

Abstract
Methicillin-resistant Staphylococcus aureus infections are a significant global health challenge in part due to the emergence of strains exhibiting resistance to nearly all classes of antibiotics. This underscores the urgent need for the rapid development of novel antimicrobials to circumvent this burgeoning problem. Previously, whole-cell screening of a library of 2,5-disubstituted thiazole compounds revealed a lead compound exhibiting potent antimicrobial activity against MRSA. The present study, conducting a more rigorous analysis of the structure-activity relationship of this compound, reveals a nonpolar, hydrophobic functional group is favored at thiazole-C2 and an ethylidenehydrazine-1-carboximidamide moiety is necessary at C5 for the compound to possess activity against MRSA. Furthermore, the MTS assay confirmed analogues 5, 22d, and 25 exhibited an improved toxicity profile (not toxic up to 40 μg/mL to mammalian cells) over the lead 1. Analysis with human liver microsomes revealed compound 5 was more metabolically stable compared to the lead compound (greater than eight-fold improvement in the half-life in human liver microsomes). Collectively the results presented demonstrate the novel thiazole derivatives synthesized warrant further exploration for potential use as future antimicrobial agents for the treatment of multidrug-resistant S. aureus infections.
Keywords: Antimicrobials, Drug-resistance, Methicillin-resistant Staphylococcus aureus (MRSA), Thiazole compounds, HEK293 Toxicity
1. Introduction
Methicillin-resistant Staphylococcus aureus (MRSA) infections remain a significant public health challenge globally. Though reports have indicated the incidence of healthcare-associated MRSA (HA-MRSA) infections have diminished [1, 2], transmission of community-associated MRSA (CA-MRSA) infections, primarily strains USA300 and USA400 [3], has continued to present major problems amongst a diverse population including healthcare workers [4], prison inmates [5, 6], military service personnel [6], contact sport athletes [7, 8], homeless individuals [9], intravenous drug users [9, 10], tattoo recipients [11], neonates [12], and young children [13, 14]. Moreover, CA-MRSA infections are typically associated with more severe morbidity and mortality than their HA-MRSA counterparts [15]. While CA-MRSA is a leading cause of skin and soft-tissue infections [16, 17], MRSA has also been associated with more complicated medical diseases including necrotizing pneumonia [18], osteomyelitis [19], and sepsis [20], leading to over 11,000 deaths annually [21].
A recent study has estimated the total annual burden upon society for treatment of CA-MRSA infections alone may exceed US$13 billion [22]. Part of the associated cost is due to failure of current antimicrobials to treat certain clinical isolates of MRSA that have developed resistance to these therapeutic agents. Indeed, clinical isolates of both CA-MRSA and HA-MRSA have been documented that exhibit resistance to an array of different antibiotic classes including the β-lactams [23], macrolides [24], quinolones [25, 26], tetracyclines [27], and lincosamides [27]. Further exacerbating the problem, strains have emerged which exhibit resistance to first-line antibiotics (such as mupirocin [27, 28] for the treatment of MRSA skin infections) and drugs deemed agents of last resort (such as linezolid [29, 30] and vancomycin [31]). Prudent use and development of effective antimicrobials is a critical step to alleviate complications and costs associated with MRSA infections. Therefore there is an urgent need for the development of novel therapeutic agents and treatment strategies to circumvent this significant global health issue.
Utilizing whole-cell screening of a library of substituted thiazoles, our research group identified a novel lead thiazole compound that possesses potent antimicrobial activity against clinically relevant isolates of MRSA, vancomycin-intermediate S. aureus (VISA), and vancomycin-resistant S. aureus (VRSA) [32]. The basic structure of the lead 1 consists of a central thiazole ring connected to two distinct moieties – a lipophilic side chain at C2 and a cationic amino group at C5. The objectives of the present study were to construct a series of analogues to the lead 1 (Table 1) with modifications to the functional groups at both the thiazole-C2 and C5 positions to more rigorously ascertain the structure-activity relationship of these compounds against a diverse array of HA-MRSA and CA-MRSA isolates, identify new derivatives exhibiting an improved toxicity profile against mammalian cells, and to enhance the metabolic stability profile of the lead 1.
Table 1.
Minimum inhibitory concentration (MIC) of thiazole compounds against methicillin-resistant Staphylococcus aureus (MRSA) ATCC 43300.
| Analogue | MIC (μg/mL) |
|---|---|
| 1 (lead) | 1.3 |
| 5 | 1.4 |
| 7 | 12.6 |
| 9 | >35.1 |
| 10 | >45.8 |
| 11 | >42.4 |
| 12 | 44.5 |
| 13 | 44.2 |
| 15 | >37.0 |
| 16 | >38.3 |
| 19 | >36.9 |
| 21b | >39.8 |
| 21c | >46.2 |
| 22a | >46.7 |
| 22b | 5.9 |
| 22c | 3.3 |
| 22d | 6.3 |
| 24 | >43.9 |
| 25 | 1.6 |
2. Chemistry
The detailed synthetic protocols and spectral data of the lead 1 (Figure 1) in addition to all intermediates have been reported elsewhere [32, 33]. All thiazole compounds were dissolved in dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO, USA) to achieve a stock 10 mM solution.
Figure 1.
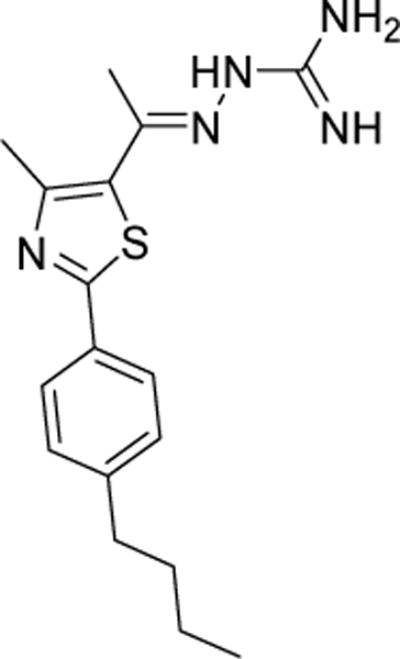
Chemical structure of the lead compound 1.
The (4-iodophenyl)thiazole derivative 3 was prepared by heating a mixture of the commercially available 4-iodothiobenzamide 2 and 3-chloro-2,4-pentanedione in absolute ethanol, as illustrated in Scheme 1. The phenylthiazolyl methyl ketone derivatives 4 and 6 were prepared via the Sonogashira cross coupling of the (4-iodophenyl)thiazole derivative 3 with commercially available 1-hexyne and 1-nonyne, respectively, in DMF using a bis(triphenylphosphine)palladium(II) dichloride catalyst, copper(I) iodide co-catalyst, and caesium carbonate base (Scheme 1). The hydrazinecarboximidamide derivatives 5 and 7 were synthesized by treatment of the phenylthiazolyl methyl ketone derivatives 4 and 6, respectively, with aminoguanidine hydrochloride in the presence of a catalytic amount of lithium chloride in absolute ethanol (Scheme 1).
Scheme 1.
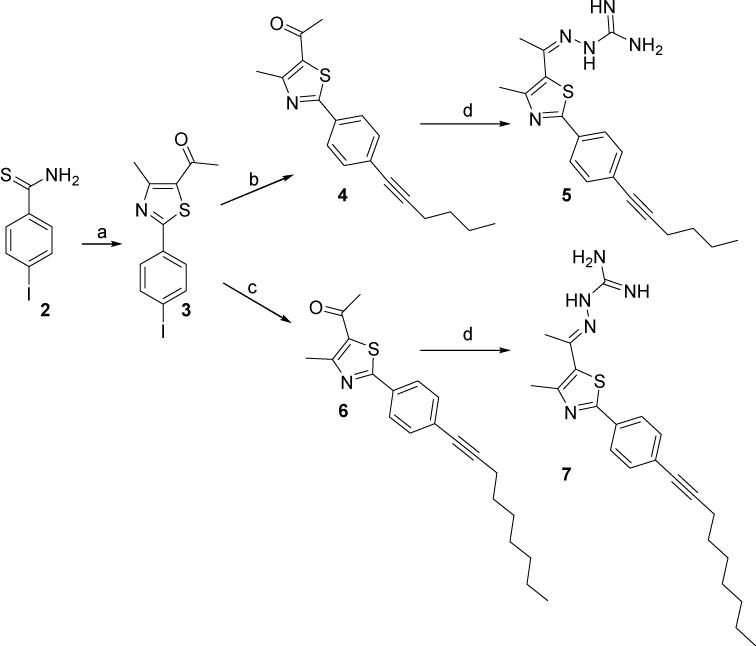
Reagents and conditions: (a) 3-chloro-2,4-pentanedione, EtOH, reflux, 24 h; (b) 1-hexyne, PdCI2(PPh3)2, Cul,Cs2CO3, DMF, sealed tube, 65 °C, 15h; (c)1-nonyne, PdCI2(PPh3)2, Cul, Cs2CO3, DMF, sealed tube, 70 °C. 15 h; (d) aminoguanidine HCl, LiCl, EtOH, reflux, 24 h.
The amide derivatives 10–13 were prepared in quantitative yields by reacting the 4-butylphenylthiazole acid chloride intermediate 8 [34] with the appropriate amines in THF, as illustrated in Scheme 2. Compound 16 was synthesized in three steps, starting with the formation of the amide derivative 9 by way of reacting the acid chloride intermediate 8 with ammonium hydroxide in THF at room temperature. The amide intermediate 9 was then heated in thionyl chloride to give the nitrile intermediate 14, which upon subsequent treatment with NaN3 in the presence of iodine gave the tetrazole-containing thiazole derivative 16 as shown in Scheme 2. The nitrile intermediate 14 was also treated with hydroxylamine hydrochloride in absolute ethanol with a catalytic amount of potassium carbonate to afford the thiazole derivative 15. The phenylthiazolyl methyl ketone derivative 18 was prepared by treatment of the commercially available 4-aminothiobenzamide 17 with 3-chloro-2,4-pentanedione in absolute ethanol.
Scheme 2.
