

Abstract
A series of new 2-methoxy-4-(5-phenyl-4,5-dihydro-1H-pyrazol-3-yl)phenolderivatives, 4–13, were synthesized and tested for their human MAO inhibitory activity. All the compounds were found to be selective and reversible toward hMAO-A except 4, a selective inhibitor of hMAO-B and 12, a nonselective inhibitor. Compound 7 was found to be a potent inhibitor of hMAO-A with Ki = 0.06 ± 0.003 μM and was having selectivity index of (SI = 1.02 × 10–5). It was found to be better than standard drug, Moclobemide (hMAO-A with Ki = 0.11 ± 0.01 μM) with selectivity index of SI = 0.049. Molecular docking simulation was carried out to understand the crucial interactions responsible for selectivity and potency.
Keywords: Human monoamine oxidase, pyrazoline, enzyme inhibitors, molecular docking simulation
Human monoamine oxidases are flavin adenine dinucleotide (FAD) containing enzymes located on the outer mitochondrial membrane. They are responsible for the metabolism of dietary amines as well as neurotransmitters. There exist two different isoforms, hMAO-A and hMAO-B, differentiated by their gene, amino acid sequence, substrate preference, and affinity toward their selective inhibitors.1,2 Serotonin, adrenaline, and noradrenaline are preferably metabolized by hMAO-A,3 while beta-phenylethylamine and benzylamine byhMAO-B,4 tyramine, dopamine, and some other important amines are common substrates for both isoforms.5 Nonselective inhibition of these isoforms leads to the serious adverse effects and hence the early MAO-inhibitor drugs were abandoned from clinical practice. In the past decade it has been demonstrated that specific inhibition of these isoforms may have potential therapeutic utility in the treatment of depression (MAO-A selective) and neurodegenerative disorders (MAO-B selective).6−8
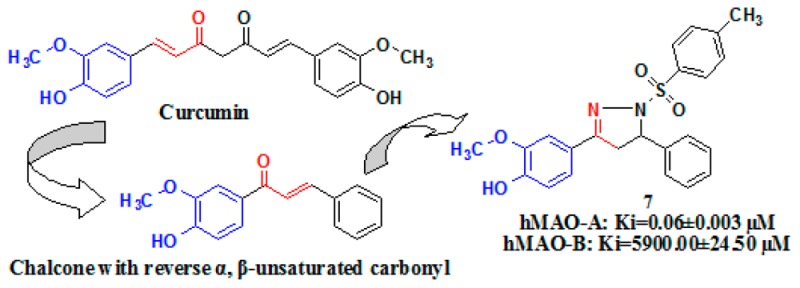
The present study explores the synthesis, hMAO-inhibitory activity, and binding mode analysis of few newer pyrazoline derivatives. The design strategy utilized the structural features in curcumin, a natural compound with proven hMAO inhibitory and antidepressant activity (Figure 1).9−14 Due to the poor bioavailability and extensive metabolism, curcumin has never been recognized as potential candidate for the treatment of depression. We prepared few chalcones that resembles aryl-α,β-unsaturated carbonyl portion of curcumin but by reversing the position of the double bond and the carbonyl group. These derivatives were then cyclized to provide a series of novel pyrazoline derivatives. They are then tested for their hMAO inhibitory activity in both the isoforms. Molecular docking studies were also carried out to understand the interactions that are crucial for their potency and selectivity.
Figure 1.
