

Abstract
Many proteases cut the PAR2 N-terminus resulting in conformational changes that activate cells. Synthetic peptides corresponding to newly exposed N-terminal sequences of PAR2 also activate the receptor at micromolar concentrations. PAR2-selective small molecules reported here induce PAR2-mediated intracellular calcium signaling at nanomolar concentrations (EC50 = 15–100 nM, iCa2+, CHO-hPAR2 cells). These are the most potent and efficient small molecule ligands to activate PAR2-mediated calcium release and chemotaxis, including for human breast and prostate cancer cells.
Keywords: Protease activated receptor 2, agonist, cyclohexylglycine, structure−activity relationships
Protease-activated receptors (PARs) are class A GPCRs, self-activated by their newly exposed N-terminus following cleavage by proteases. Protease-activated receptor 2 (PAR2) is one of four PAR subtypes and its N-terminus is cleaved by enzymes (e.g., trypsin, tryptase, factor Xa/VIIa, granzyme A) at the R36-S37 cleavage site to reveal an activating sequence S37LIGKV (human) or S37LIGRL (rodent).1−3 Some proteases cleave elsewhere, with the cysteine protease cathepsin S cleaving at G40-K41 and E56-T57.4,5 PAR2 has been implicated in disease models of, for example, arthritis,6,7 inflammatory bowel disease,8,9 asthma,10,11 cardiovascular diseases,12,13 cancers,14,15 and metabolic dysfunction.16,17 However, roles for PAR2 in human diseases remain uncertain, with both pro- and anti-inflammatory properties as well as protective and disease-promoting roles reported.1,18,19
Synthetic agonists and antagonists are therefore valuable tools for investigating PAR2 functions in vivo. Most ligands used to date to evaluate PAR2 biology have been peptides, which tend to be metabolically unstable.20,21 Synthetic hexapeptide SLIGRL-NH2, which is more potent than the native sequence SLIGKV-NH2 in activating human PAR2 on cells, has been the most widely used peptide agonist, but it is only active at micromolar concentrations.20 Extensive structure–activity relationships, performed on this peptide by replacing each amino acid with other natural/unnatural amino acids, have generated more potent peptides. For example, the N-terminal serine has been replaced by a heterocycle, such as 2-furoyl (2f);22 4-(2-methyloxazolyl), 5-isoxazolyl (Isox), 2-pyrazolyl, 2-pyridolyl, 3-pyridolyl, 2-benzofuranoyl, 2-naphthoyl, 2-benzothienyl,23 and 2-aminothiazol-4-yl (2-at).24 Heptapeptides like 2f-LIGRLO-NH2 (1, Figure 1) and X-LIGRLI-NH2 (2, with different heterocycles) were reported to be selective agonists for PAR2 with EC50 between 0.1–0.8 μM measured by intracellular calcium (iCa2+) release in various cell lines (e.g., HCT-15, HT29, 16HBE14o-).22−24 Linking a hexadecyl lipid via three polyethylene glycol (PEG) spacer units to the ornithine amine side chain (3) or at the C-terminal of 2f-LIGRL also evidently increases agonist potency to nanomolar concentrations in an iCa2+ assay on 16HBE14o- cells.25 A few nonpeptidic PAR2 agonists (4, AC-55541 and AC-264613) were also reported with potency similar to 1 (EC50 100–300 nM, iCa2+).26−28 Our goal here was to minimize size, reduce polar atoms and polar surface area, and reduce rotatable bonds yet still produce a potent, rule-of-five compliant compound with higher potency and ligand efficiency than 1–4 and greater stability in biological media.
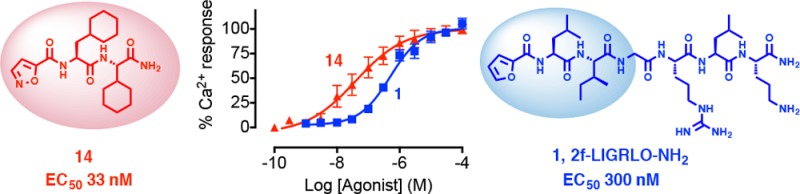
Figure 1.

