

SUMMARY
Development of cancer has been linked to chronic inflammation, particularly via interleukin-23 (IL-23) and IL-17 signaling pathways. However, the cellular source of IL-17 and underlying mechanisms by which IL-17-producing cells promote human colorectal cancer (CRC) remain poorly defined. Here, we demonstrate that innate γδT (γδT17) cells are the major cellular source of IL-17 in human CRC. Microbial products elicited by tumorous epithelial barrier disruption correlated with inflammatory dendritic cell (inf-DC) accumulation and γδT17 polarization in human tumors. Activated inf-DCs induced γδT17 cells to secrete IL-8, tumor necrosis factor alpha, and GM-CSF with a concomitant accumulation of immunosuppressive PMN-MDSCs in the tumor. Importantly, γδT17 cell infiltration positively correlated with tumor stages and other clinicopathological features. Our study uncovers an inf-DC-γδT17-PMN-MDSC regulatory axis in human CRC that correlates MDSC-meditated immunosuppression with tumor-elicited inflammation. These findings suggest that γδT17 cells might be key players in human CRC progression and have the potential for treatment or prognosis prediction.
INTRODUCTION
Colorectal cancer (CRC) is one of the most common fatal malignancies worldwide. Links between cancer and inflammation were first made by Rudolf Virchow in the nineteenth century. Accumulating evidence has demonstrated that chronic inflammation and cancer are closely linked (Balkwill et al., 2005; Balkwill and Mantovani, 2001; Coussens and Werb, 2002; Karin, 2006). Cancer-related inflammation promotes tumor development and progression through many different mechanisms, such as promoting angiogenesis and metastasis, subverting immune responses, and altering responses to chemotherapeutic agents (Mantovani et al., 2008). A persistent inflamed microenvironment can also trigger mutagenic processes that serve as cancer-initiating events. Further tumor progression is augmented by the continuous presence of inflammatory cells and cytokines, which might transform an inflamed microenvironment into an immunosuppressive milieu (Grivennikov et al., 2010). Accordingly, treatment with nonsteroidal anti-inflammatory agents has shown decreased incidence and mortality in several tumor types (Rothwell et al., 2010).
The interleukin-23 (IL-23)-IL-17 axis plays a critical role in human CRC (Grivennikov et al., 2012; Langowski et al., 2006) and T helper 17 (Th17) cell expression signatures in CRC have been shown to be associated with poor survival (Grivennikov et al., 2012; Zou and Restifo, 2010). Although Th17 cells are implicated to predominately produce IL-17 in murine colon cancer models (Grivennikov et al., 2012), the source of IL-17 in human CRC has not been defined. In addition, the underlying mechanisms by which IL-17 and its related cytokines promote human CRC development and progression remain incompletely understood.
γδT cells have been recently demonstrated the major innate source of IL-17 (γδT17) and are known to play a critical role in autoimmune and inflammatory diseases (Petermann et al., 2010; Sutton et al., 2009) such as inflammatory bowel disease (Park et al., 2010), psoriasis (Cai et al., 2011; Pantelyushin et al., 2012), dermatitis (Gray et al., 2013), and hepatitis (Wang et al., 2013). Recent studies also showed that γδT17 cells could facilitate tumor growth via promoting angiogenesis in mice (Silva-Santos, 2010; Wakita et al., 2010). However, the properties and roles of γδT17 cells in human cancer-related inflammation have not been examined. Here, we have demonstrated that tumor-infiltrating γδT17 cells, but not Th17 cells or Tc17 cells, are the major IL-17A (hereafter referred to as IL-17)-producing cells in human CRC. Disruption of epithelial barrier in colon led to bacterial invasion, which correlated with inflammatory DC (inf-DC) accumulation and activation to secrete IL-23 thus promoting γδT17 polarization. Activated γδT17 cells also secreted other cytokines including IL-8, tumor necrosis factor alpha (TNF-α), and GM-CSF, which might chemoattract polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) in the tumor and sustain their immunosuppressive activity. Tumor-infiltrating γδT17 cells positively correlated with advanced tumor clinico-pathological features. Thus we reveal an inf-DCs-γδT17-PMN-MDSCs regulatory axis in human CRC that correlates immune suppression and tumor progression.
RESULTS
γδT17 Cells Are the Predominant IL-17-Producing Cells in Human CRC
Inflammatory signature genes have been shown to be upregulated in human CRC (Reichling et al., 2005; Wood et al., 2007). We found that IL-17 was increased both at the level of transcription and protein expression (Figure S1A and S1B) in human CRC tissues. To investigate the source of IL-17, we prepared single cell suspensions from tumor and fresh paired normal tissues. We found that the major IL-17-producing cells were CD3+ T cells (Figure 1A, left) in tumor. The percentages and absolute numbers of both IL-17+ cells in CD45+ cells and CD3+ cells were substantially increased in tumor compared with paired normal tissues (Figure 1A, right). Furthermore, we found that IL-17 was produced by CD8+ T cells (Tc17), CD4+ T cells (Th17), and γδTCR+ T cells (γδT17) (Figure 1B, left) but not innate lymphoid cells (ILCs) (Figure S1C). The percentages and absolute numbers of Tc17, Th17, and γδT17 cells were substantially increased in tumor tissues compared to those in paired normal tissues (Figure 1B, right). Among all IL-17-producing cells, the percentages and absolute numbers of γδT17 cells were much more than Tc17 cells and Th17 cells in tumor (Figure 1C). ELISA analysis also revealed that γδT17 cells secreted much more IL-17 than Th17 or Tc17 cells (Figure S1D). These γδT17 cells were readily seen in tumor as revealed by immunofluorescent (IF) staining. IL-17 was colocalized with γδTCR+ cells in the tumor border (Figure 1D). Furthermore, we found that IL-17 production was greatly elevated in tumor-infiltrating γδT cells (Figure S1E), whereas interferon-γ (IFN-γ) production was not substantially altered (Figure S1F). These results indicate that γδT17 cells are the major IL-17-producing cells in human CRC.
Figure 1. γδT17 Cells Are the Major Source of IL-17 in Human CRC.
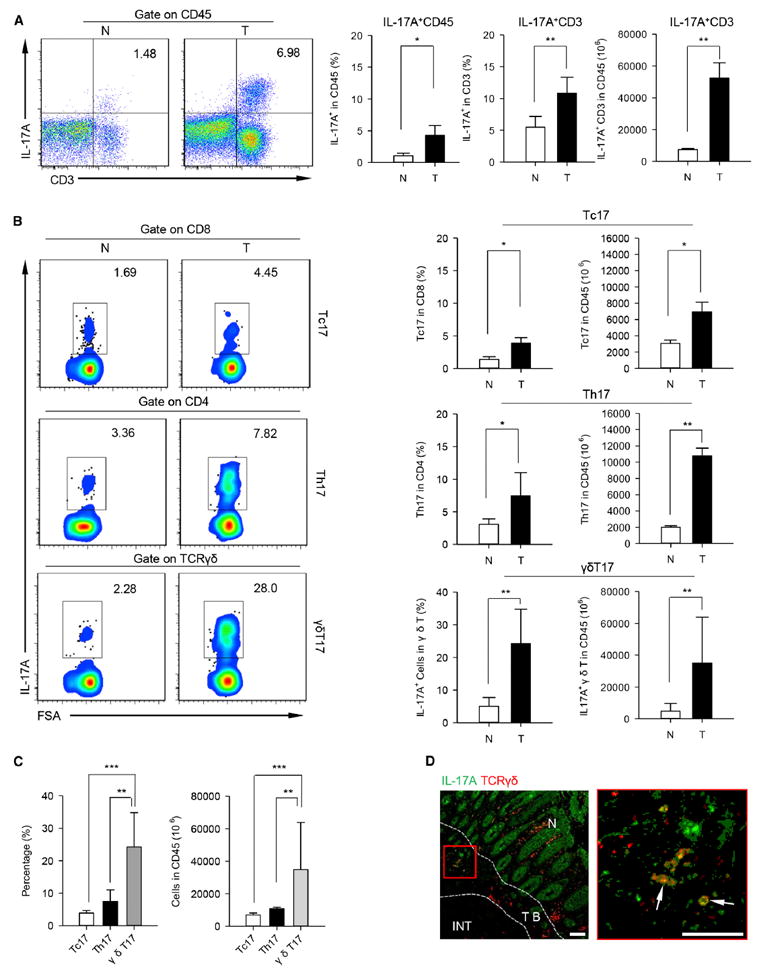
Intracellular IL-17 production assessed by flow cytometry (FCM) on single cell suspensions prepared from tumor and paired normal tissues of CRC patients. (A) Left panel shows representative flow cytometric analysis of CD3+ IL-17A+ cells in tumor and paired normal tissues. Plots were gated on CD45+ leukocytes, one of 34 independent experiments is shown. Right panel shows bar diagram that summarizes the percentages of IL-17A+ cells in the CD45+ cells (left) and CD45+ CD3+ cells (middle), as well as absolute numbers (right) of CD3+ IL-17A+ cells in CD45+ cells (1 ×106). N, normal tissue; T, tumor. Data are shown as mean ± SEM; n = 34; *p < 0.05; **p < 0.01.