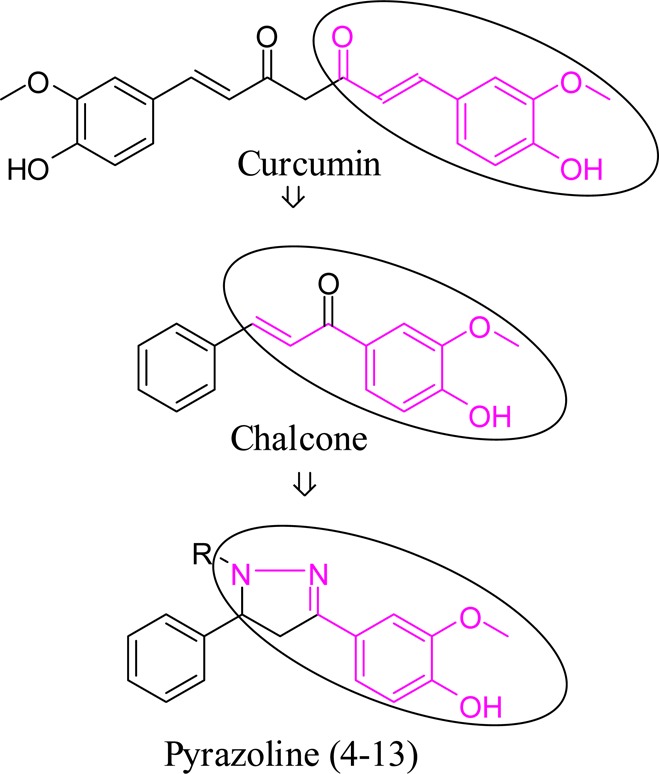
Design strategy adopted for designing MAO inhibitors.
Results and Discussion
Chemistry
The pyrazoline derivatives 4–13 were synthesized according to the reactions outlined in Schemes 1 and 2. The chalcone intermediate 3 was prepared through Claisen–Schmidt condensation of benzaldehyde with 4-hydroxy-3-methoxy acetophenone in aqueous sodium hydroxide 60% in ethanol for 48 h. The yield of intermediate 3 was found to be poor (28%), and hence, we protected the hydroxyl group of 4-hydroxy-3-methoxy acetophenone with 3,4-dihydro-α-pyran. The protected acetophenone 1 provided protected chalcone 2, which upon deprotection with p-toluene sulfonic acid produced intermediate 3 with an improved yield of 90% (Scheme 1).
Scheme 1.

Reagents and conditions: (a) PPTS, dry DCM, 3,4-dihydro-α-pyran, stirring for 12 h, rt; (b) C6H5–CHO, aq. NaOH (60%), EtOH, 48 h, rt, HCl (6 N) up to pH 2; (c) PTSA, EtOH, stirring for 12 h, rt; (d) NH2NH2.H2O, EtOH, reflux, 3 h.
Scheme 2.

Reagents and conditions: (a) C6H5COCl, pyridine, reflux 3h; (b) C6H5–NCS, EtOH, reflux, 2 h; (c) p-CH3–C6H4SO2Cl in THF, stirring for 15 min, 10–15 °C; (d) Cl-CO-OR, K2CO3, EtOH, stirring, 15–20 min, <10 °C; (e) C6H5–NH–NH2, EtOH, NaOH, reflux, 4 h; (f) NH2NH(C=X)NH2, ethanolic NaOH, reflux, 6 h; (g) NH2NH2·H2O, AcOH, reflux, 4 h.