The PAR2 peptide agonist 1 was sequentially truncated to reduce the size of the peptide, and the first three residues were replaced with Isox-Cha-Ile, a component of 4. Agonist activities for its derivatives (Table 1) were evaluated, using Chinese hamster ovary cells stably expressing human PAR2 (CHO-hPAR2), for intracellular Ca2+ release since this signaling via PAR2 has been implicated in multiple diseases.7,9,18 In these cells, synthetic peptide SLIGKV-NH2 was 10-fold or more less potent (EC50 3 μM) than 1 and 4. Peptide 7 was less potent than 8, suggesting that the extra C-terminal leucine was unnecessary. Truncating 8 to 9 did not affect agonist activity, but truncation to 10 reduced the potency to that of 1 and 4.
Table 1. PAR2 Agonist Activity in Inducing Ca2+ Release in CHO-hPAR2 Cells.
| # | compd | pEC50 ± SEM | EC50 (nM) |
|---|---|---|---|
| 1 | 2f-LIGRLO-NH2 | 6.5 ± 0.1 | 300 |
| 4 | GB110 | 6.7 ± 0.2 | 200 |
| 5 | trypsin | 8.9 ± 0.3 | 1 |
| 6 | SLIGKV-NH2 | 5.5 ± 0.1 | 3000 |
| 7 | Isox-Cha-Ile-GRL-NH2 | 6.7 ± 0.1 | 200 |
| 8 | Isox-Cha-Ile-GR-NH2 | 7.1 ± 0.2 | 80 |
| 9 | Isox-Cha-Ile-G-NH2 | 7.0 ± 0.2 | 95 |
| 10 | Isox-Cha-Ile-NH2 | 6.4 ± 0.2 | 400 |
| 11 | Isox-Cha-Chg-GRL-NH2 | 7.1 ± 0.1 | 80 |
| 12 | Isox-Cha-Chg-GR-NH2 | 7.2 ± 0.3 | 62 |
| 13 | Isox-Cha-Chg-G-NH2 | 7.7 ± 0.2 | 18 |
| 14 | Isox-Cha-Chg-NH2 | 7.5 ± 0.3 | 33 |
| 15 | H2N-Cha-Chg-GR-NH2 | <4.0 | >100000 |
| 16 | H2N-Chg-GR- NH2 | <4.0 | >100000 |
Docking of 10 into a PAR2 homology model (Supporting Information, SI), generated by sequence alignment of PAR2 with the crystal structure of nociceptin/orphanin FQ receptor (pdb code 4EA3), suggested that the binding pocket surrounding the Ile-binding site is large and can accommodate a bulkier substituent. Cyclohexylglycine (Chg) replaced Ile in compounds 7–10 to produce 11–14. Compounds 13 and 14, with the fewest residues, were more potent agonists in inducing Ca2+ release than 11 and 12 (Table 1). This was despite the presence in 11 and 12 of an arginine, which often forms strong hydrogen bonds or electrostatic interactions within proteins.
Removing Isox and Cha (15 and 16) almost abolished activity, suggesting that these groups were more important than arginine for interactions in the ligand binding site. The minimal pharmacophore involved three residues, Isox-Cha-Ile-NH2 (10) and Isox-Cha-Chg-NH2 (14, AY77), the latter being ∼10-fold more potent for inducing intracellular Ca2+ release.
Agonist 14 was next docked into the PAR2 homology model and the best fitting docked pose of the ligand was selected for further analysis. We were interested in learning what interactions between 14 and PAR2 were responsible for activating the PAR2-Ca2+ pathway. From the ligand docking experiments, the nitrogen of isoxazole was predicted to form a hydrogen bond with Y82, while the terminal amide may also interacted with residues D228 and Y156 through hydrogen bonding. In addition, one of the amide carbonyl groups of 14 was predicted to interact with Y156 (Figure 2a). The surface view of the PAR2 homology model suggested that the isoxazole, Cha, and Chg bound into three distinct pockets (Figure 2b). Compound 14 was also docked into a PAR2 homology model derived from the PAR1 crystal structure (results not shown); however, the docked pose did not agree with the PAR2 mutagenesis results unlike this model (Figures 2b–d).
Figure 2.