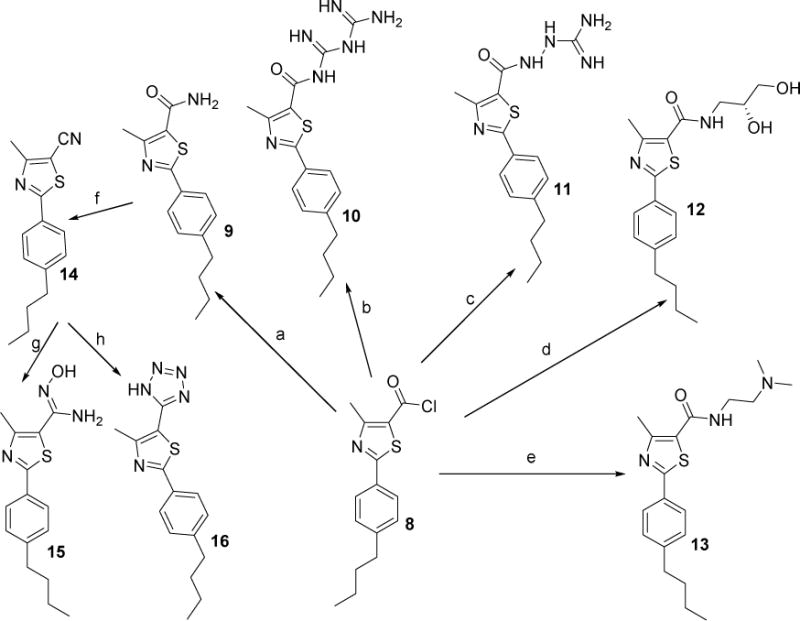
Reagents and conditions: (a) 30 % aq NH4OH, THF, rt, 24 h; (b) biguanidine hydrochloride, Et3N, THF, 24 h; (c) aminoguanidine hydrochloride, Et3N, THF, 24 h; (d) (R)-(−)-3-amino-1,2-propanediol,THF, rt, 24 h; (e) N,N-dimethylethylenediamine, THF, rt, 48 h; (f) thionyl chloride, reflux, 7 h; (g) NH2OH HCI, K2C03, EtOH, 78 °C, 24 h; (h)NaN3, l2, DMF, 120°C, 15h.
Synthesis of the hydrazinecarboximidamide derivative 19 was achieved by treatment of the phenylthiazolyl methyl ketone derivative 18 with aminoguanidine hydrochloride in the presence of a catalytic amount of lithium chloride (Scheme 3).
Scheme 3.
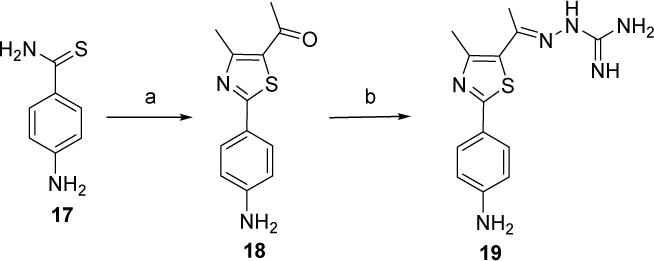
Reagents and conditions:(a) 3-chloro-2,4-pentanedione, EtOH, reflux, 20 h; (b) aminoguanidine hydrochloride, LiCl, EtOH, reflux, 24 h.
Phenylthiazole methylketone derivatives 21a–d and 24 were prepared via the Suzuki-Miyaura cross coupling of the (4-iodophenyl)thiazole derivative 3 with the commercially available phenylboronic acid derivatives 20a–d and 23, respectively, in the presence of a catalytic quantity of palladium(II) acetate and (2-biphenyl)dicyclohexylphosphine ligand, as shown in Scheme 4. Synthesis of the hydrazinecarboximidamide derivatives 22a–d and 25 was achieved by treatment of phenylthiazole methylketone derivatives 21a–d and 24, respectively, with aminoguanidine hydrochloride in the presence of lithium chloride as catalyst (Scheme 4).
Scheme 4.
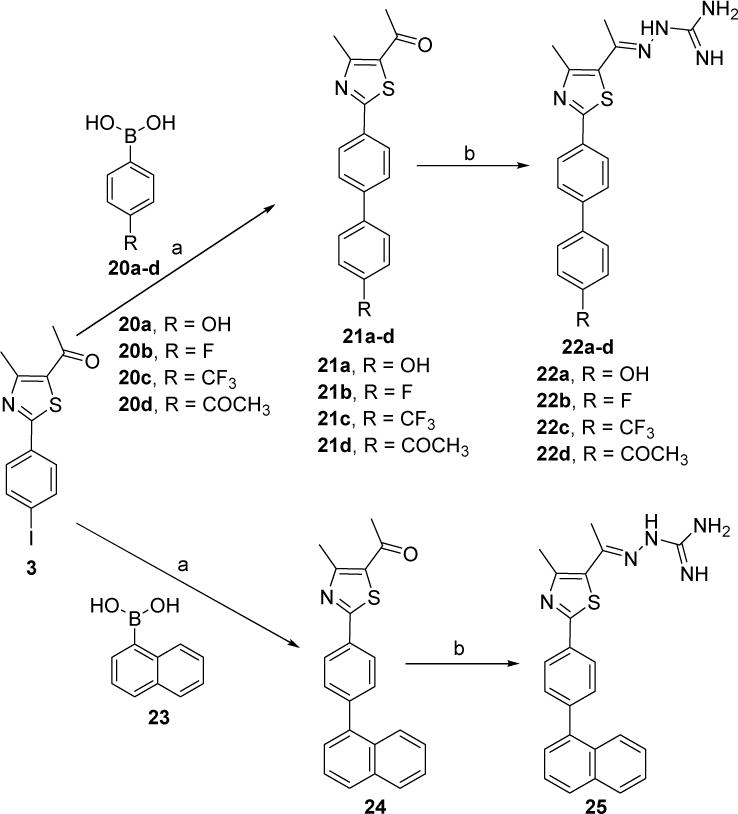
Reagents and conditions: a) Pd(OAc)2, (2-biphenyl)dicyclohexylphosphine, K3PO4, toluene, 90 °C 24 h; b) aminoguanidine hydrochloride, LiCl, EtOH, reflux, 24 h.
3. Biological Results and Discussion
3.1. Antibacterial activity of thiazole compounds and vancomycin against MRSA, VISA, and VRSA
To ascertain the structure-activity relationships of the lead thiazole compound more thoroughly, derivatives were initially constructed with modifications to the thiazole-C5 cationic moiety (keeping the lipophilic alkane side chain at thiazole-C2 intact). Substitution of the ethylidenehydrazine-1-carboximidamide of the lead 1 with moieties such as a tetrazole (16), an amide derivative (9–13), or a hydroxamidine (15) results in complete abolishment of antimicrobial activity against MRSA (minimum inhibitory concentration (MIC) > 35.1 μg/mL (Table 1)). This trend continues when the amino moiety is replaced with a ketone in derivatives 21b–c and 24. Interestingly, derivatives 22b–c and 25 (consisting of the same cationic head group as the lead 1 but with substitutions to the linear alkane side chain at thiazole-C2 identical to those in compounds 21b–c and 24) retain antimicrobial activity; among the groups studied thus far, the ethylidenehydrazine-1-carboximidamide is the only one to retain potency at this position of the structural series and we therefore retained it in all future analogs.
Modifications made to the linear alkane side chain at thiazole-C2 revealed hydrophobic, nonpolar moieties at this position are preferred for the compound to retain potent antimicrobial activity. The presence of a hydrophilic, polar group, such as an amine (19) or alcohol (22a) at this position, results in complete loss of antimicrobial activity, with both compounds possessing a MIC > 36.9 μg/mL (Table 1). Replacement of the alkane side chain with hydrophobic, polar substituents such as an acetyl group (22d, MIC = 6.3 μg/mL), a fluoride (22b), or a trifluoromethyl group (22c) results in the compounds possessing antimicrobial activity, but with a MIC higher than the parent compound. On the other hand, substitution of the alkane side chain with a nonpolar, hydrophobic moiety, such as an alkyne (5 with MIC of 1.4 μg/mL) or naphthalene (25 with MIC of 1.6 μg/mL) functional group, results in derivatives with potent antimicrobial activity (nearly identical MIC to the lead 1). Once again this confirms that a more nonpolar, hydrophobic functional group is needed at the C5 position for the thiazole compounds to possess potent antibacterial activity. This is in agreement with previously reported findings where alkane, cycloalkane, cycloalkene, and arene substitutions at thiazole-C2 resulted in compounds with stronger activity against MRSA [32]. Interestingly, extending the alkyne length from a hexyne (5) to a nonyne (7) group results in diminished anti-MRSA activity with the MIC increasing nine-fold from 1.4 μg/mL to 12.6 μg/mL. This is similar to what was previously found with lengthening of the alkane side chain at thiazole-C2; increasing the alkane side chain beyond four methylene units resulted in a drastic reduction in antimicrobial activity of the compounds. Future studies examining decreasing the alkyne side chain length at thiazole-C5 and its effect on anti-MRSA activity warrant further exploration. Additionally, repositioning the nonpolar moiety (at the ortho and meta positions of the phenyl substituent connected to C2 on the thiazole ring) would be of interest to assess if the para position plays a crucial role in the antimicrobial activity of the compounds.
After confirming that five derivatives (5, 22b–d and 25) possessed strong antimicrobial activity against a single strain of MRSA, we next assessed their activity against an array of clinically relevant multidrug-resistant HA-MRSA and CA-MRSA strains as well as vancomycin-intermediate (VISA) and vancomycin-resistant (VRSA) S. aureus isolates. All five compounds maintained their activity (with MICs identical or two-fold higher than those reported against MRSA ATCC43300) against MRSA isolates exhibiting resistance to mupirocin (NRS107), linezolid (NRS119), erythromycin (USA300), tetracycline (USA300), ciprofloxacin (USA500), clindamycin (USA500), and gentamicin (USA500) (Table 2); this indicates cross-resistance between these antibiotics and the thiazole compounds is unlikely to occur. The thiazole derivatives also exhibited potent activity against strains of MRSA (USA300 and USA400) responsible for the majority of MRSA-related skin and soft tissue infections in North America [3, 35]. Additionally, analogues 5 (MIC between 1.3–2.6 μg/mL), 22b (MIC between 2.9–5.9 μg/mL), and 25 (MIC of 1.6 μg/mL) proved to be similar in activity or better than vancomycin (MIC of 3.0 μg/mL) against two VISA isolates tested. Furthermore, while all three VRSA strains exhibited resistance to vancomycin (MIC > 190.2 μg/mL), the lead thiazole (1) and the five most potent derivatives retained their antimicrobial activity with MIC values ranging from 0.7 μg/mL (for 1) to 6.7 μg/mL (for 22c). Finding alternative therapeutic options (such as these thiazole compounds) to vancomycin and linezolid, agents of last resort for treatment of severe MRSA infections, is critical to address the burden of these challenging infections.
