain at the C3 position (26) abrogated activity in the cAMP assay (EC50 > 10 000 nM) and reduced activity in the β-arrestin assay by 2 orders of magnitude (EC50 = 209 nM). Compound 19 displayed activity similar to that of 1 in the cAMP assay but had modest activity in the β-arrestin assay (Table 1). None of the covalent analogues of 2 displayed a better activity profile than the parent in either the β-arrestin or the cAMP assay (Table 2). Although the azide (33) and the isothiocyanate (34) analogues showed reduced potencies in the cAMP assay (EC50 = 166 nM and 118 nM, respectively) compared to parent compound 2 (EC50 = 71 nM), they were some 2- to 3-fold more potent than 1 (EC50 = 324 nM) and of comparable potency to the best compound (20) in that series (EC50 = 174 nM). However, the potencies of 33 and 34 in the β-arrestin assay were reduced by 1 order of magnitude as compared to parent compound 2. Compound 36 containing the “bulkier” benzophenone functionality at the C4 position showed no activity in the cAMP assay and only residual activity in the β-arrestin assay, an activity profile justifying our decision not to pursue the benzophenone analogue of 1.
Figure 2.
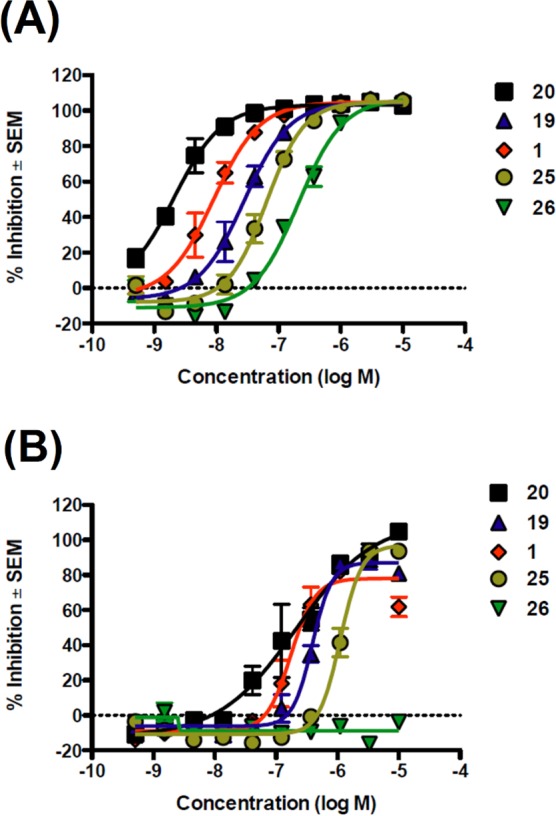
Antagonism of CP55,940-dependent β-arrestin recruitment (A) and cAMP inhibition (B) by CB1R analogues of 1 in vitro. (A) CHO-K1 PathHunter hCB1R cells were pretreated with indicated test compounds (0–10 μM) for 30 min followed by treatment with CP55,940 (EC80) for 90 min. β-Arrestin recruitment was quantified using the PathHunter assay. (B) CHO-K1 cAMP HitHunter hCB1 cells were pretreated with allosteric modulators (0–10 μM) for 30 min followed by treatment with CP55,940 (EC80) for 30 min. cAMP inhibition was quantified using the HitHunter assay. Data are presented as % inhibition compared to maximal CP55,940 effect ± SEM from two independent replicates per assay. Derived data are presented in Table 1.
Figure 3.
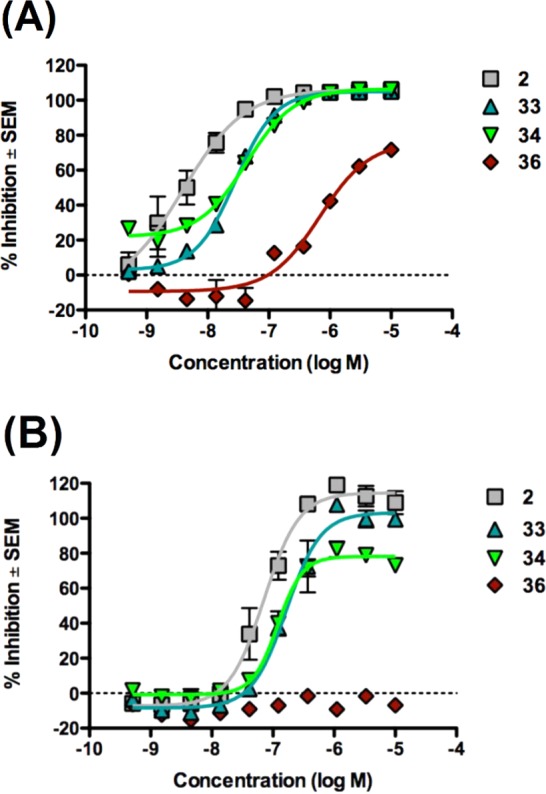
Antagonism of CP55,940-dependent β-arrestin recruitment (A) and cAMP inhibition (B) and by CB1R analogues of 2 in vitro. Effects on CP55,940-induced β-arrestin recruitment and cAMP inhibition in the presence of various analogues of 2. (A) CHO-K1 PathHunter hCB1 cells were pretreated with allosteric modulators (0–10 μM) for 30 min followed by treatment with CP55,940 for 90 min. β-Arrestin recruitment was quantified using the PathHunter assay. (B) CHO-K1 cAMP HitHunter hCB1 cells were pretreated with allosteric modulators (0–10 μM) for 30 min followed by treatment with CP55,940 for 30 min. cAMP inhibition was quantified using the HitHunter assay. Data are presented as % inhibition compared to maximal CP55,940 effect ± SEM from two independent replicates per assay. Derived data are presented in Table 2.
Table 1.

| cAMP accumulation |
β-arrestin recruitment |
|||||
|---|---|---|---|---|---|---|
| compound | X | Y | EC50 (95% CI)a | Emax (%) ± SEMb | EC50 (95% CI)a | Emax (%) ± SEMb |
| 1 | Cl | H | 324 (294–482) | 78.1 ± 4.49 | 9.05 (6.63–12.4) | 105 ± 2.10 |
| 19 | N3 | H | 389 (332–459) | 87.1 ± 3.02 | 28.2 (22.5–35.0) | 105 ± 2.07 |
| 20 | NCS | H | 174 (121–252) | 111 ± 12.1 | 2.09 (1.24–3.53) | 103 ± 1.38 |
| 25 | Cl | N3 | 1120 (962–1280) | 96.8 ± 3.75 | 64.0 (51.0–80.2) | 105 ± 2.70 |
| 26 | Cl | NCS | >10000 | –8.84 ± 1.18 | 209 (160–271) | 107 ± 3.94 |
Table 2.

To extend the functional profiling of what emerged from the data presented above as our novel lead CB1R allosteric ligand, 20, we evaluated its activity in the guanosine 5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) binding assay in mouse brain membranes. This assay reflects the functional response of GPCR ligands at the level of GDP/GTP exchange by the ternary, agonist-activated GPCR–G protein complex, an event that can modulate the activity of downstream effector proteins. The assay is considered reflective of the degree of G protein activation following GPCR agonist engagement, an event more proximal to the GPCR itself in the biosignaling cascade than is cAMP formation or G protein-independent β-arrestin signaling.71 We observed that 20 inhibited CP55,940-induced [35S]GTPγS binding to CB1R in mouse brain membranes by progressively decreasing the Emax in a concentration-dependent manner (Figure 4) with more efficacy compared to 1. When administered alone, 20 was “silent” and did not display CB1R agonism or inverse agonism in the [35S]GTPγS binding assay performed with mouse brain membranes (Figure 5). Also, 20 did not exhibit any signs of CB1R agonism or inverse agonism in hCB1 CHO cell membranes up to 1 μM. A statistically significant, but much reduced, inverse agonism compared to 1 was observed only at suprapharmacological concentration (10 μM; data not shown). This compound has been extensively studied for its CB1R NAM as well as inverse-agonist activity in CB1R-mediated downstream signaling pathways and in different cell lines, where it consistently showed lack of inverse agonism. The data are beyond the scope of this paper and will be published elsewhere.
Figure 4.
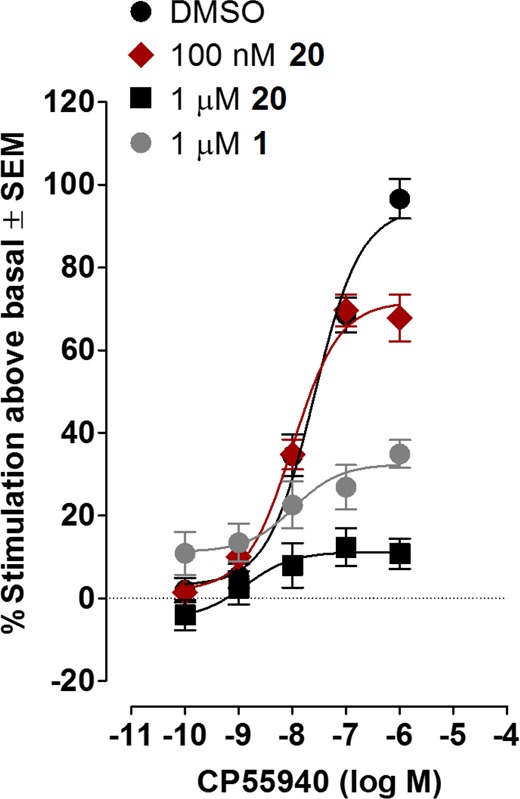
[35S]GTPγS assay depicting the NAM effect of increasing concentrations of 20. Mean % increases in [35S]GTPγS binding to mouse brain membranes induced by CP55,940 in the presence of DMSO (n = 12) or 100 nM or 1 μM of 20 (n = 6) or 1 μM of 1 (n = 4). The mean Emax value of CP55,940 with its 95% confidence interval in parentheses is 94.3% (85.8–102.8%) in the presence of DMSO, 71.7% (65.3–78.1%) in the presence of 100 nM of 20, 11.2% (5.6–16.8%) in the presence of 1 μM of 20, and 32.4% (24.0–40.8%) in the presence of 1 μM of 1. Vertical lines show SEM values.
Figure 5.
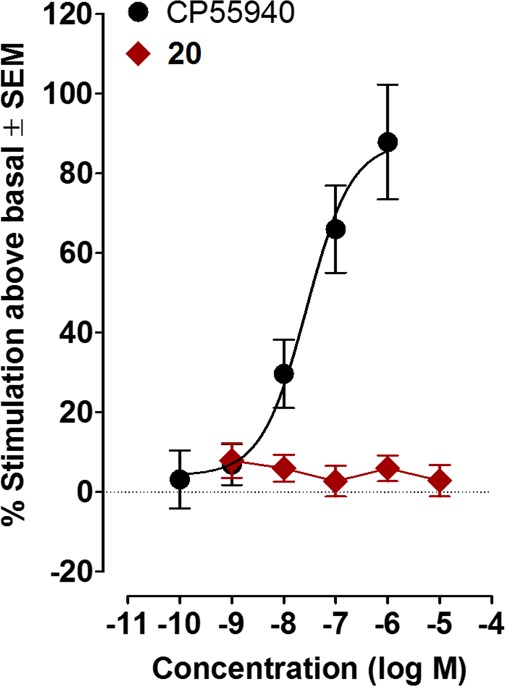
Activity of 20 and CP55,940 in the [35S]GTPγS assay. Mean % changes in [35S]GTPγS binding to mouse brain membranes elicited by CP55,940 or 20. The mean EC50 value of CP55,940 with a 95% confidence interval is 27.3 nM (7.2–103.2 nM, n = 6), and its corresponding Emax value is 87.8% (67.9 and 107.7%). Vertical lines show SEM values.
The activity profile of 20 in the [35S]GTPγS binding assay is in marked contrast to that reported for parent compounds 1 and 2, which elicit CB1R inverse-agonist activity in addition to acting as CB1R NAM.26,41,42 To the best of our knowledge, this is the first report of a potent CB1R NAM lacking inverse agonism. This functional distinction between 20 and the standard CB1R NAMs 1 and 2 carries significant translational and rational drug-design implications. As detailed elsewhere, CB1R inverse agonism has been associated with peripheral (e.g., gastrointestinal) and central (e.g., psychobehavioral) adverse events in preclinical animal models of disease and in humans.21,23 The proposition has thus been advanced that agents capable of attenuating CB1R information transmission with intrinsically limited, if any, functional potency to elicit negative-efficacy responses might display an enhanced benefit-to-risk profile as therapeutics relative to conventional CB1R antagonists/inverse agonists for diseases with a pathogenic component of CB1R hyperactivity.
