

Abstract
In the last five years, the detailed understanding of how to overcome T790M drug resistance in non-small cell lung cancer (NSCLC) has culminated in the development of a third-generation of covalent EGFR inhibitors with excellent clinical outcomes. However, the emergence of a newly discovered acquired drug resistance challenges the concept of small molecule targeted cancer therapy in NSCLC.
TARGETED CANCER THERAPY: PARADIGM SHIFT IN CANCER TREATMENT
Mutations in the gene encoding the epidermal growth factor receptor (EGFR) have been discovered to be associated with the onset and progression of non-small cell lung cancer (NSCLC). The first NSCLC trials with first-generation EGFR inhibitors gefitinib and erlotinib (Figure 1A) were disappointing with partial responses observed in only 10% of treated patients.1 Subsequent gene sequencing revealed recurrent activating mutations in the kinase domain of EGFR that account for a dramatic clinical response (70%) of tyrosine kinase inhibitor (TKI) treatment as compared to conventional chemotherapy.2 The discovery and the specific targeting of these oncogenic drivers leading to a tumor regression seemed to be a major breakthrough in targeted cancer therapy and led to a paradigm shift in cancer treatment. However, the initial hopeful perspectives did not last long, as patients acquired drug resistances within months, limiting the effective treatment with TKIs. In approximately 60% of resistant cases, the patients develop a secondary point mutation at the gatekeeper position of the kinase domain (T790M) that represents a major challenge in the treatment of NSCLC.3 The replacement of a threonine by the sterically more demanding methionine (i) increases the affinity to ATP and (ii) provokes a steric repulsion of the 4-aminoquinazoline-based inhibitors erlotinib and gefitinib, resulting in a different binding mode and significant loss of inhibitory activity (Figure 1B).4 Second-generation EGFR TKIs, including the drug afatinib (Figure 1A), sparked a glimmer of hope in overcoming T790M drug resistance, as they showed promising results in preclinical studies.5 These inhibitors incorporate a Michael acceptor to covalently target a rare cysteine (Cys797) in EGFR at the lip of the ATP-binding site. This electrophile represents the only distinctive feature as compared to EGFR Type-I inhibitors, and thus, the potential of these drugs to overcome the T790M drug resistance is directly correlated with covalent modification of the target protein (Figure 1A). These findings have renewed the interest in covalent drug design and prompted further efforts to characterize them, although covalent drugs have long been avoided in medicinal chemistry. Their nonspecific reactivity and potential for off-target reactivity that may cause tissue injury and drug-related toxicity were major concerns.6
Figure 1.
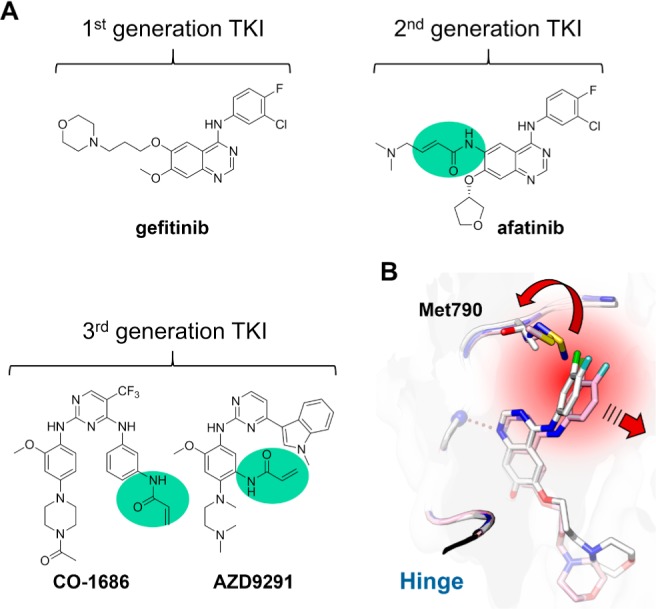
(A) Chemical structures of representative examples of the three generations of EGFR inhibitors currently used in the treatment of NSCLC. The reactive acrylamides are highlighted in green. (B) Illustration of the steric repulsion of the first-generation inhibitor gefitinib upon T790M gatekeeper mutation. The gefitinib binding pose observed with EGFR wild type (white, PDB code: 2ITY) would lead to a steric clash with the methionine side chain (blue, PDB code: 3UG1), resulting in an unfavored binding pose (pink, PDB code: 3UG2).
