

Abstract
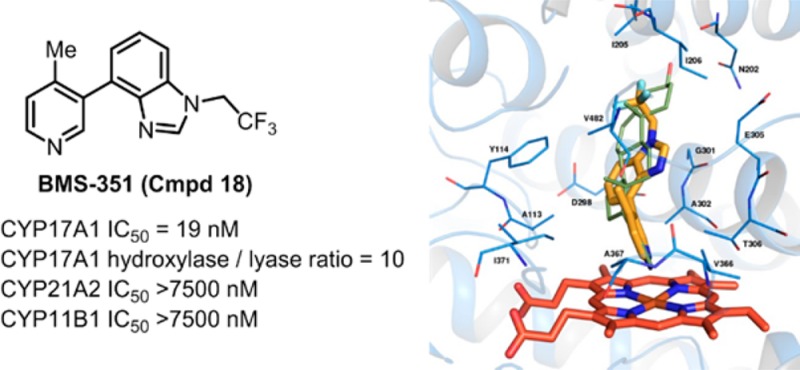
Efforts to identify a potent, reversible, nonsteroidal CYP17A1 lyase inhibitor with good selectivity over CYP17A1 hydroxylase and CYPs 11B1 and 21A2 for the treatment of castration-resistant prostate cancer (CRPC) culminated in the discovery of BMS-351 (compound 18), a pyridyl biaryl benzimidazole with an excellent in vivo profile. Biological evaluation of BMS-351 at a dose of 1.5 mg in castrated cynomolgus monkeys revealed a remarkable reduction in testosterone levels with minimal glucocorticoid and mineralcorticoid perturbation. Based on a favorable profile, BMS-351 was selected as a candidate for further preclinical evaluation.
Keywords: Prostate cancer, CYP17A1, benzimidazoles, lyase, CYP11B1, CYP21A2, cortisol
Prostate cancer (CaP) is the second leading cause of cancer death in men in the United States.1 An estimated 220,800 new cases are expected to be diagnosed in 2015 with 1 in 30 dying of this metastatic disease. Since Huggins and Hodges made the observation over 70 years ago that the growth and survival of prostate cancer is fueled by the presence of androgens,2 androgen deprivation therapy (ADT) became the first line of treatment for men afflicted by CaP.3 In particular, chemical castration with a luteinizing hormone-releasing hormone such as Lupron is a commonly accepted and effective initial method of treatment.4 Unfortunately, this therapy only reduces the circulating levels of testosterone by 90%, which originates from the testes, and it does not affect the remaining 10% of testosterone produced by the adrenal glands.5 Within 19 months, the androgen receptor will engage in a heightened sensitivity to the presence of low levels of testosterone and other androgens as well as progesterone, causing a re-emergence of cancer growth to appear and an inevitable progression to castration-resistant prostate cancer (CRPC).6 As a result of the severity of this disease, new therapies have been highly coveted for this urgent and unmet medical need.
Limited options to treat CRPC have warranted new research developments in this area. Until recently, chemotherapy with docetaxel was the only second-line therapy to improve the overall survival of prostate cancer patients followed by ketoconazole (Nizoral), an antifungal agent that can ameliorate some of the symptoms of prostate cancer through nonselective CYP inhibition (Figure 1).6,7 However, the overwhelming toxicity associated with each of these treatments relegates their use to short-term in any medicinal regimen. Recent approval of abiraterone acetate (Zytiga)8 by the FDA for postchemotherapy use represents a major breakthrough and allows for the near elimination of the remaining 10% testosterone produced by the adrenal glands that can promote and sustain CRPC. Through a unique mechanism of action, abiraterone functions by selectively targeting CYP17A1, an enzyme located in the endoplasmic reticulum of the testes and adrenals that has dual hydroxylase and lyase functionality. By its ability to inhibit CYP17A1, it can also deter the production of androgens intratumorally. Unfortunately, the irreversible nature of this steroidal inhibitor blocks both hydroxylase and lyase, thereby causing a mineralcorticoid excess through increased production of progesterone through the ACTH feedback loop,9 thus prompting coadministration of the drug with prednisone.10 The development of a selective inhibitor of the lyase functionality of CYP17A1 would help circumvent the need for corticosteroid supplementation and the occurrence of intolerable treatment-emergent events. The success of such a drug would also be contingent upon its selectivity over other steroidogenic cytochrome P450 enzymes (CYPs) such as CYP 21A2 and 11B1 to prevent glucocorticoid and mineralcorticoid excess. For example, TAK-700 (orteronel), a former candidate in Phase III clinical trials for the treatment of CRPC, worked through modest selective action against CYP17A1 lyase;11 however, it was also being coadministered with prednisone further suggesting that additional selectivity against these other CYPs is essential. Similarly, abiraterone is also an inhibitor of CYP21A2 and 11B1, which further necessitates coadministration of prednisone. In this communication, we describe the discovery of BMS-351, a potent and selective, nonsteroidal CYP17A1 lyase inhibitor with robust selectivity over steroidogenic CYPs 21A2 and 11B1.
Figure 1.