Structural comparison between 1 and 20 implies that modifications of 1 at C5 can generate CB1R NAMs that retain the affinity–efficacy profile of 1 but are devoid of or exhibit reduced inverse-agonist activity of conventional CB1R NAMs. To date, very limited SAR studies have been carried out with variations at the C5 position. This work identifies C5 position as the key site for potential modifications for generating future CB1 NAMs lacking inverse-agonist activity. This conclusion is supported by recently published mutational and computational data indicating that electrostatic interactions and van der Waals forces between the nitrogen in the piperidine ring of 1 and the CB1R aspartate residue D6.58(366) in CB1R transmembrane helix 6 is crucial for inverse agonism such that the absence of this nitrogen abrogates the inverse-agonism action of 1.42 In our hands, even with the presence of the piperidine nitrogen, 20 did not evidence inverse agonism in mouse brain membranes and hCB1 CHO cells (up to 1 μM). This result invites the notion that the increased length of the NCS group at C5 in 20 compared to the Cl group at that position in 1 might extend 20 in its CB1R binding pocket slightly beyond 1’s original docking position, potentially obviating or reducing the interaction between the C5 nitrogen on the piperidine ring of 20 and that of the CB1R D6.58(366) residue, leading to loss of inverse agonism.
Ligand Binding Studies
To profile the ligand-binding characteristics of our lead CB1R covalent ligand, 20, we first evaluated the effect of 20 on the specific binding of the orthosteric ligand [3H]CP55,940 to membranes obtained from CHO cells overexpressing hCB1R. Both 20 and 1 significantly enhanced the binding of [3H]CP55,940 to hCB1 CHO cell membranes and acted as CB1 PAM of binding. As indicated by the data shown in Figure 6, 20 produced this enhancement with significantly greater potency (lower EC50) than 1.
Figure 6.
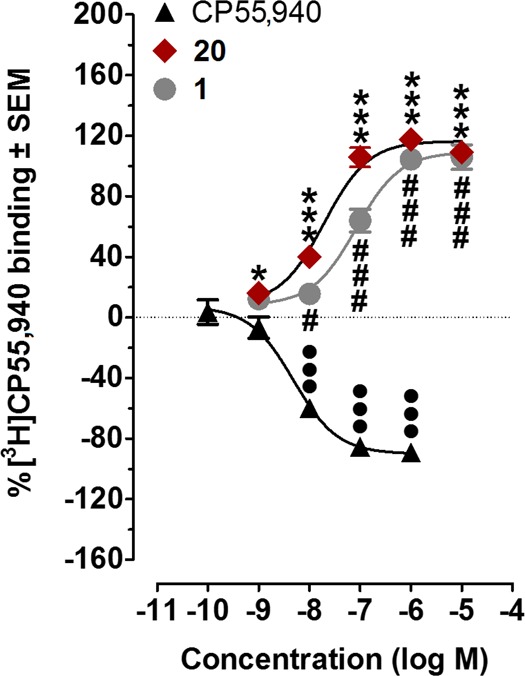
Positive allosteric modulation of [3H]CP55,940 binding by 20 and 1. Effects of 20, 1, and CP55,940 on [3H]CP55,940 binding to hCB1 CHO cell membranes (n = 6). Mean values significantly different from zero are indicated by the symbols * (for 20), # (for 1), and • (for CP55,940); one symbol = P < 0.05; three symbols = P < 0.001; Student’s one sample t test). Positive values indicate enhancement of [3H]CP55,940 binding. The mean Emax values of 20, 1, and CP55,940, with their 95% confidence limits shown in brackets, are 116.5% (108.3 and 124.6%), 109.3% (99.1 and 119.5%), and −90.1% (−81.6 and −98.6%), respectively. The corresponding EC50 values, again with 95% confidence limits shown in brackets, are 19.6 nM (10.4 and 36.9 nM), 83.0 nM (44.1 and 156.2 nM), and 4.9 nM (2.6 and 9.0 nM).
To investigate the ability of 20 to label the CB1R allosteric site(s) covalently, we carried out time-course experiments between 20 and hCB1R in membranes isolated from HEK293 cells overexpressing the receptor. We indexed the covalent association between 20 and hCB1R as the extent to which a preincubation of the isolated membranes with 20 at a concentration of 500 nM followed by extensive membrane washings with centrifugation influencing the subsequent level (Bmax) of specific [3H]CP55,940 binding to hCB1R in the washed membranes. The binding of [3H]CP55,940 to hCB1R increased in a time-dependent manner, reaching a maximum by 60 min preincubation time with isothiocyanate 20 (Figure 7). Incubation of the CB1 receptor with 20 for 90 and 120 min reduced specific binding of [3H]CP55,940. Presumably, the extended incubation time (beyond 60 min) resulted in nonspecific covalent modification of the receptor and impaired its ability to bind [3H]CP55,940. These data are consistent with the characteristic dependency of the association between covalent ligands/probes and target proteins upon the length of time the protein is incubated with the probe.60,62,47,57,58
Figure 7.
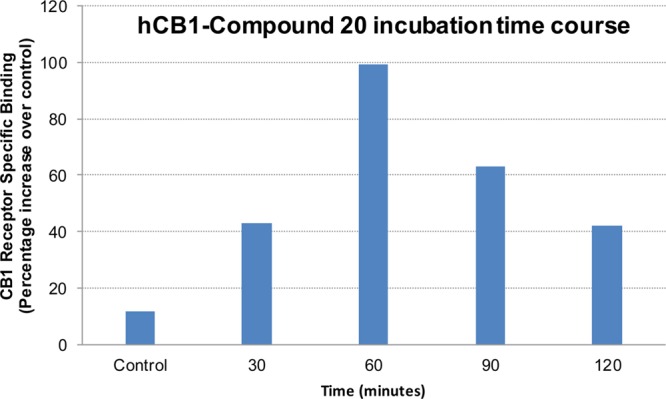
Time-course studies showing the effect of 20 preincubation on CP55,940 binding. HEK293-hCB1R membranes incubated with 500 nM of 20 for 0 (control), 30, 60, 90, and 120 min.
The data in Figure 7 provided sufficient guidance to establish experimental parameters for determining the effect of 20 on [3H]CP55,940 specific binding to hCB1R over a range of radioligand concentrations in a saturation-binding assay. Parent compound 1 was profiled in parallel as the nonderivatized control lacking the chemically reactive isothiocyanate moiety and, thus, incapable of covalently interacting with hCB1R. In accord with our previous receptor-labeling studies,45−48 we pretreated the HEK293 hCB1R membranes with either test compound (500 nM, final concn) for 60 min, subsequently washed the membranes extensively to quench this incubation, and quantified any change observed for the subsequent specific binding of [3H]CP55,940 to hCB1R (as Bmax) in the washed membranes. The saturation binding curves (Figure 8) show that preincubation with 20 increased maximal hCB1R specific binding of [3H]CP55,940 by ∼2.25-fold, whereas 1 had no effect. The combined data in Figures 7 and 8 indicate that 20 covalently labels hCB1R by virtue of its C5 isothiocyanate group.
Figure 8.
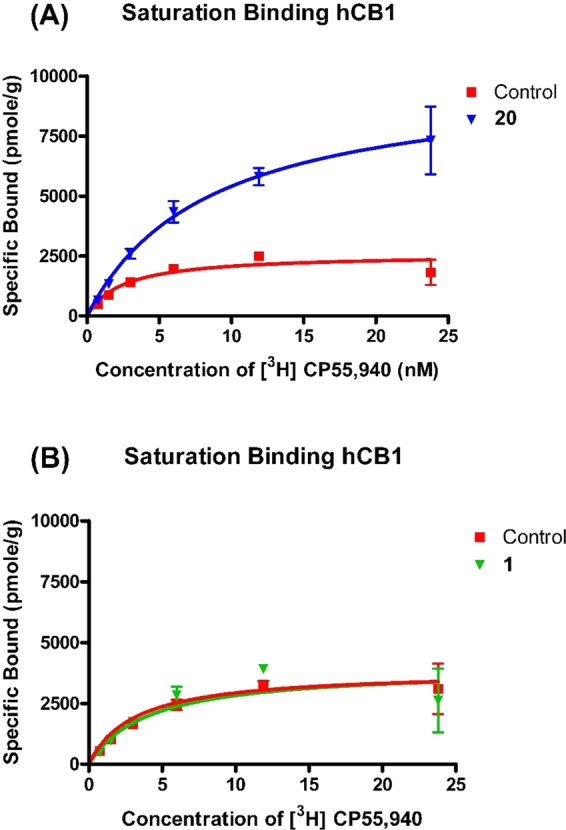
Comparative covalent labeling of 20 (A) and 1 (B) with hCB1R. Effect of (A) 20 or (B) 1 on the saturation binding of [3H] CP55,940 to HEK293-hCB1R cell membranes. Membranes were incubated with allosteric ligands (500 nM) at 30 °C for 60 min and extensively washed, followed by saturation binding of [3H]CP55,940 at concentrations ranging from 0 to 25 nM. Unbound [3H]CP55,940 was removed by washing–filtration, and the membrane-bound radioactivity was quantified. 1 did not label the receptor covalently and hence did not affect CP55,940 binding after wash, whereas 20 labeled the receptor covalently and caused ∼2.25 fold increase in CP55,940 binding, as indexed by the respective Bmax values. Data shown represent the mean ± SEM of three independent experiments performed in duplicate.
Conclusion
Adverse events associated with standard CB1R orthosteric ligands (agonists and antagonists/inverse agonists) have prompted alternative approaches in chemical pharmacology for leveraging the translational potential of drug-like small-molecules that modulate CB1R-dependent signaling. Predominant in this newer thinking for enhancing therapeutics targeting of CB1R are neutral antagonists with minimal, if any, inverse-agonist effects and allosteric ligands.23,24 Due to greater receptor selectivity and finer control over downstream signaling than standard CB1R orthosteric modulators, CB1R allosteric modulators offer opportunities for novel pharmacotherapies with potentially enhanced safety and efficacy profiles. Rational design of CB1R allosteric modulators as drugs requires greater understanding of the receptor’s allosteric binding site(s) and the molecular pharmacology of ligands that are engaged by it. As part of our continued commitment to address this need, we report our approach of derivatizing the classical CB1R allosteric modulators 1 and 2 with a chemically reactive electrophilic (NCS) or photoactivatable (azide and benzophenone) groups at the judiciously selected positions within each molecule. Functional characterization demonstrated that these novel analogues displayed the prototypical paradoxical pharmacological properties which make them PAMs of CP55,940 binding but NAM in function. With all of these analogues and in the assay systems utilized, we observed a consistent bias toward β-arrestin over cAMP-dependent signaling. Among these, 20 emerged as the most potent NAM in the cellular cAMP, G-protein-independent β-arrestin, and G protein-dependent GTPγS functional assays, and it was more potent than the parent compound 1. It was also more potent than 1 as a PAM of orthosteric ligand binding. Notably, when applied alone, 20 did not affect the constitutive activity of the receptor in the GTPγS assay, making it the first ever covalent, potent CB1R NAM without significant inverse-agonist activity, a property suggestive of a lower adverse-event risk. Compound 20 engaged the CB1R allosteric site(s) covalently, making this compound a unique and valuable probe with which to help elucidate the (sub)molecular features of ligand recognition and engagement by CB1R. Ongoing work in this regard will incorporate 20 and newer-generation CB1R allosteric covalent probes into our established LAPS paradigm in conjunction with site-specific CB1R point mutations and peptide-level LC/MS/MS for identifying experimental amino acid residues critical to CB1R allosteric ligand binding/function and mapping structural features and topology of the CB1R allosteric ligand-binding pocket(s). Because preliminary data indicate that binding of 20 to hCB1R is irreversible, the utility of this compound as a structural probe may be extended to 20–hCB1R cocrystallization studies aimed at mapping the location of the CB1R allosteric site(s) and their atomic-level features.
Methods
PathHunter CB1 β-Arrestin Assay
Chinese hamster ovary K1 (CHO-K1)-PathHunter hCB1 β-arrestin cells (DiscoveRx, Fremont, CA) were seeded at 5000 cells/well in 384-well plates 24 h before use and incubated at 37 °C, 5% CO2. Compounds were dissolved in dimethyl sulfoxide (DMSO) and diluted in optimized cell culture (OCC) media. Agonist EC80 was determined directly from an agonist dose–response curve (data not shown). The CP55,940 EC80 was 31.1 ± 0.47 nM (mean ± SEM, n = 3 independent experiments). Five microliters of allosteric modulator or vehicle solution was added to each well at the appropriate concentrations and incubated for 30 min. Five microliters of agonist was then added to each well followed by a 90 min incubation. Fifteen microliters of detection reagent was then added followed by further 60 min incubation at room temperature. Chemiluminescence was measured on a standard luminescence plate reader as relative light units (RLU). Basal RLU was defined as zero. Results were calculated as the percentage inhibition of CP55,940 maximal effect. Data were analyzed using the four-parameter variable-slope and allosteric EC50 shift nonlinear regression equations in Prism 5.0 (GraphPad, San Diego, CA). The results of this analysis are presented as Emax ± SEM, and EC50 (nM) with 95% CI.