(B) Left panel representative flow cytometric analysis of IL-17 secretion from CD8+ T cells (Tc17, top), CD4+ T cells (Th17, middle), and γδTCR+ T cells (γδT17, bottom) in tumor and paired normal tissues. Plots were gated on CD45+ CD3+ CD8+ (top), CD45+ CD3+ CD4+ (middle), or CD45+ CD3+ γδTCR+ T (bottom) cells, one of 28 independent experiments is shown. Right panel bar diagram summarizes the percentages (left) of IL-17A+ subpopulations present in the CD8+, CD4+, and γδTCR+ T cells, and absolute numbers (right) of Tc17, Th17, and γδT17 cells in CD45+ cells (1 × 106). N, normal tissue; T, tumor. Data are shown as mean ± SEM; n = 28; *p < 0.05; **p < 0.01.
(C) Summarized data show the percentages (left) and absolute numbers (right) of Tc17, Th17, and γδT17 cells in tumor tissues. Data are shown as mean ± SEM; n = 28; **p < 0.01; ***p < 0.001.
(D) Paraffin sections from CRC patients (scale bars represent 100 μM) were stained with anti-human pan-γδTCR (red) and anti-human IL-17A (green) for immunofluorescent (IF) staining. Right panel (scale bars represent 100 μM) is the magnified view indicated by the red box in the left panel and the arrows display the colocalization of γδTCR and IL-17A in tumor. One of three independent experiments is shown. TB, tumor border; INT, intratumor; N, normal tissue.
See also Figure S1.
Characteristics and Distribution of γδT17 Cells in Human CRC
We next examined the characteristics of γδT17 cells in human CRC. We found that approximately 80%–90% of γδT cells isolated from normal and tumor tissues expressed CD69, whereas fewer than 50% of γδT cells in peripheral blood (PB) from corresponding patients were CD69+ (Figure S2A). In addition to IL-17A, a small quantity of γδT17 cells also produced IL-17F (Figure S2B). It was notable that most γδT17 cells in normal and tumor tissues also produced TNF-α, but not IFN-γ (Figure 2A). Moreover, γδT17 cells produced more TNF-α in tumor than that in paired normal tissues (Figure S2C). In contrast, Th17 cells secreted low amounts of TNF-α (Figure S2D). Furthermore, γδT17 cells from normal and tumor tissues did not produce appreciable amounts of IL-22, IL-10, or IL-4 (Figure 2B).
Figure 2. Characteristics and Distribution of γδT17 Cells in Human CRC.
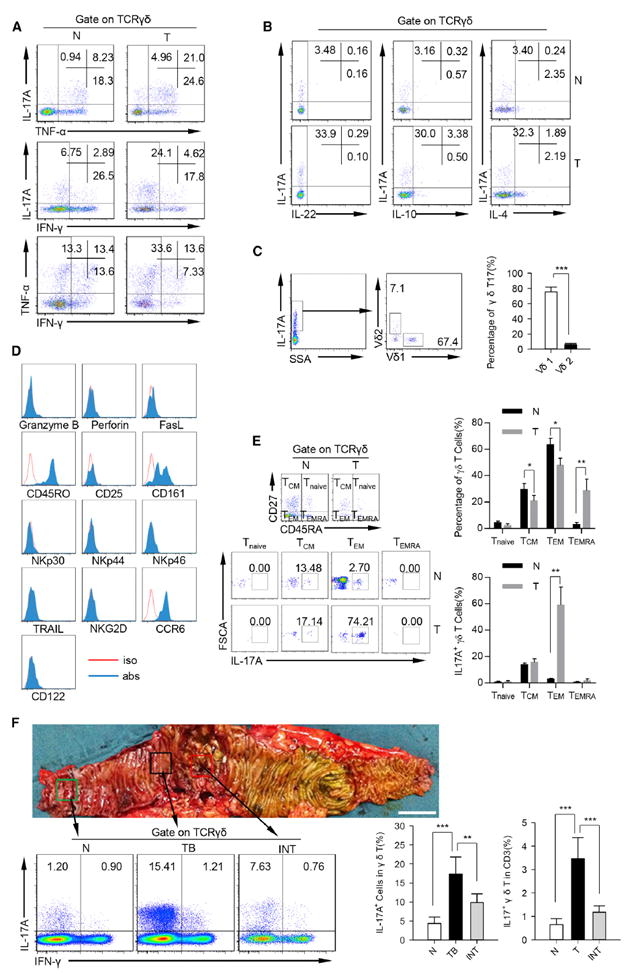
(A) Representative flow cytometric analysis of intracellular IL-17, IFN-γ, and TNF-α production by γδT cells in tumor and paired normal tissues. Plots were gated on γδTCR+ cells. One of ten independent experiments is shown. N, normal tissue; T, tumor.
(B) Representative flow cytometric analysis of IL-22, IL-10 and IL-4 production by γδT cells in tumor and paired normal tissues. Plots were gated on CD45+ CD3+ γδTCR+ cells. One of ten independent experiments is shown. N, normal tissue; T, tumor.
(C) Single cell suspensions from tumor tissues were stained with a panel of antibodies (anti-human CD45, anti-human CD3, anti-human TCRVd1, anti-human TCRVd2, and anti-human IL-17A) and analyzed by FCM. Left panel shows representative flow cytometric analysis of TCRVδ1+ cells and TCRVδ2+ cells in tumor-infiltrating γδT17 cells. Plots were gated on TCRγδ+ IL-17A+ cells. One of ten independent experiments is shown. Right panel bar diagram shows percentages of TCRVδ1+ cells and TCRVδ2+ in tumor-infiltrating γδT17 cells. Data are shown as mean ± SEM; n = 10; ***p < 0.001.
(D) Phenotype of tumor-infiltrating γδT17 cells in CRC patients were detected by FCM. Flow plots were gated on CD45+ CD3+ TCRγδ+ IL-17A+ cells. One of six independent experiments is shown.
(E) Left panel representative flow cytometric analysis of differentiation-related markers of γδT17 cells in the tumor and paired normal tissues. Up right panel shows summarized data that show the percentages of Tnaive, Tcm, Tem, and Temra in γδ T cells. Bottom right panel shows summarized data that show the percentages of IL-17A+ cells in Tnaive, Tcm, Tem, and Temra γδ T cells. Plots were gated on CD45+ CD3+ TCRγδ+ cells. One of six independent experiments is shown. N, normal tissue; T, tumor.
(F) Bottom left shows that γδT17 cells were detected by FCM. Plots were gated on CD45+ CD3+ TCRγδ+ cells. One of ten independent experiments is shown. Bottom right bar diagram shows percentages of IL-17A+ cells in γδT cells and IL-17A+ γδT cells in CD3+ cells in normal, tumor border, and intratumor tissues. N, normal tissue; TB, tumor border; INT, intratumor. Data are shown as mean ± SEM; n = 10; **p < 0.01; ***p < 0.001.
See also Figure S2.
Human γδT cells have two major Vδ gene usages (Vantourout and Hayday, 2013). The majority of γδT17 cells (67.4%–83.6%) in tumor tissues were Vδ1+ cells while fewer than 10% of γδT17 cells expressed Vδ2 (Figure 2C). Tumor-infiltrating γδT17 cells were CD45RO+, CD161+, and CCR6+ cells (Figure 2D; Figure S2E), but granzyme B, perforin, FasL, TRAIL, NKG2D, NKp30, NKp44, NKp46, CD25, and CD122 were negative. Interestingly, most γδT17 cells in normal tissues were of central memory T cell phenotype (Tcm, CD45RA− CD27+), whereas γδT17 cells in tumor tissues were effector memory T cells (Tem, CD45RA− CD27−) (Figure 2E). However, the other surface markers expression did not reveal any difference on γδT17 cells from the tumors or paired tissues (Figure S2F). To further analyze the distribution of γδT17 cells in human CRC, we prepared single cell suspensions from paired normal, tumor border, and intratumor tissues. γδT17 cells were substantially increased both in tumor border (approximately 12%–24%) and intratumor tissues (approximately 8%–13%) compared with normal tissues (approximately 1%–6%; Figure 2F). Taken together, these data suggest that tumor-infiltrating γδT17 cells are predominantly of the effector memory T cell phenotype and show unique distribution patterns in human CRC.
Tumor-Infiltrating Inflammatory DCs Trigger γδT17 Cell Polarization
The IL-23-IL-17 axis has been clearly linked to the tumor-elicited inflammation in mouse colon cancer models (Grivennikov et al., 2012). We also found that both transcripts encoding IL-23p19 and IL-23 protein (Figure 3A) were increased in human CRC. IL-23p19 was expressed by CD11c+ DCs but not CD68+ macrophages (Figure 3B) in tumor as revealed by IF staining. To further determine the cellular source of IL-23 in tumor-infiltrating myeloid cell subsets, we detected total tumor-infiltrating DCs (CD45+ Lin− HLA-DR+ CD11c+ DCs) and different DC subsets (Figure S3A) by flow cytometry (FCM). We found that both the percentages and absolute numbers of total DCs (Figure 3C) and CD14+ DCs (Figure S3B) were substantially increased in the tumor compared with paired normal tissues or PB. Previous studies demonstrated that a subpopulation of Th17-associated inf-DCs characterized as CD45+ Lin− HLA-DR+ CD11c+ CD1c+ CD16− were increased in ovarian and breast tumor ascites (Segura et al., 2013). We found that both CD1c+ CD16− and CD1c− CD16+ subpopulations were conspicuously increased in tumor tissues (Figure 3D, left). Morphological analysis showed that tumor-infiltrating inf-DCs (Figure S3C) displayed typical DC morphology with numerous dendrites, distinct from the macrophage-like morphology of inflammatory macrophages (inf-macrophages; CD45+ Lin− HLA-DR+ CD11c+ CD1c− CD16+ cells) (Figure 3D, right). In addition, tumor-infiltrating inf-DCs were positive for CD80, CD83, CD86, PD1, CD206, HLA-DR, and CD11c (Figure 3E), suggesting that these cells were immature DCs. Further study showed that both the frequency and numbers of inf-DCs were substantially elevated in tumor compared with those from paired normal tissues and PB or health donors (HD) (Figure 3F). Inf-macrophages were also substantially increased in tumor (Figure 3G). However, the percentages and numbers of inf-macrophages in PB of CRC patients and healthy donors were no different (Figure 3G). These data suggest that tumor-infiltrating inf-DCs and inf-macrophages are primary inflammatory myeloid cell subsets in human CRC.