Structures of CYP17A1 inhibitors.
Screening of our internal compound collection for steroidal mimics uncovered a series of pyridyl biaryl indazoles with remarkable activity against CYP17A1. Compound 1 was identified as our inaugural lead with a CYP17A1 IC50 = 4 nM (Figure 2). Additional profiling regarding xenobiotic CYP selectivity and select ADME properties revealed that further optimization would be needed to improve selectivity primarily against CYP1A2 and metabolic stability in human (H), mouse (M), and cyno (C) liver microsomes.12 Careful monitoring for xenobiotic CYP inhibition was necessary to reduce the potential for drug–drug interactions.
Figure 2.

Initial lead.
To guide our efforts in analogue design, we utilized a homology model based on vitamin D-25 hydroxylase since no X-ray crystal structure of CYP17A1 was available at the time of our SAR studies.13 Vitamin D-25 hydroxylase is a member of the CYP P450 superfamily of enzymes and is also known as CYP2R1. Evidence of its expression in the testes and the probability that it would be more predictive for CYP17A1 lyase activity made it an attractive system to conduct our modeling experiments. In the docking simulations, it was postulated that the pyridyl nitrogen of compound 1 would complex to the heme iron in the binding pocket and that the pyridyl biaryl ring system would be oriented parallel and in close proximity to the I-helix (Figure 3). Energy minimization of 1 led to an orthogonal orientation between the pyridyl ring and the indazole moiety. From the model, the indazole group appears to serve as a linker and allows substitution from N1 to form critical interactions with key residues in the hydrophobic pocket. We surmised that pi-stacking of the N1-phenyl group with Phe114 gave compound 1 its excellent potency. Based on this premise, additional aryl and heteroaryl groups were incorporated at the N1-position.
Figure 3.
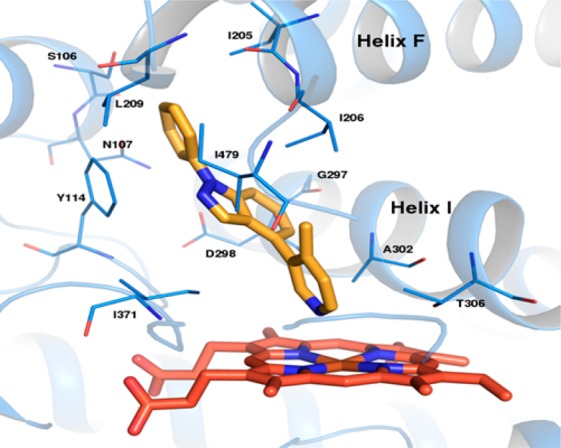
Predicted binding mode of compound 1 in a homology model derived from the CYP2R1 crystal structure 3C6G. Compound 1 is colored yellow, and heme orange. Residues contacting compound 1 are shown as lines.
The preparation of N1-substituted indazoles is found in Scheme 1. Starting from commercially available 3-bromo-4-methylpyridine (2), reaction with triisopropyl borate in THF at −78 °C, employing n-butyllithium for halogen-metal exchange, followed by an acidic workup gave boronic acid 3.14 Suzuki coupling of compound 3 with 3-bromo-2-methylaniline at 90 °C afforded biaryl 4.15 Intermediate 4 was then cyclized to indazole 5 by treatment with isoamyl nitrite, potassium acetate, and acetic acid in toluene.16 Finally, coupling of an aryl or heteroaryl halide to indazole 5 with copper(I) iodide, potassium phosphate, and (1R,2R)-cyclohexane-1,2-diamine in dioxane at 100 °C gave analogues 6–11.17
Scheme 1. Synthesis of N1-Aryl/Heteroaryl Substituted Indazoles.

Reagents and conditions: (a) triisopropyl borate, n-BuLi, toluene, THF – 78 °C, 81%; (b) Pd(PPh3)4, 3-bromo-2-methylaniline, Na2CO3, DME, EtOH, H2O, 90 °C, 92%; (c) isoamyl nitrite, KOAc, AcOH, toluene, 73%; (d) aryl/heteroaryl halide, (1R,2R)-cyclohexane-1,2-diamine, CuI, K3PO4, dioxane, 100 °C, 25–74%.