HitHunter cAMP Assay
Chinese Hamster Ovary K1 (CHO-K1)-HitHunter hCB1R cells (DiscoveRx) were seeded at 10 000 cells/well in 384-well plates 24 h before use and incubated at 37 °C under 5% CO2. Compounds were dissolved in DMSO and diluted in OCC media. Agonist EC80 was determined directly from an agonist dose–response curve (data not shown). The CP55,940 EC80 was 7.5 ± 0.15 nM (mean ± SEM, n = 3 independent experiments). Media was aspirated and replaced with 10 μL of 1:1 HBSS/HEPES:cAMP XS+Ab reagent containing 20 μM forskolin (DiscoveRx). Five microliters of test compound or vehicle solution was added to each well at the appropriate concentrations and incubated for 30 min. Five microliters of agonist was then added to each well followed by a 30 min incubation. Twenty microliters of cAMP XS+ED/CL lysis cocktail (DiscoveRx) was then added followed by 60 min incubation at room temperature. Finally, 20 μL of cAMP XS+EA reagent (DiscoveRx) was added followed by 3 h incubation at room temperature. Chemiluminescence was measured on a standard luminescence plate reader (as RLUs). Basal RLU was defined as zero. Results were calculated as the percentage inhibition of CP55,940 maximal effect. Data were analyzed using the four-parameter variable slope and allosteric EC50 shift nonlinear regression equations in GraphPad Prism 5.0 (GraphPad, San Diego, CA). The results of this analysis are presented as Emax ± SEM, and EC50 (nM) with 95% CI.
Radioligand Displacement Assay
Chinese hamster ovary (CHO) cells transfected with cDNA encoding human cannabinoid CB1 receptors were maintained at 37 °C in Dulbecco’s modified Eagle’s medium nutrient mixture F-12 HAM, supplemented with 1 mM l-glutamine, 10% fetal bovine serum, 0.6% penicillin–streptomycin, and G418 (400 μg/mL). All cells were exposed to 5% CO2 in their media, and were passaged twice a week using nonenzymatic cell dissociation solution. For membrane preparation, cells were removed from flasks by scraping, centrifuged, and then frozen as a pellet at −20 °C until required. Before use in a radioligand binding assay, cells were defrosted, diluted in Tris buffer (50 mM Tris-HCl and 50 mM Tris-base), and homogenized with a 1 mL hand-held homogenizer.
The assays were carried out with [3H]CP55,940 and Tris binding buffer (50 mM Tris-HCl, 50 mM Tris-base, 0.1% BSA, pH 7.4), total assay volume 500 μL, using the filtration procedure described previously.41,72 Binding was initiated by the addition of transfected human CB1 CHO cell membranes (50 μg of protein per well). All assays were performed at 37 °C for 60 min before termination by the addition of ice-cold Tris binding buffer, followed by vacuum filtration using a 24-well sampling manifold (Brandel Cell Harvester; Brandel Inc., Gaithersburg, MD) and Brandel GF/B filters that had been soaked in wash buffer at 4 °C for at least 24 h. Each reaction well was washed six times with a 1.2 mL aliquot of Tris binding buffer. The filters were oven-dried for 60 min and then placed in 3 mL of scintillation fluid (Ultima Gold XR, PerkinElmer, Seer Green, Buckinghamshire, UK). Radioactivity was quantified by liquid scintillation spectrometry. Specific binding was defined as the difference between the binding that occurred in the presence and absence of 1 μM unlabeled CP55,940. The concentration of [3H]CP55,940 used in our displacement assays was 0.7 nM. The compounds under investigation were stored as 10 mM stock solutions in DMSO, the vehicle concentration in all assay wells being 0.1% DMSO.
[35S]GTPγS Binding Assay
Mouse brain membranes (5 μg protein), prepared as described previously,41 were preincubated for 30 min at 30 °C with adenosine deaminase (0.5 IU/mL). The membranes were then incubated with CP55,940 or 20, or with CP55,940 ± 20, 1, or vehicle, for 60 min at 30 °C in assay buffer (50 mM Tris-HCl, 50 mM Tris-base, 5 mM MgCl2, 1 mM EDTA, 100 mM NaCl, 1 mM DTT, 0.1% BSA) in the presence of 0.1 nM [35S]GTPγS and 30 μM GDP, in a final volume of 500 μL. Binding was initiated by the addition of [35S]GTPγS. Nonspecific binding was measured in the presence of 30 μM GTPγS. The reaction was terminated by rapid vacuum filtration (50 mM Tris-HCl, 50 mM Tris-base, 0.1% BSA) using a 24-well sampling manifold (cell harvester, Brandel, Gaithersburg, MD) and GF/B filters (Whatman, Maidstone, UK) that had been soaked in buffer (50 mM Tris-HCl, 50 mM Tris-base, 0.1% BSA) for at least 24 h. Each reaction tube was washed six times with a 1.2 mL aliquot of ice-cold wash buffer. The filters were oven-dried for at least 60 min and then placed in 3 mL of scintillation fluid (Ultima Gold XR, PerkinElmer, Seer Green, Buckinghamshire, UK). Radioactivity was quantified by liquid scintillation spectrometry.
Data Analysis
Most results were calculated as percentage changes from a basal level (zero) of [3H]CP55,940 or [35S]GTPγS binding (in the presence of vehicle). GraphPad Prism 5.0 (GraphPad, San Diego, CA) was used to construct sigmoidal log concentration–response curves and to calculate values of EC50, Emax, SEM, and 95% confidence intervals. Some mean values were compared using Student’s one sample t test. P values <0.05 were considered to be significant.
Rat Brain and HEK293 Cell Membrane Preparations
Rat forebrain membranes for binding assays were prepared according to a published protocol.73 HEK293 cells overexpressing hCB1R were disrupted by cavitation in a pressure cell, and membranes were sedimented by ultracentrifugation, as described.46 The membrane pellet was resuspended in TME buffer (50 mM Tris-HCl, 5 mM MgCl2, 1 mM EDTA, pH 7.4), and membrane protein was quantified with a Bradford dye-binding method (Bio-Rad Laboratories).
[3H] CP55,940 Saturation-Binding to hCB1 in the Presence of Allosteric Ligands
Membrane preparations either from rat brain or HEK293 cells overexpressing hCB1 receptor were resuspended in TME–BSA (TME containing 0.1% BSA), and aliquots of this suspension containing 25 μg of proteins were added to each assay well. Membranes were preincubated with 1 (for 1 h) or 20 (for 0, 30, 60, 90, and 120 min) at 500 nM concentration of allosteric ligand at 30 °C with agitation. The excess ligand was removed during washes with TME–BSA and TME buffers and centrifugations at 27 000g, 30 °C. Membrane proteins were quantified with a Bradford dye binding method (Bio-Rad Laboratories). Saturation binding assays were performed with the washed membranes and [3H]CP55,940 radioligand at concentrations ranging between 0 and 25 nM. Nonspecific binding was evaluated in the presence of 5 μM unlabeled CP55,940. The assay was performed at 30 °C for 1 h with gentle agitation. The resultant material was transferred to Unifilter GF/B filter plates and the unbound ligand removed using a Packard Filtermate-96 Cell Harvester (PerkinElmerPackard, Shelton, CT). Filter plates were washed four times with ice-cold wash buffer (50 mM Tris base, 5 mM MgCl2 containing 0.5% BSA, pH 7.4). Bound radioactivity was quantified using the Packard top count scintillation counter. Nonspecific binding was subtracted from the total bound radioactivity to obtain the specific binding of [3H]CP-55,940 (represented as pmol/mg of protein). All assays were performed in triplicate, and data points were represented as the mean Bmax and Kd values, calculated by nonlinear regression using Graphpad Prism 4.0 on a Windows platform.
The assay was performed in 200 μL of TME–BSA buffer at 30 °C for 1 h with gentle agitation. Filtration and washing were performed as described above. Nonspecific binding was subtracted from the total bound radioactivity to obtain [3H]CP55,940 specific binding of (as pmol/mg protein). All assays were performed in triplicate, and data points were represented as the mean Bmax and Kd values, calculated by nonlinear regression using GraphPad Prism 5.0.
Experimental Section
All commercial chemicals and solvents were purchased from Sigma-Aldrich, Inc. (St. Louis, MO), Alfa Aesar, and Combi-blocks as reagent grade and unless otherwise specified were used without further purification. Biotage Initiator microwave system was used for the synthesis of a few of the intermediates of the final covalent probes. Reaction progress was monitored by thin-layer chromatography (TLC) using commercially prepared silica gel 60 F254 glass plates. Compounds were visualized under ultraviolet (UV) light or by staining with iodine, phosphomolybdic acid, or p-anisaldehyde reagent. Flash column chromatography was carried out on a Biotage SP1, Biotage Isolera, or Interchim purification unit using prepacked columns from Reveleris, Biotage, and Luknova. Solvents used include hexanes, ethyl acetate, acetone, methanol, and dichloromethane. Characterization of compounds and their purity were established by a combination of HPLC, TLC, mass spectrometry, and NMR analyses. NMR spectra were recorded in DMSO-d6, chloroform-d, or methanol-d4, on a Varian NMR spectrometer (1H NMR at 500 MHz and 13C NMR at 125 MHz). Chemical shifts were recorded in parts per million (δ) relative to tetramethylsilane (TMS; 0.00 ppm) or solvent peaks as the internal reference. Multiplicities are indicated as br (broadened), s (singlet), d (doublet), t (triplet), q (quartet), quin (quintet), sept (septet), or m (multiplet). Coupling constants (J) are reported in hertz (Hz). All test compounds were greater than 95% pure as determined by LC/MS analysis performed using a Agilent Technologies 1260 Infinity reverse-phase HPLC, with a dual-wavelength UV–visible detector and an Agilent Technologies 6120 Quadrupole mass spectrometer (electrospray ionization).
5-Chloro-3-ethyl-N-[4-(piperidin-1-yl)phenethyl]-1H-indole-2-carboxamide (1)
To a mixture of 16a (500 mg, 2.24 mmol) and 8 (548 mg, 2.68 mmol) in 5 mL of anhydrous NMP under an argon atmosphere and at room temperature were added HOBT (302 mg, 2.24 mmol), DIPEA (347 mg, 2.68 mmol), and EDCI (486 mg, 3.13 mmol), and the resulting mixture was stirred overnight. The reaction mixture was diluted with cold water and the crude product was extracted in ether (3×). The combined organic layer was washed with water and brine and dried (Na2SO4). Volatiles were evaporated under reduced pressure, and the crude product obtained was purified by flash column chromatography on silica gel (10–40%; EtOAc:hexanes) to give 1 as a white solid (709 mg, 77% yield). Rf = 0.7 (EtOAc/hexanes = 50/50). 1H NMR (500 MHz, chloroform-d): 9.28 (s, 1H), 7.54 (d, J = 1.5 Hz, 1H), 7.29 (d, J = 9.0 Hz, 1H), 7.20 (dd, J = 8.5 Hz, J = 2.0 Hz, 1H), 7.14 (d, J = 8.5 Hz, 2H), 6.92 (d, J = 8.5 Hz, 2H), 6.04–5.94 (m, 1H,), 3.79 (q, J = 6.5 Hz, 2H), 3.13 (t, J = 5.5 Hz, 4H), 2.89 (t, J = 6.5 Hz, 2H), 2.69 (q, J = 8.0 Hz, 2H), 1.75–1.67 (m, 4H), 1.61–1.54 (m, 2H), 1.08 (t, J = 7.5 Hz, 3H). Mass spectrum m/z 410.18 [M + H]+.
4-(Piperidin-1-yl)benzaldehyde (6)
To a solution of 5 (20 g, 161.1 mmol) and piperidine (16.47 g, 193 mmol) in 60 mL of dry NMP under an argon atmosphere was added anhydrous K2CO3 (44.5 g, 322 mmol), and the resulting solution was stirred at 135 °C for 12 h. Reaction contents were allowed to cool to room temperature and were diluted with ice cold water. Product was extracted in ether (3×), and the combined organic layer was washed with water and brine and dried over MgSO4. Solvent was evaporated under reduced pressure, and purification of the crude product by flash column chromatography (0–15%; EtOAc:hexanes) afforded 6 as a pale yellow solid (23.78 g, 78.2% yield). Rf = 0.5 (EtOAc/hexanes = 20/80). 1H NMR (500 MHz, chloroform-d): δ 9.74 (s, 1H), 7.72 (d, J = 9.5 Hz, 2H), 6.89 (d, J = 9.0 Hz, 2H), 3.45–3.37 (m, 4H), 1.74–1.63 (m, 6H). Mass spectrum m/z 190.12 [M + H]+.