Figure 3. γδT17 Cells Are Activated by Inf-DCs via IL-23.
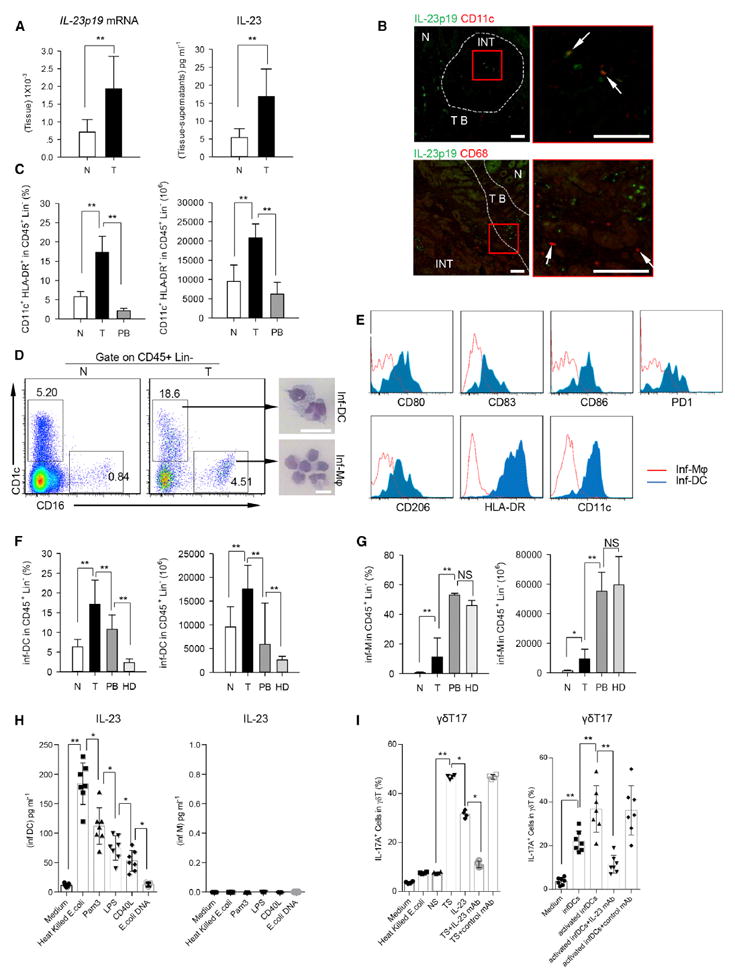
(A) Left panel shows that the relative mRNA level of IL-23p19 in tumor and paired normal tissues was determined by quantitative RT-PCR and normalized to GAPDH. Right panel shows concentrations of IL-23 in tumor and paired normal tissue-derived supernatants were detected by ELISA. N, normal tissue; T, tumor. Data are shown as mean ± SEM; n = 5; **p < 0.01.
(B) Left panel shows representative images of paraffin sections from CRC patients stained with anti-human IL-23p19 (green) and anti-human CD11c (upper, red) or anti-human CD68 (lower, red) by IF staining (scale bars represent 100 μM). Right panel represents magnified view indicated by the red box in the left panel (scale bars represent 100 μM). The arrows indicate colocalizations of CD11c and IL-23p19 intumor (upper), but not for CD68 and IL-23p19 (lower). One of three independent experiments is shown. N, normal tissue; TB, tumor border; INT, intratumor.
(C) Percentages (in CD45+ Lin− cells) and absolute numbers (in 1 × 106 CD45+ Lin− cells) of CD11c+ HLA-DR+ cells in normal tissues, paired tumor, and autologous PB were quantified by FCM. N, normal tissue; T, tumor; PB, peripheral blood. Data are shown as mean ± SEM; n = 20; **p < 0.01.
(D) Left panel shows representative flow cytometric analysis of inf-DCs (CD45+ Lin− CD1c+ CD16−) and inf-macrophages (CD45+ Lin− CD1c− CD16+) in tumor and paired normal tissues from CRC patients. Plots were gated on CD45+ Lin− cells. One of ten independent experiments is shown. Right panel shows Giemsa/May-Grunwald staining that shows morphology of tumor-infiltrating inf-DCs and inf-macrophages (scale bars represent 20 μM) sorted by fluorescence-activated cell sorting (FACS). One of six independent experiments is shown. N, normal tissue; T, tumor.
(E) Phenotypes of tumor-infiltrating inf-DCs (CD45+ Lin− CD11c+ HLA-DR+ CD1c+ CD16−) and inf-macrophages (CD45+ Lin− CD11c+ HLA-DR+ CD1c− CD16+) were detected by FCM. One of three independent experiments is shown.
(F) Percentages and absolute numbers (in 1 × 106 CD45+ Lin− cells) of inf-DCs in tumor, paired normal tissues, autologous PB and PB from healthy donors were quantified by FCM. N, normal tissue; T, tumor; PB, peripheral blood; HD, healthy donors. Data are shown as mean ± SEM; n = 20; **p < 0.01.
(G) Percentages and absolute numbers (in 1 × 106 CD45+ Lin− cells) of inf-macrophages in the same set of samples as in above (F) were quantified by FCM. N, normal tissue; T, tumor; PB, peripheral blood; HD, healthy donors. Data are shown as mean ± SEM; n = 20; NS, no statistical significance; *p < 0.05; **p < 0.01.
(H) Sorted tumor-infiltrating inf-DCs (left) and inf-macrophages (right) were treated with various ligands (heat-killed E. coli, Pam3, LPS, CD40L, E.coli DNA or Medium alone) for three days in vitro. IL-23 levels in the supernatants were detected by ELISA. Data are shown as mean ± SEM; n = 6; *p < 0.05; **p < 0.01.
(I) Left panel shows that γδT cells sorted from normal tissues were treated with heat-killed E.coli, NS, TS, IL-23, TS with IL-23 neutralizing antibody or TS with control mAb respectively for 14 days. Right panel shows that the sorted γδT cells were cocultured with resting inf-DCs or pre-activated inf-DCs (treated with heat-killed E. coli for three days) sorted from paired tumor tissues in the medium containing IL-23 neutralizing antibody or not for 2 weeks in vitro. Percentages of IL-17A+ cells in γδT cells were determined by FCM. NS, normal tissue-derived supernatants; TS, tumor-derived supernatants. Data are shown as mean ± SEM; n = 6; *p < 0.05; **p < 0.01.
See also Figures S3 and S4.
Our previous study demonstrated that γδT17 cells were activated by IL-23 in skin inflammation (Cai et al., 2011). We found that IL-23 was substantially increased both at the transcriptional and protein level in tumor-infiltrating inf-DCs (Figure S3D). Then we examined whether microbial products stimulated inf-DCs to produce IL-23. Heat-killed E. coli, Pam3, LPS, and CD40L activated inf-DCs but not inf-macrophages to produce IL-23 (Figure 3H). To determine the role of IL-23 in γδT17 cell polarization in human CRC, we stimulated γδT cells from normal tissues with IL-23 or supernatants from tumor or paired normal tissues. IL-17 production by γδT cells could be induced by supernatants derived from tumors (TS) or IL-23, but not by supernatants from paired normal tissues (NS) (Figure 3I, left). Furthermore, TS-induced IL-17 production by γδT cells could be blocked by IL-23p19 neutralizing antibody (Figure 3I, left; Figure S3E). To further decode whether inf-DCs activated by microbial products could polarize γδT17 cells, we cocultured both pre-activated and resting tumor-infiltrating inf-DCs with γδT cells isolated from normal tissues. We found that both resting and activated inf-DCs could effectively induce γδT17 cells in an IL-23-dependent manner (Figure 3I, right). However, inf-DCs activated by microbial products were more efficient at eliciting γδT17 cell polarization than resting inf-DCs. In addition, TS and activated inf-DCs could promote γδT17 cell proliferation and survival in vitro (Figures S4A–S4D). Collectively, these findings suggest that tumor-infiltrating inf-DCs activated by microbial products induce γδT17 polarization.