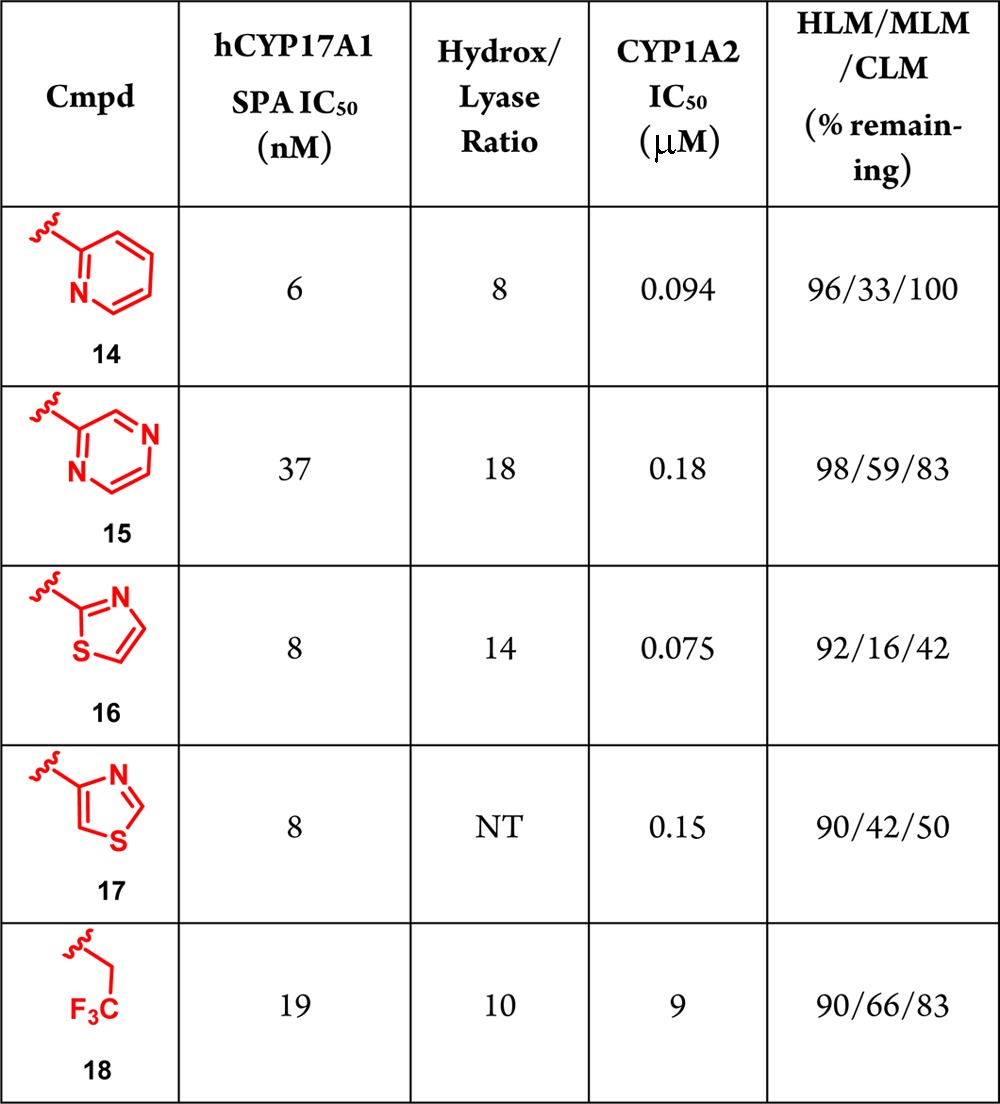
Shown in Table 1 are the results of our survey of N1-aryl and heteroaryl indazoles. Initially, a fluorine substituent was incorporated onto the N1-phenyl ring of compound 1 to determine if this change would be effective in improving metabolism. The resulting compound, 6, maintained potency and exhibited improved in vitro metabolic stability compared with indazole 1; however, the amount remaining in mouse liver microsomes remained low, and inhibition of CYP1A2 continued to be an issue. To further increase metabolic stability in mouse and cyno liver microsomes, various heterocycles were examined (7–11). The pyridyl analogue 7 led to a slight improvement in CYP17A1 potency, but gave similar metabolic stability and CYP1A2 inhibition as compound 6. Adding another nitrogen to the pyridyl ring to yield N1-pyridazyl substituted indazole 8 led to an analogue with a similar profile. In contrast, when N1 was replaced with a pyrazine (i.e., 9), cyno metabolic stability increased compared to indazole 1 with no xenobiotic CYP selectivity issues. Encouraged by these results, five-membered rings were also examined; e.g., isomeric thiazoles 10 and 11. Although CYP17A1 potency was retained, both of these compounds displayed poorer metabolic stability and CYP1A2 inhibition compared with compound 1 and the six-membered heterocycles. Overall, the addition of aryl or heteroaryl groups at N1 led to compounds with exceptional activity against CYP17A1. Unfortunately, none of the analogues exhibited the ideal profile with respect to both metabolic stability and xenobiotic CYP selectivity. The lack of promising data regarding these two criteria led us to speculate that these issues could be specific to this series.
Table 1. SAR of the Indazole Seriesa.
