

Abstract
Control of the protein synthetic machinery is deregulated in many cancers, including melanoma, in order to increase protein production. Tumor suppressors and oncogenes play key roles in protein synthesis from the transcription of rRNA and ribosome biogenesis to mRNA translation initiation and protein synthesis. Major signaling pathways are altered in melanoma to modulate the protein synthetic machinery thereby promoting tumor development. However, despite the importance of this process in melanoma development, involvement of the protein synthetic machinery in this cancer type is an underdeveloped area of study. Here, we review the coupling of melanoma development to deregulation of the protein synthetic machinery. We examine existing knowledge regarding RNA Polymerase I inhibition and mRNA translation focusing on their inhibition for therapeutic applications in melanoma. Furthermore, the contribution of amino acid biosynthesis and involvement of ribosomal proteins are also reviewed as future therapeutic strategies to target deregulated protein production in melanoma.
Keywords: melanoma, protein synthesis, ribosomal proteins, amino acids, mRNA translation
1. INTRODUCTION
Significant progress is being made to develop effective melanoma therapeutics. Most of the current progress has resulted from a better understanding of the cellular pathways altered in melanoma. In particular, regulation of the MAPK pathway has been very actively studied, which has resulted in the development of drugs targeting mutant V600EBRAF, such as vemurafenib (Flaherty et al., 2010, Larkin et al., 2014) and dabrafenib (Falchook et al., 2012, Hauschild et al., 2012), and MEK, such as trametinib (Flaherty et al., 2012, Salama and Kim, 2013). Furthermore, anti-CTLA4 and anti-PD-1 immunotherapies have improved treatment options available to melanoma patients (Hodi et al., 2010, Topalian et al., 2014). While these significantly improve short-term treatment options, they are restricted to those patients who will respond to these drugs and limited by the development of recurrent resistant disease (Johnson et al., 2014, Sullivan and Flaherty, 2013, Wagle et al., 2014). Thus, there is still a need for a better understanding of the disease in order to develop therapies that would have long-term efficacy without the development of resistance.
Knowing what cellular alterations are most important for melanoma transformation is essential to identify new targets for treating melanoma. The vast majority of melanomas have constitutive activation of the MAPK pathway by BRAF or NRAS mutations. This leads to increased cell growth, proliferation, and survival. The PI3K/AKT signaling pathway also plays an important role in melanoma, being constitutively activated in many melanomas, and is important for regulating cell proliferation and loss of regulation of apoptotic signaling (Smalley and Herlyn, 2005). Upstream of AKT, PTEN loss is observed in over 25% of melanomas and its functional deregulation is important for tumor development (Stahl et al., 2003). Increasingly, it is being realized how alterations to oncogenic and tumor suppressive factors are tied to the protein synthetic machinery. This review highlights how pathways necessary for melanoma growth and proliferation are linked to the protein synthetic machinery and how targeting protein synthesis provides promising therapeutic treatment options for melanoma.
2. PROTEIN SYNTHETIC MACHINERY FACTORS
The entire process of protein synthesis requires substantial energy for the transcription of ribosomal RNA (rRNA) by RNA polymerase I or III (Pol I or Pol III), transcription of ribosomal protein mRNA by RNA polymerase II for ribosome biogenesis, transfer RNA (tRNA) transcription by Pol III, processing of pre-rRNA by small nucleolar RNA (snoRNA) guided exo- and endonuclease activity, and mRNA translation initiation, elongation, and termination. Transcription of rRNA, necessary components of ribosomes, is carried out by Pol I and its associated factors. In general, rRNA transcription requires the formation of a pre-initiation complex consisting of Pol I, upstream binding factor (UBF), and the SL1 complex, which contains the TATA-binding protein (TBP) and three Pol I-specific TBP-associated factors (TAFs): TAFI48, TAFI63, and TAFI95/110 (White, 2005) (Fig. 1A). In addition, transcription intermediary factor IA (TIF-IA) is necessary for transcriptional activity as well as TIFIC and transcription factor IIH (TFIIH) (White, 2005) (Fig. 1A). These factors and others not detailed for the brevity of this review are important for effective rRNA synthesis and thus are potential targets for regulation of Pol I activity. Furthermore, following transcription, RNA may interact with many different RNA-binding proteins involved in steps such as maturation, transport, stability, regulation, and translation of RNA, the perturbation of which can lead to disease (Glisovic et al., 2008). For a more detailed review of RNA polymerase I and its regulation as well as RNA-binding proteins, see references (Grummt, 2003) and (Gerstberger et al., 2014) , respectively.
Figure 1. Factors associated with Pol I transcription and mRNA translation.
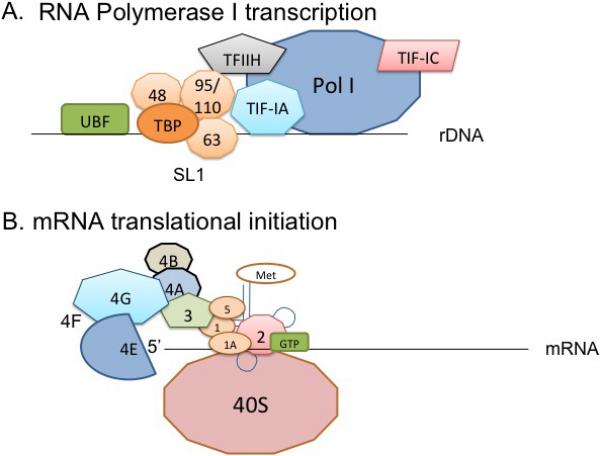
(A) Transcription of rDNA by RNA polymerase I (Pol I). Ribosomal RNA (rRNA) is transcribed from rDNA by Pol I and its associated factors. A pre-initiation complex of Pol I, upstream binding factor (UBF), and the SL1 complex forms around rDNA. The SL1 complex consists of the TATA-binding protein (TBP) and three Pol I specific TBP-associated factors (TAFs): TAFI48, TAFI63, and TAFI95/110. Transcription intermediary factor IA (TIF-IA), TIFIC, and transcription factor IIH (TFIIH) also assist in rDNA transcription. (B) Cap-dependent mRNA translation initiation. A ternary complex of initiator met-tRNA, eIF2, and GTP becomes part of the larger 43S pre-initiation complex by associating with the 40S small ribosomal subunit, eIF1, eIF1A, and eIF3. eIF3 binds to eIF4G in the eIF4F complex to recognize mRNA. The eIF4F complex comprises of eIF4E, eIF4G, and eIF4A. eIF4E recognizes the 5’ 7-methyl-guanosine cap, eIF4A performs RNA helicase activity on mRNA, and eIF4G binds to the poly(A)-binding protein (PABP) (not shown) and the 43S pre-initiation complex via eIF3. eIF4F is stabilized by eIF4B. eIF5 is recruited to trigger joining of the 60S ribosomal subunit once the mRNA start codon is recognized.
Translation of mRNA into protein requires the interaction of the ribosome with initiation, elongation, and release factors. Cap-dependent translation initiation requires the recognition of the 5’ 7-methyl-guanosine cap by the eIF4F complex. As shown in Figure 1B, this complex consists of eIF4E, which recognizes the 5’ cap, eIF4A, a RNA helicase, and eIF4G, which binds eIF3 of the pre-initiation complex, and poly(A)-binding protein (PABP) to circularize mRNA. eIF4B stabilizes the eIF4F complex by binding eIF4A and PABP (Blagden and Willis, 2011). The 43S pre-initiation complex consists of a ternary complex of initiator met-tRNA, eIF2, and GTP, the 40S small ribosomal subunit, eIF1, eIF1A, and eIF3 (Fig. 1B). The pre-initiation complex can then recognize mRNA via the binding of eIF3 to eIF4G. Scanning of mRNA then proceeds with the assistance of eIF1 and eIF1a until the AUG start codon is recognized where eIF5 is then recruited, triggering the hydrolysis of GTPs on eIF2 and eIF5B to join the 60S ribosomal subunit. eIF5A is actually an elongation factor that promotes the translation of polyproline motifs (Gutierrez et al., 2013). Thus, translation initiation proceeds with efficient functioning of the eIF4F complex and the 43S pre-initiation complex. For a detailed review of mRNA translation initiation, including cap-dependent, and IRES-mediated translation, see reference (Sonenberg and Hinnebusch, 2009) and for information on elongation and termination during protein synthesis, see reference (Dever and Green, 2012).