Epithelial Barrier Failure and Bacterial Product Release in Human CRC
Having demonstrated that the primary commensal bacteria E. coli products stimulate inf-DCs to produce IL-23, we hypothesized that epithelial barrier failure caused by tumor development might result in commensal bacterial product release in CRC, which in turn could stimulate inf-DCs for γδT17 cell polarization. Indeed, epithelium structure in the tumor was deteriorated as revealed by H&E staining (Figure 4A). We further stained the tumor tissues with tight junction proteins such as ZO-1, claudin-2, and pancytokeratin to detect the epithelial barrier integrity. Both ZO-1 and claudin-2 were markedly decreased and disordered in the tumor tissues compared with adjacent normal tissues (Figure 4B). In addition, the mRNA expression levels of transcripts encoding ZO-1, OCLN, and MUC2 were substantially decreased in the tumors compared to those in the paired normal tissues (Figure 4C). To investigate the bacterial product invasion in CRC, we rigorously washed fresh tumor and paired normal tissues and homogenized them for tissue LPS detection. LPS level was substantially increased in tumor (Figure 4D). To investigate whether commensal bacteria invaded into the tumor, we detected paraffin sections by bacterial universal 16sRNA by using in situ hybridization. We observed that bacterial 16sRNA was located in the tumor border and adjacent normal tissues (Figure 4E). Bacterial universal 16sRNA was substantially increased in tumor tissues (Figure 4F), suggesting that bacterial invasion occurred in the tumor. Similar results were observed with bacterial universal 16sRNA quantitative PCR analysis in the tumor and paired normal tissues (Figure 4G). These data suggest that tumor-elicited epithelial barrier failure might lead to commensal bacteria and bacterial product invasion, which correlates with inf-DC activation and IL-23 secretion in human CRC.
Figure 4. Epithelial Barrier Failure and Bacterial Products invasion in Human CRC.
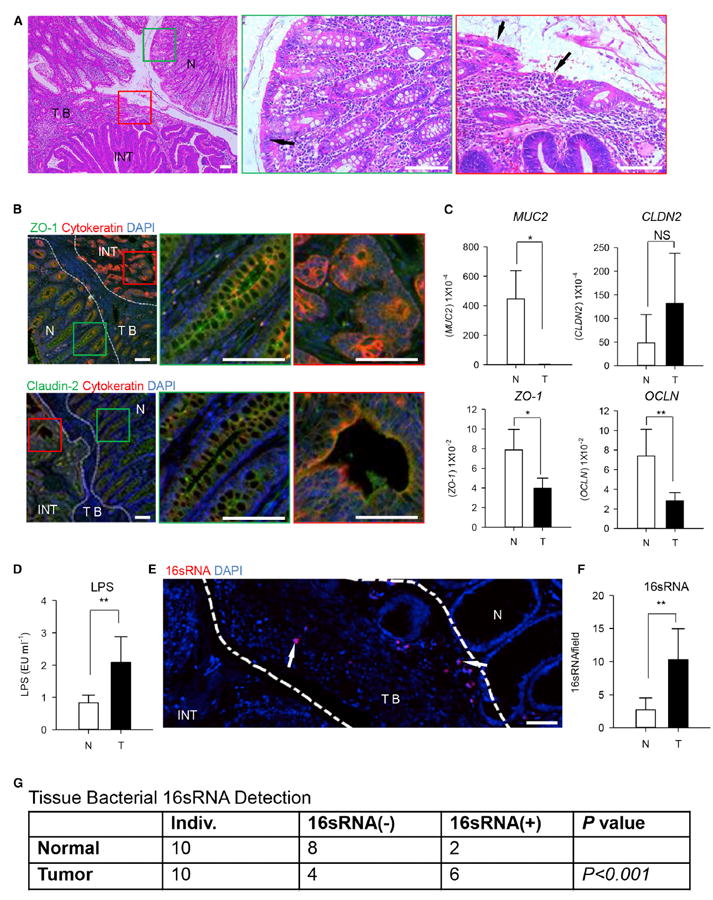
(A) Left panel shows representative histological images of the tumor and adjacent tissues by H&E staining (scale bars represent 100 μM). Middle and right panels represent magnified view indicated by the green box of the normal area (middle) and the red box of the tumor area (right) in the left panel, respectively. Arrow in the middle panel indicates the integrated epithelium structure in the normal tissue. Arrows in the right panel indicates the deteriorated epithelium structure in the tumor. NT, normal tissue; TB, tumor border; INT, intratumor.
(B) Left panel shows representative images of ZO-1 (upper, green) and claudin-2 (lower, green) expressions by IF staining in paraffin sections containing both tumor and normal tissues (scale bars represent 100 μM). Middle and right panels represent magnified view indicated by the green box of normal area (middle) and the red box of tumor area (right) in the left panel, respectively. One of three independent experiments is shown. N, normal tissue; TB, tumor border; INT, intratumor.
(C) The relative mRNA levels of MUC2, CLDN2, ZO-1, and OCLN in the tumor and paired normal tissues were determined by quantitative RT-PCR and normalized to GAPDH. N, normal tissue; T, tumor. Data are shown as mean ± SEM; n = 5; NS, no statistical significance; *p < 0.05; **p < 0.01.
(D) ELISA assay to measure LPS concentrations in supernatants from tumor and paired normal tissues. The LPS levels were normalized back to the weight of the tissue samples used. N, normal tissue; TB, tumor. Data are shown as mean ± SEM; n = 10; **p < 0.01.
(E) Representative image of bacterial 16 RNA invaded into tumor by fluorescence in situ hybridization (FISH) in paraffin sections (scale bars represent 100 μM). The arrows indicate the location of 16 RNA (red) in tumor border. N, normal tissue; TB, tumor border; INT, intratumor. One of ten independent experiments is shown.
(F) The 16 RNA+ bacteria in tumor and paired normal tissues were counted by FISH. N, normal tissue; T, tumor border. Data are shown as mean ± SEM; n = 10; **p < 0.01.
(G) The 16 RNA+ bacteria in the tumor and paired normal tissues were detected by RT-PCR; data are analyzed by Fisher’s exact test; n = 10.
γδT17 Cell-Derived Cytokines Promote the Recruitment, Proliferation, and Survival of PMN-MDSC
We next examined how γδT17 cells played a role in tumor-elicited inflammation and immune suppression in human CRC. We noted that γδT cells isolated from normal tissues could be induced to produce IL-8, TNF-α, and GM-CSF in addition to IL-17 (Figure 5A). IL-17A, IL-8, and GM-CSF production by γδT cells was partly dependent on IL-23, whereas TNF-α production was independent (Figure 5A). To further dissect whether IL-8, TNF-α, and GM-CSF were produced by γδT17 cells in the tumor tissues, we detected intracellular cytokines of tumor-infiltrating γδT17 cells by FCM. As shown in Figure 5B, more than 50% of tumor-infiltrating γδT17 cells also produced IL-8, TNF-α, and GM-CSF. IL-8+ γδT17 cells, TNF-α+ γδT17 cells and GM-CSF+ γδT17 cells were substantially increased in tumor compared with those in paired normal tissues (Figure 5C). In contrast, the production of IL-8, TNF-α, and GM-CSF in Th17 cells was marginally increased in tumor tissues (Figures S5A and S5B) but much lower than that in γδT17 cells. Moreover, CD161+ CCR6+ γδT17 cells isolated from tumor tissues produced much more IL-8, TNF-α, and GM-CSF than those cells from paired normal tissues (Figure 5D).
Figure 5. Inf-DCs Activated by Bacterial Products Stimulate γδT Cells Producing IL-17A, IL-8, TNF-α, and GM-CSF.
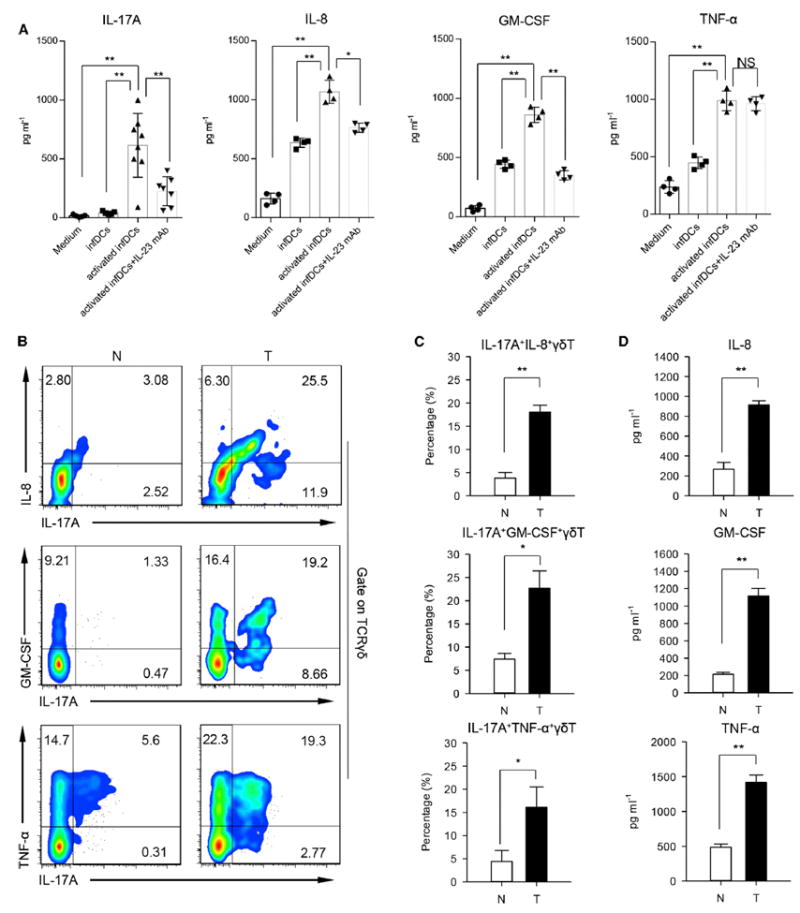
(A) γδT cells sorted from normal tissues were cocultured with resting or preactivated inf-DCs (pretreated with heat-killed E. coli for 3 days) in medium containing IL-23 neutralizing antibody or not for 3 days in transwell system. Then, γδT cells were harvested and incubated in serum-free medium for another 3 days. Concentrations of IL-17A, IL-8, TNF-α, and GM-CSF in the γδT cells supernatants were determined by ELISA. Data are shown as mean ± SEM; n = 6; NS, no statistical significance; *p < 0.05; **p < 0.01.