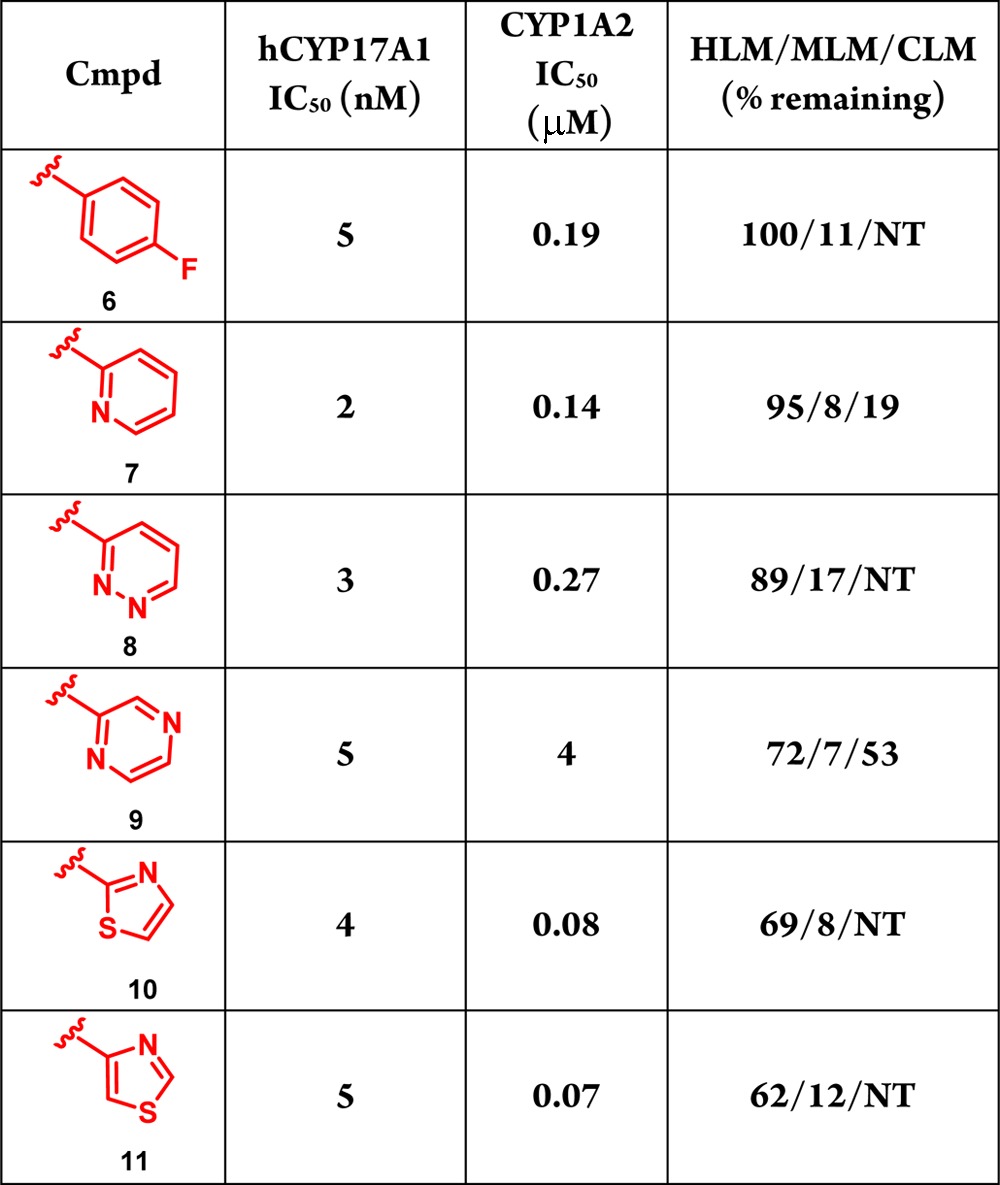
NT= not tested.
Since continuous inhibition of CYP17A1 is most likely required for efficacy, identifying a chemotype with acceptable metabolic stability became a primary focus. Based on the homology model, it appeared that exchanging the indazole for a benzimidazole would be an acceptable modification. The synthesis of these compounds was accomplished by a two-step sequence (Scheme 2). Suzuki coupling of pyridyl boronic acid 3 with 4-bromo-1H-benzo[d]imidazole (12) gave biaryl 13.12 This intermediate was then reacted with a heteroaryl halide in the presence of copper(I) iodide, cesium carbonate, and a diamine ligand in DMF at 110 °C to afford analogues 14–17.18 Alternatively, if an alkyl group was desired at N1, reaction of biaryl 13 with cesium carbonate and the alkyl triflate in DMSO furnished alkylated product 18.
Scheme 2. Synthesis of N1-Heteroaryl/Trifluoroethyl Substituted Benzimidazoles.

Reagents and conditions: (a) Pd(PPh3)4, 3-bromo-2-methylaniline, Na2CO3, DME, EtOH, H2O, 90 °C, 93%; (b) heteroaryl halide, Cul, Cs2CO3, (1R,2R)-N1,N2-dimethylcyclohexane-1,2-diamine, DMF, 110 °C, 58–68% or 2,2,2-trifluoroethyl trifluoromethanesulfonate, Cs2CO3, DMSO, rt, 81%.
The results of our SAR study employing benzimidazoles as a replacement for the indazole ring are shown in Table 2.19 Incorporation of a N1-pyridyl group, exemplified by benzimidazole 14, led to a compound of similar potency as indazole 7. More importantly, compound 14 displayed superior metabolic stability compared to 7, especially with regard to cyno liver microsomes. Replacing the N1-substituent with a pyrazine resulted in benzimidazole 15, which had moderate potency, but improved liver microsome stability across all species. The five-membered heterocycles at N1 (16 and 17) also exhibited better profiles than the corresponding indazoles 10 and 11. Although both benzimidazoles had decreased potency in this series, their activity in the CYP17A1 assay was excellent, and the stability increased in human, mouse, and cyno liver microsomes. In general, the benzimidazoles demonstrated comparable potency against CYP17A1 as the indazoles and had enhanced metabolic stability.