(E)-1-[4-(2-Nitrovinyl)phenyl]piperidine (7)
To a solution of 6 (20 g, 106 mmol) in 100 mL of anhydrous nitromethane was added NH4OAc (24.44 g, 317 mmol) under an argon atmosphere, and the resulting mixture was refluxed for 2 h. Solvent was removed under reduced pressure, and the reaction mixture was diluted with ethyl acetate and water (2:1). The organic layer was separated, the aqueous layer was extracted with ethyl acetate (3×), and the combined organic layer was washed with water and brine and dried over MgSO4. The solvent was evaporation under reduced pressure, and the crude product was purified by flash column chromatography (5%–20%; EtOAc:hexanes) to give 7 as a dark orange solid (16.10 g, 65.6% yield). Rf = 0.48 (EtOAc/hexanes = 20/80). 1H NMR (500 MHz, chloroform-d): δ 7.95 (d, J = 13.5 Hz, 1H), 7.50 (d, J = 13.5 Hz, 1H), 7.42 (d, J = 9.0 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 3.40–3.36 (m, 4H), 1.74–1.63 (m, 6H). Mass spectrum m/z 233.12 [M + H]+.
2-(4-(Piperidin-1-yl)phenyl)ethanamine (8)
To a cooled (5 °C) solution of 7 (15 g, 64.3 mmol) in 120 mL of anhydrous methanol at 5 °C was added NaBH4 (14.66 g, 387 mmol) in small portions under an argon atmosphere, and the reaction mixture was stirred for 2 h while allowing it to warm to room temperature. It was then quenched with dropwise addition of 80 mL of saturated NH4Cl. The mixture was concentrated under reduced pressure, and the crude product was partitioned in ethyl acetate and water. The organic layer was separated, the aqueous layer extracted with ethyl acetate (3×), and the combined organic layer was washed with brine and dried over Na2SO4. Evaporation of volatiles under reduced pressure gave a crude mixture which was purified by flash column chromatography (10%–40%; EtOAc:hexanes) to yield the intermediate 1-(4-(2-nitroethyl)phenyl)piperidine as a pale yellow oil (12.8 g, 85% yield). Rf = 0.42 (EtOAc/hexanes = 20/80). 1H NMR (500 MHz, chloroform-d): δ 7.07 (d, J = 9.0 Hz, 2H), 6.88 (d, J = 9.0 Hz, 1H), 4.55 (t, J = 7.5 Hz, 2H), 3.23 (d, J = 7.5 Hz, 2H), 3.17–3.10 (m, 4H), 1.73–1.66 (m, 4H), 1.61–1.53 (m, 2H). Mass spectrum m/z 233.12 [M + H]+.
To a solution of this intermediate (12.5 g, 53.40 mmol) in 100 mL of anhydrous THF and methanol (13:1) was added NiCl2·6H2O (15.22 g, 64 mmol) under an argon atmosphere, and reaction mixture was stirred for 45 min at room temperature. It was then cooled to −5 °C, and NaBH4 (12.11 g, 320 mmol) was added in small portions. The reaction was then gradually warmed to room temperature, stirred for 3 h, quenched with saturated aqueous solution of NH4Cl, and concentrated under reduced pressure. The residue was diluted with ethyl acetate and water and filtered. The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3×). The combined organic layer was washed with brine, dried (Na2SO4), and evaporated under vacuum to yield pure amine 8 (8.8 g, 81% yield). Rf = 0.81 (MeOH/DCM = 20/80). 1H NMR (500 MHz, chloroform-d): δ 7.08 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.0 Hz, 2H), 3.17–3.08 (m, 4H), 2.92 (t, J = 7.0 Hz, 2H), 2.66 (t, J = 7.0 Hz, 2H), 1.76–1.66 (m, 4H), 1.61–1.52 (m, 2H), 1.31 (br s, 2H). Mass spectrum m/z 205.16 [M + H]+.
4-Chlorobenzenediazonium Chloride (10a)
To a suspension of finely powdered 9a (2.54 g, 20 mmol) in 10 mL of 24% aq hydrochloric acid at 0 °C was added a cold aqueous solution of sodium nitrite (1.7 g, 23 mmol), and the reaction mixture was stirred for 1 h while maintaining the temperature between 0 and 5 °C. The resulting pale yellow solution of diazonium salt 10a was directly used for the next reaction.
4-Nitrobenzenediazonium Chloride (10b)
The compound was synthesized as per the procedure described for 10a using nitroaniline 9b.The reaction solution of diazonium salt 10b was directly used for the next reaction.
Ethyl-2-acetyl Pentanoate (12a)
To a flask containing 300 mL of anhydrous ethanol at 10 °C was added sodium metal (6.0 g, 261.0 mmol) portionwise under an argon atmosphere, and the mixture was stirred for 30 min to complete dissolution. To this was added ethyl acetoacetate (34.0 g, 261.0 mmol), and the resulting solution was refluxed for 30 min and allowed to cool to room temperature. This was followed by addition of iodopropane (11a, 44.44 g, 261.0 mmol) over a period of 30 min through a dropping funnel, and the reaction mixture was refluxed for 12 h. The mixture was cooled to room temperature and filtered, and the filtrate was neutralized by adding 1 N hydrochloric acid, concentrated under reduced pressure, and partitioned in ethyl acetate and water. The organic layer was separated, the aqueous layer extracted with ethyl acetate (3×), and the combined organic layer was washed with brined and dried over MgSO4. The product was purified by flash column chromatography (0%–20%; EtOAc:hexanes) to give 12a as a clear liquid (35.1 g, 78% yield). Rf = 0.45 (EtOAc/hexanes = 20/80). 1H NMR (400 MHz, chloroform-d) δ 4.20 (q, J = 7.2 Hz, 2H), 3.42 (t, J = 7.2 Hz, 1H), 2.22 (s, 3H), 1.92–1.76 (m, 2H), 1.40–1.20 (m, 5H, especially 1.28, t, J = 7.2 Hz, 3H), 0.93 (t, J = 7.2 Hz, 3H). Mass spectrum m/z 172.10 [M + H]+.
Ethyl 2-Acetyl-4-(1,3-dioxoisoindolin-2-yl)butanoate (12c)
To a 500 mL flask containing 150 mL of anhydrous acetone were added ethyl acetoacetate (10.0 g, 77.0 mmol) and K2CO3 (11.68 g, 85 mmol), and the mixture was stirred for 2 h at room temperature. To this was added 2-(3-iodopropyl)isoindoline-1,3-dione (11c; 24.21 g, 77.0 mmol) and refluxed overnight. The mixture was filtered through a Buchner funnel, and the filtrate was cooled to room temperature. Volatiles were evaporated, water was added to the crude product, and the resultant mass was acidified to pH 4. The aqueous layer was extracted in dichloromethane (2×), and the combined organic layer was dried over MgSO4 and triturated in hexane to obtain the desired compound 12c as a white solid (18.31 g, 75.1% yield). 1H NMR (500 MHz, DMSO-d6) δ 7.87–7.82 (m, 2H), 7.74–7.69 (m, 2H), 4.19 (qd, J = 7.0 Hz, 2.0 Hz, 2H), 3.71 (t, J = 7.0 Hz, 2H), 3.50 (t, J = 7.0 Hz, 1H), 2.25 (s, 3H), 1.96–1.81 (m, 2H), 1.78–1.63 (m, 2H), 1.26 (t, J = 7.0 Hz, 3H). Mass spectrum m/z 332.15 [M + H]+.
General Procedure for Synthesis of Substituted Indole-2-carboxylates
To a solution of 2-alkylated ethyl acetoacetate 12 (2.9 mmol) in 30 mL of ethanol was added NaOAc (6.12 mmol) under an argon atmosphere, and the resulting mixture was stirred at room temperature for 45 min, followed by cooling to 0 °C. Aryldiazonium salt 10 was added to the reaction along with additional NaOAc to maintain the pH at 5, and the resulting solution was stirred for 3 h while maintaining the temperature between 0 and 5 °C. The reaction was quenched by adding saturated aqueous NaHCO3 solution, and the volatiles were removed under reduced pressure. The mixture was extracted with ethyl acetate (3×), the organic layer was washed with water and brine and dried over Na2SO4. The solvent was removed under vacuum to give the crude product as red oil which was a mixture of corresponding azo and hydrazone (13 and 14) intermediates. This mixture was passed through a small column of silica and dried under high vacuum to give a thick orange-brown mass. To this mass was added 100 mL of 20% sulfuric acid in anhydrous ethanol and was refluxed for 24 h. The reaction mixture was cooled to room temperature and neutralized by adding aqueous NaHCO3 solution. Crude product was extracted with ethyl acetate (3×). The combined organic layer was washed with water and brine, dried over Na2SO4, and concentrated under vacuum to yield a crude mixture which was purified by flash column chromatography on silica gel (0%–20%; EtOAc: hexanes) to give pure desired indole esters 15.
Ethyl 5-Chloro-3-ethyl-1H-indole-2-carboxylate (15a)
The compound was synthesized as per the general procedure as a white solid (64% yield). Rf = 0.35 (EtOAc/hexanes = 20/80). 1H NMR (400 MHz, chloroform-d): δ 8.74 (br s, 1H), 7.65 (d, J = 1.2 Hz, 1H), 7.30 (d, J = 8.8 Hz, 2H), 7.25 (dd, J = 8.8 Hz, J = 1.2 Hz, 1H), 4.42 (q, J = 7.2 Hz, 2H), 3.07 (q, J = 7.2 Hz, 2H), 1.43 (t, J = 7.2 Hz, 3H), 1.26 (t, J = 7.2 Hz, 3H). Mass spectrum m/z 251.07 [M + H]+.
Ethyl 3-Ethyl-5-nitro-1H-indole-2-carboxylate (15b)
The compound was synthesized as per the general procedure as a pale white solid (57% yield). Rf = 0.35 (EtOAc/hexanes = 20/80). 1H NMR (400 MHz, chloroform-d): δ 9.02 (s, 1H), 8.69 (d, J = 2.0 Hz, 1H), 8.22 (dd, J = 8.8 Hz, J = 2.4 Hz, 1H), 7.43 (d, J = 9.6 Hz, 1H), 4.46 (q, J = 7.6 Hz, 2H), 3.16 (q, J = 7.2 Hz, 2H), 1.45 (t, J = 7.6 Hz, 3H), 1.31 (t, J = 7.2 Hz, 3H). Mass spectrum m/z 263.11 [M + H]+.
Ethyl 5-Chloro-3-(2-(1,3-dioxoisoindolin-2-yl)ethyl)-1H-indole-2-carboxylate (15c)
The compound was synthesized as per the general procedure as a white solid (71% yield). 1H NMR (500 MHz, DMSO-d6): δ 11.73 (s, 1H), 7.79 (s, 4H), 7.63 (d, J = 1.5 Hz, 1H), 7.38 (d, J = 8.5 Hz, 1H), 7.20 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 4.19 (q, J = 7.0 Hz, 2H), 3.83 (t, J = 6.0 Hz, 2H), 3.35 (t, J = 6.0 Hz, 2H), 1.27 (t, J = 6.5 Hz, 3H). Mass spectrum m/z 397.09 [M + H]+
5-Chloro-3-ethyl-1H-indole-2-carboxylic Acid (16a)
To a solution of 15a (330 mg, 1.31 mmol) in 30 mL of dioxane was added a solution of KOH (440 mg, 7.7 mol) in 3 mL of water, and the resulting solution was refluxed for 2 h. It was then cooled to room temperature, concentrated under reduced pressure, and neutralized by addition of 1 N hydrochloric acid. The precipitated acid was filtered, washed with cold water, and air-dried to give pure acid 16a (306 mg, 98% yield) as white solid. 1H NMR (500 MHz, DMSO-d6): δ 13.04 (s, 1H), 11.57 (s, 1H,), 7.71 (s, 1H), 7.40 (d, J = 9.0 Hz, 1H), 7.23 (dd, J = 9.0 Hz, 2.0 Hz, 1H), 3.03 (q, J = 7.5 Hz, 2H), 1.16 (t, J = 7.5 Hz, 3H). Mass spectrum m/z 224.04 [M + H]+.
3-Ethyl-5-nitro-1H-indole-2-carboxylic Acid (16b)
The compound was synthesized as per the procedure for 16a, as a solid (72% yield). 1H NMR (400 MHz, DMSO-d6): δ 12.14 (s, 1H), 8.67 (d, J = 2.0 Hz, 1H), 8.11 (dd, J = 8.8 Hz, 2.5 Hz, 1H), 7.54 (d, J = 8.8 Hz, 1H), 3.12 (q, J = 8.0 Hz, 2H), 1.22 (t, J = 7.6 Hz, 3H). Mass spectrum m/z 235.06 [M + H]+.