Predicted binding mode for agonist 14 in a PAR2 homology model derived from nociceptin/orphanin FQ receptor (pdb code 4EA3). (A) Agonist AY77 (14) is predicted to form multiple hydrogen bonds and hydrophobic interactions with PAR2. (B) Surface view of PAR2 model showing Isox, Cha, and Chg components occupying three distinct binding pockets. (C) iCa2+ responses induced by agonist 14 were significantly reduced in PAR2 mutants Y156A, Y326A, D228A, L307A, and Y82A compared to wild-type (WT), suggesting that these residues interact with 14. Each data point is mean ± SEM of 3–5 independent experiments. (D) The C-terminal amide NH2 of 14 forms hydrogen bonds with Y156 and D228. The cyclohexylglycine may make a hydrophobic interaction with L307 and the amide carbonyl of Cha may form a water mediated H-bond with Y326. Isoxazole may form a H-bond with Y82.
To validate the binding site and pose of 14, the agonist activity of this compound was investigated in CHO cells transfected with mutant PAR2 (Y82A, Y156A, D228A, L307A, Y326A) (Figure 2c). Mutations of Y82, Y326, D228, or Y156 to alanine resulted in loss of H-bond interactions of these residues with 14, its agonist activity in inducing iCa2+ being significantly reduced by nearly 100-fold. Additionally, a L307A mutation suggested a larger binding cavity for 14 so that Chg could not fit as snugly into the pocket, resulting in loss of activity. Residues Y82, D228, and Y156 were in close proximity (within 2.0–3.0 Å, Figure 2d) to the ligand, consistent with H-bonding, but Y326 was located further away from the proposed binding site of 14 and may not form a direct H-bond with ligand. However, 14 may interact with Y326 through water-mediated H-bonding, consistent with reduced agonist activity in the Y326A mutant cells. To our knowledge, no binding site for a PAR2 ligand has ever been clearly elucidated before. To further support the binding mode of 14, its amide NH2 was methylated to give Isox-Cha-Chg-NMe2, which was less active than 14 (data not shown) suggesting that the C-terminal NH2 may be involved in H-bonding. Compound 14 is the first small molecule agonist (MW < 500) reported to activate PAR2 at low nanomolar concentrations. In terms of ligand efficiency (PEI, Table 2), 14 is superior to other PAR2 agonists. Agonist 14 was also more stable in rat plasma than peptides 1 and 6 (Figure 3a), making it potentially more valuable for physiologically relevant studies.
Table 2. Percentage Efficiency Index (PEI).
| compd | MW (Da) | PEIa |
|---|---|---|
| 1 | 778 | 0.51 |
| 3 | 2003 | 0.35 |
| 4 | 609 | 0.82 |
| 13 | 461 | 1.52 |
| 14 | 404 | 1.73 |
PEI calculated as fraction of Ca2+ (i.e., 50% = 0.5) induced by 100 nM agonist divided by MW (kDa).29
Figure 3.

(a) Agonist 14 was stable in rat plasma after 4 h (pH 7.4, 37 °C, initial concentration 1 μM) compared to peptides 1 and 6. The plasma samples were analyzed by LCMS (n = 3). (b) Agonists 1 and 14 (AY77) activate Ca2+ release via PAR2. Experiments were performed in CHO-hPAR2 cells (solid lines) versus CHO cells (PAR2–/–, dotted lines), n ≥ 3. Each data point represents mean ± SEM.
The selectivity of agonist 14 was evaluated in CHO-hPAR2 cells compared to CHO cells without PAR2 transfection. In CHO-hPAR2 cells, agonist 14 was 10-fold more potent (EC50 33 nM) than peptide agonist 1 (EC50 300 nM) and neither of these agonists induced any iCa2+ release at 100 μM concentrations in PAR2–/–-CHO cells, revealing that they both activate Ca2+ release selectively via PAR2 (Figure 3b).
Some synthetic peptides have been shown to cross-react with PAR1 and PAR2 receptors.20 Therefore, 14 was assessed for receptor subtype selectivity in a desensitization assay using human prostate cancer cells (PC3) that expressed both PAR1 and PAR2 (SI, Figure S1).
In this desensitization experiment, cells were first exposed to 1 (a selective PAR2 agonist) at 25 μM and induced Ca2+ release, whereas there was no response to a second treatment with this same agonist (SI, Figure S1a). This indicated that the PAR2 on the cell membrane had been desensitized by the first agonist treatment and thus did not respond to the second treatment. By contrast, a PAR1 agonist (TFLLR-NH2) was able to activate the PAR2-desensitized cells causing a second calcium signal following initial treatment with 1 (SI, Figure S1b). Compound 14, at up to 10 μM, did not trigger this second calcium signal after initial PAR2 desensitization with 1 (SI, Figure S1c), consistent with 14 being selective for PAR2 over PAR1.