Table 2. SAR of the Benzimidazole Seriesa.
NT= not tested.
Having attained the initial requirements to progress this series, our attention shifted to the ultimate goal of achieving lyase selectivity. Thus, all benzimidazoles were tested against the individual hydroxylase and lyase components of CYP17A1 (Table 2).20 The results indicated good selectivity ranging from 8–18, with pyrazine 15 giving the highest hydroxylase/lyase ratio. Unfortunately, the major factor precluding further development of these compounds was potent CYP1A2 inhibition.
Because potential pi interactions from N1-heterocycles could assist in binding in the 1A2 pocket, an alkyl group was installed to disrupt this potential interaction. Attaching a trifluoroethyl group to N1 led to benzimidazole 18 (BMS-351), which demonstrated only weak inhibition of CYP1A2 and was free of any other xenobiotic CYP issues (Table 2).21 In addition, the hydroxylase/lyase selectivity ratio of 10 was notable. Combined with the favorable potency and metabolic stability for this compound, benzimidazole 18 became the program lead.
Subsequent to our studies, the crystal structure of CYP17A122 was elucidated in the presence of abiraterone and gave us some insight into how our compounds could be binding to CYP17A1. This structural data revealed that abiraterone binds to the heme iron, forming a 60 °C angle above the heme plane and packing against the central I helix with the 3β–OH interacting with Asn 202 in the F helix (Figure 4). Based on this information, docking of benzimidazole 18 in the crystal structure showed that our lead compound is predicted to bind in a similar mode as abiraterone, making electrostatic interactions with neighboring residues.
Figure 4.
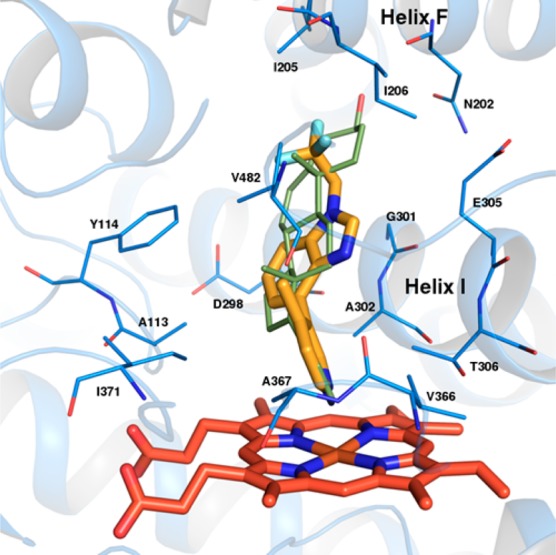
Predicted binding mode of compound 18 in the CYP17A1/abiraterone crystal structure. Compound 18 is colored yellow, and heme orange. The crystallographic pose of abiraterone is shown in green.
Compound 18 was further profiled in in vitro assays and in pharmacokinetic (PK) and pharmacodynamic (PD) studies. Since rodents lack adrenal CYP17A1, literature reports suggested that cynos would be the more relevant species to conduct these additional experiments.23 Comparison of benzimidazole 18 with abiraterone showed compound 18 was slightly less potent against CYP17A1 (Table 3). The adrenal IC50 values correlated with the CYP17A1 IC50 values for each compound. Not surprisingly, when the hydroxylase/lyase ratios were examined, compound 18 had higher selectivity than abiraterone, which was attributed to the reversible nature of our inhibitor. Furthermore, the lack of inhibition of CYP21A2 and CYP11B1 remained an additional factor differentiating the compounds since activity against these CYPs leads to mineralcorticoid excess and therefore the undesirable coadministration of prednisone. Benzimidazole 18 was determined to be inactive against both CYPs in human and cyno, whereas abiraterone was found to be a modest inhibitor. The combination of excellent human CYP17A1 potency and CYP11B1/21A2 selectivity associated with compound 18 may lead to an improved cortisol profile in human compared to abiraterone.
Table 3. Comparison of Cmpd 18 vs Abiraterone.