3-Ethyl-5-nitro-N-[4-(piperidin-1-yl) phenethyl]-1H-indole-2-carboxamide (17)
The compound was synthesized as per the procedure for 1, as a solid (82% yield). Rf = 0.7 (EtOAc/hexanes = 50/50). 1H NMR (500 MHz, chloroform-d): δ 9.97 (s, 1H), 8.58 (d, J = 2.0 Hz, 1H), 8.16 (dd, J = 9.5 Hz, J = 2.0 Hz, 1H), 7.44 (d, J = 9.5 Hz, 1H), 7.15 (d, J = 8.5 Hz, 2H), 6.93 (d, J = 8.5 Hz, 2H), 6.18–6.07 (m, 1H), 3.82 (q, J = 6.0 Hz, 2H), 3.14 (t, J = 5.5 Hz, 4H), 2.91 (t, J = 7.0 Hz, 2H), 2.79 (q, J = 7.5 Hz, 2H), 1.76–1.68 (m, 4H), 1.62–1.54 (m, 2H), 1.13 (t, J = 7.5 Hz, 3H). Mass spectrum m/z 421.22 [M + H]+.
5-Amino-3-ethyl-N-[4-(piperidin-1-yl)phenethyl]-1H-indole-2-carboxamide (18)
To a solution of 17 (600 mg, 1.43 mmol) in 30 mL of anhydrous THF and anhydrous methanol (13:1) was added NiCl2·6H2O (356 mg, 1.49 mmol) under an argon atmosphere, and reaction mixture was stirred at room temperature for 45 min. The mixture was then cooled to −5 °C to which was added NaBH4 (324 mg, 8.56 mmol) portionwise, and the reaction was gradually warmed to room temperature while stirring for 1 h. Reaction was quenched with saturated NH4Cl and concentrated under reduced pressure. The residue was diluted with ethyl acetate and water and filtered, the organic layer was separated, and the aqueous layer was extracted with ethyl acetate (3×). The combined organic layer was washed with water and brine, dried (Na2SO4), and evaporated under vacuum to yield 18 (479 mg, 86% yield). Rf = 0.8 (MeOH/DCM = 20/80). 1H NMR (500 MHz, DMSO-d6): δ 10.60 (s, 1H), 7.74 (t, J = 5.5 Hz, 1H), 7.08 (d, J = 9.0 Hz, 3H), 6.85 (d, J = 9.0 Hz, 2H), 6.69 (d, J = 2.0 Hz, 1H), 6.62 (dd, J = 9.0 Hz, 2.0 Hz, 1H), 4.56 (br s, 2H), 3.44 (q, J = 6.5 Hz, 2H), 3.06 (t, J = 5.5 Hz, 4H), 2.90 (q, J = 7.5 Hz, 2H), 2.74 (t, J = 7.5 Hz, 2H), 1.64–1.56 (m, 4H), 1.54–1.46 (m, 2H), 1.11 (t, J = 7.5 Hz, 3H). Mass spectrum m/z 390.24 [M + H]+.
5-Azido-3-ethyl-N-[4-(piperidin-1-yl)phenethyl]-1H-indole-2-carboxamide (19)
To a solution of 18 (400 mg, 1.10 mmol) in 40 mL of THF under argon atmosphere was added tert-butyl nitrite (1.6 g, 15.5 mmol) and TMSN3 (1.2 g, 10.42 mmol), and the reaction was stirred overnight at room temperature. Solvent was evaporated under reduced pressure to give crude product which was purified using flash column chromatography on silica gel (10%–40%; EtOAc:hexanes) to yield pure 19 as a brown solid (180 mg, 42.0% yield). Rf = 0.81 (MeOH/DCM = 20/80). 1H NMR (500 MHz, chloroform-d): δ 9.14 (s, 1H), 7.35 (d, J = 9.0 Hz, 1H), 7.20 (d, J = 2.0 Hz, 1H), 7.14 (d, J = 9.0 Hz, 2H), 6.95 (dd, J = 9.0 Hz, J = 2.0 Hz, 1H), 6.92 (d, J = 9.0 Hz, 2H), 5.98 (br t, J = 6.0 Hz, 1H,), 3.78 (q, J = 6.5 Hz, 2H), 3.13 (t, J = 5.5 Hz, 4H), 2.89 (t, J = 6.5 Hz, 2H), 2.70 (q, J = 8.0 Hz, 2H), 1.75–1.68 (m, 4H), 1.64–1.54 (m, 2H), 1.08 (t, J = 8.0 Hz, 3H). Mass spectrum m/z 417.23 [M + H]+.
3-Ethyl-5-isothiocyanato-N-[4-(piperidin-1-yl)phenethyl]-1H-indole-2-carboxamide (20)
To a solution of 18 (400 mg, 1.10 mmol) in 5 mL of CH2Cl2 was added di(2-pyridyl) thionocarbonate (308 mg, 1.32 mmol), and the reaction mixture was stirred at room temperature for 15 min. It was quenched with cold water and extracted with dichloromethane (3×), and the combined organic layer was washed with brine, dried on Na2SO4, and evaporated under vacuum. The resultant residue was purified on silica gel (5%–25%; EtOAc:hexanes) to give pure compound 20 (388 mg, 87% yield). Rf = 0.35 (EtOAc/hexanes = 20/80). 1H NMR (400 MHz, chloroform-d): δ 9.79 (s, 1H), 7.46 (s, 1H), 7.35 (d, J = 8.8 Hz, 1H), 7.17–7.09 (m, 3H, especially 7.14, d, J = 8.0 Hz, 2H), 6.92 (d, J = 8.0 Hz, 2H), 6.03 (br t, J = 6.4 Hz, 1H), 3.80 (q, J = 6.0 Hz, 2H), 3.13 (br t, J = 5.6 Hz, 4H), 2.90 (t, J = 6.4 Hz, 2H), 2.70 (q, J = 7.6 Hz, 2H), 1.76–1.67 (m, 4H), 1.62–1.54 (m, 2H), 1.08 (t, J = 7.6 Hz, 3H). Mass spectrum m/z 433.21 [M + H]+.
3-(2-Aminoethyl)-5-chloro-1H-indole-2-carboxylic Acid (21)
To a solution of 15c (10.0 g, 25.2 mmol) in 200 mL of ethanol was added ethanolamine (3.08 g, 50.4 mmol), and the reaction mixture was refluxed for 14 h. It was then cooled to room temperature, volatiles were removed under vacuum, and the mixture was partitioned in ethyl acetate and water. The organic layer was separated, the aqueous layer was extracted with ethyl acetate (3×), and the combined organic layer was washed with brine and dried over Na2SO4. The solvent was removed under vacuum to give the lactam intermediate as a pure white solid (4.9 mg, 88% yield). Rf = 0.2 (EtOAc/hexanes = 50/50). 1H NMR (500 MHz, DMSO-d6) δ 11.81 (s, 1H), 7.68 (d, J = 2.0 Hz, 1H), 7.66 (s, 1H), 7.39 (d, J = 9.0 Hz, 1H), 7.21 (dd, J = 7.0 Hz, 1.5 Hz, 1H), 3.54 (t, J = 4.0 Hz, 2H), 2.92 (t, J = 4.0 Hz, 2H). Mass spectrum m/z 221.04 [M + H]+.
To this intermediate (4.5 g, 20.39 mmol) in 100 mL of dioxane:H2O (4:1) was added KOH (6.87 g, 122.0 mmol) in excess, and the mixture was refluxed overnight. It was then cooled to room temperature, and the volatiles were removed under vacuum. The residue was diluted with ice cold water and acidified to pH 5 with concd hydrochloric acid to give a precipitate which was filtered and air-dried to give the desired 21 as a white solid. (4.77 g, 98% yield). Rf = 0.15 (MeOH/DCM = 20/80). 1H NMR (500 MHz, DMSO-d6) δ 11.19 (s, 1H), 8.79 (br s, 3H), 7.62 (d, J = 2.0 Hz, 1H), 7.33 (d, J = 8.5 Hz, 1H), 7.08 (dd, J = 8.5 Hz, J = 2.0 Hz, 1H), 3.18 (t, J = 6.0 Hz, 2H), 3.01 (t, J = 6.0 Hz, 2H). Mass spectrum m/z 239.05 (M + H)+.
3-(2-((tert-Butoxycarbonyl)amino)ethyl)-5-chloro-1H-indole-2-carboxylic Acid (22)
To a solution of 21 (4.0 g, 16.76 mmol) in 80 mL of THF was added Boc anhydride (3.84 g, 17.60 mmol) at 0 °C. To this were added 50 mL of aq saturated NaHCO3 solution and water (2:1), and the reaction mixture was stirred at 0 °C for 3 h and then allowed to warm up to room temperature and stirred for 24 h. Solvent was then removed under vacuum, ice cold water was added to the residue, and it was acidified to pH 5 with cold 5% aq hydrochloric acid. The resultant precipitate was filtered, and the residue was washed with cold water and air-dried to give crude product as a cream colored solid which was recrystallized in methanol to give pure desired product 22 (5.0 g, 88% yield). Rf = 0.25 (MeOH/DCM = 10/90). 1H NMR (500 MHz, DMSO-d6) δ 11.59 (s, 1H), 7.68 (s, 1H), 7.39 (d, J = 8.5 Hz, 1H), 7.20 (dd, J = 8.5 Hz, J = 1.5 Hz, 1H), 6.87 (t as br s, 1H), 3.16 (t, J = 8.5 Hz, 2H), 3.15 (t, J = 8.5 Hz, 2H), 1.31 (s, 9H). Mass spectrum m/z 339.11 [M + H]+.
tert-Butyl (2-(5-Chloro-2-((4-(piperidin-1-yl)phenethyl)carbamoyl)-1H-indol-3-yl)ethyl)carbamate (23)
The compound was synthesized by coupling 22 with 8 as per the procedure for 1, as a white solid (68% yield). Rf = 0.8 (EtOAc/hexanes = 50/50). 1H NMR (500 MHz, chloroform-d) δ 9.39 (s, 1H), 7.64 (s, 1H), 7.54 (d, J = 2.0 Hz, 1H), 7.32 (d, J = 8.5 Hz, 1H), 7.21 (dd, J = 8.5 Hz, 2.0 Hz, 1H), 7.15 (d, J = 8.0 Hz, 2H), 6.88 (d, J = 8.5 Hz, 2H), 4.92 (br s, 1H), 3.75 (dd, J = 15.5 Hz, 6.5 Hz, 2H), 3.21 (dd, J = 16.0 Hz, 6.0 Hz, 2H), 3.11 (t, J = 6.0 Hz, 4H), 3.09–3.03 (m, 2H), 2.95 (t, J = 8.0 Hz, 2H), 1.70 (quint, J = 5.5 Hz, 4H), 1.58–1.54 (m, 2H), 1.49 (s, 9H). Mass spectrum m/z 526.25 [M + H]+.
3-(2-Aminoethyl)-5-chloro-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (24)
To 100 mL of CH2Cl2 was added 23 (2.0 g, 3.81 mmol) followed by dropwise addition of 10 mL of TFA, and the reaction was stirred at room temperature for 3 h. Volatiles were then removed under vacuum, and the crude product was washed with saturated NaHCO3 and extracted in dichloromethane (3×). The combined organic layer was washed with brine, dried over Na2SO4, and evaporated under vacuum to give 24 as a white solid (1.47 g, 91% yield). Rf = 0.2 (MeOH/DCM = 20/80). 1H NMR (500 MHz, chloroform-d) δ 10.32 (t as br s, 1H), 9.85 (s, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.34 (d, J = 8.5 Hz, 1H), 7.18 (dd, J = 9.0 Hz, 2.0 Hz, 1H), 7.13 (d, J = 9.0 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 3.72 (q, J = 6.5 Hz, 2H), 3.12 (t, J = 5.5 Hz, 4H), 2.94–2.84 (m, 6H), 1.76–1.66 (m, 4H), 1.62–1.52 (m, 2H), 1.35 (s, 2H). Mass spectrum m/z 426.20 [M + H]+.
3-(2-Azidoethyl)-5-chloro-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (25)
To a 10 mL solution of NaN3 (2.07 g, 31.9 mmol) in H2O was added trifluoromethanesulfonyl anhydride (3.0 g, 10.63 mmol) in DCM at 0 °C and stirred for 2 h while maintaining the temperature. The organic layer was separated, the aqueous layer was extracted with DCM (2×), and the organic layers were combined to afford TfN3. In a separate round-bottom flask, a solution of 24 (1.0 g, 2.35 mmol) in 20 mL of H2O:methanol (1:20) was treated with K2CO3 (2.6 g, 18.83 mmol) and CuSO4 (751 mg, 4.71 mmol). To this mixture was added the above TfN3 solution, and it was stirred at room temperature for 18 h. Volatiles were then removed under vacuum, and the residue was dissolved in DCM, washed with water and brine, and dried over Na2SO4. The organic layer was concentrated under vacuum, and the crude product was purified on silica gel (0%–20%; EtOAc:hexanes) to obtain 25 as a pure compound (679 mg, 64%). Rf = 0.7 (EtOAc/hexanes = 50/50). 1H NMR (500 MHz, chloroform-d) δ 11.49 (s, 1H), 8.16–8.06 (m, 1H), 7.75 (s, 1H), 7.43 (d, J = 8.5 Hz, 1H), 7.21 (d, J = 8.5 Hz, 1H), 7.09 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.0 Hz, 2H), 3.56–3.42 (m, 4H), 3.24 (t, J = 7.0 Hz, 2H), 3.13–3.01 (m, 4H), 2.76 (t, J = 7.0 Hz, 2H), 1.67–1.55 (m, 4H), 1.55–1.44 (m, 2H). Mass spectrum m/z 452.19 [M + H]+.