Compounds 4–13 were synthesized through the reactions outlined in Scheme 2. Compound 4 was obtained by the reaction of intermediate 3 with excess hydrazine hydrate in ethanol.15 Reaction of benzoyl chloride with compound 4 in pyridine provided compound 5.16 Compound 6 was obtained by refluxing compound 4 with phenyl isothiocyanate in ethanol.17 Reaction of p-toluene sulphonyl chloride with compound 4 in tetrahydrofuran provided compound 7.16 Compounds 8 and 9 were obtained by the reaction of compound 4 with ethyl chloroformate and phenyl chloroformate, respectively, in the presence of potassium carbonate in ethanol.15 Refluxing intermediate 3 with phenylhydrazine in ethanol provided compound 10.18 Treatment of intermediate 3 with hydrazine hydrate in the presence of acetic acid under reflux afforded compound 11.19 Compound 12 was prepared by the reaction of intermediate 3 with two equivalents of thiosemicarbazide in the presence of sodium hydroxide in ethanol.17 Compound 13 was prepared by the reaction of intermediate 3 with two equivalents of amino guanidine hydrochloride in the presence of aqueous sodium hydroxide in ethanol.17
All the final compounds, 4–13, have been characterized using 1H NMR, 13C NMR, and ESI-MS spectra. In 1H NMR, the three protons present at fourth and fifth position of the pyrazoline ring exhibited splitting pattern characteristic of AMX system and displayed three doublet of doublets (dd). Chemical shifts for HA proton appeared at δ 2.74–3.748 ppm (JAM = 16–18 Hz and JAX = 3.13–8.8 Hz). For HM proton the chemical shift appeared at δ 3.35–3.94 ppm (JMA = 12–18 Hz and JMX = 4.4–12 Hz) and for HX proton at δ 4.74–5.99 ppm (JXM = 11.4–12 Hz and JXA = 4.0–4.8 Hz). The proton of phenolic −OH appeared at δ 9.19–10.01 ppm, as singlet/broad peak. The methoxy (Ar-OCH3) protons have appeared at around δ 3.764–3.84 ppm; for compound 9 the −CH2 proton appeared as sharp peak at δ 4.0–4.06 ppm as a quartet. The proton for compound 6, the −NH–, has appeared at δ 10.04 ppm .The methyl proton for compounds 7(Ar–CH3) and 11(−CO–CH3) appeared as singlet at δ 2.268 and 2.293 ppm, respectively. For compound 9 the methyl (−CH2–CH3) proton appeared between δ 1.07–1.10 ppm as a triplet.
13C NMR has been recorded for all the compounds (4–13), the peaks for pyr-C3, C4, and C5 are in the range of δ 148.36–156.30, 41.46–56.35, and 60.97–64.02 ppm, respectively. The peaks for Ar–OCH3 are in the range of δ 55.88–56.75 ppm. Compound 7 Ar-CH3 peak appeared at δ 21.26 ppm. Compound 11 displayed −CO–CH3 peak at δ 26.81 ppm. Compound 9 displayed −CH2–CH3 and −CH2–CH3 peaks at δ 26.57 and 61.27 ppm, respectively. Compounds 5, 8, 9, and 11 displayed the −C=O peak at δ 165.14, 155.73, 154.18, and 167.55 ppm, respectively. Compounds 6 and 12 displayed the −C=S peak at δ 173.75 and 176.68 ppm, respectively. Compound 13 displayed H2N–C=NH peak at δ 155.18 ppm. ESI-mass spectrum spectra of all the final compounds displayed the (M + 1)+ as base peak, except compound 10, which displayed (M – 1)− as a base peak. Experimental procedures and spectral data are presented in the Supporting Information.
Biochemistry
All the synthesized compounds 4–13 were screened for their hMAO inhibitory activity20,21 using recombinant human MAO isoforms. MAO inhibitory activity was determined by measuring the production of H2O2 from p-tyramine, the common substrate for both hMAO-A and hMAO-B, using the Amplex-Red MAO assay kit. The compounds and reference inhibitors did not react directly with the Amplex-Red reagent indicating that they do not interfere with the measurements. The compounds were also confirmed as they did not interact with resorufin by treating the maximum concentration of compounds with various concentrations of resorufin in order to detect if the fluorescence signal is the same with or without our compounds in the medium. No significant quenching of resorufin was observed.
The experimental data concerning the inhibition of hMAO-A and hMAO-B inhibition by the novel compounds 4–13 were presented in Table 1, together with their selectivity indices. The specificity index was expressed as SI = Ki(MAO-A)/Ki(MAO-B). Protein was determined according to Bradford22 using bovine serum albumin as the standard. Eight compounds, 5–11 and 13, were found to inhibit hMAO-A selectively and reversibly, while compound 4 was found to be selective toward hMAO-B and compound 12 nonselective toward both the isoform. The most potent hMAO-A inhibitor in this series was compound 7, which is carrying a tosyl group at 1N position of pyrazoline ring. Compound 7 inhibited hMAO-A potently and selectively with the Ki value of 0.06 ± 0.003 μM and SIhMAO-B of 1.02 × 10–5, which is better than Moclobemide, a well-known selective MAO-A inhibitor (Ki value of 0.11 ± 0.01 μM and SIhMAO-B of 0.049, Table 1). A Lineweaver–Burk plot for compound 7 is presented in the Supporting Information. Compound 4, which is carrying no substitution at the 1N position of pyrazoline ring, inhibited hMAO-B selectively and competitively with the Ki value of 0.35 ± 0.02 μM, which is lower than the Ki value of selegiline, a well-known selective MAO-B inhibitor (Ki value of selegiline for hMAO-B was estimated as 0.38 ± 0.01 μM). However, the SI value calculated for compound 4 (SIhMAO-B = 14.34, Table 1) was not found to be as good as the one calculated for Selegiline (SIhMAO-B = 44.44, Table 1).