| Cmpd 18 |
Abiraterone |
|||
|---|---|---|---|---|
| In vitro assay | Human | Cyno | Human | Cyno |
| CYP17A1 SPA | 19 | 4 | 11 | 2 |
| IC50 (nM) | ||||
| Hydrox/Lyase | 10 | 11 | 3 | 2 |
| Adrenal SPA IC50 (nM) | 20 | 2 | 21 | 3 |
| CYP21A2 IC50 (nM) | >7500 | >7500 | 1050 | 149 |
| CYP11B1 IC50 (nM) | >7500 | >7500 | 872 | 2460 |
A PK/PD study was conducted on benzimidazole 18, and the results are summarized in Table 4. Consistent with its high intrinsic permeability in Caco-2 cells (400 nm/s for A → B and 300 nm/s for B → A), compound 18 was well absorbed following oral administration from solution formulations at the doses denoted in Table 4. Thus, excellent oral bioavailability (Fpo) was achieved. Based on the PK study conducted at 4 mg/kg in cyno, compound 18 exhibited a moderate Cmax and demonstrated a low peak/trough ratio of 16, which is desirable in order to avoid reaching the IC50 values of CYP17A1 hydroxylase, CYP21A2, and CYP11B1 that would lead to adverse effects. Dosing BID would enable 24 h coverage, while maintaining a flat PK profile. A PD study executed at 1 mg/kg led to >90% decrease in testosterone. With only 80 nM of free drug circulating, benzimidazole 18 was found to be highly potent with little drug needed to initiate and sustain a PD response.
Table 4. Pharmacokinetic Parameters in Cyno for Cmpd 18a.
| po dose (mg kg–1) | 4 |
| iv dose (mg kg–1) | 2 |
| Cmax (μM), po | 3.0 ± 1.1 |
| Tmax (h), po | 1.3 ± 0.7 |
| AUC (μM·h), po | 12.3 ± 4.0 |
| Cl, (mL min–1 kg–1), iv | 19.0 ± 4.8 |
| Vss (L/kg), iv | 4.4 ± 1.9 |
| F (%) | 97 ± 12 |
Values are reported as mean ± standard deviation.
Benzimidazole 18 and abiraterone were evaluated in a 1-day castrated cyno PK/PD study (Figures 5 and 6). This study was initiated at 1.5 mg/kg BID for compound 18 and 42 mg/kg BID for abiraterone. Dosing for compound 18 and abiraterone was administered at time 0 and 8 h. The results of the PK study are shown in Figure 5. The Cmax was attained at approximately 1 h for both compounds and also at 9.5 h due to the second dose given at 8 h.24 BID dosing enabled 24 h coverage, while maintaining a moderate Cmax with a low peak/trough ratio. This dosing regimen achieved comparable exposure of compound 18 to abiraterone.
Figure 5.
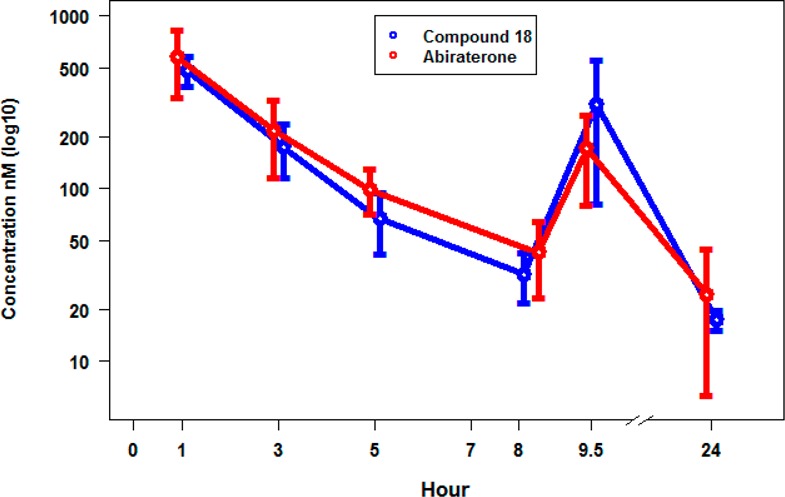
One-day castrated cyno PK data for compound 18 (blue) (1.5 mg/kg BID) vs abiraterone (red) (42 mg/kg BID). Average ± standard deviation (nM) shown at the time points indicated during 1–24 h, n = 4.
Figure 6.
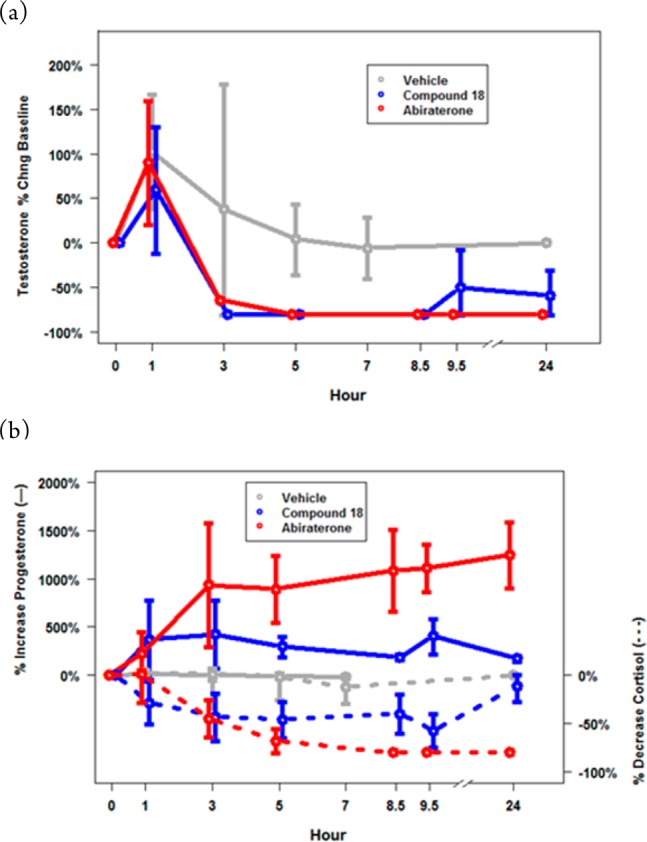
One-day castrated cyno PD data for compound 18 (1.5 mg/kg BID) vs abiraterone (42 mg/kg BID); (a) T average ± standard deviation of percentage change from baseline at time points indicated during 1–24 h and (b) elevation/reduction P and C dual-axis, respectively, n = 4.