Many processes regulate mRNA translation initiation. eIF2 of the ternary complex consists of α, β, and γ subunits. The GTP bound to eIF2 is hydrolyzed during translation initiation. Under normal unstressed conditions GDP is exchanged for GTP via the guanine nucleotide exchange factor (GEF) eIF2B. However, various stresses can activate kinases that phosphorylate eIF2α causing eIF2 to bind to eIF2B and prevent dissociation, which prevents GEF activity and inhibits translation initiation (Gebauer and Hentze, 2004) (Fig. 2A). The eIF4F complex is also subjected to regulation via post-translational modifications. eIF4E-binding proteins (4EBPs) bind to eIF4E to inhibit mRNA translation (Pause et al., 1994). Kinases such as mTOR phosphorylate 4EBPs preventing interaction with eIF4E, which allows eIF4E to bind to eIF4G thus permitting translation initiation (Gebauer and Hentze, 2004, Brunn et al., 1997) (Fig. 2B). In addition to 4EBP1, mTOR also regulates p70S6K. Activation of p70S6K by mTOR phosphorylation leads to the degradation of PDCD4, a protein that interferes with eIF4A/eIF4G interaction (Dorrello et al., 2006) and the activation of eIF4B (Blagden and Willis, 2011) (Fig. 2B). mTOR inactivation prevents target phosphorylation leading to decreased translation initiation (Ma and Blenis, 2009). MNK1/2, activated via the MAPK pathway phosphorylates and activates eIF4E and eIF4B (Blagden and Willis, 2011, Topisirovic et al., 2004) (Fig. 2B). Thus, eIF2 of the ternary complex and the eIF4F complex are key regulatory points. Regulation of translation initiation by cellular processes or for therapeutic efforts centers on these factors. For a more comprehensive review of mRNA translation initiation and its regulatory components, see reference (Gebauer and Hentze, 2004).
Figure 2. mRNA translational regulation.
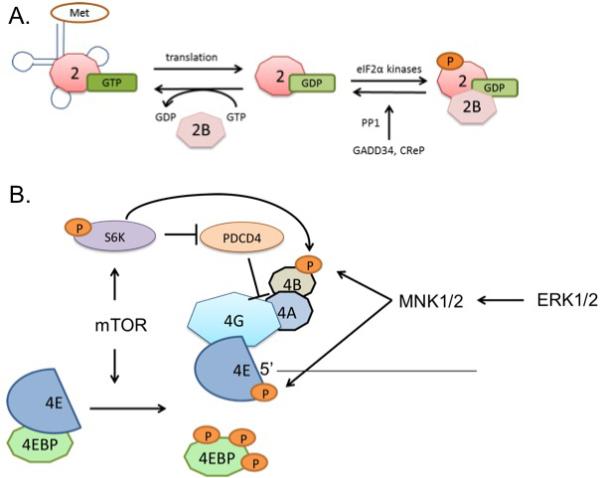
(A) GTP from the ternary complex of initiator met-tRNA, eIF2, and GTP is hydrolyzed into GDP during translation initiation. GEF activity of eIF2B typically exchanges GDP with GTP to continue translation. However, the α subunit of eIF2 can be phosphorylated by kinases activated by various stresses, such as amino acid starvation, ER stress, heme depletion, and viral infection. The phosphorylation causes a tighter binding of eIF2 to eIF2B, inhibiting GEF activity and thus translation initiation. GADD34 and CReP counter the kinase activity by bringing phosphorylated eIF2α to PP1 for dephosphorylation to enhance translation. (B) The eIF4F complex is regulated by MAPK and PI3K/AKT/mTOR pathways. MAPK activity through MNK1/2 phosphorylates eIF4E and eIF4B, enhancing translation. mTOR phosphorylates 4EBP and p70S6K (S6K) to promote translation.
3. MELANOMA SIGNALING PATHWAYS COUPLED TO THE PROTEIN SYNTHETIC MACHINERY
The entire process to produce protein requires coordination of many different components from the transcription of rRNA genes via RNA polymerase I for ribosome biogenesis to mRNA translation and its associated initiation and elongation factors. Several signaling pathways commonly perturbed in melanoma have been linked to protein production suggesting an essential requirement in melanoma development (Fig. 3A).
Figure 3. Targeting translation in melanoma.
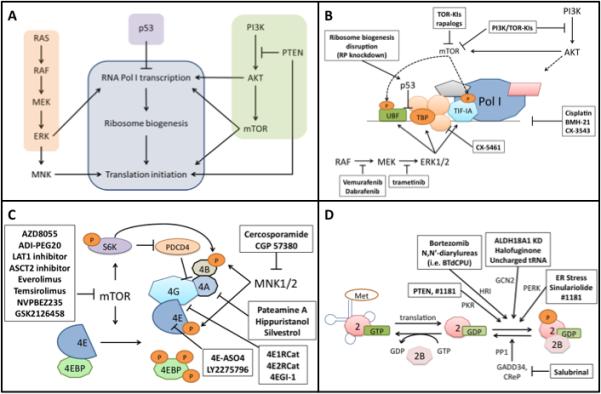
(A) Diagram of how the MAPK pathway, PI3K pathway, and p53 modulate protein synthetic machinery. (B) Pol I transcription can be regulated by many different mechanisms. This figure depicts the regulatory methods most relevant to melanoma. UBF, TBP, and TIF-IA can all be regulated by the MAPK pathway via ERK1/2. The PI3K/AKT/mTOR pathway regulates transcription through the phosphorylation of UBF and TIF-IA. AKT also regulates Pol I transcription through mTORC1-independent mechanisms. p53 is able to impair Pol I transcription by inhibiting SL1 binding to UBF. Nonspecific inhibitors of Pol I transcription include cisplatin, BMH-21, and CX-3543 which disrupt transcription through activity on genomic DNA. Drugs targeting signaling pathways can also exert effect through disruption of Pol I transcription. BRAF and MEK inhibitors impair ERK1/2 ability to activate UBF, TBP, and TIF-IA. TOR kinase inhibitors (TOR-KIs) and rapalogs impair the ability of mTOR to activate UBF and TIF-IA. PI3K/TOR-KIs also prevent feedback activation of PI3K by mTOR inhibition and prevent mTORC1-independent Pol I activation by AKT. CX-5461 acts independently of mTOR and MAPK pathways by preventing SL1 complex association. (C) eIF4F complex inhibition. mTOR regulates eIF4F activity through 4EBP1 and p70S6K. Rapalogs, TOR-KIs, PI3K/TOR-KIs, and amino acid starvation can all inhibit mTOR activity, thus reducing mRNA translation. Regulation of translation through the MAPK pathway can be inhibited at the point of MNK1/2 by cercosporamide. Multiple drugs targeting the eIF4F complex are currently under investigation as potential cancer therapeutics. These include eIF4A inhibitors, inhibitors of eIF4E/eIF4G interaction, eIF4E cap-binding activity, and antisense oligonucleotide inhibitors of eIF4E. Furthermore, the biogenesis of 60S and 40S subunits necessary for mRNA translation can be impaired via ribosomal protein (RP) knockdown, or various Pol I disruptors. (D) Ternary complex inhibition. Activation of any of the eIF2α kinases causes phosphorylation of eIF2α, leading to sequestration of eIF2B, preventing GEF activity needed to continue translation. Many stresses can lead to kinase activation. Amino acid starvation or the mimicking of uncharged tRNA can activate GCN2. ER stress is a common activator of PERK. N,N’-diarylureas are activators of HRI, and PTEN activity independent of PI3K/AKT can lead to PKR activation. In addition, inhibitors of eIF2α phosphatase activity such as salubrinal can block eIF2B function.