The functions of PAR2 agonists were further evaluated in human breast cancer cells (MDA-MB-231), known to express PAR2.30 Activation of PAR2 induced by agonists 1 or 14 at 100 nM caused chemotactic migration that was attenuated by treating with an anti-PAR2 antibody (SAM11). This is consistent with the observed cell migration being PAR2-specific, the small agonist 14 being as effective and as potent as the larger peptide agonist 1 in activating PAR2 in this assay (Figure 4).
Figure 4.

PAR2-activated chemotaxis in MDA-MB-231 breast cancer cells. Compounds 1 and 14 (100 nM) induced chemotaxis in a transwell migration assay. Preincubating cells with PAR2 antibody SAM11 (1 μg/mL) 30 min prior to agonist addition resulted in inhibition of chemotaxis. The chemotactic index is the ratio of agonist to random migration (24 h) through a 5 μm membrane, mean ± SEM, ***p < 0.001.
Small molecules described here (e.g., 13, 14) are the most potent PAR2 agonists reported to induce Ca2+ release. Receptor modeling in combination with human PAR2 mutants transfected into CHO cells were used to probe the mechanism of Ca2+ release induced by 14. Putative interactions, predicted from modeling studies, between 14 and PAR2 residues Y82, Y156, D228, L307, and Y326 were experimentally supported by PAR2 mutagenesis data. Effects of 14 were also measured on cancer cells, including chemotaxis of breast cancer cells and Ca2+ desensitization of prostate cancer cells. Compound 14 is a potent, effective, and selective small molecule (MW 404) agonist for PAR2 with the highest ligand efficiency yet known. It is more stable than peptides in rat plasma, activates PAR2-mediated Ca2+ signaling with high potency (EC50 33 nM, CHO-hPAR2), and has agonist activity on two human cancer cell lines. Compound 14 is a valuable new tool for interrogating PAR2 physiology3 and a new pharmacophore to exploit for creating potent agonists and antagonists as prospective drug candidates that selectively modulate PAR2.
Experimental Procedures
General Methods
Reagents were purchased from Sigma-Aldrich or Chem-Impex Int., Inc. Compound purity was assessed by NMR spectroscopy, rpHPLC, and high-resolution mass spectra (HRMS). All assayed compounds were ≥95% pure, as determined by rpHPLC (UV detection at 214, 230, and 254 nm). Other experimental methods and characterization data of all compounds, except 13 and 14, are reported in Supporting Information. Synthetic compounds 13 and 14 are characterized below.
Solid-Phase Synthesis
All compounds were synthesized on solid phase by standard Fmoc SPPS procedures using Rink amide resin (100–300 mg, 0.70 mmol/g), purified by reversed phase HPLC to >95% purity, and characterized by high resolution MS and NMR spectroscopy.
Isox-Cha-Chg-G-NH2 (13)
tR = 9.1 min. HRMS: [MNa]+ 484.2530 (calc. for C23H36N5O5+) 484.2533 (found). 1H NMR (600 MHz, DMSO-d6), δ 0.83–1.22 (m, 10H), 1.31 (m, 1H), 1.51–1.75 (m, 13H), 3.54–3.70 (2 sets of dd, J = 5.7 Hz, 2H), 4.11 (t, J = 7.9 Hz, 1H), 4.57 (m, 1H), 7.05 (br s, 1H), 7.17 (d, J = 1.9 Hz, 1H), 7.19 (br s, 1H), 8.00 (d, J = 8.2 Hz, 1H), 8.14 (t, J = 5.7 Hz, 1H), 8.75 (d, J = 1.9 Hz, 1H), 8.99 (d, J = 8.2 Hz, 1H). 13C NMR (150 MHz, DMSO-d6), δ 25.5, 25.6, 25.7, 25.8, 26.0, 28.2, 29.0, 31.7, 33.2, 33.6, 38.5, 41.7, 50.2, 57.6, 106.1, 151.7, 155.4, 162.3, 170.6, 170.8, 171.5.