Table 1. Experimentally determined Ki Values for the Newly Synthesized 2-Methoxy-4-(5-phenyl-4,5-dihydro-1H-pyrazol-3-yl)phenol Derivatives for hMAO Isoforms A and B.
| calcd Ki value (μM) |
exptl Ki values (μM)b |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| compda | MAO-A (2BXR) | MAO-B (2BYB) | SI | MAO-A | MAO-B | SIc | inhibition type | reversibility | MAO selectivity |
| 4 | 5.38 | 0.5 | 11.96 | 5.02 ± 0.25 | 0.35 ± 0.02 | 14.343 | competitive | reversible | B |
| 5 | 4.13 | 50.5 | 0.08 | 4.25 ± 0.21 | 41.05 ± 3.06 | 0.104 | competitive | reversible | A |
| 6 | 0.59 | 15.7 | 0.04 | 0.55 ± 0.03 | 15.90 ± 1.22 | 0.035 | competitive | reversible | A |
| 7 | 0.05 | 180842.9 | 0.00 | 0.06 ± 0.003 | 5900.00 ± 324.50 | 1.02 × 10–5 | competitive | reversible | A |
| 8 | 0.24 | 4.1 | 0.06 | 0.29 ± 0.02 | 4.00 ± 0.29 | 0.073 | competitive | reversible | A |
| 9 | 0.42 | 143.5 | 0.0029 | 0.44 ± 0.03 | 201.25 ± 12.40 | 0.002 | competitive | reversible | A |
| 10 | 2.58 | 15.9 | 0.16 | 2.16 ± 0.15 | 27.95 ± 1.90 | 0.077 | competitive | reversible | A |
| 11 | 3.17 | 32.7 | 0.10 | 3.87 ± 0.18 | 33.50 ± 2.86 | 0.116 | competitive | reversible | A |
| 12 | 1.755 | 3.5 | 0.50 | 2.58 ± 0.15 | 2.78 ± 2.00 | 0.928 | competitive | reversible | nonselective |
| 13 | 19.24 | 134.5 | 0.14 | 9.55 ± 0.58 | 201.35 ± 14.45 | 0.047 | competitive | reversible | A |
| curcumin | 0.602 | 16.40 | 0.036 | 0.71 ± 0.04 | 21.50 ± 1.19 | 0.033 | competitive | reversible | A |
| selegiline | 16.89 ± 1.24 | 0.38 ± 0.01 | 44.448 | suicide inhibitor | irreversible | B | |||
| moclobemide | 0.11 ± 0.01 | 2.24 ± 0.02 | 0.049 | competitive | reversible | A | |||
Racemic compounds were used for the experiments.
Each value represents the mean ± SEM of three independent experiments.
Selectivity index. It was calculated as Ki(MAO-A)/Ki(MAO-B). Selectivity toward MAO-A increases as the corresponding SI decreases, while selectivity toward MAO-B isoform increases as the corresponding SI increases.
All the compounds inhibited the hMAO isoforms reversibly, and reversibility was assessed using a centrifugation-ultra filtration method previously reported23 (data presented in Supporting Information). Irreversible inhibitors of MAO previously have been used as clinical drugs, but this type of inhibition has been shown to induce significant toxic effects. Competitive reversible inhibitors have less influence in the enzyme recovery after withdrawal since the ingested tyramine is able to displace the inhibitor from the MAO active site and be metabolized in the normal way by peripheral enzyme in gut and liver.24 Besides, the slow and variable enzyme recovery following the withdrawal of irreversible inhibitors is a disadvantage in clinical use since the turnover rate for MAO biosynthesis in the human brain seems to require about 40 days. Thus, newly synthesized reversible MAO inhibitors may have a value in this aspect.