3.1 MAP kinase pathway in Pol I transcription
The MAPK pathway is an important regulator of rRNA transcription and thus, is coupled to protein synthesis. UBF, TIF-IA, and TBP are essential components of the pre-initiation complex necessary for transcription initiation of rDNA by Pol I (White, 2005). UBF and TIF-IA stimulation of Pol I transcription is ERK1/2-dependently activated (Stefanovsky et al., 2001, Stefanovsky and Moss, 2008, Zhao et al., 2003, Hannan et al., 2013) and TBP promoter activity is MEK-dependently induced (Johnson et al., 2000, Johnson et al., 2003) (Fig. 3B). Increased TBP expression could then increase SL1 abundance as the SL1 complex contains TBP, and thus could promote Pol I transcription (Hannan et al., 2013). In addition, ERK1/2 can phosphorylate and stabilize c-MYC (Sears et al., 1999), which in turn can increase Pol I transcription (Hannan et al., 2013). Over-activation of the MAPK pathway is commonly observed in melanomas, however it is also prevalent in other cancers such as colon cancer and serous ovarian cancer (Dhillon et al., 2007). Thus, MAPK pathway activation in these cancers can enhance rRNA transcription, which is necessary for ribosome production and consequently protein synthesis.
3.2 PI3 kinase pathway in Pol I transcription
The PI3K/AKT/mTOR pathway also ties into multiple Pol I transcription processes. PTEN loss is observed in many cancers including breast, lung, glioblastoma, and melanoma (Leslie and Downes, 2004). Decreasing PTEN expression, as found in many melanomas, enhances RNA Pol I-dependent transcription through the PI3K/AKT/mTOR pathway (Zhang et al., 2005) (Fig. 3A). In glioblastoma cell lines, increasing PTEN expression disrupted SL1 binding to rDNA promoters (Zhang et al., 2005). AKT can activate Pol I transcription at multiple stages, including initiation, elongation, and cotranscriptional processing and it cooperates with c-MYC to promote Pol I transcription (Chan et al., 2011). Part of its effect on rRNA synthesis is independent of mTORC1 (Chan et al., 2011). mTOR predominantly acts through its targets 4EBP1 and p70S6K to affect mRNA translation initiation (Gebauer and Hentze, 2004) but it can also affect Pol I transcription. Through p70S6K, it regulates phosphorylation of UBF (Hannan et al., 2003), and phosphorylation and activation of TIF-IA (Mayer et al., 2004), enhancing Pol I transcription (Fig. 3B). The PI3K pathway is associated with protein synthetic machinery and is frequently activated in many cancers such as breast, endometrium, colon (Yuan and Cantley, 2008), and melanoma thus, it is an important regulator of this process in cancer.
3.3 Regulation of translation initiation factors
mRNA translation initiation factors are linked to melanoma growth and proliferation. For example, eIF3c downregulation causes cell cycle arrest, reduced cell proliferation, and cell death of mouse melanoma cells (Emmanuel et al., 2013). mRNA levels of a negative regulator of translation, eIF3f, are decreased in many melanomas and its overexpression induces apoptosis and inhibits cell proliferation, likely by enhancing ribosome degradation (Shi et al., 2006). Furthermore, EIF3F loss of heterozygosity (LOH) was found in 75% to 92% of melanomas resulting in decreased EIF3F gene copy number compared to normal tissues (Doldan et al., 2008). Collectively, this implies that eIF3, the initiation factor required for interaction between the eIF4F complex and the 43S pre-initiation complex, is altered in melanoma to promote mRNA translation. Although IRES trans-acting factors (ITAFs) can also play an important role in cancer (Faye and Holcik, 2014), this review focuses on cap-dependent translation and the 43S pre-initiation complex.
mRNA recruitment to the 40S ribosome subunit is the rate limiting step of mRNA translation and eIF4F is an essential component of this activity, suggesting that it plays a key role in translational control (Duncan et al., 1987). EIF4A1 mRNA, but not mRNA from other eIF4 members, is overexpressed in human melanoma cells (Eberle et al., 1997). eIF4E and eIF4G are upregulated in many cancers (Silvera et al., 2010). Over 2 decades ago, it was observed that eIF4E is present in limiting amounts in cells and overexpression of eIF4E caused the transformation of fibroblasts (Lazaris-Karatzas et al., 1990). Furthermore, its overexpression can selectively alter the translation of cancer-related mRNA transcripts (Larsson et al., 2007). Recently, the eIF4F complex has been identified as an important downstream target of both the PI3K/AKT/mTOR and MAPK pathways.
The PI3K/AKT/mTOR and MAPK pathways can regulate eIF4F at various levels. eIF4E is necessary for oncogenic transformation by PI3K and AKT (Ruggero et al., 2004). In multiple melanoma cell lines, the MAPK pathway downregulated microRNA miR-768-3p leading to eIF4E upregulation and increased protein synthesis (Jiang et al., 2014). Recently, persistent formation of the eIF4F complex was associated with resistance to anti-BRAF, anti-MEK, and combination therapies (Boussemart et al., 2014). Inhibiting the eIF4F complex synergized with mutant BRAF inhibition (Boussemart et al., 2014). Furthermore, expressing V600EBRAF in melanocytes increased eIF4E phosphorylation and protein synthesis (Croft et al., 2014). Thus, the effect of V600EBRAF can act through mRNA translation initiation. The MAPK pathway is also linked to translation initiation through ERK1/2 phosphorylation of MNK1/2. These serine/threonine kinases phosphorylate eIF4E at Ser209, which promotes cellular transformation (Topisirovic et al., 2004). The MAPK and PI3K/AKT/mTOR pathways regulate the initiation factor eIF4B. MNK1 from the MAPK pathway and p70S6K1 from the PI3K/AKT/mTOR pathway both phosphorylate eIF4B, causing increased association with eIF3 and increased eIF4A activity (Blagden and Willis, 2011) (Fig. 3C). These results provide evidence that signaling pathways essential to melanoma growth and survival can act through translational initiation processes to exert their effect.
3.4 Elongation factors
Factors that facilitate mRNA translation elongation have also been studied in melanoma development. Inhibition of hypusination, the conversion of a lysine residue to hypusine, of the elongation factor eIF5A has been found to be impaired in murine melanoma growth (Jasiulionis et al., 2007). eIF5A2 is a target of PI3K/AKT that promotes melanoma cell invasion and MMP-2 activity and its expression is correlated with melanoma thickness while inversely correlated to survival (Khosravi et al., 2014). In addition, the eukaryotic elongation factor eEF2 was identified as a potential serum biomarker in melanoma (Suzuki et al., 2010). The MAPK and PI3K/AKT pathways cooperate to activate eEF2 by inhibiting eEF2K phosphorylation of eEF2, which promotes mRNA translation elongation (Wang et al., 2014b). Involvement of elongation factors in melanoma development provides another avenue for therapeutic targeting, but this research area needs more development.