Isox-Cha-Chg-NH2 (14)
tR = 9.4 min. HRMS: [MH]+ 405.2496 (calc. for C21H33N4O4+) 405.2498 (found). 1H NMR (600 MHz, DMSO-d6), δ 0.82–1.20 (m, 9H), 1.29 (m, 1H), 1.50–1.59 (m, 5H), 1.59–1.71 (m, 9H), 4.08 (t, J = 7.7 Hz, 1H), 4.53 (m, 1H), 7.00 (br s, 1H), 7.14 (d, J = 1.9 Hz, 1H), 7.36 (br s, 1H), 7.77 (d, J = 9.4 Hz, 1H), 8.73 (d, J = 1.7 Hz, 1H), 8.97 (d, J = 8.5 Hz, 1H). 13C NMR (150 MHz, DMSO-d6), δ 25.9, 26.0, 26.1, 26.2, 26.4, 28.4, 29.6, 32.1, 33.6, 34.1, 39.1, 51.4, 57.3, 106.6, 152.2, 155.8, 162.8, 171.5, 173.0.
Cell Culture, Expression Constructs, and Transfections
Methods for preparation of Chinese hamster ovary (CHO), PC3, and MDA-MB-231 cancer cells are described in the Supporting Information.
Intracellular Calcium Release Assay
Cells were plated at ∼5 × 104 cells/well in a 96-well clear-bottom black-wall assay plate (Corning) and incubated overnight at 37 °C. Before assay, the medium was removed, and cells were incubated with dye-loading buffer (12 mL HBSS buffer, 4 μM Fluo-3 AM, 25 μL of Pluronic acid F-127, 1% FBS) for 1 h at 37 °C. Cells were washed once (HBSS buffer containing 2.5 mM probenecid and 20 mM HEPES, pH 7.4). Stock solutions of compounds in DMSO were diluted with HBSS buffer to give assay concentrations. Agonist activity was measured at a range of concentrations of test compound (50 μL) added to cells in 50 μL of HBSS buffer. Intracellular Ca2+ was monitored for 300 s (excitation 495 nm, emission 520 nm) after injecting compound. Duplicate measurements for each data point were reported as means ± SEM from multiple experiments. Changes in fluorescence (% response) were plotted against log[compound]. Half maximal effective concentrations (EC50) were derived from concentration–response curves using a nonlinear regression curve fitting (Graphpad Prism v6). Calcimycin (A23187, Invitrogen) was used to measure maximum fluorescence; individual results were normalized.
Rat Plasma Stability Assay
Compounds in DMSO (5 μL, 100 μM) were added to a 1:1 mixture of PBS and rat plasma (495 μL) at 37 °C. At intervals of 0, 5, 15, 30, 60, 120, 180, and 240 min, 50 μL was removed and added to 150 μL of MeCN. Samples were centrifuged (2500 rpm, 5 min) and supernatant (50 μL) analyzed by UPLC-MS. Data were plotted as % peak area of samples at t = 0 min.
Transwell Cell Migration
A transwell polycarbonate filter insert with 5 μm membrane was used to determine cell migration. Both sides of the membrane were coated with collagen (1 mg/mL, 50 μL) for 2 min before air-drying. Cells were lifted with nonenzymatic cell dissociation solution and resuspended in 0.1% BSA serum-free L-15. Cells were seeded at 2.5 × 105 cells/insert and allowed to incubate (3 h, 37 °C). Then, 700 μL of 0.1% BSA serum-free L-15 with 100 nM of PAR2 agonist was added to the bottom chamber and incubated for 24 h. For inhibition of chemotaxis, SAM11 (1 μg/mL) was preincubated for 30 min prior to adding agonist. Cells on top of the membrane were removed with a cotton swab and fixed in 4% PFA. The insert was washed 2× in PBS and stained with 1% crystal violet. Migrated cells on the underside of the membrane were counted using a Nikon Ti–U inverted brightfield microscope.
Molecular Modeling
A homology model of human PAR2 was generated by alignment with Nociceptin/orphanin FQ receptor structure (pdb code 4EA3). Methods used for generating the PAR2 homology model and docking ligands are in Supporting Information.
Acknowledgments
We thank the ACRF Imaging Facility (Brisbane) for use of microscopes and Prof. K. K. Khanna (QIMR-Berghofer Institute, Brisbane) for MDA-MB-231 cells.