The compounds are unique in having curcumin aryl head at the third position of pyrazoline when compared with earlier reported pyrazolines.25,26 As they are designed based on the structure of curcumin, the comparison was made with reference to curcumin and standard used rather than earlier reported pyrazolines.
Molecular Docking Simulation
Molecular docking simulation was carried out to understand the effect of chirality and other structural features that determine the potency and selectivity. The 5C carbon of pyrazoline is chiral in nature, and two isomers are possible. In the presented work we did not make any attempt to separate the two isomers, and the simulation approach has been employed to check the effect of chirality on activity and selectivity. The approach has been adopted by other groups as well as our groups in our earlier communications.15−18,28 Many researchers preferred to use the PDB of MAO cocrystallized with noncovalent ligands (2Z5X, 2V60, 2BK3);29,30 we also made an attempt, but the experimental and predicted values did not correlate well. Hence, we adopted earlier protocol reported by our group with 2BXR and 2BYB.
Docking results were in good agreement with the experimentally determined values (Table 1). Average of estimated Ki value of R- and S-isomer has been considered for comparison with the experimental Ki value (Ki values of individual isomers are presented in Supporting Information); R-isomers were found to be more potent than S-isomers in selectively inhibiting hMAO-A, except for compound 5. The magnitude of the difference was calculated as 1.04–4.61-fold. Hence, for comparison with the experimental value, averages of R- and S-isomers were calculated for each isoform. These values were found to be in good agreement with the experimentally determined values.
In order to understand the factors that contribute toward the selectivity, the docked complexes of compounds 4 and 7 were studied.
Interaction of Compound 7 with hMAO-A
As per calculated Ki value, R-isomer was 4-fold more potent than S-isomer (See Supporting Information). They also differ in orientation within the catalytic pocket of hMAO-A. R-isomer: it oriented in such a way that the substitution at 1N position (Ring C) oriented toward pocket 1 (Aromatic cage lined by FAD, Tyr407 and Tyr444). This places the phenyl ring (Ring B) at fifth position of pyrazoline in pocket 2 (lined by Ile180, Ile335, Leu337, Met350, and Phe352) and the phenyl ring (Ring A) at third position of pyrazolinein pocket 3 (lined by Gly74, Arg206, Ile207, Phe208, Glu216, and Trp441). There is an H-bonding interaction between the hydroxy oxygen of ring A with carbonyl (−C=O) oxygen of Ile207 (Figure 2). S-isomer: it also oriented itself in a way similar to R-isomer placing Ring C and Ring B in pocket 1 and pocket 2, respectively. Due to this, Ring A is almost placed in the central core where pyrazoline resides and could not be able to occupy pocket 3 completely. Also, there is no H-bonding interactions, and this may be the reason why R-isomer is much better (4-fold) than S-isomer (Figure 2).
Figure 2.

Interaction of compound 7: (a) R-isomer; (b) S-isomer with hMAO-A (PDB: 2BXR) active site. H-bonds are shown as green dots.
Interaction of Compound 4 with hMAO-B
Both R-isomer and S-isomer have calculated Ki values close to each other (see Supporting Information). Analysis of their docked complexes revealed that they also oriented in a similar fashion within the catalytic pocket of hMAO-B. Both the isomers kept their Ring A in an aromatic cage formed by FAD, Tyr435, and Tyr398. This has kept the pyrazoline ring at the center and projected Ring B toward the entrance of the active site cavity. The hydroxy group of both isomers established H-bonding interactions with the active site residues (Figure 3).
Figure 3.

Interaction of Compound 4: (a) R-isomer; (b) S-isomer with hMAO-B (PDB: 2BYB) active site. H-bonds are shown as green dots.