Steroid measurements were taken at the time points indicated in Figure 6. Both compounds showed good PD effects in castrated cynos with BID dosing regimens. We were pleased to find benzimidazole 18 achieved an 80% reduction in testosterone (T) (p = 0.01), which was similar to abiraterone at almost all time points (Figure 6a). Furthermore, both CYP17A1 inhibitors reached T levels below the lower limits of quantification (LLQ = 1.2 ng/dL). Notably, compound 18 diverges in profile from abiraterone with regard to progesterone (P) and cortisol (C) (Figure 6b). Although administration of drug caused hormone levels to reach a maximal increase in T and P or decrease in C after 1 h (likely due to stress), levels of P and C from compound 18 returned to baseline levels after 24 h while sustaining a desirable >50% decrease in T. However, abiraterone continued to have a significant effect on P and C at 24 h; levels of P continued to increase to 1247% (p < 0.0001) and cortisol continued to decrease to −80% (p < 0.0001). Based on this data, compound 18 exhibits a superior profile to abiraterone by maintaining a maximal response against T throughout the study and a minimal effect on P and C, allowing baseline levels to be achieved at 24 h. This data correlates well with the in vitro data (Table 3), which indicates that abiraterone inhibits CYP21A2 and CYP11B1 to a greater extent than compound 18 amounting to larger changes in P and C levels. It is expected that the differences will be equally pronounced in human given the greater potency of abiraterone against CYP11B1 and CYP21A2 and the lack of activity of benzimidazole 18 toward these steroidogenic CYPs. The in vitro data for abiraterone suggests a greater disturbance in glucocorticoid levels and implies that administration of abiraterone would lead to similarly unfavorable perturbations in P and C levels in human. Compound 18 has the potential to exhibit minimal disruption to P and C levels in human, while abrogating T to castrate levels with minimal dosage of drug.
In conclusion, SAR studies in the indazole and benzimidazole series culminated in the discovery of benzimidazole 18, a potent inhibitor of CYP17A1 lyase activity with exquisite selectivity relative to CYP11B1 and CYP21A2. This preclinical profile could lead to improved cortisol control in human compared to abiraterone. A PK/PD study in castrated monkey corroborated the in vitro data and provided further evidence that compound 18 may differentiate from other known treatments in humans. Overall, benzimidazole 18 (BMS-351) emerges as an outstanding preclinical candidate to treat CRPC and is likely to minimize the side effects of current therapies due to its exceptional selectivity.
Acknowledgments
We thank the Department of Discovery Synthesis for the large scale synthesis of BMS-351. We also acknowledge the Departments of Lead Discovery & Optimization and Discovery Analytical Sciences for ADMET and compound characterization support. We thank Debra Wescott, Maria Helfrich, and Beth Roberts from Clinical Pathology for analysis of serum samples from cyno PK/PD studies.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00310.
Full experimental procedures and characterization data for compound 18; representative procedure and characterization data for compounds 1–17; statistical analysis of compounds tested and in vivo studies; procedures for biological assays, pharmacokinetic experiments, and in vivo efficacy experiments (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- National Cancer Institute website. Cancer Topics: Prostate Cancer. http://www.cancer.gov/cancertopics/types/prostate (accessed 2015. May 28).