(B) Representative flow cytometric analysis of intracellular IL-8, GM-CSF, TNF-α, and IL-17A production by γδT cells in tumor and paired normal tissues. Plots were gated on TCRγδ+ cells. One of six independent experiments is shown. N, normal tissue; T, tumor.
(C) Percentages of IL-17A+ IL-8+ γδT cells, IL-17A+ GM-CSF+ γδT cells, and IL-17A+ TNF-α+ γδT cells in γδT cells were quantified by FCM. Data are shown as mean ± SEM; n = 6;*p < 0.05; **p < 0.01.
(D) IL-8, GM-CSF, and TNF-α in the sorted CD161+ CCR6+ γδT cells supernatants were detected by ELISA. Data are shown as mean ± SEM; n = 6; **p < 0.01.
See also Figure S5.
Recent studies have shown that TNF-α (Sade-Feldman et al., 2013; Zhao et al., 2012), IL-17A (He et al., 2010), and GM-CSF (Kapanadze et al., 2013) are involved in the accumulation of tumor-infiltrating MDSCs in mice. MDSCs are one of the major immunosuppressive cell subsets in the tumor microenvironment (Gabrilovich et al., 2012). To investigate the impact of γδT17 cells and secreted cytokines on MDSCs in human CRC, we detected MDSCs in tumor and paired fresh normal tissues by FCM. We found that PMN-MDSCs characterized as CD45+ Lin− HLA-DR− CD11b+ CD33+ CD66b+ (Figures S6A–S6D) were predominant and greatly increased in tumors compared to those in paired normal tissues (Figure 6A, upper). In contrast, M-MDSCs were scarce in tumor tissues (Figure 6A, lower; Figure S6D). The detailed gating strategy was shown in Figures S6A–S6C. Sorted tumor-infiltrating PMN-MDSCs displayed typical polymorphonuclear granulocyte morphology (Figure 6B). These PMN-MDSCs produced much more arginase-I (Figure 6C) and ROS (Figure 6D) compared with autologous PMNs. Furthermore, PMN-MDSCs isolated from tumor potently inhibited the proliferation of activated autologous T cell (Figures 6E and 6F) and IFN-γ production (Figure 6G). However, PMN from healthy donors did not show any suppressive activity (Figures S6E and S6F). We also compared the frequencies and numbers of M-MDSCs and PMN-MDSCs in tumor with paired normal tissues and PB, as well as health donor PB. We found that the frequency of both M-MDSCs (Figure 6H, left) and PMN-MDSCs (Figure 6I, left) was substantially increased in tumor compared with paired normal tissues, but regarding the absolute number, only PMN-MDSCs were greatly increased. Besides, PMN-MDSCs were abundant and about 80-fold more than M-MDSCs in tumor tissues (Figure 6J).
Figure 6. γδT17 Cells Promote the Migration, Proliferation, and Survival of PMN-MDSCs.
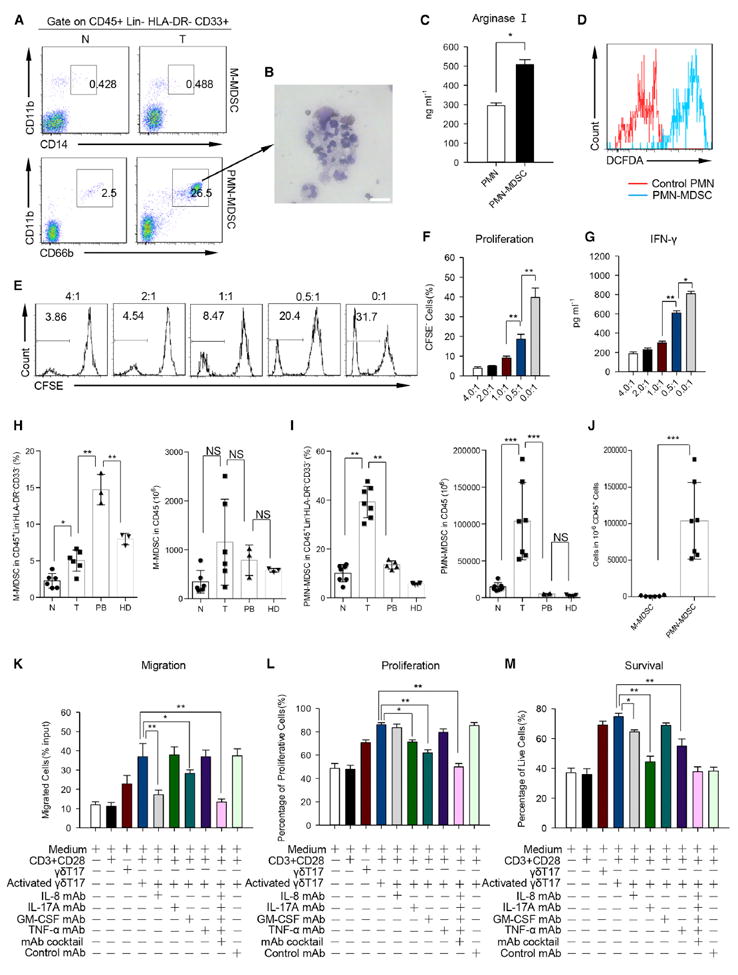
(A) Representative flow cytometric analysis of M-MDSC (upper) and PMN-MDSC (lower) in tumor and paired normal tissues from CRC patients. Plots were gated on CD45+ Lin− HLA-DR− CD33+ cells. One of six independent experiments is shown.
(B) Giemsa/May-Grunwald staining shows morphology of tumor-infiltrating PMN-MDSC cells sorted by FACS (scale bars represent 15 μm). One of three independent experiments is shown.
(C) PMN-MDSC cells sorted from tumor tissues and paired PMN from PB of same patient were cultured in vitro for 6 hr. Concentrations of arginase I in the supernatants were detected by ELISA. Data are shown as mean ± SEM; n = 6; *p < 0.05.
(D) Similar to (C), ROS levels in PMN-MDSC or PMN were measured by staining with 5,6-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (DCFDA). One of three independent experiments is shown.
(E and F) Sorted PMN-MDSC were cocultured with CFSE-pre-labeled CD3+ T cells isolated from autologous PB at different ratios (PMN-MDSC/CD3+ T cells in 4:1; 2:1; 1:1; 0.5:1 and 0:1) in the presence of anti-CD3 and anti-CD28 mAbs. CD3+ T cell proliferation was evaluated on day 6 by FCM. One of five independent experiments is shown (E). Bar diagram summarizes the percentages of proliferated cells (CFSElo) in CD3+ T cells (F). Data are shown as mean ± SEM; n = 5; **p < 0.01.
(G) PMN-MDSC from tumor tissues were cocultured with CD3+ T cells from autologous PB for 12 hr. Concentrations of IFN-γ in the supernatants were detected by ELISA. Data are shown as mean ± SEM; n = 6; *p < 0.05; **p < 0.01.
(H and I) Percentages and absolute numbers (in 1 × 106 CD45+ Lin− HLA-DR− CD33− cells) of M-MDSCs (H) and PMN-MDSCs (I) in tumor (n = 6), normal paired tissues (n = 6), PB (H): n = 3; (I): n = 5) from CRC patients and PB (H): n = 3; (I): n = 5) from healthy donors (HD) were quantified by FCM. N, normal tissue; T, tumor; PB, autologous peripheral blood; HD, healthy donors. Data are shown as mean ± SEM; NS, no statistical significance; *p < 0.05; **p < 0.01; ***p < 0.001.
(J) Absolute numbers (in 1 × 106 CD45+ Lin− HLA-DR− CD33− cells) of M-MDSCs and PMN-MDSCs in the same sets of tumor tissues. Data are shown as mean ± SEM; n = 7; ***p < 0.001.
(K, L, and M) Sorted tumor-infiltrating PMN-MDSCs (upper well) and CD161+ CCR6+ γδT cells or in vitro polarized γδT17 cells (lower well) were cocultured in transwell plate for 6 hr, 24 hr, or 72 hr respectively. For migration assay (K), cells in the lower well were collected and counted. For proliferation assay (L), sorted tumor-infiltrating PMN-MDSCs were prelabeled with CFSE, the proliferation of PMN-MDSCs was detected by FCM after 24 hr coculture. For survival assay (M), PI was added in the cocultured medium for the last 15 min, PI+ PMN-MDSC cells were examined by FCM. Data are shown as mean ± SEM; n = 6; *p < 0.05; **p < 0.01. Similar results were obtained by coculture in vitro polarized γδT17 cells with tumor-infiltrating PMN-MDSCs in the same condition as above.
See also Figure S6.