Conclusion
Ten compounds (4–13) were synthesized with structural features of curcumin in which eight compounds were found to be potent, selective, competitive, and reversible inhibitor of hMAO-A. Compound 7 was found to be more potent than curcumin and also the standard drug moclobemide. Molecular docking simulation studies revealed that the R-isomer of compound 7 was 4-fold better than the S-isomer.
Acknowledgments
Author was thankful to Birla Institute of Technology for providing financial support as a prestigious Institute Fellowship and acknowledges the services of Dr. Reddy’s Institute of Life Science, Hyderabad, and Central Instrumentation Laboratory-(CIL), Guru Jambeshwar University of Science and Technology, Hisarfor, spectral characterization.
Glossary
Abbreviations
- MAO
monoamine oxidase
- hMAO
human MAO
- rMAO
rat MAO
- SAR
structure–activity relationship
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00326.
Experimental procedures for synthesis of compounds, biochemical assay, and molecular docking. Spectral characterization data (1H NMR, 13C NMR, and ESI-MS) for all the final compounds along with spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- YOUDIM M. B. H.; COLLINS G. G. S.; SANDLER M.; JONES A. B. B.; PARE C. M. B.; NICHOLSON W. J. Biological Sciences: Human Brain Monoamine Oxidase: Multiple Forms and Selective Inhibitors. Nature 1972, 236 (5344), 225–228. 10.1038/236225b0. [DOI] [PubMed] [Google Scholar]
- Collins G. G. S.; Sandler M.; Williams E. D.; Youdim M. B. H. Multiple Forms of Human Brain Mitochondrial Monoamine Oxidase. Nature 1970, 225 (5235), 817–820. 10.1038/225817a0. [DOI] [PubMed] [Google Scholar]
- Johnston J. P. Some Observations Upon a New Inhibitor of Monoamine Oxidase in Brain Tissue. Biochem. Pharmacol. 1968, 17, 1285–1297. 10.1016/0006-2952(68)90066-X. [DOI] [PubMed] [Google Scholar]
- Knoll J.; Magyar K. Some Puzzling Pharmacological Effects of Monoamine Oxidase Inhibitors. Adv. Biochem. Psychopharmacol. 1971, 5, 393–408. [PubMed] [Google Scholar]
- O’Carroll A. M.; Fowler C. J.; Phillips J. P.; Tobbia I.; Tipton K. F. The Deamination of Dopamine by Human Brain Monoamine Oxidase - Specificity for the Two Enzyme Forms in Seven Brain Regions. Naunyn-Schmiedeberg's Arch. Pharmacol. 1983, 322, 198–202. 10.1007/BF00500765. [DOI] [PubMed] [Google Scholar]
- Da Prada M.; Keller H. H.; Kettler R. Comparison of the New MAO-A Inhibitors Moclobemide, Brofaromine and Toloxatone with Tranylcypromine in an Animal Experiment: Significance for Clinical Practice. Psychiatr. Prax. 1989, 16 (Suppl 1), 18–24. [PubMed] [Google Scholar]
- Lavian G.; Finberg J. P.; Youdim M. B. The Advent of a New Generation of Monoamine Oxidase Inhibitor Antidepressants: Pharmacologic Studies with Moclobemide and Brofaromine. Clin. Neuropharmacol. 1993, 16 (Suppl 2), S1–S7. [PubMed] [Google Scholar]