5-Chloro-3-(2-isothiocyanatoethyl)-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (26)
To a solution of 24 (1.0 g, 2.353 mmol) in 50 mL of CH2Cl2 was added di(2-pyridyl) thionocarbonate (656 mg, 2.82 mmol), and the reaction mixture was stirred at room temperature for 15 min. It was quenched with cold water and extracted with dichloromethane (3×), and the combined organic layer was washed with brine, dried on Na2SO4, and evaporated under vacuum. The resultant residue was purified on silica gel (5%–25%; EtOAc:hexanes) to give pure compound 26 (956 mg, 87% yield). Rf = 0.35 (EtOAc/hexanes = 20/80). 1H NMR (500 MHz, DMSO-d6) δ 11.52 (s, 1H), 8.17 (t, J = 5.5 Hz, 1H), 7.81 (d, J = 2.0 Hz, 1H), 7.44 (d, J = 8.5 Hz, 1H), 7.22 (ddd, J = 8.5 Hz, 2.0 Hz, 1.0 Hz, 1H), 7.10 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 3.82 (t, J = 7.0 Hz, 2H), 3.48 (q, J = 7.0 Hz, 2H), 3.38 (t, J = 7.0 Hz, 2H), 3.07 (t, J = 5.5 Hz, 4H), 2.76 (t, J = 7.0 Hz, 2H), 1.65–1.56 (m, 4H), 1.56–1.46 (m, 2H). Mass spectrum m/z 468.16 [M + H]+.
2-Bromo-6-(pyrrolidin-1-yl)pyridine (29)
A mixture of 27 (10.0 g, 42.2 mmol) and 28 (13.87 mL, 169 mmol) was stirred for 1 h at room temperature. The reaction mixture was quenched with 100 mL of saturated NaHCO3 solution and diluted with 100 mL of dichloromethane. The organic layer was separated, washed with water and brine, and dried (MgSO4) and concentrated under reduced pressure. The resulting crude product was crystallized from methanol (50 mL) to afford the desired product 29 (8.5 g, 37.4 mmol, 89% yield) as a white solid. 1H NMR (500 MHz, methanol-d4) δ: 7.22 (t, J = 8.0 Hz, 1H), 6.64 (d, J = 7.5 Hz, 1H), 6.23 (d, J = 8.0 Hz, 1H), 3.43 (t, J = 6.5 Hz, 4H), 2.04–1.94 (m, 4H). Mass spectrum m/z 228.01 [M + H]+.
2-(3-Nitrophenyl)-6-(pyrrolidin-1-yl)pyridine (30)
To a solution of 29 (2 g, 8.81 mmol) and 3-nitrophenylboronic acid (1.62 g, 9.69 mmol) in 1,2-dimethoxyethane (10 mL) were added Ba(OH)2 (3.32 g, 19.37 mmol) and water (4 mL) under argon atmosphere. The contents were degassed, Pd(Ph3P)4 (0.305 g, 0.26 mmol) was added, and the resulting mixture was irradiated with microwaves at 150 °C for 15 min. It was then diluted with 200 mL of ethyl acetate and water (1:1), and the organic layer was separated, washed with water and brine, and dried over MgSO4. Volatiles were concentrated under reduced pressure to give crude product which was purified by silica gel chromatography (20–70%; EtOAc:hexanes) to give 30 (2.05 g, 86% yield) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ: 8.57 (t, J = 1.5 Hz, 1H), 8.50 (td, J = 8.0 Hz, J = 1.5 Hz, 1H), 8.23 (ddd, J = 8.0 Hz, J = 2.5 Hz, J = 1.0 Hz, 1H), 7.74 (t, J = 8.0 Hz, 1H), 7.63 (dd, J = 8.0 Hz, J = 7.0 Hz, 1H), 7.28 (d, J = 7.0 Hz, 1H), 6.51 (d, J = 8.0 Hz, 1H), 3.49 (t, J = 6.5 Hz, 4H), 2.04–1.94 (quin, J = 3.0 Hz, 4H). Mass spectrum m/z 270.1 [M + H]+.
3-(6-(Pyrrolidin-1-yl)pyridin-2-yl)aniline (31)
To a solution of 30 (5 g, 18.57 mmol) in methanol (100 mL) was added catalytic amounts of Raney-nickel, and the resultant mixture was stirred for 3 h under hydrogen atmosphere. Raney-nickel was filtered off and, filtrate was concentrated and purified by silica gel chromatography (20–70%; EtOAc:hexanes) to give 31 (4.25 g, 96% yield) as a whitish solid. 1H NMR (500 MHz, chloroform-d) δ: 7.52–7.48 (m, 1H), 7.46 (t, J = 2.0 Hz, 1H), 7.45–7.41 (m, 1H), 7.29 (s, 1H), 7.23 (t, J = 8.0 Hz, 1H), 6.99 (d, J = 7.0 Hz, 1H), 6.72 (ddd, J = 5.5 Hz, J = 2.5 Hz, J = 1.0 Hz, 1H), 3.75 (br s, 2H), 3.57 (t, J = 6.5 Hz, 4H), 2.08–1.80 (m, 4H). Mass spectrum m/z 240.2 [M + H]+.
1-(4-Azidophenyl)-3-(3-(6-(pyrrolidin-1-yl)pyridin-2-yl)phenyl)urea (33)
To a solution of triphosgene (70 mg, 0.21 mmol) in toluene (10 mL) was added 31 (130 mg, 0.54 mmol) followed by Et3N (2.4 mL, 23.6 mmol) under inert atmosphere, and the mixture was heated to 70 °C for 3 h under argon atmosphere. The reaction was concentrated under reduced pressure and carried forward to the next step without any purification. To a solution of this isocyanate intermediate in DCM (4 mL) was added 4-azidoaniline 32 (72 mg, 0.167 mmol) followed by Et3N (2.4 mL, 23.6 mmol), and the mixture was stirred at 0 °C for 6 h. The reaction mixture was diluted in 20 mL of DCM and water (1:1). The organic layer was separated, washed with water, dried (Na2SO4), and concentrated under reduced pressure. The crude product was purified by silica gel chromatography (10–50%; EtOAc:hexanes) to afford the desired product 33 (150 mg, 75% yield) as white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.78 (s, 1H), 8.75 (s, 1H), 8.07 (t, J = 2.0 Hz, 1H), 7.63 (dt, J = 7.5 Hz, 1.5 Hz, 1H), 7.56 (dd, J = 8.5 Hz, 7.0 Hz, 2H), 7.52 (d, J = 9.0 Hz, 2H) 7.34 (t, J = 7.5 Hz, 1H), 7.08–7.03 (m, 3H), 6.42 (d, J = 9.0 Hz, 1H), 3.54–3.42 (m, 4H), 2.04–1.92 (m, 4H). Mass spectrum m/z 400.18 [M + H]+.
1-(4-Isothiocyanatophenyl)-3-(3-(6-(pyrrolidin-1-yl)pyridin-2-yl)phenyl)urea (34)
To a solution of 33 (25 mg, 0.06 mmol) in 5 mL of benzene under an argon atmosphere was added triphenylphosphine (34.2 mg, 0.12 mmol), and the reaction mixture was refluxed for 4 h. The reaction mixture was cooled to room temperature, 1 mL of CS2 was added to this, and the reaction mix was stirred at 40 °C for 12 h. Volatiles were evaporated under reduced pressure to obtain crude product which was purified using flash column chromatography (10%–40%; EtOAc:hexanes) to obtain pure 34 as a white solid (17 mg, 74% yield). Rf = 0.78 (MeOH/DCM = 20/80). 1H NMR (500 MHz, DMSO-d6) δ 8.96 (s, 1H), 8.84 (s, 1H), 8.07 (t, J = 2.0 Hz, 1H), 7.64 (d, J = 7.5 Hz, 1H), 7.59–7.51 (m, 4H, esp. 7.54, d, J = 9.5 Hz, 2H), 7.38 (d, J = 9.0 Hz, 2H), 7.35 (t, J = 7.0 Hz, 1H), 7.06 (d, J = 7.5 Hz, 1H), 6.42 (d, J = 8.5 Hz, 1H), 3.54–3.42 (m, 4H), 2.04–1.92 (m, 4H). Mass spectrum m/z 416.15 [M + H]+.
1-(4-Benzoylphenyl)-3-(3-(6-(pyrrolidin-1-yl)pyridin-2-yl)phenyl)urea (36)
To a solution of triphosgene (23 mg, 0.77 mmol) in toluene (2 mL) was added 35 (40 mg, 0.16 mmol) followed by Et3N (0.78 mL, 7.8 mmol) under inert atmosphere, and the mixture was heated to 70 °C for 3 h under argon atmosphere. The reaction was concentrated under reduced pressure and carried forward to the next step without any purification.
To this intermediate dissolved in THF (10 mL) was added 31 (39 mg, 0.167 mmol) followed by Et3N (0.78 mL, 7.8 mmol), and the mixture was stirred at 0 °C for 1 h and then at room temperature for 5 h. The reaction mixture was concentrated under reduced pressure, and crude product was recrystallized from methanol to give desired product 36 (60 mg, 78% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 9.19 (s, 1H), 8.92 (s, 1H), 8.10 (t, J = 2.0 Hz, 1H), 7.75 (d, J = 9.0 Hz, 2H), 7.71 (dd, J = 8.0 Hz, 1.5 Hz, 2H), 7.69–7.63 (m, 4H), 7.61–7.53 (m, 4H), 7.37 (t, J = 8.0 Hz, 1H), 7.07 (d, J = 7.5 Hz, 1H), 6.43 (d, J = 8.5 Hz, 1H), 3.54–3.45 (m, 4H), 2.03–1.93 (m, 4H). Mass spectrum m/z 463.21 [M + H]+.
Acknowledgments
The work was supported by National Institutes of Health grants DA027113 and EY024717 to G.A.T. and DA09158 to A.M. A portion of this work was submitted in 2011 by A. Kulkarni in partial fulfillment of M.S. degree requirements from Northeastern University, Boston, MA.
Glossary
Abbreviations Used
- AEA
arachidonoylethanol amine
- 2-AG
2-arachidonoylglycerol
- BSA
bovine serum albumin
- cAMP
cyclic adenosine monophosphate
- CB1R
cannabinoid 1 receptor
- CB2R
cannabinoid 2 receptor
- CHO
Chinese hamster ovary cells
- CI
confidence interval
- CO2
carbon dioxide
- CNS
central nervous system
- DMSO
dimethyl sulfoxide
- DCM
dichloromethane
- DIPEA
N,N-diisopropylethylamine
- DPT
di-2-pyridyl thionocarbonate
- EDTA
ethylenediaminetetraacetic acid
- EtOAc
ethyl acetate
- Et3N
triethylamine
- EDCI
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- GDP
guanosine diphosphate
- GTP
guanosine triphosphate
- GPCRs
G-protein-coupled receptors
- [35S]GTPγS
guanosine 5′-O-(3-[35S]thio)triphosphate
- HEK293
human embryonic kidney 293 cells
- HPLC
high performance liquid chromatography
- HTS
high-throughput screening
- Hz
hertz
- HOBT
hydroxybenzotriazole
- K2CO3
potassium carbonate
- KOH
potassium hydroxide
- LAPS
ligand-assisted protein structure
- LCMS
liquid chromatography mass spectrometry
- LiAlH4
lithium aluminum hydride
- MgSO4
magnesium sulfate
- MHz
megahertz
- MeOH
methanol
- NaBH4
sodium borohydride
- NAMs
negative allosteric modulators
- Na2SO4
sodium sulfate
- NaOAc
sodium acetate
- NaHCO3
sodium bicarbonate
- NaNO2
sodium nitrite
- NiCl2·6H2O
nickel chloride hexahydrate
- NH4OAc
ammonium acetate
- NH4Cl
ammonium chloride
- NMR
nuclear magnetic resonance spectroscopy
- NMP
N-methylpyrrolidinone
- OCC
optimized cell culture
- RLU
relative light units
- THF
tetrahydrofuran
- TLC
thin-layer chromatography
- SAR
structure–activity relationship
- SEM
standard error of mean
- Tris
tris(hydroxymethyl)aminomethane
- TMSN3
trimethylsilyl azide
- UV
ultraviolet
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b01303.