We next examined whether the accumulation of PMN-MDSCs was the result of γδT17 cell activation. To this end, we used an in vitro coculture transwell system to assess the contribution of γδT17 cells in tumor-infiltrating PMN-MDSC migration, proliferation, and survival. We found that activated γδT17 cells isolated from tumor tissues could effectively promote tumor-infiltrating PMN-MDSC migration within 6 hr in IL-8 and GM-CSF-dependent manner (Figure 6K). In addition, tumor-infiltrating PMN-MDSCs were substantially expanded within 24 hr when they were cocultured with paired γδT17 cells (Figure 6L). The enhanced PMN-MDSC proliferation mediated by γδT17 cells could be blocked by IL-17A and GM-CSF neutralizing antibodies. More strikingly, γδT17 cells substantially promoted PMN-MDSC survival, and this effect was substantially blocked by IL-8, IL-17A, and TNF-α neutralizing antibodies (Figure 6M). In contrast, γδT17 cells from normal tissues showed less activity to stimulate PMN-MDSC migration, proliferation, and survival (Figures S6G–S6I). Collectively, these data show that γδT17 cells, via IL-17A, IL-8, GM-CSF, and TNF-α, promote the accumulation and expansion of PMN-MDSC to facilitate immunosuppression in vitro.
γδT17 Cells Impact Advanced Clinicopathological Features in Human CRC
To investigate the implication of γδT17 cells to clinical features of human CRC, we collected and analyzed clinical data from 117 CRC patients (Table S1). We found that the frequency of tumor-infiltrating γδT17 cells was substantially and positively correlated with advancing TNM stages (Figure 7A). Similar results were observed when the absolute numbers of γδT17 cells in one million CD45+ cells were analyzed (Figure 7B). In contrast, the frequency of Th17 or Tc17 cells did not show overall correlation with TNM stages, although Th17 cells did increase in stage III human CRC (Figures S7A and S7B). The percentages of tumor-infiltrating γδT17 cells were also positively associated with other clinicopathological features, including tumor size, tumor invasion, lymphatic and vascular invasion, lymph node metastasis, and serum CEA levels (Figure 7C). Another cohort of patients was also examined for a correlation between γδT17 cells and inf-DCs, PMN-MDSCs, and IL-23 and IL-17 levels in the tumor. We found that the percentages of tumor-infiltrating γδT17 cells were positively associated with tumor-infiltrating inf-DCs, PMN-MDSCs, and IL-23 and IL-17 levels in tumor tissues (Figure 7D). In addition, IL-23 expression was also correlated with inf-DCs accumulation and total IL-17 protein expression (Figure S7C). These findings suggest that tumor-infiltrating γδT17 cells are associated with tumor invasiveness and progression. Thus the frequency and numbers of tumor-infiltrating γδT17 cells might be a prognostic factor in human CRC.
Figure 7. The Associations of Tumor-Infiltrating γδT17 Cells with Clinicopathological Features of Human CRC.
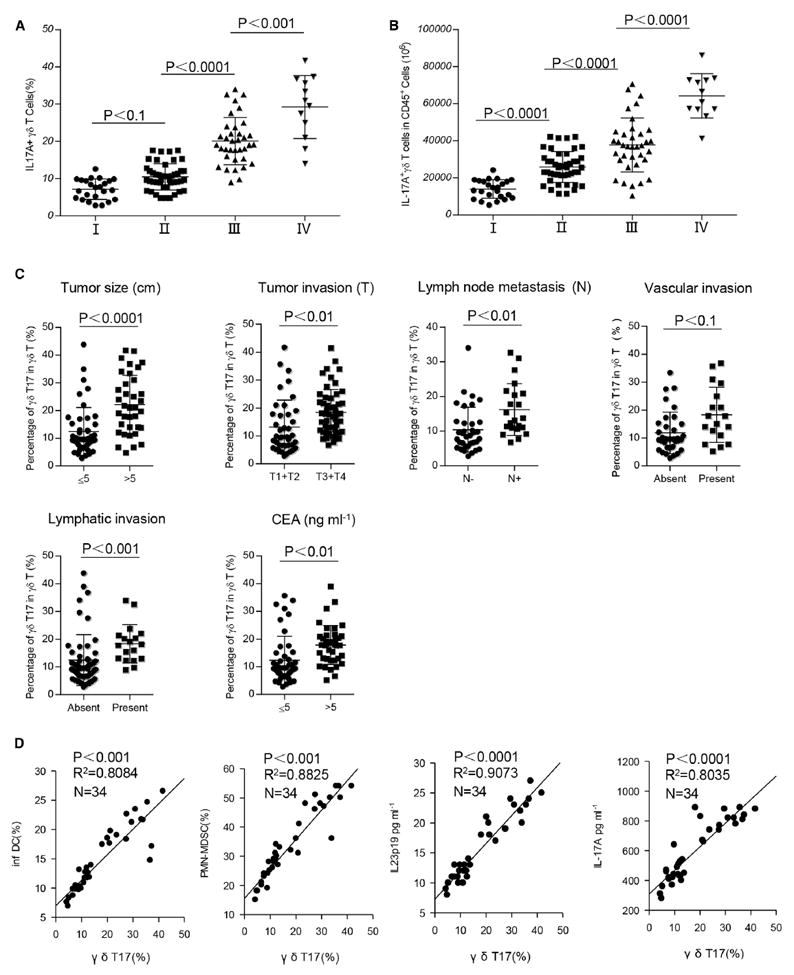
(A and B) γδT17 cells percentages and absolute numbers in CD45+ CD3+ γδTCR+ cells were analyzed by FCM. The potential correlations between the percentages or numbers of γδT17 cells and TNM stages were analyzed. (A) Percentages and (B) absolute numbers (in 1 × 106 CD45+ cells) of γδT17 cells were positively correlated with TNM stages. Data are shown as mean ± SEM; n = 117.
(C) Potential correlations of γδT17 cell percentages with other clinicopathological features. Data are shown as mean ± SEM; **p < 0.01, NS, no statistical significance. CEA, carcinoembryonic antigen.
(D) Potential correlations of γδT17 cell percentages with other immune cell subsets, IL-23 or IL-17A concentrations in tumors. Tumor-infiltrating γδT17 cells in CD45+ CD3+ γδTCR+ cells, inf-DCs in CD45+ Lin− CD11c+ HLA-DR+ cells, and PMN-MDSCs in CD45+ Lin− CD33+ HLA-DR− cells were defined with specific staining and analyzed by FCM. The concentrations of IL-17 and IL-23 in tumor tissue-derived supernatants were measured by ELISA. The correlations between γδT17 cells and inf-DCs, γδT17 cells and PMN-MDSCs, γδT17 cells and IL-23 level, γδT17 cells and IL-17A level were evaluated respectively.
See also Figure S7.
DISCUSSION
In this study, we clearly demonstrate that innate γδT cells are the major source of IL-17 in human CRC. γδT17 cell activation is triggered by IL-23, which is highly expressed in human CRC tissues. The source of IL-23 is mainly from tumor-infiltrating inf-DCs, which is activated by microbial pathogen invasion as a consequence of tumorous epithelial barrier deterioration. These γδT17 cells not only secrete large amounts of IL-17 but also other cytokines including IL-8, GM-CSF, and TNF-α. More importantly, our in vitro studies demonstrate that tumor-infiltrating γδT17 cells chemoattract PMN-MDSCs and further expand and provide survival advantage for them to maintain immune suppressive activity via secretion of these cytokines. We also demonstrate a strong and positive correlation between tumor-infiltrating γδT17 cells and advanced clinicopathological features including TNM stages, tumor sizes, and lymphatic and vascular invasions, all of which are signatures of poor clinical outcome. Taken together, these findings suggest that innate γδT17 cells contribute to human CRC development and progression.
γδT17 cells play a critical role in the pathogenesis of several inflammation-associated diseases in mice (Cai et al., 2011; Gray et al., 2013; Pantelyushin et al., 2012; Park et al., 2010; Petermann et al., 2010; Shichita et al., 2009; Sutton et al., 2009). The role of γδT17 cells and their characteristics in human inflammation-associated diseases are emerging (Caccamo et al., 2011; Fenoglio et al., 2009; Kenna et al., 2012). In murine colon cancer models, abT cells are considered to be the major IL-17 producer (Grivennikov et al., 2012; Wu et al., 2009). However, it was unclear whether IL-17 is also mainly produced by Th17 cells in human CRC. Despite two recent studies showing that both IL-17 transcription and protein expression are substantially increased in human CRC tissues (Chung et al., 2013; Tosolini et al., 2011), it was assumed that Th17 cells were the source of IL-17, but this was not directly proved. We have used flow cytometric analysis and have demonstrated that IL-17 is almost exclusively secreted by CD3+ cells including Th17, Tc17, and γδT17 cells, but not ILCs. In addition, γδT17 cells are the major IL-17 producer both in percentages and absolute numbers. This conclusion is also supported by ELISA data. Thus, our study formally establishes γδT17 cells as the major IL-17-producing cells in human CRC.
The murine γδT17 cells share many characteristics with other IL-17-producing cell subsets (Hedrick et al., 2009), including constitutive expression of CCR6 and CD161. A recent study has shown that human PB IL-17+ Vγ9Vδ2 T cells have a predominant terminally differentiated phenotype (CD45RA+ CD27−) and express granzyme B, TRAIL, FasL, and CD161 (Caccamo et al., 2011). In our study, human tumor-infiltrating γδT17 cells have a predominant effector memory phenotype (CD45RA− CD27−) and express CCR6 and CD161, but not granzyme B, perforin, FasL, TRAIL, NKG2D, NKp30, NKp44, NKp46, CD25, or CD122. In contrary to the PB IL-17+ Vγ9Vδ2 T cells, the dominant tumor-infiltrating γδT17 cells are Vδ1+ cells. Moreover, tumor-infiltrating γδT17 cells produce both IL-17 and TNF-α, but not IFN-γ in tumor tissues. These data suggest that human tumor-infiltrating γδT17 cells are a subset of multifunctional IL-17-producing cells with an effector memory phenotype and limited cytotoxicity.