WAS THE FAILURE OF THE SECOND-GENERATION OF EGFR INHIBITORS IN T790M DRUG-RESISTANT PATIENTS PREDICTABLE?
Despite initial promising data for the second-generation EGFR inhibitors, their efficacy in patients was insufficient. A consideration of the structures of these drugs led investigators to ask if the failure of these drugs to efficiently target T790M drug resistance could have been foreseen, especially since they were derived from first-generation aminoquinazolines that were originally designed to inhibit the wild type form of EGFR. Accordingly, on-target toxicity occurred during treatment and led to severe side effects such as skin rash and diarrhea, thereby limiting the clinically achievable concentration.7 The required high drug dosage can be attributed to insufficient in vivo potency. Although covalent inhibitors form an irreversible modification, the initial step is a reversible interaction with the target protein to form a noncovalent drug–target complex. The subsequent covalent bond formation can only occur from the stabilized complex. The reduced stabilization in consequence of, e.g., the sterically demanding T790M mutation, as observed for 4-aminoquinazoline-based second-generation EGFR inhibitors, leads to a more pronounced dissociation of the drug–EGFR target complex. This event lowers the rate of covalent bond formation and results in reduced clinical efficacy. After initial enthusiasm, it became clear that modifying a weak inhibitor with a reactive electrophile was not sufficient to achieve in vivo efficacy (Figure 2A).4
Figure 2.

Binding mode of covalent EGFR tyrosine kinase inhibitors. The binding equilibrium indicates, whether the binding of ligand (L) and receptor (R) is favored. (A) The emergence of the T790M gatekeeper mutation induces steric hindrance of 4-aminoquinazolines such as afatinib with the methionine side chain (highlighted in red) and promotes the dissociation of the reversible ligand and receptor complex [LR]. Therefore, covalent bond formation (highlighted in yellow) of second-generation inhibitors with the receptor, yielding the covalent adduct L–R, cannot sufficiently occur (PDB code: 4G5P). (B) Third-generation TKIs, as exemplified by the structural analogue WZ4002, avoid the steric conflict with Met790 and therefore achieve complete receptor occupancy (PDB code: 3IKA). (C) The C797S mutation mediates resistance to irreversible drugs since the less nucleophilic serine side chain cannot undergo covalent bond formation (highlighted in red) at physiological conditions (model based on PDB code: 3IKA).
CO-1686 AND AZD9291: REASONS FOR THEIR CLINICAL SUCCESS
The urgent need for an efficient therapy in NSCLC patients that suffer from T790M drug resistance encouraged a reassessment of the development of EGFR inhibitors. In order to efficiently target EGFR-T790M, the receptor must be considered as an entire new protein rather than a mutant form of the wild type. The identified oncogenic drivers as well as the drug resistance mutation together with their well-understood underlying mechanisms provided a unique opportunity for rational approaches to develop new drugs to overcome the T790M drug resistance. Considering the advances the field has gained so far from first- and second-generation EGFR inhibitors in the clinics, the following features were crucial to achieve sufficient clinical efficacy: (i) a novel scaffold to specifically target the T790M gatekeeper mutant variant that can avoid a steric clash with Met790, while (ii) sparing wild type inhibition and being mutant-selective, and (iii) a reactive substituent to alkylate Cys797 in EGFR to overcome T790M drug resistance by achieving maximum drug-target residence time. A new generation of covalent EGFR inhibitors including CO-1686 (rociletinib) and AZD9291 (osimertinib) has been developed by applying these considerations (Figure 1A). Both drugs contain a distinctive aminopyrimidine scaffold as a hinge binding element, and they avoid the steric interference with the mutant methionine gatekeeper residue. Moreover, CO-1686 and AZD9291 incorporate an acrylamide as a Michael acceptor (Figure 2B). These novel inhibitors showed dramatic in vivo efficacy and progressed to human clinical trials, which enrolled patients suffering from advanced NSCLC driven by EGFR-activating mutations and who had relapsed into disease progression after previous treatment with existing EGFR TKIs. Both drugs showed impressive response rates of about 60% in patients harboring the acquired T790M drug-resistant mutation. The median progression-free survival for T790M-positive patients treated with rociletinib was 13.1 months and 9.6 months after administering osimertinib.8,9