SMILES data (CSV)
The authors declare no competing financial interest.
Supplementary Material
References
- Pertwee R. G.; Howlett A. C.; Abood M. E.; Alexander S. P.; Di Marzo V.; Elphick M. R.; Greasley P. J.; Hansen H. S.; Kunos G.; Mackie K.; Mechoulam R.; Ross R. A. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and their Ligands: Beyond CB(1) and CB(2). Pharmacol. Rev. 2010, 62, 588–631. 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P.; Batkai S.; Kunos G. The Endocannabinoid System as an Emerging Target of Pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P.; Kunos G. Modulating the Endocannabinoid System in Human Health and Disease-Successes and Failures. FEBS J. 2013, 280, 1918–1943. 10.1111/febs.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M.; Bab I.; Biro T.; Cabral G. A.; Dey S. K.; Di Marzo V.; Konje J. C.; Kunos G.; Mechoulam R.; Pacher P.; Sharkey K. A.; Zimmer A. Endocannabinoid Signaling at the Periphery: 50 Years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. 10.1016/j.tips.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane W. A.; Dysarz F. A. III; Johnson M. R.; Melvin L. S.; Howlett A. C. Determination and Characterization of a Cannabinoid Receptor in Rat Brain. Mol. Pharmacol. 1988, 34, 605–613. [PubMed] [Google Scholar]
- Matsuda L. A.; Lolait S. J.; Brownstein M. J.; Young A. C.; Bonner T. I. Structure of a Cannabinoid Receptor and Functional Expression of the Cloned cDNA. Nature 1990, 346, 561–564. 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- Munro S.; Thomas K. L.; Abu-Shaar M. Molecular Characterization of a Peripheral Receptor for Cannabinoids. Nature 1993, 365, 61–65. 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Galiegue S.; Mary S.; Marchand J.; Dussossoy D.; Carriere D.; Carayon P.; Bouaboula M.; Shire D.; Le Fur G.; Casellas P. Expression of Central and Peripheral Cannabinoid Receptors in Human Immune Tissues and Leukocyte Subpopulations. Eur. J. Biochem. 1995, 232, 54–61. 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- Cabral G. A.; Raborn E. S.; Griffin L.; Dennis J.; Marciano-Cabral F. CB2 Receptors in the Brain: Role in Central Immune Function. Br. J. Pharmacol. 2008, 153, 240–251. 10.1038/sj.bjp.0707584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn E. J.; Deng L.; Thakur G. A.; Vemuri K.; Zvonok A. M.; Lai Y. Y.; Makriyannis A.; Hohmann A. G. Prophylactic Cannabinoid Administration Blocks the Development of Paclitaxel-Induced Neuropathic Nociception during Analgesic Treatment and Following Cessation of Drug Delivery. Mol. Pain 2014, 10, 27. 10.1186/1744-8069-10-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L.; Guindon J.; Vemuri V. K.; Thakur G. A.; White F. A.; Makriyannis A.; Hohmann A. G. The Maintenance of Cisplatin- and Paclitaxel-Induced Mechanical and Cold Allodynia Is Suppressed by Cannabinoid CB(2) Receptor Activation and Independent of CXCR4 Signaling in Models of Chemotherapy-Induced Peripheral Neuropathy. Mol. Pain 2012, 8, 71. 10.1186/1744-8069-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson J. L.; Gentry K. R.; Dengler E. C.; Wallace J. A.; Kerwin A. A.; Armijo L. M.; Kuhn M. N.; Thakur G. A.; Makriyannis A.; Milligan E. D. Intrathecal Cannabilactone CB(2)R Agonist, AM1710, Controls Pathological Pain and Restores Basal Cytokine Levels. Pain 2012, 153, 1091–1106. 10.1016/j.pain.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn E. J.; Thakur G. A.; Wood J. A.; Zvonok A. M.; Makriyannis A.; Hohmann A. G. Pharmacological Characterization of AM1710, a Putative Cannabinoid CB2 Agonist from the Cannabilactone Class: Antinociception Without Central Nervous System Side-Effects. Pharmacol., Biochem. Behav. 2011, 98, 493–502. 10.1016/j.pbb.2011.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanolkar A. D.; Lu D.; Ibrahim M.; Duclos R. I. Jr.; Thakur G. A.; Malan T. P. Jr.; Porreca F.; Veerappan V.; Tian X.; George C.; Parrish D. A.; Papahatjis D. P.; Makriyannis A. Cannabilactones: A Novel Class of CB2 Selective Agonists with Peripheral Analgesic Activity. J. Med. Chem. 2007, 50, 6493–6500. 10.1021/jm070441u. [DOI] [PubMed] [Google Scholar]
- Wilkerson J. L.; Gentry K. R.; Dengler E. C.; Wallace J. A.; Kerwin A. A.; Kuhn M. N.; Zvonok A. M.; Thakur G. A.; Makriyannis A.; Milligan E. D. Immunofluorescent Spectral Analysis Reveals the Intrathecal Cannabinoid Agonist, AM1241, Produces Spinal Anti-Inflammatory Cytokine Responses in Neuropathic Rats Exhibiting Relief from Allodynia. Brain Behav. 2012, 2, 155–177. 10.1002/brb3.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S.; Thatte J.; Buzard D. J.; Jones R. M. Therapeutic Utility of Cannabinoid Receptor Type 2 (CB(2)) Selective Agonists. J. Med. Chem. 2013, 56, 8224–8256. 10.1021/jm4005626. [DOI] [PubMed] [Google Scholar]
- Dhopeshwarkar A.; Mackie K. CB2 Cannabinoid Receptors as a Therapeutic Target-What Does the Future Hold?. Mol. Pharmacol. 2014, 86, 430–437. 10.1124/mol.114.094649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S. L.; Thakur G. A.; Makriyannis A. Cannabinergic Ligands. Chem. Phys. Lipids 2002, 121, 3–19. 10.1016/S0009-3084(02)00143-3. [DOI] [PubMed] [Google Scholar]
- Pertwee R. G. Emerging Strategies for Exploiting Cannabinoid Receptor Agonists as Medicines. Br. J. Pharmacol. 2009, 156, 397–411. 10.1111/j.1476-5381.2008.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur G. A.; Tichkule R.; Bajaj S.; Makriyannis A. Latest Advances in Cannabinoid Receptor Agonists. Expert Opin. Ther. Pat. 2009, 19, 1647–1673. 10.1517/13543770903436505. [DOI] [PubMed] [Google Scholar]
- Moreira F. A.; Grieb M.; Lutz B. Central Side-Effects of Therapies Based on CB1 Cannabinoid Receptor Agonists and Antagonists: Focus on Anxiety and Depression. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 133–144. 10.1016/j.beem.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Moreira F. A.; Crippa J. A. The Psychiatric Side-Effects of Rimonabant. Rev. Bras. Psiquiatr. 2009, 31, 145–153. 10.1590/S1516-44462009000200012. [DOI] [PubMed] [Google Scholar]
- Janero D. R. Cannabinoid-1 Receptor (CB1R) Blockers as Medicines: Beyond Obesity and Cardiometabolic Disorders to Substance Abuse/Drug Addiction with CB1R Neutral Antagonists. Expert Opin. Emerging Drugs 2012, 17, 17–29. 10.1517/14728214.2012.660916. [DOI] [PubMed] [Google Scholar]
- Kunos G.; Osei-Hyiaman D.; Batkai S.; Sharkey K. A.; Makriyannis A. Should Peripheral CB(1) Cannabinoid Receptors Be Selectively Targeted for Therapeutic Gain?. Trends Pharmacol. Sci. 2009, 30, 1–7. 10.1016/j.tips.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman J.; Delatte M. S.; Paronis C. A.; Vemuri K.; Thakur G. A.; Makriyannis A. Some Effects of CB1 Antagonists with Inverse Agonist and Neutral Biochemical Properties. Physiol. Behav. 2008, 93, 666–670. 10.1016/j.physbeh.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. R.; Baillie G. L.; Thomas A.; Stevenson L. A.; Easson M.; Goodwin R.; McLean A.; McIntosh L.; Goodwin G.; Walker G.; Westwood P.; Marrs J.; Thomson F.; Cowley P.; Christopoulos A.; Pertwee R. G.; Ross R. A. Allosteric Modulation of the Cannabinoid CB1 receptor. Mol. Pharmacol. 2005, 68, 1484–1495. 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- Horswill J. G.; Bali U.; Shaaban S.; Keily J. F.; Jeevaratnam P.; Babbs A. J.; Reynet C.; Wong-Kai-In P. PSNCBAM-1, A Novel Allosteric Antagonist at Cannabinoid CB1 Receptors with Hypophagic Effects in Rats. Br. J. Pharmacol. 2007, 152, 805–814. 10.1038/sj.bjp.0707347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamplona F. A.; Ferreira J.; Menezes de Lima O. Jr.; Duarte F. S.; Bento A. F.; Forner S.; Villarinho J. G.; Bellocchio L.; Wotjak C. T.; Lerner R.; Monory K.; Lutz B.; Canetti C.; Matias I.; Calixto J. B.; Marsicano G.; Guimaraes M. Z.; Takahashi R. N. Anti-Inflammatory Lipoxin A4 is an Endogenous Allosteric Enhancer of CB1 Cannabinoid Receptor. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 21134–21139. 10.1073/pnas.1202906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee M.; Vitiello S.; Bellocchio L.; Hebert-Chatelain E.; Monlezun S.; Martin-Garcia E.; Kasanetz F.; Baillie G. L.; Panin F.; Cathala A.; Roullot-Lacarriere V.; Fabre S.; Hurst D. P.; Lynch D. L.; Shore D. M.; Deroche-Gamonet V.; Spampinato U.; Revest J. M.; Maldonado R.; Reggio P. H.; Ross R. A.; Marsicano G.; Piazza P. V. Pregnenolone Can Protect the Brain from Cannabis Intoxication. Science 2014, 343, 94–98. 10.1126/science.1243985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. I.; Lewis R. J. Emerging Opportunities for Allosteric Modulation of G-Protein Coupled Receptors. Biochem. Pharmacol. 2013, 85, 153–162. 10.1016/j.bcp.2012.09.001. [DOI] [PubMed] [Google Scholar]
- Fay J. F.; Farrens D. L. A Key Agonist-Induced Conformational Change in the Cannabinoid Receptor CB1 Is Blocked by the Allosteric Ligand Org 27569. J. Biol. Chem. 2012, 287, 33873–33882. 10.1074/jbc.M112.352328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. J.; Christopoulos A.; Lindsley C. W. Allosteric Modulators of GPCRs: A Novel Approach for the Treatment of CNS Disorders. Nat. Rev. Drug Discovery 2009, 8, 41–54. 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro H. A.; Howard J. L.; Pollard G. T.; Carroll F. I. Positive Allosteric Modulation of the Human Cannabinoid (CB) Receptor by RTI-371, A Selective Inhibitor of the Dopamine Transporter. Br. J. Pharmacol. 2009, 156, 1178–1184. 10.1111/j.1476-5381.2009.00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priestley R. S.; Nickolls S. A.; Alexander S. P.; Kendall D. A. A Potential Role for Cannabinoid Receptors in the Therapeutic Action of Fenofibrate. FASEB J. 2015, 29, 1446–1455. 10.1096/fj.14-263053. [DOI] [PubMed] [Google Scholar]
- Laprairie R. B.; Bagher A. M.; Kelly M. E.; Denovan-Wright E. M. Cannabidiol is a Negative Allosteric Modulator of the Type 1 Cannabinoid Receptor. Br. J. Pharmacol. 2015, 172, 4790. 10.1111/bph.13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur G. A.; Kulkarni P. M.. Preparation of Phenylindole Derivatives for Use as CB1 Cannabinoid Receptor Modulators. WO2013103967A1, Jul 11, 2013.
- Ignatowska-Jankowska B. M.; Baillie G. L.; Kinsey S.; Crowe M.; Ghosh S.; Owens R. A.; Damaj I. M.; Poklis J.; Wiley J. L.; Zanda M.; Zanato C.; Greig I. R.; Lichtman A. H.; Ross R. A. A Cannabinoid CB1 Receptor-Positive Allosteric Modulator Reduces Neuropathic Pain in the Mouse with No Psychoactive Effects. Neuropsychopharmacology 2015, 10.1038/npp.2015.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamage T. F.; Ignatowska-Jankowska B. M.; Wiley J. L.; Abdelrahman M.; Trembleau L.; Greig I. R.; Thakur G. A.; Tichkule R.; Poklis J.; Ross R. A.; Pertwee R. G.; Lichtman A. H. In-vivo Pharmacological Evaluation of the CB1-Receptor Allosteric Modulator Org-27569. Behav. Pharmacol. 2014, 25, 182–185. 10.1097/FBP.0000000000000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y.; Qiu Y.; Jing L.; Thorn D. A.; Zhang Y.; Li J. X. Behavioral Effects of the Cannabinoid CB1 Receptor Allosteric Modulator ORG27569 in Rats. Pharmacol. Res. Perspect. 2014, 2 (6), e00069. 10.1002/prp2.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur G. A.; Tichkule R. B.; Kulkarni P. M.; Kulkarni A. R.. Allosteric modulators of the cannabinoid 1 receptor. WO/2015/027160A2, Aug 22, 2014.