IL-23 secreted by myeloid cells is considered to be a promoter in the progression of CRC in mice and humans (Langowski et al., 2006). Our previous study showed that myeloid cells secreted IL-23 to promote inflammation in mouse psoriasis model (Cai et al., 2011). Here, we find that IL-23 is substantially increased in human CRC tumor tissues, consistent with previous studies (Langowski et al., 2006). Previous studies in CPC-APC mouse colon cancer model have shown that IL-23 is primarily secreted by macrophages (Grivennikov et al., 2012). On the contrary, studies in human showed that both DCs and macrophages could produce IL-23 (Cai et al., 2011; Langowski et al., 2006), highlighting the differences between humans and mice. We find that tumor-infiltrating inf-DCs are the major source of IL-23. Upon bacterial product stimulation, tumor-infiltrating inf-DCs, but not inf-macrophages, produce IL-23. Deregulation of tight junctions and cell-cell contacts has been previously described in advanced human adenocarcinoma (Wang et al., 2011). In addition, gut microbiota has been suggested to contribute to inflammation-associated cancer and to the regulation of the host immune system (Hooper et al., 2012). In our study, bacterial 16sRNA is detected in situ in tumor and tumorous LPS amounts are also substantially increased compared with normal tissues. Moreover, consistent with previous studies (Grivennikov et al., 2012), tumorous epithelial barrier failure in human CRC is demonstrated by histology analysis and the decrease and disorder of tight junction proteins. These data suggest that inf-DC accumulation and activation in human CRC might be a result of microbial product invasion caused by barrier deterioration, which in turn links to tumor-elicited inflammation via γδT17 cell activation.
γδT17 cells are multifunctional inflammatory cells that can produce various cytokines (Cai et al., 2011; Pantelyushin et al., 2012). Tumor-infiltrating γδT17 cells in human CRC not only produce large amounts of IL-17 but also secrete IL-8, TNF-α, and GM-CSF. Moreover, the production of IL-8 and GM-CSF in γδT17 cells is partly inf-DC- and IL-23-dependent. In contrast, Th17 cells secrete marginal amounts of these cytokines. Previous studies have shown that TNF-α (Sade-Feldman et al., 2013; Zhao et al., 2012), IL-17 (He et al., 2010), and GM-CSF (Kapanadze et al., 2013) are involved in the mobilization and recruitment of tumor-infiltrating MDSCs in mice. Indeed, we find that PMN-MDSCs, but not M-MDSCs, are preferentially accumulated in the tumor microenvironment and positively correlate with tumor-infiltrating γδT17 cells in human CRC. Further in vitro studies have demonstrated that tumor-infiltrating γδT17 cells also promote PMN-MDSC proliferation and survival via IL-17A, IL-8, TNF-α, and GM-CSF. Our findings are consistent with previous preclinical studies demonstrating that IL-17 recruits Gr-1+ CD11b+ MDSCs to promote tumor growth (He et al., 2010). In addition, our findings suggest that γδT17 cells promote recruited MDSCs for enhanced survival and further expansion. Although it is difficult to determine whether these PMN-MDSCs are from PB neutrophils, PMN-MDSCs in human CRC have elevated arginase-I and ROS production, and potent immune suppression of T cell proliferation and function. Taken together, we speculate that γδT17 cells might convert tumor-elicited inflammation to an immunosuppressive environment by promoting PMN-MDSC accumulation and expansion in the tumor area. Subsequently, these events govern human CRC development and progression.
In summary, our study has revealed innate γδT17 cells to be key regulators in human CRC-elicited inflammation and immunosuppression. This appears to be initiated through tumorous epithelial barrier disruption leading to microbial product release that correlates with inf-DC accumulation and activation, thus promoting γδT17 cell polarization. Activated γδT17 cells secrete large amounts of cytokines including IL-17, TNF-α, IL-8, and GM-CSF, which might mobilize and recruit PMN-MDSCs to establish a potent immunosuppressive environment for promoting tumor progression and immune evasion. It is worth noting that these in vitro studies are correlative and descriptive due to the nature of human subject studies. Nevertheless, our findings suggest that γδT17 cells might be a prognostic factor of human CRC, and eradication of these cells might have a potential for effective treatment.
EXPERIMENTAL PROCEDURES
Clinical Specimens
Colorectal tumor (T, homogeneous cellularity, without foci of necrosis), paired normal intestine tissues (N), and some fresh PB were obtained from 154 patients with colorectal adenocarcinoma who underwent surgical resection at the Second Affiliated Hospital, Zhejiang University School of Medicine. To study the distribution of γδT17 cells, we collected intratumoral (INT) tissues or tissues from tumor border (TB) and paired normal tissues from the same patients. Autologous PB was collected before surgery. Normal autologous tissue was obtained from a macroscopically normal part of the excised intestine, at least 5 cm away from the tumor. None of the patients had received radiotherapy or chemotherapy before operation. Control PB samples were obtained from 28 healthy donors from the Zhejiang Blood Center, all of whom were negative for antibodies against hepatitis C virus, hepatitis B virus, HIV, and syphilis. All samples were anonymously coded in accordance with local ethical guidelines (as stipulated by the Declaration of Helsinki), and written informed consent was obtained and the protocol was approved by the Review Board of the Second Affiliated Hospital of Zhejiang University School of Medicine.
Cell Isolation and Culture
Freshly excised tissues were cut into small pieces and then digested in RPMI 1640 medium containing 2% FBS, type IV collagenase (1 mg/ml), and hyal-uronidase (10 ng/ml) for 2–3 hr at 37°C. Inf-DCs, inf-macrophages, total γδT cells, CD161+ CCR6+ γδT17 cells, CD161+ CCR6+ Th17 cells, CD161+ CCR6+ Tc17 cells, Th17 cells, Tc17 cells, and PMN-MDSCs in single cell suspensions were incubated with various antibody cocktails (Table S2) and then sorted by Aria II cell sorter (BD Biosciences). PB lymphocytes were isolated after centrifugation on a Ficoll gradient followed by cell sorting. The purity of all sorted cells was greater than 90%.
Flow Cytometry
For extracellular staining of immune markers, we prepared single cell suspensions by mechanic dispersion and enzymatic digestion of normal and tumor tissues. We preincubated fresh tissue cells (1 × 106/ml) in a mixture of PBS, 2% fetal calf serum, and 0.1% (w/v) sodium azide with FcgIII/IIR-specific antibody to block nonspecific binding and stained with different combinations of fluorochrome-coupled antibodies (Table S2). The antibody cocktails for inf-DCs, inf-macrophages, and M-MDSCs lineage assay were CD3, CD15, CD19, CD20, CD56, CD235a, or CD3, CD19, CD20, CD56, CD235a; for PMN-MDSCs assay was CD3, CD14, CD16, CD19, CD20, CD56, CD235a, or CD3, CD19, CD20, CD56, CD235a. For intracellular staining, we followed the manufacturer’s protocol after 6 hr incubation in the presence of Leukocyte Activation Cocktail (BD PharMingen). Fluorescence data were collected on a FACSCanto II system (BD Biosciences) and analyzed with FlowJo software (Tree Star).
RNA Extraction and Gene Expression by qPCR
Tissue and cell RNAs were extracted with the RNeasy Micro Kit (QIAGEN) and reverse transcribed into cDNA by oligo-dT primer (Invitrogen) and Superscript First Strand Synthesis System (Invitrogen). cDNA was analyzed by real-time PCR with SYBR Green I Master Mix (Roche) with the StepOnePlus instrument (Applied Biosystems) for the target genes (Table S3). Data were presented as arbitrary units and calculated as 2(Ct(GAPDH–gene of interest)).
In Vitro γδT17 Cell Polarization
γδT cells isolated from normal tissues were cultured in the conditional medium containing heat-killed E. coli, supernatants derived from normal tissues (NS), supernatants derived from tumor tissues (TS), or recombinant human IL-23 (10 μg/ml, R&D Systems). IL-23 neutralizing antibody (Clone 24901, 0.1μg/ml,) was added when needed. At day 14, IL-17+ cells were detected by FCM.
ELISA Assay and LPS Detection
IL-23, IL-17A, IL-8, TNF-α, GM-CSF, and IFN-γ were measured by ELISA kits (R&D Systems). Arginase I levels were also determined by an ELISA kit (BioVendor). Tissue LPS levels were analyzed in duplicate wells in 96-well plates according to the manufacturer’s instructions with Limulus Amoebocyte Lysate (LAL) assay QCL-1000 (Lonza, ValaisSwitzerland). The LPS levels were normalized back to the weight of the tissue samples used.
T Cell Proliferation Assay
For PMN-MDSCs or PMN-mediated T cell suppression experiments, sorted tumor-infiltrating PMN-MDSCs or PMN from health donors were cocultured with autologous CFSE labeled T cell (2 × 104) at 4:1, 2:1, 1:1, and 0:1 in the medium containing CD3 (10 μg/ml, R&D Systems) and CD28 (10 μg/ml, R&D Systems). At day 6, cells were harvested, and CFSE− CD3+ T cells were detected by FCM.