Table 2.
Minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) of thiazole compounds 1, 5, 22b–d, 25 and vancomycin against seven methicillin-resistant (MRSA), three vancomycin-intermediate (VISA), and three vancomycin-resistant Staphylococcus aureus (VRSA) strains.
| S. aureus strain | 1 | 5 | 22b | 22c | 22d | 25 | Vancomycin | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||
| MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | |
| NRS107 (MRSA) |
2.6 | 2.6 | 1.4 | 1.4 | 2.9 | 5.9 | 3.3 | 6.7 | 6.3 | 12.5 | 1.6 | 1.6 | <1.5 | <1.5 |
| NRS119 (MRSA) |
2.6 | 2.6 | 1.4 | 1.4 | 5.9 | 11.7 | 6.7 | 6.7 | 6.3 | 12.5 | 1.6 | 1.6 | <1.5 | <1.5 |
| NRS194 (MRSA) |
1.3 | 1.3 | 1.4 | 1.4 | 5.9 | 11.7 | 6.7 | 13.3 | 6.3 | 6.3 | 1.6 | 1.6 | 0.7 | 0.7 |
| USA300 (MRSA) |
1.3 | 1.3 | 1.4 | 1.4 | 5.9 | 5.9 | 6.7 | 6.7 | 6.3 | 6.3 | 1.6 | 1.6 | 0.7 | 0.7 |
| USA 400 (MRSA) |
1.3 | 1.3 | 1.4 | 1.4 | 5.9 | 11.7 | 6.7 | 13.3 | 6.3 | 6.3 | 1.6 | 1.6 | 0.7 | 0.7 |
| USA500 (MRSA) |
1.3 | 1.3 | 1.4 | 1.4 | 5.9 | 5.9 | 3.3 | 3.3 | 6.3 | 6.3 | 1.6 | 3.2 | 0.7 | 0.7 |
| ATCC 43300 (MRSA) |
1.3 | 1.3 | 1.4 | 1.4 | 5.9 | 5.9 | 3.3 | 3.3 | 6.3 | 6.3 | 1.6 | 1.6 | 0.7 | 0.7 |
| NRS1 (VISA) |
1.3 | 2.6 | 0.7 | 0.7 | 2.9 | 2.9 | 3.3 | 3.3 | 6.3 | 6.3 | 1.6 | 1.6 | 3.0 | 3.0 |
| NRS19 (VISA) |
1.3 | 2.6 | 1.4 | 1.4 | 5.9 | 5.9 | 3.3 | 3.3 | 12.5 | 12.5 | 1.6 | 1.6 | <1.5 | <1.5 |
| NRS37 (VISA) |
2.6 | 2.6 | 1.4 | 1.4 | 2.9 | 5.9 | 3.3 | 3.3 | 3.1 | 6.3 | 1.6 | 1.6 | 3.0 | 3.0 |
| VRS1 (VRSA) |
0.7 | 0.7 | 1.4 | 1.4 | 2.9 | 2.9 | 6.7 | 6.7 | 3.1 | 3.1 | 3.2 | 3.2 | >190.2 | >190.2 |
| VRS4 (VRSA) |
0.7 | 1.3 | 1.4 | 2.8 | 2.9 | 2.9 | 6.7 | 6.7 | 1.6 | 3.1 | 3.2 | 3.2 | >190.2 | >190.2 |
| VRS5 (VRSA) |
0.7 | 1.3 | 2.8 | 2.8 | 2.9 | 2.9 | 6.7 | 6.7 | 6.3 | 6.3 | 3.2 | 3.2 | >190.2 | >190.2 |
Subsequent to establishing that the lead compound and the five most active analogues exhibited potent antimicrobial activity against a diverse spectrum of CA-MRSA, HA-MRSA, VISA, and VRSA isolates, we next turned our attention to assessing whether these compounds were bacteriostatic or bactericidal. Antimicrobial agents that are bactericidal, as opposed to their bacteriostatic counterparts, are thought to help patients recover more rapidly from infections, resulting in a better clinical outcome [36]. To assess if the thiazole compounds were bacteriostatic or bactericidal, the minimum bactericidal concentration (MBC) was determined. As Table 2 presents, all six thiazole compounds tested exhibited MBC values that were identical to or two-fold higher than their MIC values. The results mimic those of vancomycin, a known bactericidal antibiotic, indicating the thiazole compounds are bactericidal.
3.2. Time-kill analysis of most potent thiazole analogues against MRSA USA300
To confirm the thiazole compounds are in fact bactericidal agents against MRSA, a time-kill analysis was performed. MRSA USA300 cells in late logarithmic growth were treated with 3 × MIC of the lead thiazole (1), the two most potent derivatives (5 and 25), or vancomycin. Interestingly, a simple substitution at thiazole-C2 from an alkane/alkyne (1/5) to the more conformationally-restricted naphthalene analogue (25), results in a dramatic shift in the rate of bacterial killing by the thiazole compounds. As Figure 2 demonstrates, compounds 1 and 5 completely eradicate MRSA growth within 4 hours while compound 25 requires 10 hours to achieve the same effect. Though all three compounds possess nearly identical MIC values, the structural modifications made at thiazole-C2 significantly affect the rate of bacterial killing observed for each compound against MRSA.
Figure 2.
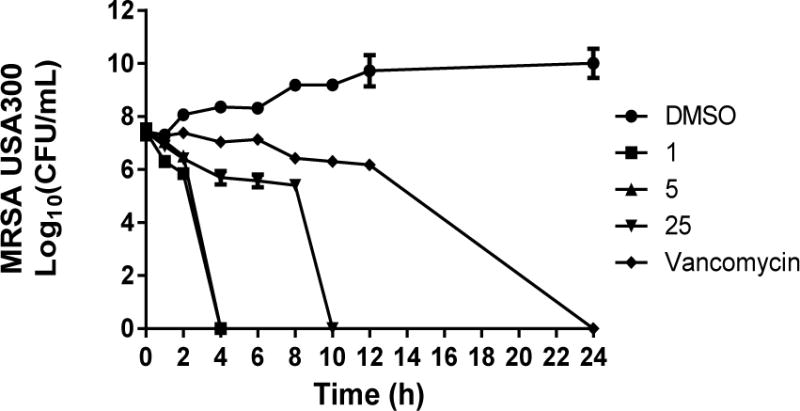
Time-kill analysis of thiazole compounds 1, 5, 25, and vancomycin against methicillin-resistant Staphylococcus aureus (MRSA USA300) over a 24 hour incubation period at 37 °C. DMSO served as a control. The error bars represent standard deviation values obtained from triplicate samples used for each compound/antibiotic studied.
While all three thiazole compounds exhibit the ability to eliminate MRSA growth completely within 10 hours, vancomycin requires 24 hours to achieve the same result. This is similar to what has been reported elsewhere regarding vancomycin’s slow bactericidal activity [37]. Rapid bactericidal activity is considered to be a critical factor in slowing the emergence of bacterial resistance to an antimicrobial agent and is important clinically in preventing an infection from spreading [36]. Additionally, bactericidal agents have been shown both clinically and through in vivo studies to be superior to bacteriostatic agents for the treatment of certain invasive diseases such as endocarditis [38]. Thus these thiazole compounds may have the potential to be utilized in a wide array of clinically important MRSA diseases from skin and soft tissue infections to systemic infections such as endocarditis.
3.3. Toxicity analysis of potent thiazole derivatives against mammalian cells
Selective toxicity is an important property that both approved antibiotics and novel antimicrobial compounds must possess. The ability for antimicrobial agents to exhibit their activity on the target microorganism while not causing harm to host (mammalian) tissues is important to ascertain early in the drug discovery process. Previously, the lead thiazole (1) was found to be nontoxic to human cervical (HeLa) cells at a concentration of 11 μg/mL [32]. A principal objective of the present study was to develop new analogues of the lead that exhibited an improved/more selective toxicity profile. To assess this, the lead compound and five most potent derivatives against MRSA (5, 22b–d and 25) were screened against a human embryonic kidney (HEK293) cell line using the MTS assay. Figure 3 presents the results garnered. At a concentration of 40 μg/mL, the lead 1 and compounds 22b and 22c proved to be toxic to mammalian cells. However, three of the novel analogues – compounds 5 (alkynyl side chain), 22d (p-acetylbenzyl), and 25 [p-(1-naphthyl)] – exhibit an improved toxicity profile compared to the lead 1 at the tested concentration. This concentration (40 μg/mL) represents a 25- (for 25) to 28-fold (for 5) difference between the MIC values determined against MRSA for these compounds. Thus there is a significant improvement in the toxicity profile of these novel analogues when compared to the lead compound.
Figure 3.
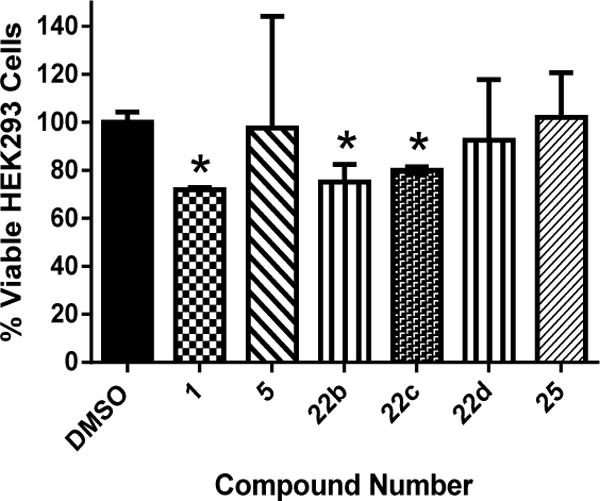
Percent viable mammalian cells (measured as average absorbance ratio (test agent relative to DMSO)) for cytotoxicity analysis of thiazole compounds 1, 5, 22b–d, and 25 at 40 μg/mL against HEK293 cells using the MTS 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H- tetrazolium) assay. DMSO was used as a negative control to determine a baseline measurement for the cytotoxic impact of each compound. The absorbance values represent an average of a minimum of three samples analyzed for each compound. Error bars represent standard deviation values for the absorbance values. A unpaired t-test, P ≤ 0.05, demonstrated statistical difference between the values obtained for compounds 1, 22b and 22c relative to the cells treated with DMSO.