- Youdim M. B. H.; Weinstock M. Therapeutic Applications of Selective and Non-Selective Inhibitors of Monoamine Oxidase A and B That Do Not Cause Significant Tyramine Potentiation. NeuroToxicology 2004, 25, 243–250. 10.1016/S0161-813X(03)00103-7. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Ku B. S.; Yao H. Y.; Lin Y. H.; Ma X.; Zhang Y. H.; Li X. J. Antidepressant Effects of Curcumin in the Forced Swim Test and Olfactory Bulbectomy Models of Depression in Rats. Pharmacol., Biochem. Behav. 2005, 82, 200–206. 10.1016/j.pbb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Kulkarni S. K.; Bhutani M. K. Antidepressant Activity of Curcumin: Involvement of Serotonin and Dopamine System. Psychopharmacology 2008, 201, 435–442. 10.1007/s00213-008-1300-y. [DOI] [PubMed] [Google Scholar]
- Rajeswari A.; Sabesan M. Inhibition of Monoamine Oxidase-B by the Polyphonic Compound, Curcumin and Its Metabolite Tetrahydrocurcumin, in a Model of Parkinson’s Disease Induced by MPTP Neurodegenration in Mice.. Inflammopharmacology 2008, 16, 96–99. 10.1007/s10787-007-1614-0. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Luo J.; Zhang M.; Yao W.; Ma X.; Yu S. Y. Effects of Curcumin on Chronic, Unpredictable, Mild, Stress-Induced Depressive-like Behaviour and Structural Plasticity in the Lateral Amygdala of Rats. Int. J. Neuropsychopharmacol. 2014, 17, 793–806. 10.1017/S1461145713001661. [DOI] [PubMed] [Google Scholar]
- Wang R.; Xu Y.; Wu H. L.; Li Y. B.; Li Y. H.; Guo J. B.; Li X. J. The Antidepressant Effects of Curcumin in the Forced Swimming Test Involve 5-HT1 and 5-HT2 Receptors. Eur. J. Pharmacol. 2008, 578, 43–50. 10.1016/j.ejphar.2007.08.045. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Ku B. S.; Yao H. Y.; Lin Y. H.; Ma X.; Zhang Y. H.; Li X. J. The Effects of Curcumin on Depressive-like Behaviors in Mice. Eur. J. Pharmacol. 2005, 518, 40–46. 10.1016/j.ejphar.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Vishnu Nayak B.; Ciftci-Yabanoglu S.; Jadav S. S.; Jagrat M.; Sinha B. N.; Ucar G.; Jayaprakash V. Monoamine Oxidase Inhibitory Activity of 3,5-Biaryl-4,5-Dihydro-1H-Pyrazole-1-Carboxylate Derivatives. Eur. J. Med. Chem. 2013, 69, 762–767. 10.1016/j.ejmech.2013.09.010. [DOI] [PubMed] [Google Scholar]
- Sahoo A.; Yabanoglu S.; Sinha B. N.; Ucar G.; Basu A.; Jayaprakash V. Towards Development of Selective and Reversible Pyrazoline Based MAO-Inhibitors: Synthesis, Biological Evaluation and Docking Studies. Bioorg. Med. Chem. Lett. 2010, 20 (1), 132–136. 10.1016/j.bmcl.2009.11.015. [DOI] [PubMed] [Google Scholar]
- Jagrat M.; Behera J.; Yabanoglu S.; Ercan A.; Ucar G.; Sinha B. N.; Sankaran V.; Basu A.; Jayaprakash V. Pyrazoline Based MAO Inhibitors: Synthesis, Biological Evaluation and SAR Studies. Bioorg. Med. Chem. Lett. 2011, 21 (14), 4296–4300. 10.1016/j.bmcl.2011.05.057. [DOI] [PubMed] [Google Scholar]
- Ozdemir Z.; Kandilci H. B.; Gumusel B.; Calis U.; Bilgin a A. Synthesis and Studies on Antidepressant and Anticonvulsant Activities of Some 3-(2-Furyl)-Pyrazoline Derivatives. Eur. J. Med. Chem. 2007, 42 (3), 373–379. 10.1016/j.ejmech.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Nepali K.; Singh G.; Turan A.; Agarwal A.; Sapra S.; Kumar R.; Banerjee U. C.; Verma P. K.; Satti N. K.; Gupta M. K.; Suri O. P.; Dhar K. L. A Rational Approach for the Design and Synthesis of 1-Acetyl-3,5-Diaryl-4,5-dihydro(1H)pyrazoles as a New Class of Potential Non-Purine Xanthine Oxidase Inhibitors. Bioorg. Med. Chem. 2011, 19, 1950–1958. 10.1016/j.bmc.2011.01.058. [DOI] [PubMed] [Google Scholar]