4. TARGETING PROTEIN SYNTHESIS FOR MELANOMA THERAPY
Many protein synthesis inhibitors (PSIs) tested clinically in the past targeted the elongation step, impairing peptidyl transferase function, translocation, or aminoacyl-tRNA binding (Vazquez, 1979, Pestka, 1977). These inhibitors demonstrated nonspecific toxicity by blocking global protein synthesis in normal cells (Malina et al., 2012). This led many researchers to disregard the mRNA translation apparatus as a viable treatment option. For example, in a phase II study of homoharringtonine, a PSI that blocks protein synthesis by blocking the A-site in the peptidyl-transferase center (Gurel et al., 2009), patients with various solid tumors including melanoma, sarcoma, breast, head & neck, and colorectal carcinomas neither completely nor partially responded to the agent (Ajani et al., 1986). However, novel routes to inhibit translation more effectively with less toxicity are being developed, leading to a new wave of therapies, which are discussed below.
4.1 RNA polymerase I in cancer
RNA polymerase I activity, and to a larger extent ribosome biogenesis, may be linked to cancer development. Increased numbers of nucleoli were identified as a marker of aggressive tumors (Derenzini et al., 2009). Nucleolar changes were first observed in 1896 when cancer cells were observed to have large nucleoli (Pianese, 1896). It was later determined that the nucleolus of all malignant cells are larger in proportion to nuclear size (MacCarty, 1936). Perry et al. reported that many melanomas contained macronucleoli (Perry et al., 1986). Furthermore, malignant melanoma tumor thickness correlated with increased numbers of nucleolar organizer regions (NORs), which are regions containing chromosomal DNA and protein involved in ribosomal synthesis (Barzilai et al., 1998). Since nucleolar size is closely related to its function in ribosome biogenesis (Derenzini et al., 1998, Hernandez-Verdun, 2006), enlarged nucleoli in melanoma suggests a potential link between increased ribosome biogenesis, and to a larger extent mRNA translation, and melanoma. Pol I transcription and ribosome biogenesis are aberrantly regulated in cancer (Hannan et al., 2013, Bywater et al., 2013) and the MAPK and PI3K/AKT/mTOR pathways are linked to these processes (Fig. 3A). Whether increased ribosome biogenesis constitutes a dependency in melanoma, and a weakness to be therapeutically exploited, needs further investigation.
4.2 RNA polymerase I inhibition
Many approved anticancer drugs affect rRNA biogenesis by indirectly impairing RNA polymerase I function (Hein et al., 2013). In one study of 36 drugs in clinical use, 21 were found to have rRNA synthesis inhibitory activities, either by inhibiting rRNA transcription or rRNA processing, including alkylating agents such as cisplatin and oxaliplatin and DNA intercalating agents such as doxorubicin, mitoxantrone, and actinomycin D (Burger et al., 2010). Inhibition of ribosome biogenesis by chemotherapeutic drugs may contribute to the efficacy of particular therapeutic regimens (van Sluis and McStay, 2014). However, since melanoma should be dependent on ribosome biogenesis, it would be expected that a drug affecting rRNA synthesis like cisplatin should be effective. Cisplatin inhibits synthesis of rRNA by forming DNA adducts (Jamieson and Lippard, 1999, Rosenberg, 1985) causing cytotoxicity due to poor DNA repair (Chaney et al., 2005) and displacing UBF as well as RNA Pol I to the periphery of the nucleolus (Jordan and Carmo-Fonseca, 1998). Unfortunately, even though it is effective in several cancers, and used as a first-line therapy in cancers such as testicular, ovarian, cervical, head & neck, and small-cell lung cancer (Basu and Krishnamurthy, 2010), it is not so in melanoma (Lee et al., 1995). This is likely because two genes needed to remove cisplatin-induced DNA damage, ERCC1 and XPF, are induced by cisplatin in melanoma (Li and Melton, 2012) and not necessarily because melanoma is not dependent on ribosome biogenesis. To combat cisplatin resistance, trials have tested its effectiveness in combination with other more effective melanoma drugs. For instance, cisplatin combined with IFN-alpha and IL2 can produce response rates of 50-60% and complete response in 20% of melanoma patients (Anderson et al., 1995). However, not all combinations improve responses as cisplatin and tamoxifen was only effective in 26% of cases (Middleton et al., 2000). Nevertheless, clinical trials are ongoing to test the effectiveness of cisplatin in combination with ipilimumab, ADI-PEG 20, AM0010, and LY2801653 in melanoma patients (NCT01409174, NCT01665183, NCT02009449, and NCT01285037). Thus, combinations of existing chemotherapeutic drugs may be effective for treating melanoma due in part to an effect on rRNA biogenesis.
Most anticancer drugs affecting rRNA synthesis are likely too toxic to be ideal clinical treatment options. Even if cisplatin in combination with therapies such as IFN-alpha and IL2 improves patient outcome, it is not an ideal agent to target the protein synthetic mechanisms due to toxicity associated with general DNA damage. BMH-21 is another drug that non-specifically targets DNA by intercalating into DNA to inhibit rDNA transcription (Colis et al., 2014, van Sluis and McStay, 2014). In melanoma, a mouse xenograft model showed that BMH-21 significantly decreased tumor growth (Peltonen et al., 2014), however, it causes widespread DNA damage making it toxic. Other chemotherapeutic drugs have encountered similar issues. Even though oxaliplatin (Raymond et al., 1998), doxorubicin (Sivam et al., 1995), mitoxantrone (Fujimoto and Ogawa, 1982), and actinomycin D (Kroon et al., 2014) can have therapeutic efficacy in preclinical studies in melanoma, this has not had high therapeutic response rates in clinical trials (Lutzky, 2006, Ellerhorst et al., 1999, Arseneau et al., 1986). Other treatment options to disrupt rDNA transcription by directly or indirectly inhibiting Pol I transcription might be better approaches (Fig. 3B). CX-5461, a drug developed by Cylene Pharmaceuticals, disrupts pre-initiation complex formation on the rDNA promoter, thus inhibiting Pol I transcription, and was demonstrated to decrease melanoma, pancreatic, and lymphoma growth in vivo (Drygin et al., 2011, Bywater et al., 2012, van Sluis and McStay, 2014). CX-5461 activates p53, induces autophagy and senescence, and is tolerated by normal cells (Drygin et al., 2011, Bywater et al., 2012). Although not yet tested clinically in melanoma patients, it would be interesting to see if this drug could overcome problems encountered with nonspecific inhibitors of Pol I transcription. Furthermore, another small molecule, CX-3543, can disrupt nucleolar G-quadruplex complexes, inhibiting Pol I transcription, which can cause apoptosis in cancer cells and was demonstrated to decrease breast as well as pancreatic tumor growth (Drygin et al., 2009). CX-3543 has completed phase II trials to assess its efficacy in neuroendocrine and carcinoid tumors (NCT00780663). While existing drugs targeting Pol I transcription have not been effective for treating melanoma, the next generation of specific inhibitors of Pol I transcription may be better options.
4.3 mRNA translation inhibition
Translation of mRNA to protein involves initiation, elongation, termination, and ribosome recycling. Initiation is the most common point to target translational regulation, where recruitment of mRNA to the 40S ribosomal subunit is the rate-limiting step (Ingolia et al., 2009, Sonenberg and Hinnebusch, 2009). Increased translational activity during melanoma development involves initiation factors essential for melanoma growth. Many initiation factors are being investigated as potential therapeutic targets. Best known are eIF4F, necessary for cap-dependent translation initiation, and eIF2 of the ternary complex (Figs. 3C and 3D).