With respect to the nonspecific reactivity and off-target reactivity that occur with covalent drugs, the aforementioned clinical studies showed no significant off-target toxicity. The dosage of CO-1686 could even be increased up to 1000 mg twice daily without observing a maximum tolerated dose.8 AZD9291 was dosed up to 240 mg daily and no dose-limiting toxic effects could be identified, indicating that an incorporated acrylamide warhead may represent a compromise with respect to reactivity and toxicity.9 These recent findings will further strengthen the development of covalent drugs and maybe diminish the major concerns about their off-target potential. Only very few patients displayed any drug-related side effects associated with EGFR-WT toxicity, demonstrating high mutant selectivity and reduced wild type inhibition.8,9 The most common dose-limiting adverse event for CO-1686 was hyperglycemia, which interestingly is caused by a noncovalent metabolite, which exhibits an inhibitory effect against the insulin-like growth factor 1 receptor (IGF1R) and the insulin receptor (INSR) tyrosine kinases leading to increased glucose and insulin levels.10 Dose reduction and metformin therapy as an antidiabetic medication brought hyperglycemia under control.8 These recent successes highlight that the knowledge the field has gained in recent years with respect to the relevance of oncogenic drivers, effective targeted inhibition, and acquired resistance mechanisms allows for the rational development of drugs active against a specific target protein, once a driver mutation is discovered. Targeting EGFR-dependent NSCLC represents an example of how successful personalized cancer therapy can be implemented. The covalent third-generation of EGFR inhibitors even demonstrated the successful treatment of an acquired drug resistance, giving hope to a subpopulation of patients harboring these mutations.
LIMITATIONS OF TARGETED THERAPIES
The success of targeted therapy is offset by limitations, as TKI treatment of T790M-positive patients led again to new resistances within months, indicating that all responders will eventually acquire some sort of drug resistance upon targeted treatment. In the case of AZD9291 treatment, remarkable 40% of the resistant cases developed a mutation that substitutes a less nucleophilic serine residue for the reactive cysteine (C797S). This point mutation prevents a covalent bond formation with the available covalent drugs, as a serine side chain is unlikely to undergo a Michael addition at physiological conditions, leading to a substantial loss of efficacy (Figure 2C).11 This resistance mutation has also been observed in the Bruton’s tyrosine kinase (BTK) that bears an analogous cysteine to EGFR,12 indicating a common resistance mechanism for covalent kinase inhibitors. Cysteine point mutations, therefore, may constitute a recurring liability for a broad range of covalent drugs in the future.
Considering these critical limitations, the concept of targeted cancer therapy raises the question about the prospects of this treatment. Recurrent and diverse mechanisms of drug resistance further decrease the number of patients that would benefit from a certain therapy. In fact, the population of patients is thereby split into several subgroups that all require a distinctive therapy. In this context, strong efforts will be required to unravel the emerging mechanisms of resistance as well as to develop innovative targeted drugs. Despite these challenges, targeted cancer therapy is a valuable strategy for NSCLC patients with defined clinical and molecular biomarkers. Although still too few in number, these patients profit significantly with a prolonged life expectancy as well as improved quality of life resulting from reduced side effects when compared to conventional chemotherapy. Current efforts in the identification of oncogenic drivers represent a further major aspect and allow for the intensive investigation and development of new targeted drugs. The availability of a defined set of targeted therapeutics to address given targets and their respective resistance mutations would not only allow for more flexible treatments but would also be directed toward the treatment of cancer as a chronic disease. Interestingly, treatment regimes that are fine-tuned based on the mutation status, are state-of-the-art in the treatment of HIV.
However, the outcomes from the clinics strengthen the conclusion that there will be the need for alternative strategies for the beneficial treatment of cancer beyond targeted therapies.
Glossary
ABBREVIATIONS
- ATP
Adenosine triphosphate
- BTK
Bruton tyrosine kinase
- EGFR
epidermal growth factor receptor
- HIV
human immunodeficiency virus
- IGF1R
insulin-like growth factor 1 receptor
- INSR
insulin receptor
- NSCLC
non-small cell lung cancer
- TKI
tyrosine kinase inhibitor
- WT
wild type
Author Contributions
‡ These authors contributed equally to this work. The manuscript was written through contributions of all authors.
Views expressed in this editorial are those of the authors and not necessarily the views of the ACS.
The authors declare no competing financial interest.