3.4. Metabolic stability analysis of compound 5
Previously, microsomal stability analysis of the lead 1 revealed this compound was metabolized fairly rapidly (intrinsic clearance rate of 80.3 μL/min/mg and half-life of 28.8 minutes) via a NADPH-mediated process (such as via the cytochrome P450 system) [32]. As compound 5 demonstrated nearly identical antimicrobial activity to the lead compound, we were curious to assess if the substitution of an alkane side chain with an alkyne at thiazole-C2 would enhance the metabolic stability of the compound, preventing its conversion to potentially inactive metabolites. Using pooled human liver microsomes, 5, similar to the parent compound, was found to be metabolized via a NADPH-mediated process (intrinsic clearance rate of 3.7 μL/min/mg as compared to 0.0 μL/min/mg in the absence of the cofactor, NADPH) (Table 3). Interestingly, the slower clearance rate correlates with an improved half-life for compound 5 (as compared to the lead compound) that exceeds 4 hours. This marked improvement in the metabolic stability of the thiazole compound is important as it has the potential to positively impact the pharmacokinetic profile of this compound, reduce the frequency of doses needed to be administered for treatment (fewer doses leads to improved patient compliance), while also ensuring the active drug circulates within the patient’s system to assist with treating and clearing an infection. Additionally, compounds that are metabolically stable are less susceptible to experiencing issues pertaining to toxicity and drug-drug interactions caused by metabolites [39]. The metabolic stability analysis combined with the enhanced toxicity profile of compound 5 (as compared to the lead) warrants further analysis of this compound as a potential novel antibiotic for the treatment of MRSA infections.
Table 3.
Evaluation of metabolic stability of thiazole compound 5, verapamil, and warfarin in human liver microsomes.
| Compound/Drug Tested | NADPH-dependent CLint1 (μL/min/mg) |
NADPH-dependent T1/22 (min) |
NADPH-free CLint (μL/min/mg) |
NADPH-free T1/2 (min) |
|---|---|---|---|---|
| 5 | 3.7 | >240 | 0.0 | >240 |
| Verapamil | 213 | 10.8 | 0.0 | >240 |
| Warfarin | 0.0 | >240 | 0.0 | >240 |
CLint = microsomal intrinsic clearance
T1/2 = half-life
4. Conclusion
We present herein a novel series of 2,5-disubstituted thiazole compounds exhibiting potent activity against clinically relevant isolates of MRSA, VISA, and VRSA. A rigorous analysis of the structure-activity relationship of these analogues reveals the ethylidenehydrazine-1-carboximidamide head group (at thiazole-C5) and a nonpolar, hydrophobic moiety (at thiazole-C2) are critical for the thiazole compound’s antibacterial action. Three derivatives with substitutions at thiazole-C2 (an alkyne, p-acetylbenzene, and p-naphthalene) demonstrate an improved toxicity profile against mammalian cells compared to the lead compound. Furthermore, the alkyne substitution results in a compound that is more stable to metabolism as assessed via human liver microsomes. Collectively, the results present critical information necessary for further analysis and development of these thiazole compounds as novel antimicrobial agents for use in treatment of infections caused by multidrug-resistant S. aureus.
5. Material and methods
1H NMR spectra were recorded in CDCl3 or DMSO-d6 using a 300 MHz spectrometer. Chemical shifts are reported in units of ppm on the delta (δ) scale and coupling constants (J) are reported in units of Hz. The following splitting abbreviations are used: s = singlet, d = doublet, t = triplet and m = multiplet. All melting points were recorded using capillary tubes on a Mel-Temp apparatus and are not corrected. Mass spectral analyses were performed at the Purdue University Campus-Wide Mass Spectrometry Center. Reagents and solvents were purchased from commercial vendors and were used as received without further purification, unless otherwise stated.
5.1. Procedure for synthesis of thiazole compounds and intermediates
5.1.1. 1-(2-(4-Iodophenyl)-4-methylthiazol-5-yl)ethanone (3)
4-Iodothiobenzamide (2, 3.80 mmol) and α-chloropentanedione (0.611 mg, 4.56 mmol) were added to absolute ethanol (50 mL). The reaction mixture was heated at reflux for 24 h. After removal of solvent under reduced pressure, the residue was purified by silica gel chromatography using hexanes–ethyl acetate (7:3) to provide the desired compound as light orange solid (0.800 g, 62%): mp 105–106 °C. 1H NMR (300 MHz, CDCl3) δ 7.81 (d, J = 6.6 Hz, 2 H), 7.70 (d, J = 6.6 Hz, 2 H), 2.77 (s, 3 H), 2.56 (s, 3 H).
5.1.2. 1-(2-(4-(Hex-1-yn-1-yl)phenyl)-4-methylthiazol-5-yl)ethanone (4)
1-(2-(4-Iodophenyl)-4-methylthiazol-5-yl)ethanone (3, 0.5 g, 1.45 mmol), 1-hexyne (0.373 g, 7.73 mmol), cesium carbonate (0.947 g, 2.91 mmol), dichloro-bis(triphenylphosphine)palladium(II) (0.051 g, 0.072 mmol) and CuI (0.027 g, 0.145 mmol) were dissolved in DMF (6 mL). The reaction mixture was purged with argon for 20 min. The sealed tube was closed, placed in an oil bath and stirred at 65 °C for 15 h. The reaction mixture was filtered through celite, and the celite was washed with chloroform (50 mL). The organic phase was washed with 1% hydrochloric acid (30 mL), water (3 × 40 mL) and brine (30 mL). The organic layer was dried over Na2SO4, concentrated and purified by silica gel flash column chromatography using hexanes–ethyl acetate (8:2) to provide the desired compound as yellow syrup (0.400 g, 92.5%): IR (film) 1945, 1675, 1111, 819, 666 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.90 (d, J = 8.5 Hz, 2 H), 7.46 (d, J = 8.3 Hz, 2 H), 2.76 (s, 3 H), 2.56 (s, 3 H), 2.45 (t, J = 6.9 Hz, 2 H), 1.59 (m, 4 H), 0.97 (t, J = 7.3 Hz, 3 H); ESIMS m/z (rel intensity) 298 (MH+, 100).
5.1.3. (Z)-2-(1-(2-(4-(Hex-1-yn-1-yl)phenyl)-4-methylthiazol-5-yl)ethylidene) hydrazinecarboximidamide (5)
The thiazole derivative 4 (200 mg, 0.673 mmol) was dissolved in absolute ethanol (10 mL), and aminoguanidine hydrochloride (0.088 mg, 0.808 mmol) and a catalytic amount of LiCl (5 mg) were added. The reaction mixture was heated at reflux for 24 h. The solvent was evaporated under reduced pressure. The crude product was purified by crystallization from 70% methanol and then recrystallized from methanol to afford the desired compound as a yellow solid (80 mg, 46%): mp 253–254 °C. IR (KBr) 3329, 2227, 1678, 1143, 836, 657 cm−1; 1H NMR (DMSO-d6) δ 11.35 (br s, 1 H), 7.88 (d, J = 8.1 Hz, 2 H), 7.69 (br s, 3 H), 7. 49 (d, J = 8.1 Hz, 2 H), 2.60 (s, 3 H), 2.44 (s, 3 H), 2.41 (t, J = 7.1 Hz, 2 H), 1.48 (m, 4 H), 0.93 (t, J = 7.1 Hz, 3 H); ESIMS m/z (rel intensity) 354 (MH+, 100); HRESIMS calcd for C19H24N5S 354.1509 (MH+), found 354.1514; HPLC purity 98.07% (1% TFA in MeOH:H2O – 85:15).
5.1.4. 1-(4-Methyl-2-(4-(non-1-yn-1-yl)phenyl)thiazol-5-yl)ethan-1-one (6)
The thiazole derivative 3 (750 mg, 2.19 mmol), 1-nonyne (1.44 mL, 8.76 mmol), PdCl2(PPh3)2 (76.6 mg, 0.11mmol), copper(I) iodide (41.6 mg, 0.22 mmol) and Cs2CO3 (1.42 g, 4.38 mmol) were added to a sealed tube under argon for 10 min, and then DMF (7.5 mL) was added. The tube was once again evacuated and purged with argon for 5 min and heated to 70 °C for 15 h. The tube was allowed to cool to room temperature, and the solids were removed by filtration and the filter cake was extracted with additional CHCl3 (50 mL). The combined filtrate and extracts were concentrated under vacuum and extracted with EtOAc (2 × 50 mL) and washed with water (2 × 50 mL). After evaporation of the solvent under reduced pressure, the residue was collected and purified by flash chromatography (SiO2, hexanes-EtOAc, 8.8:1.2) to yield the desired compound 6 as a dark green oil (772 mg, 100%); 1H NMR (300 MHz, CDCl3) δ 7.9 (t, J = 6.8 Hz, 2 H), 7.46 (d, J = 8.4 Hz, 2 H), 2.76 (s, 3 H), 2.55 (s, 3 H), 2.44 (t, J = 7.1 Hz, 3 H), 1.63 (t, J = 7.7 Hz, 2 H), 1.30 (m, J = 3.3 Hz, 9 H), 0.9 (q, J = 6.2 Hz, 4 H).
5.1.5. (E)-2-(1-(4-Methyl-2-[4-(non-1-yn-1-yl)phenyl]thiazol-5-yl)ethylidene)hydrazinecarboximidamide (7)
The thiazole derivative 6 (140 mg, 0.41 mmol), aminoguanidine hydrochloride (90.85 mg, 0.83 mmol), and a catalytic amount of LiCl (5 mg) were added to absolute ethanol (10 mL). The reaction mixture was heated at reflux for 24 h. After evaporation of the solvent under reduced pressure, the crude residue was extracted with CHCl3/MeOH (90:10, 2 × 40 mL) and washed with water (2 × 50 mL) and brine (40 mL). The extracts were pooled together, dried over Na2SO4 and stripped of solvents under reduced pressure. The residue was suspended in CHCl3/hexanes (50:50, 50 mL) and filtered through Whatman filter paper to afford the desired product (7) as a yellow-white solid (200 mg, 100%): mp 255–260 °C dec, 1H NMR (300 MHz, DMSO-d6) δ 7.88 (d, J = 8.3 Hz, 3 H), 7.49 (d, J = 8.4 Hz, 3 H), 2.60 (s, 3 H), 2.49 (q, J = 1.7 Hz, 5 H), 1.26 (s, 12 H), 0.85 (s, 4 H); ESIMS m/z (rel intensity) 395 (M+, 57).