4.3.1 Targeting the eIF4F complex
Multiple drugs targeting the eIF4F initiation complex to inhibit mRNA translation initiation have been developed and are at various stages of preclinical testing (Malina et al., 2011, Malina et al., 2012). Inhibitors of the eIF4A subunit include pateamine A, hippuristanol, and silvestrol (Fig. 3C). Pateamine A sequesters eIF4A onto nonspecific RNA preventing its normal functioning (Bordeleau et al., 2005, Low et al., 2005). Des-amino pateamine A (DMDA-PatA), an analog of pateamine A, led to significant tumor regression of melanoma, colon, non-small cell lung cancer, and leukemia mouse models (Kuznetsov et al., 2009). While DMDA-PatA has mRNA translational inhibitory activity, reported effects on DNA synthesis inhibition (Kuznetsov et al., 2009) and RNA transcription (Romo et al., 2004) could also contribute to efficacy. Like pateamine A, silvestrol sequesters free eIF4A nonspecifically on RNA (Bordeleau et al., 2008). Silvestrol induced G2 cell cycle arrest and decreased AKT and mTOR phosphorylation in melanoma cells (Chen and Swanson). Hippuristanol is an allosteric inhibitor of eIF4A that prevents eIF4A from binding to RNA (Bordeleau et al., 2006), but has largely been unstudied in melanoma. Other inhibitors of the eIF4F complex target eIF4E and its interaction with eIF4G or the mRNA 5’ cap (Fig. 3C). 4EGI-1 is a cap-dependent mRNA translation inhibitor that disrupts the eIF4E/eIF4G association and enhances 4EBP1 association with eIF4E (Moerke et al., 2007). 4EGI-1 inhibited xenograft melanoma and breast cancer tumor growth with negligible toxicity (Chen et al., 2012). 4E1RCat is a molecule identified in a screen to bind to eIF4G to hinder eIF4F assembly (Cencic et al., 2011b) and 4E2RCat was identified to prevent eIF4E interaction with eIF4G (Cencic et al., 2011a). Both inhibit cap-dependent translation and reverse drug resistance in a lymphoma model, but neither molecule has been studied in melanoma. Since the MAPK and PI3K pathways activate translational initiation through eIF4F, these drugs could be used to disrupt the downstream pathway signaling in melanoma cells.
4.3.1.1 Targeting the mRNA 5’ cap
Since eIF4E binds the 5’ cap of mRNA, cap analogs are eIF4F therapeutic inhibitors able to impair mRNA translation initiation. 5’ cap mimics such as ribavirin have been effective in preclinical studies (Fouchier et al., 1996); however, these may not be ideal translation inhibitors since these caps are needed for multiple RNA processes and could lead to toxic side effects (Malina et al., 2011). Processes requiring 5’ cap involvement include nuclear export, splicing, mRNA 3’-end formation, nonsense-mediated decay, and miRNA processing (Malina et al., 2012). Furthermore, cap analogs cannot freely cross phospholipid bilayers (Malina et al., 2012), complicating the clinical use of these drugs. Thus, therapies targeting the eIF4F complex rather than the cap-binding site might be a better therapeutic approach.
4.3.1.2 Targeting MNK1/2
Directly downstream of p38 and ERK in the MAPK pathway are the MNK1/2 proteins (Buxade et al., 2008, Waskiewicz et al., 1997), which are serine/threonine kinases that phosphorylate eIF4E on Ser209 (Pyronnet et al., 1999) and eIF4B (Blagden and Willis, 2011), which could be targeted to inhibit mRNA translation. eIF4E phosphorylation at Ser209 leads to cellular transformation (Topisirovic et al., 2004, Wendel et al., 2007) mediated through the MAPK pathway. As such, these kinases are potentially important therapeutic targets (Hou et al., 2012), but few specific inhibitors of MNK1/2 have been developed (Hou et al., 2012). Of those being studied, cercosporamide decreased B16 melanoma pulmonary metastases growth and colon carcinoma xenograft tumor growth by blocking MNK phosphorylation of eIF4E (Konicek et al., 2011). Another agent, CGP 57380, decreased growth of multiple V600EBRAF melanoma cell lines (Boussemart et al., 2014) (Fig. 3C). Interestingly, vemurafenib-resistant melanoma cell lines were more resistant to MNK inhibitor CGP 57380 (Boussemart et al., 2014), suggesting that vemurafenib activity could need MNK-dependent inhibition of translation initiation. Thus, MNK1/2, downstream of the MAPK pathway, is a viable target for those melanomas in which the MAPK pathway is constitutively activated.
4.3.1.3 Targeting eIF4E using antisense oligonucleotides
eIF4E antisense oligonucleotides (ASOs) could also be used as cancer therapeutics to inhibit mRNA translation (Fig. 3C). 4E-ASO4 in breast and prostate cancer mouse xenograft models decreased cancer cell growth with negligible systemic toxicity (Graff et al., 2007). A phase I clinical trial using LY2275796, an ASO targeting eIF4E, showed no effect on tumor development, including in melanoma, mesothelioma, head & neck carcinoma, colon, and lung cancer patients (Hong et al., 2011). The authors suggested that future trials should examine LY2275796 in combination with other agents (Hong et al., 2011). Lack of efficacy may have occurred for a number of reasons; eIF4E knockdown clinically may not be as robust as in preclinical models, knockdown could result in a cytostatic rather than cytotoxic response (Hong et al., 2011), IRES-mediated translation might compensate for impaired eIF4F complex, or the complex nature of translation initiation regulation could result in bypass of this impairment. Thus, ASOs might not be the right approach to target eIF4E in melanoma or in other cancer types.
4.3.1.4 Disruption of eIF4F signaling by targeting mTOR
An alternative to targeting the eIF4F complex itself for mRNA translation inhibition is to inhibit or activate regulators of the complex, such as the mTOR pathway. mTOR is a well-known regulator of growth factor signaling, stress responses, and amino acid sensing. mTOR is one component of the rapamycin and nutrient-sensitive mTORC1 complex, as well as the growth factor-sensitive and nutrient-insensitive mTORC2 complex (Guertin and Sabatini, 2007). mTORC1 targets regulators of mRNA translation, and thus, will be the focus of this section. Numerous drugs are being developed or are approved by the FDA to target mTOR. mTOR inhibitors include rapamycin analogs (rapalogs), TOR-kinase inhibitors (TOR-KI), and dual PI3K/TOR-KI (Fig. 3C). The rapalog temsirolimus is FDA-approved for advanced renal cell carcinoma (RCC) treatment (Kwitkowski et al., 2010) and completed a Phase III trial in mantle cell lymphoma (Hess et al., 2009), while the rapalog everolimus is FDA-approved for kidney cancer, breast cancer, subependymal giant cell astrocytomas, and pancreatic neuroendocrine tumors (Lebwohl et al., 2013). Rapamycin and rapalogs such as temsirolimus, and everolimus have been evaluated in melanoma. Everolimus (Rao et al., 2006) but not temsirolimus (Margolin et al., 2005) has shown some positive results in melanoma as a single agent. Phase II combination studies of everolimus with sorafenib or with temozolomide, paclitaxel, and carboplatin failed to show significant improvement (Margolin et al., 2012, Dronca et al., 2014, Hauke et al., 2013). These therapies have limitations because rapamycin and rapalogs only partially inhibit mTOR kinase activity (Malina et al., 2011). Rapamycin can suppress p70S6K phosphorylation but cannot suppress 4EBP1 phosphorylation entirely (Choo et al., 2008). In addition, a p70S6K-IRS1 negative feedback loop exists whereby inhibitors of mTOR will activate the PI3K pathway (Harrington et al., 2005). Even more worrisome for melanoma is the existence of an mTORC1-MAPK feedback loop in which everolimus activated the MAPK pathway in human tumors that included melanoma (Carracedo et al., 2008). Thus, rapamycin and rapalogs have a significant disadvantage that not only prevents them from completely inhibiting translation initiation but also activates an important melanoma cell survival pathway. An effective agent would need to avoid these drawbacks.