Glossary
ABBREVIATIONS
- 2f
2-furoyl
- Cha
cyclohexylalanine
- Chg
cyclohexylglycine
- CHO
Chinese hamster ovary cells
- 16HBE14o-
SV40-transformed human bronchial epithelial cells
- H-bond
hydrogen bond
- HBSS
Hank’s balanced salt solution
- HCT-15
human colon carcinoma cells
- HEPES
(4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HT29
human colonic adenocarcinoma cells
- iCa2+
intracellular calcium
- Isox
5-isoxazole
- MDA-MB-231
human breast adenocarcinoma cells
- MW
molecular weight
- PAR2
protease activated receptor 2
- PC3
human prostate cancer cells
- PEG
polyethylene glycol
- PEI
percent efficiency index
- TM
transmembrane
- WT
wild-type
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00429.
Desensitization experimental results, general experimental methods, compound characterization data (NMR, HPLC, HRMS), and methods used for generating PAR2 homology model (PDF)
Author Contributions
M.K.Y. and D.P.F. conceived experiments, analyzed results, and wrote the paper. M.N.A., Y.H., and J.D.H. transfected CHO cells with PAR2 mutants. M.K.Y., J.Y.S., W.X., J.L., L.L., and R.R. performed all experiments.
D.P.F. acknowledges NHMRC for grants 1047759, 1000745, 569595 and an SPRF fellowship (1027369); the Queensland Government for a CIF grant; and ARC for DP130100629 and CE140100011 grants and the ARC Centre of Excellence in Advanced Molecular Imaging. M.A. received an Australian Post-Graduate Award. J.D.H. holds ARC Future Fellowship FT120100917.
The authors declare the following competing financial interest(s): D.P.F., L.L., M.K.Y., J.Y.S., and R.R. are named inventors on US Patent 8,927,503 and are co-authors on this manuscript. The patent is owned by the University of Queensland and concerns agonists and antagonists of PAR2.
Supplementary Material
References
- Adams M. N.; Ramachandran R.; Yau M. K.; Suen J. Y.; Fairlie D. P.; Hollenberg M. D.; Hooper J. D. Structure, function and pathophysiology of protease activated receptors. Pharmacol. Ther. 2011, 130, 248–282. 10.1016/j.pharmthera.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Rothmeier A. S.; Ruf W. Protease-activated receptor 2 signaling in inflammation. Semin. Immunopathol. 2012, 34, 133–149. 10.1007/s00281-011-0289-1. [DOI] [PubMed] [Google Scholar]
- Yau M. K.; Liu L.; Fairlie D. P. Toward drugs for protease-activated receptor 2 (PAR2). J. Med. Chem. 2013, 56, 7477–7497. 10.1021/jm400638v. [DOI] [PubMed] [Google Scholar]
- Elmariah S. B.; Reddy V. B.; Lerner E. A.; Cathepsin S. Signals via PAR2 and Generates a Novel Tethered Ligand Receptor Agonist. PLoS One 2014, 9, e99702. 10.1371/journal.pone.0099702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P.; Lieu T.; Barlow N.; Metcalf M.; Veldhuis N. A.; Jensen D. D.; Kocan M.; Sostegni S.; Haerteis S.; Baraznenok V.; Henderson I.; Lindstrom E.; Guerrero-Alba R.; Valdez-Morales E. E.; Liedtke W.; McIntyre P.; Vanner S. J.; Korbmacher C.; Bunnett N. W. Cathepsin S causes inflammatory pain via biased agonism of PAR2 and TRPV4. J. Biol. Chem. 2014, 289, 27215–27234. 10.1074/jbc.M114.599712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell W. R.; Lockhart J. C.; Kelso E. B.; Dunning L.; Plevin R.; Meek S. E.; Smith A. J.; Hunter G. D.; McLean J. S.; McGarry F.; Ramage R.; Jiang L.; Kanke T.; Kawagoe J. Essential role for proteinase-activated receptor-2 in arthritis. J. Clin. Invest. 2003, 111, 35–41. 10.1172/JCI16913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman R. J.; Cotterell A. J.; Barry G. D.; Liu L.; Suen J. Y.; Vesey D. A.; Fairlie D. P. An antagonist of human protease activated receptor-2 attenuates PAR2 signaling, macrophage activation, mast cell degranulation, and collagen-induced arthritis in rats. FASEB J. 2012, 26, 2877–2887. 10.1096/fj.11-201004. [DOI] [PubMed] [Google Scholar]