5.1.6. 2-(4-Butylphenyl)-4-methylthiazole-5-carboxamide (9)
Acid chloride 8 [34] (0.200 g, 0.682 mmol) was dissolved in THF (20 mL) and then 30% aq NH4OH (10 mL) was added. The reaction mixture was stirred at room temperature for 24 h. The THF was removed on a rotary evaporator and the crude product was extracted with EtOAc (2 × 25 mL) and washed with water (2 × 20 mL) and brine (20 mL). The combined organic layer was dried over Na2SO4 and concentrated to afford the amide 9 (0.185 g) in quantitative yield: mp 160–161 °C. IR (KBr) 3245, 1691, 1611, 1121, 846, 665 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.84 (d, J = 8.2 Hz, 2 H), 7.26 (d, J = 8.2 Hz, 2 H), 5.79 (br s, 2 H), 2.74 (s, 3 H), 2.66 (t, J = 7.5 Hz, 2 H), 1.63 (m, 2 H), 1.39 (m, 2 H), 0.94 (t, J = 7.3 Hz, 3 H); APCIMS m/z (rel intensity) 275 (MH+, 100); HPLC purity 97.89% (1% TFA in MeOH:H2O – 90:10).
5.1.7. 2-(4-Butylphenyl)-N-(N-carbamimidoylcarbamimidoyl)-4-methylthiazole-5-carboxamide (10)
Acid chloride 8 (0.200 g, 0.682 mmol) was dissolved in THF (20 mL) and then biguanidine hydrochloride (0.467 g, 3.41 mmol) followed by triethylamine (0.344 g, 3.41 mmol) were added. The reaction mixture was stirred at room temperature for 24 h. The THF was removed on a rotary evaporator and the crude product was extracted with EtOAc (2 × 30 mL) and washed with water (2 × 20 mL) and brine (20 mL). The combined organic layer was dried over Na2SO4, concentrated and purified by silica gel flash column chromatography using chloroform-methanol (9.5:0.5) to provide the desired compound as a yellow solid (0.070 g, 30%): mp 150–151 °C. IR (KBr) 3312, 1694, 1655, 1148, 823, 666 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.89 (br s, 2 H), 7.25 (br s, 2 H), 2.80 (s, 3 H), 2.67 (t, J = 7.2 Hz, 2 H), 1.60 (m, 2 H), 1.38 (m, 2 H), 0.94 (t, J = 7.1 Hz, 3 H); ESIMS m/z (rel intensity) 359 (MH+, 65), 341 (MH+-NH, 68); HRESIMS calcd for C17H23N6OS m/z 359.1248 (MH+3), found 359.1251; HPLC purity 95.16% (1% TFA in MeOH:H2O–90:10).
5.1.8. 2-(2-(4-Butylphenyl)-4-methylthiazole-5-carbonyl)hydrazinecarboximidamide (11)
Acid chloride 8 (0.200 g, 0.682 mmol) was dissolved in THF (20 mL) and then amino guanidine hydrochloride (0.377 g, 3.41 mmol) followed by triethylamine (0.344 g, 3.41 mmol) were added. The reaction mixture was stirred at room temperature for 24 h. The THF was removed on a rotary evaporator and the crude product was extracted with EtOAc (2 × 30 mL) and washed with water (2 × 20 mL) and brine (20 mL). The combined organic layer was dried over Na2SO4, concentrated and purified by silica gel flash column chromatography using hexanes–ethyl acetate (4:6) to provide the desired compound as a yellow solid. (0.090 g, 40%): mp 174–175 °C. IR (KBr) 3322, 1698, 1658, 1462, 1155, 856, 665 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.87 (d, J = 8.0 Hz, 2 H), 7.24 (d, J = 8.0 Hz, 2 H), 2.78 (s, 3 H), 2.67 (t, J = 7.6 Hz, 2 H), 1.63 (m, 2 H), 1.39 (m, 2 H), 0.94 (t, J = 7.3 Hz, 3 H); ESIMS m/z (rel intensity) 332 (MH+, 100); HRESIMS calcd for C16H22N5OS m/z 332.1112 (MH+), found 332.1210; HPLC purity 96.57% (1% TFA in MeOH:H2O – 90:10).
5.1.9. (R)-2-(4-Butylphenyl)-N-(2,3-dihydroxypropyl)-4methylthiazole-5-carboxamide (12)
A mixture of the acid chloride derivative 8 (0.25 g, 0.9 mmol) and (R)-(−)-3-amino-1,2-propanediol (0.154 g, 1.7 mmol) in THF (15 mL) were stirred at room temperature for 48 h. The THF was removed under reduced pressure and the resulting crude oil was extracted with chloroform (2 × 50 mL) and the extract was washed with water (2 × 50 mL) and brine (50 mL). The combined extracts were dried over Na2SO4 (10 g), filtered and concentrated under vacuum. The residue was purified by flash column chromatography (SiO2, CHCl3-MeOH, 9.4:0.6) to afford the product 12 as dark red crystals (0.17 g, 57%): mp 93–95 °C. 1H NMR (300 MHz, CDCl3) δ 7.82 (d, J = 8.2 Hz, 2 H), 7.24 (d, J = 7.2 Hz, 4 H), 6.32 (s, 1 H), 3.89 (d, J = 5.0 Hz, 1 H), 3.65 (m, J = 5.2 Hz, 4 H), 2.73 (s, 3 H), 2.63 (t, J = 7.6 Hz, 2 H), 1.61 (t, J = 7.9 Hz, 2 H), 1.35 (q, J = 7.5 Hz, 2 H), 0.92 (t, J = 6.1 Hz, 3 H); ESIMS m/z (rel intensity) 349 (MH+, 100).
5.1.10. 2-(4-Butylphenyl)-N-[(dimethylamino)methyl]-4-methylthiazole-5-carboxamide (13)
A mixture of acid chloride derivative 8 (0.4 g, 1.44 mmol) and N,N-dimethylethylenediamine (0.63 mL, 5.7 mmol) in THF (15 mL) were stirred at room temperature for 48 h. The THF was removed under reduced pressure and the resulting residue was purified by flash chromatography (SiO2, CHCl3-MeOH, 9.3:0.7) to afford the product 13 as a pink solid (0.123 g, 24%): mp 79–81 °C. 1H NMR (300 MHz, CDCl3) δ 7.83 (d, J = 8.2 Hz, 2 H), 7.25 (d, J = 9.7 Hz, 4 H), 3.49 (t, J = 5.8 Hz, 2 H), 2.72 (s, 3 H), 2.63 (t, J = 7.6 Hz, 2 H), 2.55 (t, J = 5.9 Hz, 2 H), 2.3 (s, 6 H), 1.61 (t, J = 7.8, 2 H), 1.36 (t, J = 7.4, 2 H), 0.92 (t, J = 7.3 Hz, 3 H); ESIMS m/z (rel intensity) 346 (MH+, 100).
5.1.11. 2-(4-Butylphenyl)-4-methylthiazole-5-carbonitrile (14)
Amide 9 (0.400 g, 1.45 mmol) was dissolved in thionyl chloride (20 mL) and the solution was heated to reflux for 7 h. Thionyl chloride was removed under reduced pressure, EtOAc (30 mL) was added and the mixture was washed with saturated aqueous NaHCO3 (2 × 15 mL) and water (2 × 15 mL). The organic layer was dried over Na2SO4, concentrated and purified by silica gel flash column chromatography using hexanes–ethyl acetate (9:1) to provide the desired compound as yellow syrup (0.300 g, 81%): IR (KBr) 2246, 1456, 1122, 841, 665 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.83 (d, J = 8.1 Hz, 2 H), 7.27 (d, J = 8.1 Hz, 2 H), 2.67 (s, 3 H), 2.64 (m, 2 H), 1.63 (m, 2 H), 1.38 (m, 2 H), 0.94 (t, J = 7.3 Hz, 3 H); ESIMS m/z (rel intensity) 256 (M+, 33), 213 (M+-C3H7, 100).
5.1.12. (Z)-2-(4-Butylphenyl)-N′-hydroxy-4-methylthiazole-5-carboximidamide (15)
A mixture of the thiazole derivative 14 (0.19 g, 0.74 mmol), hydroxylamine hydrochloride (0.07 g, 1 mmol) and K2CO3 (0.102 g, 0.74 mmol) in absolute EtOH (15 mL) was stirred at room temperature for 1 h and then heated at reflux overnight. The EtOH was removed under reduced pressure and the resulting crude residue was purified by flash column chromatography (SiO2, hexanes-EtOAc, 9:1) to afford the product 15 as an off white to light yellow solid (35.5 mg, 17%): mp 135–137 °C. 1H NMR (300 MHz, CDCl3) δ 7.81 (d, J = 8.05 Hz, 2 H), 7.25 (t, J = 5.7 Hz, 2 H), 2.63 (t, J = 7.6 Hz, 5 H), 1.59 (q, J = 7.4 Hz, 2 H), 1.34 (p, J = 11.2 Hz, 2 H), 0.92 (t, J = 7.28 Hz, 3 H); ESIMS m/z (rel intensity) 290 (M+, 100).
5.1.13. 2-(4-Butylphenyl)-4-methyl-5-(1H-tetrazol-5-yl)thiazole (16)
I2 (20 mg) was added to a mixture of nitrile (14, 0.2 g, 0.781 mmol) and NaN3 (0.076 g, 1.17 mmol) and the mixture was stirred at 120 °C for 15 h. After completion of the reaction, EtOAc (15 mL) and 4 M HCl (10 mL) were added and the mixture was stirred vigorously for 10 min. The organic layer was separated and the aqueous layer was extracted with EtOAc (2 × 10 mL). The combined organic layer was washed with brine (4 × 15 mL), dried over Na2SO4, concentrated and purified by silica gel flash column chromatography using hexane-ethyl acetate (5:5) to provide the desired compound as a light yellow solid (0.085 g, 37%): mp 170–171 °C. IR (KBr) 1825, 1415, 1098, 844, 664 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.84 (d, J = 8.1 Hz, 2 H), 7.22 (d, J = 8.1 Hz, 2 H), 2.85 (s, 3 H), 2.64 (t, J = 7.5 Hz, 2 H), 1.63 (m, 2 H), 1.35 (m, 2 H), 0.93 (t, J = 7.3 Hz, 3 H); ESIMS m/z (rel intensity) 300 (MH+, 100); HRESIMS calcd for C15H18N5S m/z 300.1267 (MH+), found 300.1270; HPLC purity 98.25% (1% TFA in MeOH:H2O −90:10).