Immunofluorescent Staining and Morphological Analysis
Paraffin-embedded and formalin-fixed samples were cut into 5 μM sections, which were then processed for IF staining or H&E staining. Immunofluorescence was performed with antibodies to pan-TCRγδ, IL-17A, IL-23p19, CD11c, CD68, ZO-1, Claudin-2, and pan-Cytokeratin. Secondary antibodies were Alexa Fluor 488- or 647-conjugated goat anti–mouse IgG or Dylight 488- or 649-conjugated goat anti-rabbit IgG (Invitrogen). Images were acquired with a confocal microscopy (Zeiss LSM 710, Carl Zeiss). For morphological analysis, sorted inf-DCs, inf-macrophages, or PMN-MDSCs were subjected to cytospin and stained with May-Grunwald/Giemsa. Pictures were taken with a CFW-1308C color digital camera (Scion Corporation) on a Leica DM 4000 B microscope.
Statistical Analysis
Results were expressed as means ± SEM. Statistical analysis was performed with GraphPad Prism software version 6.1. The statistical significance of differences between groups was determined by the Student’s t test. Correlations between parameters were assessed with the Pearson correlation analysis and linear regression analysis, as appropriate. All data were analyzed with two-tailed tests unless otherwise specified, and a p value < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported by grants from the Science and Technology Department of Zhejiang Province (2011c13034~1, J.H.), the Ministry of Health of Zhejiang Province (2012ZDA021, J.H.), Natural Science Foundation of Zhejiang Province (Y2110034, FMQ; Y2100414, ZT; Z2100366, J.H. and Y2090386, X.W.) and the NIH (R01CA150947 and P01CA163223, J.Y.). This work was also funded by the Zhejiang Provincial Program for the Cultivation of High-level Innovative Health Talents (J.H.), the Zhejiang Province Key Discipline of Traditional Chinese Medicine (2012-XK-A27, J.H.) and Key Innovation Discipline of Medicine (2011-CX11, J.H.)
Footnotes
AUTHOR CONTRIBUTIONS
P.W. participated in the design and coordination of the research project, collected and processed the specimen, performed flow cytometry, analyzed the data, and contributed in the manuscript writing. D.W. performed some flow cytometry, cell sorting, ELISAs, and immunofluorescent assay. C.N. participated in the collection and process of the specimen and performed H&E staining and some in vitro experiments. J.Y. performed gene detections and FISH assay.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures, three tables, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2014.03.013.
References
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–545. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Caccamo N, La Mendola C, Orlando V, Meraviglia S, Todaro M, Stassi G, Sireci G, Fournié JJ, Dieli F. Differentiation, phenotype, and function of interleukin-17-producing human Vγ9Vδ2 T cells. Blood. 2011;118:129–138. doi: 10.1182/blood-2011-01-331298. [DOI] [PubMed] [Google Scholar]
- Cai Y, Shen X, Ding C, Qi C, Li K, Li X, Jala VR, Zhang HG, Wang T, Zheng J, Yan J. Pivotal role of dermal IL-17-producing γδ T cells in skin inflammation. Immunity. 2011;35:596–610. doi: 10.1016/j.immuni.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, Vernes JM, Jiang Z, Meng YG, Peale FV, et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med. 2013;19:1114–1123. doi: 10.1038/nm.3291. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenoglio D, Poggi A, Catellani S, Battaglia F, Ferrera A, Setti M, Murdaca G, Zocchi MR. Vdelta1 T lymphocytes producing IFN-gamma and IL-17 are expanded in HIV-1-infected patients and respond to Candida albicans. Blood. 2009;113:6611–6618. doi: 10.1182/blood-2009-01-198028. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Rami′rez-Valle F, Xu Y, Wu S, Wu Z, Karjalainen KE, Cyster JG. Deficiency in IL-17-committed Vγ4(+) γδ T cells in a spontaneous Sox13-mutant CD45.1(+) congenic mouse substrain provides protection from dermatitis. Nat Immunol. 2013;14:584–592. doi: 10.1038/ni.2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491:254–258. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He D, Li H, Yusuf N, Elmets CA, Li J, Mountz JD, Xu H. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J Immunol. 2010;184:2281–2288. doi: 10.4049/jimmunol.0902574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick MN, Lonsdorf AS, Shirakawa AK, Richard Lee CC, Liao F, Singh SP, Zhang HH, Grinberg A, Love PE, Hwang ST, Farber JM. CCR6 is required for IL-23-induced psoriasis-like inflammation in mice. J Clin Invest. 2009;119:2317–2329. doi: 10.1172/JCI37378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapanadze T, Gamrekelashvili J, Ma C, Chan C, Zhao F, Hewitt S, Zender L, Kapoor V, Felsher DW, Manns MP, et al. Regulation of accumulation and function of myeloid derived suppressor cells in different murine models of hepatocellular carcinoma. J Hepatol. 2013;59:1007–1013. doi: 10.1016/j.jhep.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Kenna TJ, Davidson SI, Duan R, Bradbury LA, McFarlane J, Smith M, Weedon H, Street S, Thomas R, Thomas GP, Brown MA. Enrichment of circulating interleukin-17-secreting interleukin-23 receptor-positive γ/δ T cells in patients with active ankylosing spondylitis. Arthritis Rheum. 2012;64:1420–1429. doi: 10.1002/art.33507. [DOI] [PubMed] [Google Scholar]
- Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, Basham B, McClanahan T, Kastelein RA, Oft M. IL-23 promotes tumour incidence and growth. Nature. 2006;442:461–465. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- Pantelyushin S, Haak S, Ingold B, Kulig P, Heppner FL, Navarini AA, Becher B. Rorγt+ innate lymphocytes and γδ T cells initiate psoriasiform plaque formation in mice. J Clin Invest. 2012;122:2252–2256. doi: 10.1172/JCI61862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SG, Mathur R, Long M, Hosh N, Hao L, Hayden MS, Ghosh S. T regulatory cells maintain intestinal homeostasis by suppressing γδ T cells. Immunity. 2010;33:791–803. doi: 10.1016/j.immuni.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann F, Rothhammer V, Claussen MC, Haas JD, Blanco LR, Heink S, Prinz I, Hemmer B, Kuchroo VK, Oukka M, Korn T. γδ T cells enhance autoimmunity by restraining regulatory T cell responses via an interleukin-23-dependent mechanism. Immunity. 2010;33:351–363. doi: 10.1016/j.immuni.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichling T, Goss KH, Carson DJ, Holdcraft RW, Ley-Ebert C, Witte D, Aronow BJ, Groden J. Transcriptional profiles of intestinal tumors in Apc(Min) mice are unique from those of embryonic intestine and identify novel gene targets dysregulated in human colorectal tumors. Cancer Res. 2005;65:166–176. [PubMed] [Google Scholar]
- Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, Meade TW. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010;376:1741–1750. doi: 10.1016/S0140-6736(10)61543-7. [DOI] [PubMed] [Google Scholar]
- Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M. Tumor necrosis factor-a blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation. Immunity. 2013;38:541–554. doi: 10.1016/j.immuni.2013.02.007. [DOI] [PubMed] [Google Scholar]
- Segura E, Touzot M, Bohineust A, Cappuccio A, Chiocchia G, Hosmalin A, Dalod M, Soumelis V, Amigorena S. Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity. 2013;38:336–348. doi: 10.1016/j.immuni.2012.10.018. [DOI] [PubMed] [Google Scholar]
- Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, Iwaki T, Okada Y, Iida M, Cua DJ, et al. Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15:946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- Silva-Santos B. Promoting angiogenesis within the tumor microenvi-ronment: the secret life of murine lymphoid IL-17-producing gammadelta T cells. Eur J Immunol. 2010;40:1873–1876. doi: 10.1002/eji.201040707. [DOI] [PubMed] [Google Scholar]
- Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman WH, Pagés F, Galon J. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011;71:1263–1271. doi: 10.1158/0008-5472.CAN-10-2907. [DOI] [PubMed] [Google Scholar]
- Vantourout P, Hayday A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat Rev Immunol. 2013;13:88–100. doi: 10.1038/nri3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakita D, Sumida K, Iwakura Y, Nishikawa H, Ohkuri T, Chamoto K, Kitamura H, Nishimura T. Tumor-infiltrating IL-17-producing gammadelta T cells support the progression of tumor by promoting angiogenesis. Eur J Immunol. 2010;40:1927–1937. doi: 10.1002/eji.200940157. [DOI] [PubMed] [Google Scholar]
- Wang X, Tully O, Ngo B, Zitin M, Mullin JM. Epithelial tight junctional changes in colorectal cancer tissues. ScientificWorldJournal. 2011;11:826–841. doi: 10.1100/tsw.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Sun R, Wei H, Tian Z. High-mobility group box 1 (HMGB1)-Toll-like receptor (TLR)4-interleukin (IL)-23-IL-17A axis in drug-induced damage-associated lethal hepatitis: Interaction of γδ T cells with macrophages. Hepatology. 2013;57:373–384. doi: 10.1002/hep.25982. [DOI] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, Wu H, Xu X, Erben U, Wu P et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest. 2012;122:4094–4104. doi: 10.1172/JCI64115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Restifo NP. T(H)17 cells in tumour immunity and immunotherapy. Nat Rev Immunol. 2010;10:248–256. doi: 10.1038/nri2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.