- Hansen K. K.; Sherman P. M.; Cellars L.; Andrade-Gordon P.; Pan Z.; Baruch A.; Wallace J. L.; Hollenberg M. D.; Vergnolle N. A major role for proteolytic activity and proteinase-activated receptor-2 in the pathogenesis of infectious colitis. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 8363–8368. 10.1073/pnas.0409535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohman R. J.; Cotterell A. J.; Suen J.; Liu L.; Do A. T.; Vesey D. A.; Fairlie D. P. Antagonism of protease-activated receptor 2 protects against experimental colitis. J. Pharmacol. Exp. Ther. 2012, 340, 256–265. 10.1124/jpet.111.187062. [DOI] [PubMed] [Google Scholar]
- Cocks T. M.; Fong B.; Chow J. M.; Anderson G. P.; Frauman A. G.; Goldie R. G.; Henry P. J.; Carr M. J.; Hamilton J. R.; Moffatt J. D. A protective role for protease-activated receptors in the airways. Nature 1999, 398, 156–160. 10.1038/18223. [DOI] [PubMed] [Google Scholar]
- Cocks T. M.; Moffatt J. D. Protease-activated receptor-2 (PAR2) in the airways. Pulm. Pharmacol. Ther. 2001, 14, 183–191. 10.1006/pupt.2001.0285. [DOI] [PubMed] [Google Scholar]
- Damiano B. P.; Cheung W. M.; Santulli R. J.; Fung-Leung W. P.; Ngo K.; Ye R. D.; Darrow A. L.; Derian C. K.; de Garavilla L.; Andrade-Gordon P. Cardiovascular responses mediated by protease-activated receptor-2 (PAR-2) and thrombin receptor (PAR-1) are distinguished in mice deficient in PAR-2 or PAR-1. J. Pharmacol. Exp. Ther. 1999, 288, 671–678. [PubMed] [Google Scholar]
- Antoniak S.; Rojas M.; Spring D.; Bullard T. A.; Verrier E. D.; Blaxall B. C.; Mackman N.; Pawlinski R. Protease-activated receptor 2 deficiency reduces cardiac ischemia/reperfusion injury. Arterioscler., Thromb., Vasc. Biol. 2010, 30, 2136–2142. 10.1161/ATVBAHA.110.213280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryden L.; Grabau D.; Schaffner F.; Jonsson P. E.; Ruf W.; Belting M. Evidence for tissue factor phosphorylation and its correlation with protease-activated receptor expression and the prognosis of primary breast cancer. Int. J. Cancer 2010, 126, 2330–2340. 10.1002/ijc.24921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam D. K.; Dang D.; Zhang J.; Dolan J. C.; Schmidt B. L. Novel animal models of acute and chronic cancer pain: a pivotal role for PAR2. J. Neurosci. 2012, 32, 14178–14183. 10.1523/JNEUROSCI.2399-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badeanlou L.; Furlan-Freguia C.; Yang G.; Ruf W.; Samad F. Tissue factor-protease-activated receptor 2 signaling promotes diet-induced obesity and adipose inflammation. Nat. Med. 2011, 17, 1490–1497. 10.1038/nm.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J.; Iyer A.; Liu L.; Suen J. Y.; Lohman R. J.; Seow V.; Yau M. K.; Brown L.; Fairlie D. P. Diet-induced obesity, adipose inflammation, and metabolic dysfunction correlating with PAR2 expression are attenuated by PAR2 antagonism. FASEB J. 2013, 27, 4757–4767. 10.1096/fj.13-232702. [DOI] [PubMed] [Google Scholar]