- Baillie G. L.; Horswill J. G.; Anavi-Goffer S.; Reggio P. H.; Bolognini D.; Abood M. E.; McAllister S.; Strange P. G.; Stephens G. J.; Pertwee R. G.; Ross R. A. CB(1) Receptor Allosteric Modulators Display Both Agonist and Signaling Pathway Specificity. Mol. Pharmacol. 2013, 83, 322–338. 10.1124/mol.112.080879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shore D. M.; Baillie G. L.; Hurst D. H.; Navas F. 3rd; Seltzman H. H.; Marcu J. P.; Abood M. E.; Ross R. A.; Reggio P. H. Allosteric Modulation of a Cannabinoid G Protein-Coupled Receptor: Binding Site Elucidation and Relationship to G Protein Signaling. J. Biol. Chem. 2014, 289, 5828–5845. 10.1074/jbc.M113.478495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur A.; Samaniego P.; Thakur G. A.; Makriyannis A.; Abood M. E. Mapping the Structural Requirements in the CB1 Cannabinoid Receptor Transmembrane Helix II for Signal Transduction. J. Pharmacol. Exp. Ther. 2008, 325, 341–348. 10.1124/jpet.107.133256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K. H.; Mahmoud M. M.; Kendall D. A. Allosteric Modulator ORG27569 Induces CB1 Cannabinoid Receptor High Affinity Agonist Binding State, Receptor Internalization, and Gi Protein-Independent ERK1/2 Kinase Activation. J. Biol. Chem. 2012, 287, 12070–12082. 10.1074/jbc.M111.316463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei Y.; Mercier R. W.; Anday J. K.; Thakur G. A.; Zvonok A. M.; Hurst D.; Reggio P. H.; Janero D. R.; Makriyannis A. Ligand-Binding Architecture of Human CB2 Cannabinoid Receptor: Evidence for Receptor Subtype-Specific Binding Motif and Modeling GPCR Activation. Chem. Biol. 2008, 15, 1207–1219. 10.1016/j.chembiol.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picone R. P.; Khanolkar A. D.; Xu W.; Ayotte L. A.; Thakur G. A.; Hurst D. P.; Abood M. E.; Reggio P. H.; Fournier D. J.; Makriyannis A. (−)-7′-Isothiocyanato-11-hydroxy-1′,1′-dimethylheptylhexahydrocannabinol (AM841), a High-Affinity Electrophilic Ligand, Interacts Covalently with a Cysteine in Helix Six and Activates the CB1 Cannabinoid Receptor. Mol. Pharmacol. 2005, 68, 1623–1635. 10.1124/mol.105.014407. [DOI] [PubMed] [Google Scholar]
- Janero D. R.; Yaddanapudi S.; Zvonok N.; Subramanian K. V.; Shukla V. G.; Stahl E.; Zhou L.; Hurst D.; Wager-Miller J.; Bohn L. M.; Reggio P. H.; Mackie K.; Makriyannis A. Molecular-Interaction and Signaling Profiles of AM3677, a Novel Covalent Agonist Selective for the Cannabinoid 1 Receptor. ACS Chem. Neurosci. 2015, 6, 1400. 10.1021/acschemneuro.5b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymanski D. W.; Papanastasiou M.; Melchior K.; Zvonok N.; Mercier R. W.; Janero D. R.; Thakur G. A.; Cha S.; Wu B.; Karger B.; Makriyannis A. Mass Spectrometry-Based Proteomics of Human Cannabinoid Receptor 2: Covalent Cysteine 6.47(257)-Ligand Interaction Affording Megagonist Receptor Activation. J. Proteome. Res. 2011, 10, 4789–4798. 10.1021/pr2005583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makriyannis A. 2012 Division of Medicinal Chemistry Award Address. Trekking the Cannabinoid Road: A Personal Perspective. J. Med. Chem. 2014, 57, 3891–3911. 10.1021/jm500220s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichert D.; Gmeiner P. Covalent Molecular Probes for Class A G Protein-Coupled Receptors: Advances and Applications. ACS Chem. Biol. 2015, 10, 1376–1386. 10.1021/acschembio.5b00070. [DOI] [PubMed] [Google Scholar]
- Davie B. J.; Sexton P. M.; Capuano B.; Christopoulos A.; Scammells P. J. Development of a Photoactivatable Allosteric Ligand for the M1 Muscarinic Acetylcholine Receptor. ACS Chem. Neurosci. 2014, 5, 902–907. 10.1021/cn500173x. [DOI] [PubMed] [Google Scholar]
- Davie B. J.; Valant C.; White J. M.; Sexton P. M.; Capuano B.; Christopoulos A.; Scammells P. J. Synthesis and Pharmacological Evaluation of Analogues of Benzyl Quinolone Carboxylic Acid (BQCA) Designed To Bind Irreversibly to an Allosteric Site of the M(1) Muscarinic Acetylcholine Receptor. J. Med. Chem. 2014, 57, 5405–5418. 10.1021/jm500556a. [DOI] [PubMed] [Google Scholar]
- Pace N. J.; Weerapana E. Diverse Functional Roles of Reactive Cysteines. ACS Chem. Biol. 2013, 8, 283–296. 10.1021/cb3005269. [DOI] [PubMed] [Google Scholar]
- Tahtaoui C.; Balestre M. N.; Klotz P.; Rognan D.; Barberis C.; Mouillac B.; Hibert M. Identification of the Binding Sites of the SR49059 Nonpeptide Antagonist into the V1a Vasopressin Receptor Using Sulfydryl-Reactive Ligands and Cysteine Mutants as Chemical Sensors. J. Biol. Chem. 2003, 278, 40010–40019. 10.1074/jbc.M301128200. [DOI] [PubMed] [Google Scholar]
- Keenan C. M.; Storr M. A.; Thakur G. A.; Wood J. T.; Wager-Miller J.; Straiker A.; Eno M. R.; Nikas S. P.; Bashashati M.; Hu H.; Mackie K.; Makriyannis A.; Sharkey K. A. AM841, a Covalent Cannabinoid Ligand, Powerfully Slows Gastrointestinal Motility in Normal and Stressed Mice in a Peripherally Restricted Manner. Br. J. Pharmacol. 2015, 172, 2406–2418. 10.1111/bph.13069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fichna J.; Bawa M.; Thakur G. A.; Tichkule R.; Makriyannis A.; McCafferty D. M.; Sharkey K. A.; Storr M. Cannabinoids Alleviate Experimentally Induced Intestinal Inflammation by Acting at Central and Peripheral Receptors. PLoS One 2014, 9, e109115. 10.1371/journal.pone.0109115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Xu W.; Vadivel S. K.; Fan P.; Makriyannis A. High Affinity Electrophilic and Photoactivatable Covalent Endocannabinoid Probes for the CB1 Receptor. J. Med. Chem. 2005, 48, 6423–6429. 10.1021/jm050272i. [DOI] [PubMed] [Google Scholar]
- Blankman J. L.; Cravatt B. F. Chemical Probes of Endocannabinoid Metabolism. Pharmacol. Rev. 2013, 65, 849–871. 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K. H.; Mahmoud M. M.; Samala S.; Lu D.; Kendall D. A. Profiling Two Indole-2-carboxamides for Allosteric Modulation of the CB1 Receptor. J. Neurochem. 2013, 124, 584–589. 10.1111/jnc.12115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawston E. E.; Connor M.; Di Marzo V.; Silvestri R.; Glass M. Distinct Temporal Fingerprint for Cyclic Adenosine Monophosphate (cAMP) Signaling of Indole-2-carboxamides as Allosteric Modulators of the Cannabinoid Receptors. J. Med. Chem. 2015, 58, 5979. 10.1021/acs.jmedchem.5b00579. [DOI] [PubMed] [Google Scholar]
- German N.; Decker A. M.; Gilmour B. P.; Gay E. A.; Wiley J. L.; Thomas B. F.; Zhang Y. Diarylureas as Allosteric Modulators of the Cannabinoid CB1 Receptor: Structure–Activity Relationship Studies on 1-(4-Chlorophenyl)-3-{3-[6-(pyrrolidin-1-yl)pyridin-2-yl]phenyl}urea (PSNCBAM-1). J. Med. Chem. 2014, 57, 7758–7769. 10.1021/jm501042u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana L.; Ali H. I.; Olszewska T.; Ahn K. H.; Damaraju A.; Kendall D. A.; Lu D. Optimization of Chemical Functionalities of Indole-2-carboxamides To Improve Allosteric Parameters for the Cannabinoid Receptor 1 (CB1). J. Med. Chem. 2014, 57, 3040–3052. 10.1021/jm5000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud M. M.; Ali H. I.; Ahn K. H.; Damaraju A.; Samala S.; Pulipati V. K.; Kolluru S.; Kendall D. A.; Lu D. Structure–Activity Relationship Study of Indole-2-carboxamides Identifies a Potent Allosteric Modulator for the Cannabinoid Receptor 1 (CB1). J. Med. Chem. 2013, 56, 7965–7975. 10.1021/jm4009828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piscitelli F.; Ligresti A.; La Regina G.; Coluccia A.; Morera L.; Allara M.; Novellino E.; Di Marzo V.; Silvestri R. Indole-2-carboxamides as Allosteric Modulators of the Cannabinoid CB(1) Receptor. J. Med. Chem. 2012, 55, 5627–5631. 10.1021/jm201485c. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Raj K. K.; Bailey M.; Alling T.; Parish T.; Rawat D. S. Antimycobacterial Activity Evaluation, Time-Kill Kinetic and 3D-QSAR Study of C-(3-aminomethyl-cyclohexyl)-methylamine Derivatives. Bioorg. Med. Chem. Lett. 2013, 23, 1365–1369. 10.1016/j.bmcl.2012.12.083. [DOI] [PubMed] [Google Scholar]
- Benington F.; Morin R. D.; Clark L. C. Jr. Mescaline Analogs. V. p-Dialkylamino-β-phenethylamines and 9-(β-Aminoethyl)julolidine. J. Org. Chem. 1956, 21, 1470–1472. 10.1021/jo01118a038. [DOI] [Google Scholar]
- Meyer M. D.; Kruse L. I. Ergoline Synthons: Synthesis of 3,4-Dihydro-6-methoxybenz[cd]indol-5(1H)-one (6-methoxy-Uhle’s ketone) and 3,4-Dihydrobenz[cd]indol-5(1H)-one (Uhle’s ketone) via a Novel Decarboxylation of Indole-2-carboxylates. J. Org. Chem. 1984, 49, 3195–3199. 10.1021/jo00191a028. [DOI] [Google Scholar]
- Isoda T.; Hayashi K.; Tamai S.; Kumagai T.; Nagao Y. Efficient Synthesis of Isothiocyanates Based on the Tandem Staudinger/Aza-Wittig Reactions and Mechanistic Consideration of the Tandem Reactions. Chem. Pharm. Bull. 2006, 54, 1616–1619. 10.1248/cpb.54.1616. [DOI] [PubMed] [Google Scholar]
- Chen L.; Jin L.; Zhou N. An Update of Novel Screening Methods for GPCR in Drug Discovery. Expert Opin. Drug Discovery 2012, 7, 791–806. 10.1517/17460441.2012.699036. [DOI] [PubMed] [Google Scholar]
- Tian X.; Kang D. S.; Benovic J. L. Beta-arrestins and G Protein-Coupled Receptor Trafficking. Handb. Exp. Pharmacol. 2014, 219, 173–186. 10.1007/978-3-642-41199-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strange P. G. Use of the GTPγS ([35S]GTPγS and Eu-GTPγS) Binding Assay for Analysis of Ligand Potency and Efficacy at G Protein-Coupled Receptors. Br. J. Pharmacol. 2010, 161, 1238–1249. 10.1111/j.1476-5381.2010.00963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R. A.; Gibson T. M.; Stevenson L. A.; Saha B.; Crocker P.; Razdan R. K.; Pertwee R. G. Structural Determinants of the Partial Agonist-Inverse Agonist Properties of 6′-Azidohex-2′-yne-Δ8-tetrahydrocannabinol at Cannabinoid Receptors. Br. J. Pharmacol. 1999, 128, 735–743. 10.1038/sj.bjp.0702836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd P. R.; Hardy J. A.; Oakley A. E.; Edwardson J. A.; Perry E. K.; Delaunoy J. P. A Rapid Method for Preparing Synaptosomes: Comparison, with Alternative Procedures. Brain Res. 1981, 226, 107–118. 10.1016/0006-8993(81)91086-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.