- Chimenti F.; Maccioni E.; Secci D.; Bolasco A.; Chimenti P.; Granese A.; Carradori S.; Alcaro S.; Ortuso F.; Yáñez M.; Orallo F.; Cirilli R.; Ferretti R.; La Torre F. Synthesis, Stereochemical Identification, and Selective Inhibitory Activity against Human Monoamine Oxidase-B of 2-Methylcyclohexylidene-(4-Arylthiazol-2- Yl)hydrazones. J. Med. Chem. 2008, 51, 4874–4880. 10.1021/jm800132g. [DOI] [PubMed] [Google Scholar]
- Yáñez M.; Fraiz N.; Cano E.; Orallo F. Inhibitory Effects of Cis- and Trans-Resveratrol on Noradrenaline and 5-Hydroxytryptamine Uptake and on Monoamine Oxidase Activity. Biochem. Biophys. Res. Commun. 2006, 344 (2), 688–695. 10.1016/j.bbrc.2006.03.190. [DOI] [PubMed] [Google Scholar]
- Bradford M. M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Chimenti F.; Carradori S.; Secci D.; Bolasco A.; Bizzarri B.; Chimenti P.; Granese A.; Yáñez M.; Orallo F. Synthesis and Inhibitory Activity against Human Monoamine Oxidase of N1-Thiocarbamoyl-3,5-Di(hetero)aryl-4,5-Dihydro-(1H)-Pyrazole Derivatives. Eur. J. Med. Chem. 2010, 45, 800–804. 10.1016/j.ejmech.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Youdim M. B. H.; Bakhle Y. S. Monoamine Oxidase: Isoforms and Inhibitors in Parkinson’s Disease and Depressive Illness. Br. J. Pharmacol. 2006, 147 (S1), S287–S296. 10.1038/sj.bjp.0706464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secci D.; Bolasco A.; Chimenti P.; Carradori S. The State of the Art of Pyrazole Derivatives as Monoamine Oxidase Inhibitors and Antidepressant/anticonvulsant Agents. Curr. Med. Chem. 2011, 18 (33), 5114–5144. 10.2174/092986711797636090. [DOI] [PubMed] [Google Scholar]
- Secci D.; Carradori S.; Bolasco A.; Bizzarri B.; D’Ascenzio M.; Maccioni E. Discovery and Optimization of Pyrazoline Derivatives as Promising Monoamine Oxidase Inhibitors. Curr. Top. Med. Chem. 2012, 12 (20), 2240–2257. 10.2174/156802612805220057. [DOI] [PubMed] [Google Scholar]
- Chimenti F.; Fioravanti R.; Bolasco A.; Manna F.; Chimenti P.; Secci D.; Rossi F.; Turini P.; Ortuso F.; Alcaro S.; Cardia M. C. Synthesis, Molecular Modeling Studies and Selective Inhibitory Activity against MAO of N1-Propanoyl-3,5-Diphenyl-4,5-Dihydro-(1H)-Pyrazole Derivatives. Eur. J. Med. Chem. 2008, 43 (10), 2262–2267. 10.1016/j.ejmech.2007.12.026. [DOI] [PubMed] [Google Scholar]
- Chimenti F.; Bolasco A.; Secci D.; Chimenti P.; Granese A.; Carradori S.; Yáñez M.; Orallo F.; Ortuso F.; Alcaro S. Investigations on the 2-Thiazolylhydrazyne Scaffold: Synthesis and Molecular Modeling of Selective Human Monoamine Oxidase Inhibitors. Bioorg. Med. Chem. 2010, 18, 5715–5723. 10.1016/j.bmc.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Desideri N.; Bolasco A.; Fioravanti R.; Proietti Monaco L.; Orallo F.; Yáñez M.; Ortuso F.; Alcaro S. Homoisoflavonoids: Natural Scaffolds with Potent and Selective Monoamine Oxidase-B Inhibition Properties. J. Med. Chem. 2011, 54, 2155–2164. 10.1021/jm1013709. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.