- Suen J. Y.; Cotterell A.; Lohman R. J.; Lim J.; Han A.; Yau M. K.; Liu L.; Cooper M. A.; Vesey D. A.; Fairlie D. P. Pathway Selective Antagonism Of Proteinase Activated Receptor 2. Br. J. Pharmacol. 2014, 171, 4112–4124. 10.1111/bph.12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg M. D.; Mihara K.; Polley D.; Suen J. Y.; Han A.; Fairlie D. P.; Ramachandran R. Biased signalling and proteinase-activated receptors (PARs): targeting inflammatory disease. Br. J. Pharmacol. 2014, 171, 1180–1194. 10.1111/bph.12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maryanoff B. E.; Santulli R. J.; McComsey D. F.; Hoekstra W. J.; Hoey K.; Smith C. E.; Addo M.; Darrow A. L.; Andrade-Gordon P. Protease-activated receptor-2 (PAR-2): structure-function study of receptor activation by diverse peptides related to tethered-ligand epitopes. Arch. Biochem. Biophys. 2001, 386, 195–204. 10.1006/abbi.2000.2207. [DOI] [PubMed] [Google Scholar]
- Devlin M. G.; Pfeiffer B.; Flanagan B.; Beyer R. L.; Cocks T. M.; Fairlie D. P. Hepta and octapeptide agonists of protease-activated receptor 2. J. Pept. Sci. 2007, 13, 856–861. 10.1002/psc.912. [DOI] [PubMed] [Google Scholar]
- McGuire J. J.; Saifeddine M.; Triggle C. R.; Sun K.; Hollenberg M. D. 2-furoyl-LIGRLO-amide: a potent and selective proteinase-activated receptor 2 agonist. J. Pharmacol. Exp. Ther. 2004, 309, 1124–1131. 10.1124/jpet.103.064584. [DOI] [PubMed] [Google Scholar]
- Barry G. D.; Suen J. Y.; Low H. B.; Pfeiffer B.; Flanagan B.; Halili M.; Le G. T.; Fairlie D. P. A refined agonist pharmacophore for protease activated receptor 2. Bioorg. Med. Chem. Lett. 2007, 17, 5552–5557. 10.1016/j.bmcl.2007.08.026. [DOI] [PubMed] [Google Scholar]
- Boitano S.; Flynn A. N.; Schulz S. M.; Hoffman J.; Price T. J.; Vagner J. Potent agonists of the protease activated receptor 2 (PAR2). J. Med. Chem. 2011, 54, 1308–1313. 10.1021/jm1013049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn A. N.; Hoffman J.; Tillu D. V.; Sherwood C. L.; Zhang Z.; Patek R.; Asiedu M. N.; Vagner J.; Price T. J.; Boitano S. Development of highly potent protease-activated receptor 2 agonists via synthetic lipid tethering. FASEB J. 2013, 27, 1498–1510. 10.1096/fj.12-217323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry G. D.; Suen J. Y.; Le G. T.; Cotterell A.; Reid R. C.; Fairlie D. P. Novel agonists and antagonists for human protease activated receptor 2. J. Med. Chem. 2010, 53, 7428–7440. 10.1021/jm100984y. [DOI] [PubMed] [Google Scholar]
- Gardell L. R.; Ma J. N.; Seitzberg J. G.; Knapp A. E.; Schiffer H. H.; Tabatabaei A.; Davis C. N.; Owens M.; Clemons B.; Wong K. K.; Lund B.; Nash N. R.; Gao Y.; Lameh J.; Schmelzer K.; Olsson R.; Burstein E. S. Identification and characterization of novel small-molecule protease-activated receptor 2 agonists. J. Pharmacol. Exp. Ther. 2008, 327, 799–808. 10.1124/jpet.108.142570. [DOI] [PubMed] [Google Scholar]
- Seitzberg J. G.; Knapp A. E.; Lund B. W.; Mandrup Bertozzi S.; Currier E. A.; Ma J. N.; Sherbukhin V.; Burstein E. S.; Olsson R. Discovery of potent and selective small-molecule PAR-2 agonists. J. Med. Chem. 2008, 51, 5490–5493. 10.1021/jm800754r. [DOI] [PubMed] [Google Scholar]
- Abad-Zapatero C.; Metz J. T. Ligand efficiency indices as guideposts for drug discovery. Drug Discovery Today 2005, 10, 464–469. 10.1016/S1359-6446(05)03386-6. [DOI] [PubMed] [Google Scholar]
- Su S.; Li Y.; Luo Y.; Sheng Y.; Su Y.; Padia R. N.; Pan Z. K.; Dong Z.; Huang S. Proteinase-activated receptor 2 expression in breast cancer and its role in breast cancer cell migration. Oncogene 2009, 28, 3047–3057. 10.1038/onc.2009.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.