5.1.14. 1-(4-Methyl-2-(4-aminophenyl)thiazol-5-yl)ethanone (18)
4-Aminothiobenzamide (0.6 g, 3.80 mmol) and α-chloropentanedione (0.611 mg, 4.56 mmol) were added to absolute ethanol (50 mL). The reaction mixture was heated at reflux for 24 h. After removal of solvent under reduced pressure, the residue was purified by silica gel chromatography using hexanes–ethyl acetate (6:4) to provide the desired compound as light brown solid (0.920 g, 97%): mp 204–205 °C. IR (KBr) 3334, 1745, 1637, 1145, 865, 666 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.89 (d, J = 8.5 Hz, 2 H), 7.07 (d, J = 8.5 Hz, 2 H), 2.66 (s, 3 H), 2.53 (s, 3 H).
5.1.15. (E)-2-(1-(4-Methyl-2-(4-aminophenyl)thiazol-5-yl)ethylidene) hydrazinecarboximidamide (19)
The thiazole derivative 18 (0.250 g, 0.954 mmol) was dissolved in absolute ethanol (50 mL), and aminoguanidine hydrochloride (0.125 g, 1.14 mmol) and a catalytic amount of LiCl (15 mg) were added. The reaction mixture was heated at reflux for 24 h. The solvent was evaporated under reduced pressure. The crude product was purified by crystallization from 70% methanol and then recrystallized from methanol to afford the desired compound as a yellow solid (0.175 g, 58%): mp > 280 °C. IR (KBr) 3402, 1705, 1665, 1156, 826, 665 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 11.44 (br s, 1 H), 7.81 (d, J = 8.4 Hz, 2 H), 7.75 (br s, 3 H), 7.00 (d, J = 8.4 Hz, 2 H), 2.58 (s, 3 H), 2.41 (s, 3 H); ESIMS m/z (rel intensity) 289 (MH+, 100); HRESIMS calcd for C13H17N6S m/z 289.1123 (MH), found 289.1120; HPLC purity 96.58% (1% TFA in MeOH:H2O – 90:10).
5.1.16. 1-(2-(4′-Hydroxy-[1,1′-biphenyl]-4-yl)-4-methylthiazol-5-yl)ethanone (21a)
Iodide 3 (0.172 g, 0.5 mmol), the 4-hydroxyphenyl boronic acid (20a, 0.205 g, 1.5 mmol), tripotassium monophosphate (0.424 g, 2 mmol) and (2-biphenyl)dicylohexylphosphine (18 mg) were dissolved in dry toluene (15 mL) and the solution was purged with argon for 20 min. Pd(OAc)2 (25 mg, 0.24 mmol) was added to the reaction mixture and the reaction mixture was heated at 90 °C under argon for 24 h. Ethyl acetate (40 mL) was added to reaction mixture, which was then washed with water (2 × 25 mL). The combined organic layer was dried over Na2SO4, concentrated and purified by flash column chromatography (EtOAc:hexanes 3:7 to 0.9:9.1) to afford the desired compound as yellow solid (0.150 g, 93%): mp 212–214 °C. IR (KBr) 3356, 1745, 1123, 856, 665 cm−1; 1H NMR (300 MHz, CDCl3) δ 7.86 (d, J = 8.3 Hz, 2 H), 7.51 (d, J = 8.3 Hz, 2 H), 7.38 (d, J = 8.5 Hz, 2 H), 6.79 (d, J = 8.6 Hz, 2 H), 2.64 (s, 3 H), 2.45 (s, 3 H); ESIMS m/z (rel intensity) 309 (M+, 100).
5.1.17. 1-(2-(4′-Fluoro-[1,1′-biphenyl]-4-yl)-4-methylthiazol-5-yl)ethanone (21b)
Iodide 3 (0.172 g, 0.5 mmol), 4-fluorophenyl boronic acid (20b, 0.209 1.5 mmol), tripotassium monophosphate (0.424 g, 2 mmol) and (2-biphenyl)dicylohexylphosphine (18 mg) were dissolved in dry toluene (15 mL) and the solution was purged with argon for 20 min. Pd(OAc)2 (25 mg, 0.24 mmol) was added to the reaction mixture and the reaction mixture was heated at 90 °C under argon for 24 h. Ethyl acetate (40 mL) was added to reaction mixture, which was then washed with water (2 × 25 mL). The combined organic layer was dried over Na2SO4, concentrated and purified by flash column chromatography (EtOAc:hexanes 3:7 to 0.9:9.1) to afford the desired compound as an off-white solid (0.140 g, 90%): mp 127–128 °C. IR (KBr) 2956, 1689, 1123, 819, 659 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.05 (d, J = 7.6 Hz, 2 H), 7.64 (m, 4 H), 7.23 (d, J = 7.5 Hz, 2 H), 2.79 (s, 3 H), 2.58 (s, 3 H); ESIMS m/z (rel intensity) 312 (M+, 100); HPLC purity 98.50% (1% TFA in MeOH:H2O – 90:10).
5.1.18. 1-(4-Methyl-2-(4′-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)thiazol-5-yl)ethanone (21c)
Iodide 3 (0.172 g, 0.5 mmol), 4-trifluromethylphenyl boronic acid (20c, 0.228 g, 1.5 mmol), tripotassium monophosphate (0.424 g, 2 mmol) and (2-biphenyl)dicylohexylphosphine (18 mg) were dissolved in dry toluene (15 mL) and the solution was purged with argon for 20 min. Pd(OAc)2 (25 mg, 0.24 mmol) was added to the reaction mixture and the reaction mixture was heated at 90 °C under argon for 24 h. Ethyl acetate (40 mL) was added to reaction mixture, which was then washed with water (2 × 25 mL). The combined organic layer was dried over Na2SO4, concentrated and purified by flash column chromatography (EtOAc:hexanes 3:7 to 0.9:9.1) to afford the desired compound as an off-white solid (0.150 g, 80%): mp 116–117 °C. IR (KBr) 2959, 1938, 1650, 1111, 819, 659 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.09 (d, J = 8.3 Hz, 2 H), 7.72 (m, 6 H), 2.80 (s, 3 H), 2.58 (s, 3 H); ESIMS m/z (rel intensity) 361 (M+, 100); HPLC purity 98.75% (1% TFA in MeOH:H2O – 90:10).
5.1.19. (E)-2-(1-(2-(4′-Hydroxy-[1,1′-biphenyl]-4-yl)-4-methylthiazol-5-yl)ethylidene)hydrazinecarboximidamide (22a)
Compound 21a (0.150 g, 0.485 mmol) was dissolved in absolute ethanol (50 mL) and aminoguanidine hydrochloride (0.064 g, 0.588 mmol) and a catalytic amount of LiCl (10 mg) were added. The reaction mixture was heated at reflux for 24 h. The solvent was evaporated under reduced pressure. The crude product was purified by crystallization from 70% methanol, and then recrystallized from methanol to afford the desired compound as a light yellow solid (0.115 g, 66%): mp 252–254 °C. IR (KBr) 3398, 1695, 1655, 1142, 855, 665 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 9.73 (s, 1 H), 8.02 (d, J = 8.3 Hz, 2 H), 7.74 (d, J = 8.4 Hz, 2 H), 7.59 (d, J = 8.6 Hz, 2 H), 6.88 (d, J = 8.6 Hz, 2 H), 2.70 (s, 3 H), 2.56 (s, 3 H); ESIMS m/z (rel intensity) 366 (MH+, 100); HRESIMS calcd for C19H20N5OS 366.1045 (MH+), found 366.1048; HPLC purity 96.11% (1% TFA in MeOH:H2O – 90:10).
5.1.20. (E)-2-(1-(2-(4′-Fluoro-[1,1′-biphenyl]-4-yl)-4-methylthiazol-5-yl)ethylidene)hydrazinecarboximidamide (22b)
Compound 21b (0.1 g, 0.321 mmol) was dissolved in absolute ethanol (50 mL) and aminoguanidine hydrochloride (0.064 g, 0.588 mmol) and a catalytic amount of LiCl (10 mg) were added. The reaction mixture was heated at reflux for 24 h. The solvent was evaporated under reduced pressure. The crude product was purified by crystallization from 70% methanol, and then recrystallized from methanol to afford the desired compound as a light yellow solid (0.077 g, 65%): mp 273–274 °C. IR (KBr) 3308, 1687, 1645, 1146, 823, 666 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 11.73 (br s, 1 H), 8.00 (d, J = 8.3 Hz, 2 H) 7.81 (m, 8 H), 7.32 (m, 2 H), 2.62 (s, 3 H), 2.44 (s, 3 H); ESIMS m/z (rel intensity) 368 (MH+, 100); HRESIMS calcd for C19H19FN5S m/z 368.1245 (MH+), found 368.1251; HPLC purity 95.78% (1% TFA in MeOH:H2O – 90:10).
5.1.21. (E)-2-(1-(4-Methyl-2-(4′-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)thiazol-5-yl)ethylidene)hydrazinecarboximidamide (22c)
Compound 21c (0.1 g, 0.277 mmol) was dissolved in absolute ethanol (50 mL) and aminoguanidine hydrochloride (0.064 g, 0.588 mmol) and a catalytic amount of LiCl (10 mg) were added. The reaction mixture was heated at reflux for 24 h. The solvent was evaporated under reduced pressure. The crude product was purified by crystallization from 70% methanol, and then recrystallized from methanol to afford the desired compounds as yellow solid (0.088 g, 76%): mp 269–270 °C. IR (KBr) 3583, 3307, 1678, 1145, 821, 665 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 11.78 (br s, 1 H), 8.05 (d, J = 8.4 Hz, 2 H), 7.98 (d, J = 8.2 Hz, 2 H), 7.90 (m, 8 H), 2.62 (s, 3 H), 2.45 (s, 3 H); ESIMS m/z (rel intensity) 418 (MH+, 100); HRESIMS calcd for C20H19F3N5S m/z 418.1423 (MH+), found 418.1420; HPLC purity 96.10% (1% TFA in MeOH:H2O – 90:10).
5.1.22. (E)-2-(1-(2-(4′-Acetyl-[1,1′-biphenyl]-4-yl)-4-methylthiazol-5-yl)ethylidene)hydrazinecarboximidamide (22d)
Compound 11d (0.150 g, 0.472 mmol) was dissolved in absolute ethanol (50 mL) and aminoguanidine hydrochloride (0.064 g, 0.588 mmol) and a catalytic amount of LiCl (10 mg) were added. The reaction mixture was heated at reflux for 24 h. The solvent was evaporated under reduced pressure. The crude product was purified by crystallization from 70% methanol, and then recrystallized from methanol to afford the desired compound as a light yellow solid (0.062, 35%): mp >280 °C. IR (KBr) 3402, 1715, 1655, 1446 1125, 856, 665 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 11.68 (br s, 1 H), 8.10 (d, J = 8.2 Hz, 4 H), 7.91 (m, 6 H), 7.80 (d, J = 8.2 Hz, 2 H), 2.72 (s, 3 H), 2.58 (s, 3 H), 2.40 (s, 3 H); ESIMS m/z (rel intensity) 392 (MH+, 100); HRESIMS calcd for C21H22N5OS m/z 392.1165 (MH+), found 392.1169.