TOR-KIs were designed to overcome the drawbacks of rapalogs by blocking both the mTOR complexes to more completely inhibit 4EBP1 and p70S6K phosphorylation, thereby inhibiting mRNA translation. TOR-KIs show better efficacy compared to rapalogs (Malina et al., 2012, Livingstone et al., 2010). Of these, AZD8055, which is being investigated in a Phase I trial for hepatocellular carcinoma (NCT00999882), synergizes with vemurafenib to inhibit vemurafenib resistant melanoma cell lines (Shi et al., 2011). While these drugs completely inhibit mTOR kinase activity, it does not address the negative feedback loop leading to activation of PI3K following mTOR inhibition. However, a class of compounds called dual specificity PI3K/TOR-KI that includes NVPBEZ235, GSK2126458, and PI-103 can block both mTOR and PI3K, leading to blocking of PI3K reactivation following p70S6K inhibition (Malina et al., 2012) (Figs. 3B and 3C). Unfortunately, these inhibitors have off-target effects (Malina et al., 2012), which is problematic. Nevertheless, some PI3K/TOR-KIs have demonstrated promising results in melanoma. NVPBEZ235, which is in clinical trials for kidney cancer (NCT01453595), prostate cancer (NCT01634061), and pancreatic neuroendocrine tumors (NCT01628913), synergized with vemurafenib to inhibit growth of vemurafenib-resistant melanoma cells (Shi et al., 2011). NVPBEZ235 combined with AZD6244, a MEK inhibitor, showed synergism in melanoma cell lines (Aziz et al., 2010) and caused tumor regression and improve survival in a genetically engineered mouse model (Roberts et al., 2012). A phase Ib clinical trial investigating NVPBEZ235 with the MEK inhibitor MEK162 was recently completed (NCT01337765) to determine the maximal tolerable dose. GSK2126458, which is in a clinical trial for advanced solid tumors and lymphoma (NCT00972686), was able to inhibit trametinib-resistant tumor growth and, in combination with dabrafenib and trametinib, significantly improve efficacy (Villanueva et al., 2013). Further studies combining GSK2126458 with the MEK inhibitor GSK1120212 induced apoptosis and enhanced growth inhibition of BRAF-resistant melanoma cells (Khalili et al., 2012, Greger et al., 2012), but a Phase I trial using this combination was terminated due to lack of efficacy (NCT01248858). In addition, PI-103 treatment in xenograft models of glioblastoma, prostate, breast, colon, and ovarian demonstrated therapeutic activity (Raynaud et al., 2007), however, although PI-103 decreased cultured melanoma cell viability, treatment in a mouse model led to immunosuppression, promoted tumor growth, and inhibited apoptosis (Lopez-Fauqued et al., 2010). Whether these negative results are due to limitations of PI3K/TOR-KI specificity or due to an inability to effectively inhibit translation remains to be determined. Incidentally, resistance to PI3K/TOR-KIs can occur due to increased eIF4E and c-MYC levels (Ilic et al., 2011) and combination therapies may be needed to resolve resistance development. In summary, the mTOR pathway is a promising target to disrupt translation initiation, but each drug targeting this pathway has particular limitations. Overcoming these issues would be required to effectively inhibit translation initiation to treat melanoma by targeting the mTOR pathway.
4.3.2 TARGETING eIF2α
Another target to inhibit translation initiation is eIF2. This initiation factor is essential for start codon recognition and the formation of the 43S ribosomal complex (Gebauer and Hentze, 2004). The guanine nucleotide exchange factor complex eIF2B normally exchanges GDP for GTP on eIF2 so that translation initiation can continue (Webb and Proud, 1997). However, when eIF2α is phosphorylated at Ser51 (Ser52 in humans) it irreversibly binds to eIF2B inhibiting GEF activity of eIF2B, thus inhibiting mRNA translation (Krishnamoorthy et al., 2001, Kimball, 1999) (Fig. 2A). It must be noted that eIF2α phosphorylation can also increase translation of some inefficiently translated mRNAs to induce pro-survival pathways (Koromilas, 2014). PKR-related endoplasmic reticulum kinase (PERK), RNA-dependent protein kinase (PKR), general control non-derepressible 2 kinase (GCN2), and heme-regulated inhibitor kinase (HRI) are four kinases that phosphorylate eIF2α (Fig. 3D). Phosphorylation of eIF2α is an important regulatory step in translation initiation that warrants more study in melanoma. Increased eIF2α expression occurs in benign and malignant melanomas (Rosenwald et al., 2003) and blocking eIF2α phosphorylation can cause malignant transformation of fibroblasts (Donze et al., 1995) suggesting a tumor suppressor role. Modulation of eIF2α phosphorylation is important for regulating translation initiation making it a potentially important therapeutic target.
4.3.2.1 PKR-Related endoplasmic reticulum kinase (PERK)
PERK is one eIF2α kinase that may have a role in melanoma growth. PERK is activated by endoplasmic reticulum (ER) stress, which can be induced by proteasome inhibitors. Vemurafenib (V600EBRAF inhibitor) treatment can activate PERK by causing GRP78 to dissociate from PERK, initiating an ER stress response leading to eIF2α phosphorylation (Ma et al., 2014). Thus, V600EBRAF inhibition exerts some of its therapeutic effect through disruption of mRNA translation. Activating eIF2α kinases may provide a unique mechanism to inhibit translation initiation. Sinulariolide and #1181 are two small molecules that activate PERK (Fig. 3D). Sinulariolide is a molecule derived from coral that activates the PERK/ eIF2α-mediated apoptotic pathway, suppressed hepatocellular carcinoma growth, and suppressed proliferation and migration of melanoma cells (Chen et al., 2013, Li et al., 2013). #1181 is a molecule shown to inhibit melanoma and breast xenograft tumor growth without apparent toxicity causing phosphorylation of eIF2α likely through PERK or PKR (Chen et al., 2012). Although these results are encouraging, PERK can also facilitate cell survival or tumor formation (Koromilas, 2014), as was seen in fibrosarcoma and mouse embryonic fibroblast cell lines (Kazemi et al., 2007) as well as in a mouse minute mutant model (Hart et al., 2012). Thus, activating eIF2α phosphorylation by targeting PERK has potential to impair melanoma growth, however the pro-survival effects of PERK must be taken into consideration.
4.3.2.2 RNA-dependent protein kinase (PKR)
PKR is another eIF2α kinase able to modulate eIF2α phosphorylation, and thus, mRNA translation initiation. PKR is an interferon (IFN) induced kinase activated by double-stranded RNA binding typically in response to a viral infection (Mounir et al., 2009). Expression of a nonfunctional dominant-negative mutant PKR in fibroblasts caused malignant transformation, suggesting PKR may function as a tumor suppressor (Koromilas et al., 1992). Inability to phosphorylate eIF2α could lead to translation initiation without any brake mediated by PKR activity. Furthermore, inactivation of PTEN, which occurs frequently in melanoma, caused a reduction of eIF2α phosphorylation in human melanoma cells (Mounir et al., 2009). Restoration of functional PTEN in PTEN-null glioblastoma or prostate cancer cells led to eIF2α phosphorylation through PKR function, linking the PTEN tumor suppressor to protein synthesis inhibition independent of its role modulating PI3K activity (Mounir et al., 2009) (Fig. 3D). Consequently, therapeutic activation of PKR function might be beneficial to PTEN mutant melanomas, but since many signaling pathways activated in melanoma are coupled to translation, it might be beneficial for treating all melanomas.