References
- Sharma S. V.; Bell D. W.; Settleman J.; Haber D. A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- Pao W.; Miller V.; Zakowski M.; Doherty J.; Politi K.; Sarkaria I.; Singh B.; Heelan R.; Rusch V.; Fulton L.; Mardis E.; Kupfer D.; Wilson R.; Kris M.; Varmus H. EGF receptor gene mutations are common in lung cancers from ″never smokers″ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 13306–13311. 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. A.; Arcila M. E.; Rekhtman N.; Sima C. S.; Zakowski M. F.; Pao W.; Kris M. G.; Miller V. A.; Ladanyi M.; Riely G. J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalczyk A.; Klüter S.; Rode H. B.; Simard J. R.; Grütter C.; Rabiller M.; Rauh D. Structural insights into how irreversible inhibitors can overcome drug resistance in EGFR. Bioorg. Med. Chem. 2008, 16, 3482–3488. 10.1016/j.bmc.2008.02.053. [DOI] [PubMed] [Google Scholar]
- Li D.; Ambrogio L.; Shimamura T.; Kubo S.; Takahashi M.; Chirieac L. R.; Padera R. F.; Shapiro G. I.; Baum A.; Himmelsbach F.; Rettig W. J.; Meyerson M.; Solca F.; Greulich H.; Wong K. K. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barf T.; Kaptein A. Irreversible protein kinase inhibitors: balancing the benefits and risks. J. Med. Chem. 2012, 55, 6243–6262. 10.1021/jm3003203. [DOI] [PubMed] [Google Scholar]
- Miller V. A.; Hirsh V.; Cadranel J.; Chen Y. M.; Park K.; Kim S. W.; Zhou C.; Su W. C.; Wang M.; Sun Y.; Heo D. S.; Crino L.; Tan E. H.; Chao T. Y.; Shahidi M.; Cong X. J.; Lorence R. M.; Yang J. C. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012, 13, 528–538. 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- Sequist L. V.; Soria J. C.; Goldman J. W.; Wakelee H. A.; Gadgeel S. M.; Varga A.; Papadimitrakopoulou V.; Solomon B. J.; Oxnard G. R.; Dziadziuszko R.; Aisner D. L.; Doebele R. C.; Galasso C.; Garon E. B.; Heist R. S.; Logan J.; Neal J. W.; Mendenhall M. A.; Nichols S.; Piotrowska Z.; Wozniak A. J.; Raponi M.; Karlovich C. A.; Jaw-Tsai S.; Isaacson J.; Despain D.; Matheny S. L.; Rolfe L.; Allen A. R.; Camidge D. R. Rociletinib in EGFR-mutated non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1700–1709. 10.1056/NEJMoa1413654. [DOI] [PubMed] [Google Scholar]
- Jänne P. A.; Yang J. C.; Kim D. W.; Planchard D.; Ohe Y.; Ramalingam S. S.; Ahn M. J.; Kim S. W.; Su W. C.; Horn L.; Haggstrom D.; Felip E.; Kim J. H.; Frewer P.; Cantarini M.; Brown K. H.; Dickinson P. A.; Ghiorghiu S.; Ranson M. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. 10.1056/NEJMoa1411817. [DOI] [PubMed] [Google Scholar]
- Simmons A. D.; Jaw-Tsai S.; Haringsma H. J.; Allen A.; Harding T. C. Abstract 793: Insulin-like growth factor 1 (IGF1R)/insulin receptor (INSR) inhibitory activity of rociletinib (CO-1686) and its metabolites in nonclinical models. Cancer Res. 2015, 75, 793–793. 10.1158/1538-7445.AM2015-793. [DOI] [Google Scholar]
- Thress K. S.; Paweletz C. P.; Felip E.; Cho B. C.; Stetson D.; Dougherty B.; Lai Z.; Markovets A.; Vivancos A.; Kuang Y.; Ercan D.; Matthews S. E.; Cantarini M.; Barrett J. C.; Jänne P. A.; Oxnard G. R. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyach J. A.; Furman R. R.; Liu T. M.; Ozer H. G.; Zapatka M.; Ruppert A. S.; Xue L.; Li D. H.; Steggerda S. M.; Versele M.; Dave S. S.; Zhang J.; Yilmaz A. S.; Jaglowski S. M.; Blum K. A.; Lozanski A.; Lozanski G.; James D. F.; Barrientos J. C.; Lichter P.; Stilgenbauer S.; Buggy J. J.; Chang B. Y.; Johnson A. J.; Byrd J. C. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]