5.1.23. 1-(4-Methyl-2-(4-(naphthalen-1-yl)phenyl)thiazol-5-yl)ethanone (24)
Iodide 3 (0.172 g, 0.5 mmol), 1-naphalene boronic acid (23, 258 g, 1.5 mmol), tripotassium monophosphate (0.424 g, 2 mmol) and (2-biphenyl)dicylohexylphosphine (18 mg) were dissolved in dry toluene (15 mL) and the solution was purged with argon for 20 min. Pd(OAc)2 (25 mg, 0.24 mmol) was added to the reaction mixture and the reaction mixture was heated at 90 °C under argon for 24 h. Ethyl acetate (40 mL) was added to reaction mixture, which was then washed with water (2 × 25 mL). The combined organic layer was dried over Na2SO4, concentrated and purified by flash column chromatography (EtOAc: hexane 3:7 to 0.9:9.1) to afford the desired compound as a light brown solid (0.170 g, 99%): mp 141–142 °C. IR (KBr) 2287, 1670, 1006, 801, 663 cm−1; 1H NMR (300 MHz, CDCl3) δ 8.11 (d, J = 6.5 Hz, 2 H), 7.90 (m, 3 H), 7.61 (m, 7 H), 2.82 (s, 3 H), 2.60 (s, 3 H); ESIMS m/z (rel intensity) 344 (MH+, 100); HRESIMS calcd for C22H18NOS m/z 344.1123 (MH+), found 344.1125.
5.1.24. (E)-2-(1-(4-Methyl-2-(4-(naphthalen-1-yl)phenyl)thiazol-5-yl)ethylidene)hydrazinecarboximidamide (25)
Compound 24 (0.1 g, 0.291 mmol) was dissolved in absolute ethanol (50 mL) and aminoguanidine hydrochloride (0.064 g, 0.588 mmol) and a catalytic amount of LiCl (10 mg) were added. The reaction mixture was heated at reflux for 24 h. The solvent was evaporated under reduced pressure. The crude product was purified by crystallization from 70% methanol, and then recrystallized from methanol to afford the desired compound as a yellow solid (0.057, 49 %): mp 241–242 °C. IR (KBr) 3299, 2300, 1675, 1618, 1142, 800, 664 cm−1; 1H NMR (300 MHz, DMSO-d6) δ 11.63 (br s, 1 H), 8.08 (m, 5 H), 7.65 (d, J = 5.1 Hz, 1 H); 7.61 (m, 9 H), 2.64 (s, 3 H), 2.44 (s, 3 H); ESIMS m/z (rel intensity) 400 (MH+, 100); HRESIMS calcd for C23H22N5S m/z 400.1323 (MH+), found 400.1327; HPLC purity 95.55% (1% TFA in MeOH:H2O – 85:15).
5.2. Biological characterization of thiazole compounds
5.2.1. Bacterial strains and reagents
Clinical isolates of MRSA, VISA, and VRSA were obtained through the Network of Antimicrobial Resistance in Staphylococcus aureus (NARSA) program. In addition, MRSA ATCC 43300 was obtained from the American Type Cultural Collection (Manassas, VA, USA). Vancomycin hydrochloride powder was purchased commercially (Gold Biotechnology Inc., St. Louis, MO, USA) and dissolved in DMSO to prepare a 10 mM stock solution.
5.2.2. Determination of minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC) against MRSA, VISA, and VRSA strains
The MICs of the thiazole compounds and vancomycin against seven clinical isolates of MRSA, three clinical isolates of VISA, and three clinical isolates of VRSA were determined using the broth microdilution method in accordance with the recommendations contained in the CLSI guidelines [40]. Bacteria were prepared in phosphate-buffered saline (PBS) to achieve a McFarland standard of 0.5. The solution was subsequently diluted 1:300 in Mueller-Hinton broth (MHB) to reach a starting inoculum of 1 × 105 colony-forming units (CFU/mL). Bacteria were then transferred to a 96-well microtiter plate. Thiazole compounds and vancomycin were added (in triplicate) to wells in the first row of the microtiter plate and then serially diluted along the vertical axis. The plate was incubated at 37 °C for 18–20 hours before the MIC was determined as the lowest concentration where visible growth of bacteria was not observed.
The MBC was determined by plating 5 μL from wells on the 96-well microtiter plate (where the MIC was determined), where no growth was observed, onto Tryptic soy agar (TSA) plates. The TSA plates were then incubated at 37 °C for 18–20 hours before the MBC was determined. The MBC was classified as the concentration where ≥99% reduction in bacterial cell count was observed.
5.2.3. Time-kill analysis of thiazole compounds 1, 5, and 25 and vancomycin against MRSA
MRSA USA300 cells in late logarithmic growth phase were diluted to ~1 × 108 colony-forming units (CFU/mL) and exposed to concentrations equivalent to 3 × MIC (in triplicate) of thiazole compounds 1, 5, and 25 and vancomycin in MHB. 20 μL samples were collected after 0, 2, 4, 6, 8, 10, 12, and 24 hours of incubation at 37 °C and subsequently serially diluted in PBS. Bacteria were then transferred to TSA plates and incubated at 37 °C for 18–20 hours before viable CFU/mL was determined.
5.2.4. In vitro cytotoxicity analysis
Compounds 1, 5, 22b–d and 25 were assayed at concentrations of 5 μg/mL, 10 μg/mL, 20 μg/mL, and 40 μg/mL against a human embryonic kidney (HEK293) cell line to determine the potential toxic effect to mammalian cells in vitro. Cells were cultured in Dulbeco’s modified Eagle’s medium (Sigma-Aldrich, St. Louis, MO, USA) with 10% fetal bovine serum (USA Scientific, Inc.) at 37 °C with 5% CO2. Controls received DMSO alone at a concentration equal to that in drug-treated cell samples. The cells were incubated with the compounds in a 96-well plate at 37 °C and 5% CO2 for 2 hours prior to addition of the assay reagent MTS 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (Promega, Madison, WI, USA). Absorbance readings (at OD490) were taken using a kinetic microplate reader (Molecular Devices, Sunnyvale, CA, USA). The quantity of viable cells after treatment with each compound was expressed as a percentage of the viability of DMSO-treated control cells.
5.2.5. Microsomal stability analysis
The metabolic stability analysis of analogue 5 was performed as described previously[32]. Compound 5 was incubated in duplicate with human liver microsomes at 37 °C. The reaction contained microsomal protein in 100 mM potassium phosphate, 2 mM NADPH, 3 mM MgCl2, pH 7.4. A control was run for each test agent omitting NADPH to detect NADPH-free degradation. At 0, 10, 20, 40, and 60 minutes, an aliquot was removed from each experimental and control reaction and mixed with an equal volume of ice-cold Stop Solution (methanol containing haloperidol, diclofenac, or other internal standard). Stopped reactions were incubated at least ten minutes at −20 °C, and an additional volume of water was added. The samples were centrifuged to remove precipitated protein, and the supernatants were analyzed by LC/MS/MS to quantitate the remaining parent. Data were converted to % remaining by dividing by the time zero concentration value. Data were fit to a first-order decay model to determine half-life. Intrinsic clearance was calculated from the half-life and the protein concentrations as follows:
5.2.6. Statistical analysis
All statistical analysis was performed using the unpaired t-test (P < 0.05) utilizing GraphPad Prism 6 software. Data for both the time-kill assay and toxicity analysis of the tested compounds are presented as mean ± standard deviation (as depicted by the error bars).
Highlights.