4.3.2.3 Heme-regulated inhibitor kinase (HRI)
HRI is a kinase that is activated upon heme deficiency, causing eIF2α phosphorylation and inhibition of protein synthesis (Chen, 2007). N,N’-diarylureas, particularly BTdCPU, activate HRI, causing eIF2α phosphorylation and inhibition of ternary complex accumulation leading to decreased proliferation of human melanoma, lung, breast, and prostate cancer cells and growth of xenograft breast tumors (Chen et al., 2011) (Fig. 3D). Bortezomib, a 26S proteasome inhibitor, caused rapid phosphorylation of eIF2α in B16F10 melanoma cells via HRI (Yerlikaya and DoKudur, 2010). Bortezomib is FDA approved for treatment of multiple myeloma and mantle cell lymphoma, and is a potential treatment option for metastatic melanoma (Shahshahan et al., 2011). It is being tested in combination with other agents such as ABT-737, sorafenib, and dacarbazine for better efficacy killing melanoma cells (Reuland et al., 2012, Kumar et al., 2013, Poklepovic et al., 2013). Thus, the therapeutic activation of HRI, like PERK and PKR activation, is a promising option to therapeutically inhibit melanoma development.
4.3.2.4 General control non-derepressible 2 kinase (GCN2)
GCN2 is also able to phosphorylate eIF2α, thus impairing mRNA translation initiation. GCN2 senses nutrient starvation and is critical for metabolic homeostasis of cancer cells (Ye et al., 2010) (Fig. 3D). Since tumor cells are typically under nutrient deprivation, GCN2 may be an appropriate therapeutic target since tumor cells would be more sensitive to nutrient disruption than normal cells, leading to triggering of GCN2 activation (Ye et al., 2010). Uncharged tRNAs bind GCN2 causing autophosphorylation at T899 (Dong et al., 2000), which activates GCN2 leading to eIF2α phosphorylation (Berlanga et al., 1999) (Fig. 3D). Halofuginone, an inhibitor of prolyl-tRNA synthetase activity can activate GCN2 function by duping the cell into thinking it is nutrient starved (Keller et al., 2012). It was shown to inhibit melanoma bone metastases as well as reduce brain metastases and is reported to decrease bladder carcinoma, glioma, prostate cancer, hepatocellular carcinoma, Wilms tumor, brain, and pancreatic cancer growth (Juarez et al., 2012). Disruption of proline biosynthesis activated GCN2, causing eIF2α phosphorylation and decreased protein synthesis (Kardos et al., 2015). Therapeutic strategies to activate GCN2 include amino acid starvation, use of inhibitors of tRNA synthetases, and mimics of uncharged tRNAs (Fig. 3D). Since cancer cells have higher demands for nutrients compared to normal cells, this eIF2α kinase may be one approach to activate phosphorylation for therapeutic application while causing minimal nonspecific toxicities.
4.3.2.5 eIF2α phosphatases
eIF2α phosphorylation can also be modulated, to disrupt mRNA translation, by phosphatases that reverse the actions of the four eIF2α kinases. Salubrinal is a molecule that inactivates the dephosphorylation of eIF2α by promoting the disassembly of protein phosphatase 1 (PP1) with constitutive repressor of eIF2α phosphorylation (CReP) or GADD34 (Chen and Perrine, 2013) (Fig. 3D). This maintains eIF2α phosphorylation leading to reduced protein production. Salubrinal is currently in a Phase II clinical trial to test its effectiveness in combination with carfilzomib for multiple myeloma treatment (NCT01775553). Therapeutic treatments maintaining phosphorylation by preventing phosphatase action are attractive therapeutic approaches for melanoma treatment but more work is needed in this area to realize its full potential.
5. AMINO ACID SYNTHESIS INHIBITION
Another way to perturb mTOR and/or GCN2 signaling for melanoma therapy to disrupt protein synthesis is by disrupting amino acid biosynthesis. Disruption of amino acid biosynthesis can potentially affect cancer cells, including melanoma, disproportionally compared to normal cells because of the intense nutrient stress experienced by cancer cells. A slight disruption of nutrient availability in cancer cells that normal cells could tolerate could trigger mTOR inactivation and GCN2 activation in melanoma, leading to inhibition of translation (Fig. 4). Thus, amino acid synthesis and bioavailability are possible therapeutic strategies for melanoma treatment.
Figure 4. Amino acid synthesis inhibition.
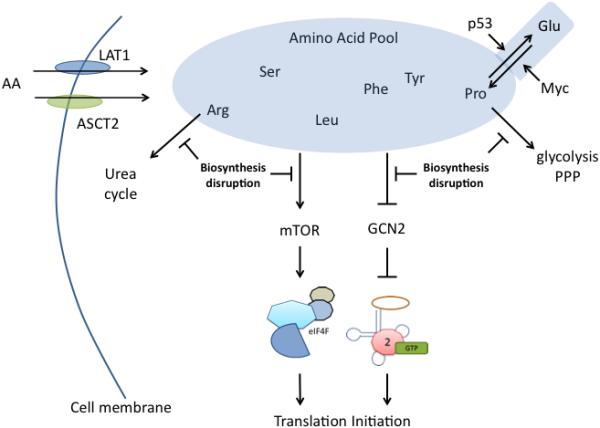
Diagram illustrating pathways targeting amino acid biosynthesis or transport for therapeutic applications. LAT1 and ASCT2 are two amino acid transporters important in melanoma. Depletion of intracellular amino acid pools, whether by disrupting transport or biosynthesis, could alter mTOR and GCN2 signaling, inhibiting mRNA translation initiation. Many amino acids regulate cellular processes other than protein synthesis. Two examples are arginine and proline. Arginine is used in the urea cycle and proline can contribute to glycolysis and the pentose phosphate pathway. Lastly, tumor suppressors and oncogenes regulate amino acid biosynthetic pathways as evidenced by the role of p53 and MYC in proline biosynthesis.
5.1 Arginine
As a therapeutic target for disrupting protein production, arginine has been at the center of amino acid research in melanoma. Clinical trials have focused on sensitivity of melanomas to arginine deprivation. Arginine is a substrate for nitric oxide synthase (NOS), and a component of the urea cycle. A majority of melanomas do not express argininosuccinate synthetase (ASS) so they rely on exogenous arginine (Savaraj et al., 2010), and as such, are susceptible to arginine depletion. For instance, arginine depletion was shown to induce rapid death of B16-F10 murine melanoma because cells failed to exit the cell cycle into a quiescent state as occurred with nontransformed cells (Scott et al., 2000). Arginine degradation depletes the substrate pool for proinflammatory nitric oxide and activates GCN2 in T-cells (Rodriguez et al., 2007). Human melanoma cells were highly sensitive to arginine deiminase (ADI) and exhibited reduced ASS gene expression (Sugimura et al., 1992). Arginine degradation using ADI leads to growth inhibition and cell death in melanoma but not normal cells (Ensor et al., 2002). Pegylated ADI (ADI-PEG20) has antitumor activity in melanoma (Ensor et al., 2002). ADI-PEG20 has been evaluated by Polaris Inc., formerly Phoenix Pharmacologics, and was granted orphan drug status by the FDA (Shen and Shen, 2006). In addition to melanoma, hepatocellular carcinoma, mesothelioma, prostate, and renal cancer are arginine auxotrophic, due to variable loss of ASS (Delage et al., 2010) and are also being tested in clinical trials (NCT01287585, NCT02029690, NCT01497925). A Phase I/II clinical trial showed 6 of 24 metastatic melanoma patients responded (5 partial, 1 complete response) and the drug was well tolerated (Ascierto et al., 2005). Latest Phase I/II clinical trial results of 31 advanced melanoma patients showed 9 patients achieved stable disease with 2 durable responses (Ott et al., 2013). Furthermore, a clinical trial is ongoing combining ADI-PEG20 with cisplatin (NCT01665183). Preclinical studies of ADI-PEG20 and cisplatin have shown additive and synergistic drug interactions with no increased toxicity (Phillips et al., 2013). Even though arginine depletion can activate GCN2 and inhibit mTOR signaling, it also activates MEK and ERK (Savaraj et al., 2010), which could lead to negative side-effects. It remains to be determined whether the effect of arginine deprivation is mediated by mTOR and GCN2 but limiting arginine availability can impair melanoma cell survival.