- a Huggins C. Effect of orchiectomy and irradiation on cancer of the prostate. Ann. Surg. 1942, 115, 1192–1200. 10.1097/00000658-194206000-00030. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Huggins C.; Hodges C. V. Studies on prostatic cancer. I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941, 1, 293–297. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Garcia J. A. Targeting the adrenal gland in castration-resistant prostate cancer: A case for orteronel, a selective CYP17A1 17,20-lyase inhibitor. Curr. Oncol. Rep. 2013, 15, 105–112. 10.1007/s11912-013-0300-1. [DOI] [PubMed] [Google Scholar]
- Gomella L. G. Effective testosterone suppression for prostate cancer: Is there a best castration therapy?. Rev. Urol. 2009, 11, 52–60. [PMC free article] [PubMed] [Google Scholar]
- Labrie F. Adrenal androgens and intracrinology. Semin. Reprod. Med. 2004, 22, 299–309. 10.1055/s-2004-861547. [DOI] [PubMed] [Google Scholar]
- Eichholz A.; Ferraldeschi R.; Attard G.; de Bono J. S. Putting the brakes on continued androgen receptor signaling in castration-resistant prostate cancer. Mol. Cell. Endocrinol. 2012, 360, 68–75. 10.1016/j.mce.2011.09.038. [DOI] [PubMed] [Google Scholar]
- Altavilla A.; lacovelli R.; Procopio G.; Alesini D.; Risi E.; Campenni G. M.; Palazzo A.; Cortesi E. Medical strategies for treatment of castration resistant prostate cancer (CRPC) docetaxel resistant. Cancer Biol. Ther. 2012, 13, 1001–1008. 10.4161/cbt.21188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M. N.; Goodin S.; DiPaola R. S. Abiraterone in prostate cancer: a new angle to an old problem. Clin. Cancer Res. 2012, 18, 1848–1854. 10.1158/1078-0432.CCR-11-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink G.Neuroendocrine Feedback Control Systems: An Introduction. In The Handbook of Neuroendocrinology, 1st ed.; Fink G., Pfaff D. W., Levine J. E., Eds.; Elsevier Ltd.: London, 2012; pp 55–72. [Google Scholar]
- Cersosimo R. J. New agents for the management of castration-resistant prostate cancer. Ann. Pharmacother. 2012, 46, 1518–1528. 10.1345/aph.1R169. [DOI] [PubMed] [Google Scholar]
- Kaku T.; Hitaka T.; Ojida A.; Matsunaga N.; Adachi M.; Tanaka T.; Hara T.; Yamaoka M.; Kusaka M.; Okuda T.; Asahi S.; Furuya S.; Tasaka A. Discovery of orteronel (TAK-700), a naphthylmethylimidazole derivative, as a highly selective 17,20-lyase inhibitor with potential utility in the treatment of prostate cancer. Bioorg. Med. Chem. 2011, 19, 6383–6399. 10.1016/j.bmc.2011.08.066. [DOI] [PubMed] [Google Scholar]
- Compounds tested for metabolic stability were incubated with human, mouse, or cyno liver microsomes for 10 min at a concentration of 0.5 μM.
- Strushkevich N.; Usanov S. A.; Plotinkov A. N.; Jones G.; Park H.-W. Structural Analysis of CYP2R1 in Complex with Vitamin D3. J. Mol. Biol. 2008, 380, 95–106. 10.1016/j.jmb.2008.03.065. [DOI] [PubMed] [Google Scholar]
- Li W.; Nelson D. P.; Jensen M. S.; Hoerrner R. S.; Cai D.; Larsen R. D.; Reider P. J. An improved protocol for the preparation of 3-pyridyl- and some arylboronic acids. J. Org. Chem. 2002, 67, 5394–5397. 10.1021/jo025792p. [DOI] [PubMed] [Google Scholar]
- Moleele S. S.; Michael J. P.; de Koning C. B. Tetralones as precursors for the synthesis of 2,2′-disbustituted 1, 1′-binaphthyls and related compounds. Tetrahedron 2008, 64, 10573–10580. 10.1016/j.tet.2008.08.079. [DOI] [Google Scholar]
- Velaparthi U.; Frennesson D. B.; Saulnier M. G.; Austin J. F.; Huang A.; Balog J. A.; Vyas D. M.. Azaindazole Compounds. US Patent No. 8,673,922, March 18, 2014.
- Klapars A.; Antilla J. C.; Huang X.; Buchwald S. L. A general and efficient copper catalyst for the amidation of aryl halides and the N-arylation of nitrogen heterocycles. J. Am. Chem. Soc. 2001, 123, 7727–7729. 10.1021/ja016226z. [DOI] [PubMed] [Google Scholar]
- Antilla J. C.; Baskin J. M.; Barder T. E.; Buchwald S. L. Copper-diamine catalyzed N-arylation of pyrroles, pyrazoles, indazoles, imidazoles, and triazoles. J. Org. Chem. 2004, 69, 5578–5587. 10.1021/jo049658b. [DOI] [PubMed] [Google Scholar]
- Huang A.Preparation of substituted benzimidazole and imidazopyridine compounds useful as CYP17A1 modulators. WO 12044537, A1, April 5, 2012.
- The two activities were run with different enzyme preparations, and therefore, the hydroxylase/lyase selectivity ratios are estimates. See Supporting Information for details.
- Additional xenobiotic CYP IC50 data for BMS-351 can be found in Supporting Information.
- DeVore N. M.; Scott E. E. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 2012, 482, 116–120. 10.1038/nature10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka M.; Hara T.; Hitaka T.; Kaku T.; Takeuchi T.; Takahashi J.; Asahi S.; Miki H.; Tasaka A.; Kusaka M. Orteronel (TAK-700), a novel non-steroidal 17,20-lyase inhibitor: Effects on steroid synthesis in human and monkey adrenal cells and serum steroid levels in cynomolgus monkeys. J. Steroid Biochem. Mol. Biol. 2012, 129, 115–128. 10.1016/j.jsbmb.2012.01.001. [DOI] [PubMed] [Google Scholar]
- The increase in T and P and decrease in C 1 h after administration of drug or vehicle is likely due to stress from the gavage, which can affect hormone levels.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.