A novel series of phenylthiazole derivatives active against MRSA is presented
Hydrophobic, nonpolar moieties are favored at the thiazole-C2 position
The most potent compounds completely eradicate MRSA growth in vitro within 4 hours
Three derivatives exhibit an improved toxicity profile over the lead compound
Compound 5 displays an improved metabolic stability profile over the lead compound
Acknowledgments
The authors would like to thank the Network of Antimicrobial Resistance in Staphylococcus aureus (NARSA) program supported under NIAID/NIH Contract # HHSN272200700055C for providing MRSA strains used in this study. ASM is supported by the Center of Special Studies, Bibliotheka Alexandria, Egypt.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hadler JL, Petit S, Mandour M, Cartter ML. Trends in invasive infection with methicillin-resistant Staphylococcus aureus, Connecticut, USA, 2001–2010. Emerg Infect Dis. 2012;18:917–924. doi: 10.3201/eid1806.120182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wyllie D, Paul J, Crook D. Waves of trouble: MRSA strain dynamics and assessment of the impact of infection control. J Antimicrob Chemother. 2011;66:2685–2688. doi: 10.1093/jac/dkr392. [DOI] [PubMed] [Google Scholar]
- 3.McDougal LK, Steward CD, Killgore GE, Chaitram JM, McAllister SK, Tenover FC. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: Establishing a national database. Journal of Clinical Microbiology. 2003;41:5113–5120. doi: 10.1128/JCM.41.11.5113-5120.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kassis C, Hachem R, Raad II, Perego CA, Dvorak T, Hulten KG, Frenzel E, Thomas G, Chemaly RF. Outbreak of community-acquired methicillin-resistant Staphylococcus aureus skin infections among health care workers in a cancer center. American Journal of Infection Control. 2011;39:112–117. doi: 10.1016/j.ajic.2010.04.220. [DOI] [PubMed] [Google Scholar]
- 5.Malcolm B. The rise of methicillin-resistant staphylococcus aureus in U.S. correctional populations. J Correct Health Care. 2011;17:254–265. doi: 10.1177/1078345811401363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aiello AE, Lowy FD, Wright LN, Larson EL. Meticillin-resistant Staphylococcus aureus among US prisoners and military personnel: review and recommendations for future studies. Lancet Infectious Diseases. 2006;6:335–341. doi: 10.1016/S1473-3099(06)70491-1. [DOI] [PubMed] [Google Scholar]
- 7.Cohen PR. Cutaneous community-acquired methicillin-resistant Staphylococcus aureus infection in participants of athletic activities. Southern Medical Journal. 2005;98:596–602. doi: 10.1097/01.SMJ.0000163302.72469.28. [DOI] [PubMed] [Google Scholar]
- 8.Kazakova SV, Hageman JC, Matava M, Srinivasan A, Phelan L, Garfinkel B, Boo T, McAllister S, Anderson J, Jensen B, Dodson D, Lonsway D, McDougal LK, Arduino M, Fraser VJ, Killgore G, Tenover FC, Cody S, Jernigan DB. A clone of methicillin-resistant Staphylococcus aureus among professional football players. New England Journal of Medicine. 2005;352:468–475. doi: 10.1056/NEJMoa042859. [DOI] [PubMed] [Google Scholar]
- 9.Gilbert M, MacDonald J, Gregson D, Siushansian J, Zhang K, Elsayed S, Laupland K, Louie T, Hope K, Mulvey M, Gillespie J, Nielsen D, Wheeler V, Louie M, Honish A, Keays G, Conly J. Outbreak in Alberta of community-acquired (USA300) methicillin-resistant Staphylococcus aureus in people with a history of drug use, homelessness or incarceration. Canadian Medical Association Journal. 2006;175:149–154. doi: 10.1503/cmaj.051565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Craven DE, Rixinger AI, Goularte TA, Mccabe WR. Methicillin-Resistant Staphylococcus-Aureus Bacteremia Linked to Intravenous Drug-Abusers Using a Shooting Gallery. American Journal of Medicine. 1986;80:770–776. doi: 10.1016/0002-9343(86)90614-5. [DOI] [PubMed] [Google Scholar]
- 11.Methicillin-resistant Staphylococcus aureus skin infections among tattoo recipients–Ohio, Kentucky, and Vermont, 2004–2005. MMWR Morb Mortal Wkly Rep. 2006;55:677–679. [PubMed] [Google Scholar]
- 12.Regev-Yochay G, Rubinstein E, Barzilai A, Carmeli Y, Kuint J, Etienne J, Blech M, Smollen G, Maayan-Metzger A, Leavitt A, Rahav G, Keller N. Methicillin-resistant Staphylococcus aureus in neonatal intensive care unit. Emerg Infect Dis. 2005;11:453–456. doi: 10.3201/eid1103.040470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herold BC, Immergluck LC, Maranan MC, Lauderdale DS, Gaskin RE, Boyle-Vavra S, Leitch CD, Daum RS. Community-acquired methicillin-resistant Staphylococcus aureus in children with no identified predisposing risk. JAMA. 1998;279:593–598. doi: 10.1001/jama.279.8.593. [DOI] [PubMed] [Google Scholar]
- 14.Karamatsu ML, Thorp AW, Brown L. Changes in community-associated methicillin-resistant Staphylococcus aureus skin and soft tissue infections presenting to the pediatric emergency department: comparing 2003 to 2008. Pediatr Emerg Care. 2012;28:131–135. doi: 10.1097/PEC.0b013e318243fa36. [DOI] [PubMed] [Google Scholar]
- 15.Tristan A, Ferry T, Durand G, Dauwalder O, Bes M, Lina G, Vandenesch F, Etienne J. Virulence determinants in community and hospital meticillin-resistant Staphylococcus aureus. Journal of Hospital Infection. 2007;65:105–109. doi: 10.1016/S0195-6701(07)60025-5. [DOI] [PubMed] [Google Scholar]
- 16.Odell CA. Community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA) skin infections. Curr Opin Pediatr. 2010;22:273–277. doi: 10.1097/MOP.0b013e328339421b. [DOI] [PubMed] [Google Scholar]
- 17.Stryjewski ME, Chambers HF. Skin and soft-tissue infections caused by community-acquired methicillin-resistant Staphylococcus aureus. Clinical Infectious Diseases. 2008;46(Suppl 5):S368–377. doi: 10.1086/533593. [DOI] [PubMed] [Google Scholar]
- 18.Gillet Y, Issartel B, Vanhems P, Fournet JC, Lina G, Bes M, Vandenesch F, Piemont Y, Brousse N, Floret D, Etienne J. Association between Staphylococcus aureus strains carrying gene for Panton-Valentine leukocidin and highly lethal necrotising pneumonia in young immunocompetent patients. Lancet. 2002;359:753–759. doi: 10.1016/S0140-6736(02)07877-7. [DOI] [PubMed] [Google Scholar]
- 19.Bocchini CE, Hulten KG, Mason EO, Gonzalez BE, Hammerman WA, Kaplan SL. Panton-Valentine leukocidin genes are associated with enhanced inflammatory response and local disease in acute hematogenous Staphylococcus aureus osteomyelitis in children. Pediatrics. 2006;117:433–440. doi: 10.1542/peds.2005-0566. [DOI] [PubMed] [Google Scholar]
- 20.David MZ, Daum RS. Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev. 2010;23:616–687. doi: 10.1128/CMR.00081-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.King MD, Waldron DR, Barber DL, Larson MM, Saunders GK, Troy GC, Zimmerman-Pope N, Ward DL. Effect of nephrotomy on renal function and morphology in normal cats. Vet Surg. 2006;35:749–758. doi: 10.1111/j.1532-950X.2006.00219.x. [DOI] [PubMed] [Google Scholar]
- 22.Lee BY, Singh A, David MZ, Bartsch SM, Slayton RB, Huang SS, Zimmer SM, Potter MA, Macal CM, Lauderdale DS, Miller LG, Daum RS. The economic burden of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA) Clin Microbiol Infect. 2013;19:528–536. doi: 10.1111/j.1469-0691.2012.03914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chambers HF. Community-associated MRSA–resistance and virulence converge. N Engl J Med. 2005;352:1485–1487. doi: 10.1056/NEJMe058023. [DOI] [PubMed] [Google Scholar]
- 24.Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB, Talan DA. Methicillin-resistant S-aureus infections among patients in the emergency department. New England Journal of Medicine. 2006;355:666–674. doi: 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- 25.Frazee BW, Lynn J, Charlebois ED, Lambert L, Lowery D, Perdreau-Remington F. High prevalence of methicillin-resistant Staphylococcus aureus in emergency department skin and soft tissue infections. Ann Emerg Med. 2005;45:311–320. doi: 10.1016/j.annemergmed.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 26.Fridkin SK, Hageman JC, Morrison M, Sanza LT, Como-Sabetti K, Jernigan JA, Harriman K, Harrison LH, Lynfield R, Farley MM. Methicillin-resistant Staphylococcus aureus disease in three communities. N Engl J Med. 2005;352:1436–1444. doi: 10.1056/NEJMoa043252. [DOI] [PubMed] [Google Scholar]
- 27.Han LL, McDougal LK, Gorwitz RJ, Mayer KH, Patel JB, Sennott JM, Fontana JL. High frequencies of clindamycin and tetracycline resistance in methicillin-resistant Staphylococcus aureus pulsed-field type USA300 isolates collected at a Boston ambulatory health center. Journal of Clinical Microbiology. 2007;45:1350–1352. doi: 10.1128/JCM.02274-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel JB, Gorwitz RJ, Jernigan JA. Mupirocin Resistance. Clinical Infectious Diseases. 2009;49:935–941. doi: 10.1086/605495. [DOI] [PubMed] [Google Scholar]
- 29.Locke JB, Morales G, Hilgers M, G CK, Rahawi S, Jose Picazo J, Shaw KJ, Stein JL. Elevated linezolid resistance in clinical cfr-positive Staphylococcus aureus isolates is associated with co-occurring mutations in ribosomal protein L3. Antimicrobial agents and chemotherapy. 2010;54:5352–5355. doi: 10.1128/AAC.00714-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson P, Andrews JA, Charlesworth R, Walesby R, Singer M, Farrell DJ, Robbins M. Linezolid resistance in clinical isolates of Staphylococcus aureus. The Journal of antimicrobial chemotherapy. 2003;51:186–188. doi: 10.1093/jac/dkg104. [DOI] [PubMed] [Google Scholar]
- 31.Hiramatsu K. Vancomycin-resistant Staphylococcus aureus: a new model of antibiotic resistance. Lancet Infectious Diseases. 2001;1:147–155. doi: 10.1016/S1473-3099(01)00091-3. [DOI] [PubMed] [Google Scholar]
- 32.Mohammad H, Mayhoub AS, Ghafoor A, Soofi M, Alajlouni RA, Cushman M, Seleem MN. Discovery and Characterization of Potent Thiazoles versus Methicillin- and Vancomycin-Resistant Staphylococcus aureus. Journal of medicinal chemistry. 2014;57:1609–1615. doi: 10.1021/jm401905m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayhoub AS, Khaliq M, Botting C, Li Z, Kuhn RJ, Cushman M. An investigation of phenylthiazole antiflaviviral agents. Bioorg Med Chem. 2011;19:3845–3854. doi: 10.1016/j.bmc.2011.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayhoub AS, Khaliq M, Kuhn RJ, Cushman M. Design, synthesis, and biological evaluation of thiazoles targeting flavivirus envelope proteins. Journal of medicinal chemistry. 2011;54:1704–1714. doi: 10.1021/jm1013538. [DOI] [PubMed] [Google Scholar]
- 35.King MD, Humphrey BJ, Wang YF, Kourbatova EV, Ray SM, Blumberg HM. Emergence of community-acquired methicillin-resistant Staphylococchus aureus USA 300 clone as the predominant cause of skin and soft-tissue infections. Ann Intern Med. 2006;144:309–317. doi: 10.7326/0003-4819-144-5-200603070-00005. [DOI] [PubMed] [Google Scholar]
- 36.Alder J, Eisenstein B. The Advantage of Bactericidal Drugs in the Treatment of Infection. Curr Infect Dis Rep. 2004;6:251–253. doi: 10.1007/s11908-004-0042-1. [DOI] [PubMed] [Google Scholar]
- 37.Deresinski S. Vancomycin in combination with other antibiotics for the treatment of serious methicillin-resistant Staphylococcus aureus infections. Clinical Infectious Diseases. 2009;49:1072–1079. doi: 10.1086/605572. [DOI] [PubMed] [Google Scholar]
- 38.Finberg RW, Moellering RC, Tally FP, Craig WA, Pankey GA, Dellinger EP, West MA, Joshi M, Linden PK, Rolston KV, Rotschafer JC, Rybak MJ. The importance of bactericidal drugs: future directions in infectious disease. Clinical Infectious Diseases. 2004;39:1314–1320. doi: 10.1086/425009. [DOI] [PubMed] [Google Scholar]
- 39.Rydzewski RM. Real World Drug Discovery: A Chemist’s Guide to Biotech and Pharmaceutical Research. Elsevier; Slovenia: 2008. [Google Scholar]
- 40.Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically—Seventh Edition: Approved Standard M7-A7. 7. Wayne, PA: 2011. [Google Scholar]