5.2 Proline
Proline biosynthesis has attracted recent attention as a therapeutic target for melanoma to disrupt protein production. Compared to melanocytes, proline biosynthesis is elevated in melanoma cells (De Ingeniis et al., 2012). Proline is synthesized from glutamate (Fig. 4) and requires the concerted effort of pyrroline-5-carboxylate synthase (P5CS) and pyrroline-5-carboxylate reductase (PYCR) converting L-glutamate to L-P5C to L-proline with the reverse reactions carried out by pyrroline-5-carboxylate dehydrogenase (P5CDH) and proline dehydrogenase (PRODH), respectively. Recent work suggests that enzymes in the proline biosynthesis pathway may contribute to cancer development. PYCR protein expression is upregulated in melanoma (De Ingeniis et al., 2012) and is MYC-dependently increased (Liu et al., 2012). PRODH, catalyzing the reverse reaction from proline to P5C is a tumor suppressor (Liu et al., 2008, Liu et al., 2006), a p53 target gene (Polyak et al., 1997), and is inhibited by MYC (Liu et al., 2012). ALDH18A1, the gene encoding P5CS, which synthesizes P5C from glutamate, is MYC-dependently induced in T-cells (Wang et al., 2011), Burkitt lymphoma, and prostate cancer (Liu et al., 2012). ALDH4A1, the gene encoding P5CDH that catalyzes the reverse reaction of P5C to glutamate, was identified as a p53-inducible gene (Yoon et al., 2004). Collectively, these results suggest proline synthesis is vital for oncogenesis, where a tumor suppressor maintains proline catabolism and proline synthesis is driven by an oncogene (Fig. 4). Recently, ALDH18A1 was targeted with siRNA to disrupt proline biosynthesis, which decreased melanoma tumor growth by activating GCN2 and decreasing protein synthesis (Kardos et al., 2015). Likewise, overexpression of PRODH to promote proline catabolism led to apoptotic cell death in colon cancer cells (Donald et al., 2001). These results suggest that reduction of proline, whether via inhibiting its biosynthesis or inducing its degradation has a detrimental effect on cancer cells, possibly by disrupting translation initiation. In addition to its effects on eIF2α, targeting proline biosynthesis has the potential to disrupt cancer cell metabolism. Proline biosynthesis has been linked to glycolysis and the pentose phosphate pathway (PPP) through what is termed the proline cycle whereby PYCR oxidation of NAD(P)H to NAD(P)+ during the conversion of P5C to proline can regenerate NAD(P)+ for glycolysis or the PPP (Phang et al., 2012, Hagedorn and Phang, 1986). Thus, targeting proline biosynthesis might inhibit multiple processes highly relied upon by cancer cells for growth, including protein production, but would have a negligible effect on normal cells.
5.3 Other amino acids
In addition to arginine and proline, the biosynthesis of many other amino acids could be useful therapeutic targets for disrupting protein production in melanoma cells. Since mTOR and GCN2 sense nutrient availability, it is likely that the presence or absence of any amino acid might trigger activation or repression of these pathways (Fig. 4). In fact, GCN2 should be activated by any uncharged tRNA (Wek et al., 1995, Zhang et al., 2002). While it is unclear whether depletion of each amino acid in cancer cells can activate GCN2, several amino acids have been studied in relation to an effect on melanoma development. Serine, leucine, tyrosine, and phenylalanine all have an important role in melanoma. The serine pathway is promoted by phosphoglycerate dehydrogenase (PHGDH) expression, an enzyme that catalyzes the first step of serine biosynthesis and is located in a region of chromosome 1p commonly amplified in melanoma as well as other cancers such as breast cancer (Possemato et al., 2011). PHGDH is amplified in melanoma and its knockdown can inhibit melanoma cell survival (Mullarky et al., 2011). It is interesting that in the same screen that identified PHGDH as a metabolic gene necessary for in vivo tumorigenesis, PYCR1 from the proline biosynthesis pathway was also found (Possemato et al., 2011), highlighting the importance of these pathways in cancer. Leucine is known to be important for mTOR signaling (Lynch, 2001). Leucine deprivation in melanoma cells causes apoptosis leading to decreased cell and tumor growth (Sheen et al., 2011). Intracellular levels of tyrosine and phenylalanine were higher in a melanoma cell line than normal cells suggesting the former are dependent on these amino acids (Fu and Meadows, 2007). Tyr and Phe restriction inhibits invasion, proliferation, apoptosis, and enhances chemotherapeutic efficacy in melanoma (Fu and Meadows, 2007). Whether mRNA translation initiation inhibition from GCN2 activation is an integral component of these observed effects from serine, leucine, tyrosine, or phenylalanine remains to be determined but each case highlights the potential therapeutic value for targeting amino acid biosynthesis for the treatment of melanoma and likely for other cancer types.
5.4 Amino acid transport
Disruption of amino acid biosynthesis is only one of multiple routes to reduce intracellular amino acid levels to inhibit protein production. Amino acid availability can be disrupted by deprivation or by siRNA targeting of biosynthetic enzymes to inactivate mTOR and activate GCN2, however an alternative approach to deplete intracellular amino acids is to target amino acid transport. Expression of the L-type amino acid transporter 1 (LAT1) and the neutral amino acid transporter ASC amino-acid transporter 2 (ASCT2) are increased in human melanomas and are also found at high levels in lung, prostate, and breast cancer (Wang et al., 2014a) (Fig. 4). ASCT2 inhibition led to decreased melanoma growth through inhibition of mTORC1 signaling dependent on leucine and glutamine (Wang et al., 2014a). In another study using a canine model, highest LAT1 mRNA levels in malignant melanoma tissues correlated with increased distant metastases (Fukumoto et al., 2013). Two LAT1 inhibitors, BCH and LPM, enhanced inhibitory growth activities of various anti-cancer drugs in this model (Fukumoto et al., 2013), highlighting its therapeutic potential. Thus, amino acid synthesis and transport offers unique approaches to disrupt melanoma development. As sensors of amino acid levels, mTOR and GCN2 could both be perturbed through disruption of amino acid synthesis or transport, impairing ternary complex and eIF4F functioning necessary for translation initiation (Fig. 4). Furthermore, targeting amino acid biosynthesis can disrupt specific metabolic processes dependent upon the particular pathway being targeted. Most normal cells do not have the demanding requirement for a constant supply of amino acids and high metabolic activity occurring in cancer cells. Thus, targeting amino acid biosynthesis may be one approach to overcome issues involved with targeting a single pathway while avoiding complications of high toxicity.
6. TARGETING RIBOSOMAL PROTEINS
Proteins constituting the ribosome itself could also be therapeutic targets in cancer to disrupt the production of proteins (Fig. 5A). The eukaryotic ribosome contains approximately 79 ribosomal proteins (RPs) and 4 ribosomal RNAs (rRNAs) in its 80S structure comprised of the 60S and 40S subunits (Ben-Shem et al., 2011) (Fig. 5A). Ribosome biogenesis is an intricate process that requires considerable energy (Warner et al., 2001) involving the transcription of rRNA, tRNA, and mRNA, RNA processing, nuclear assembly, and export of subunits. As such, regulation of the ribosome is essential to avoid wasting of cellular energy. Ribosome production, and in effect protein synthesis, can be directly regulated by oncogenes and tumor suppressors (Ruggero and Pandolfi, 2003). Since the rate of protein synthesis is proportional to the rate of cell proliferation and growth (Baxter and Stanners, 1978, Rudra and Warner, 2004, Thomas, 2000), cancer cells must rely heavily on protein synthesis. This reliance on protein synthesis can be attributed not only to disruption of translational control at the level of mRNA translation but also to accelerated ribosome biogenesis (Schneider and Sonenberg, 2007) (Fig. 5A). Recently, a link between ribosome biogenesis and the tumor suppressor p53 was discovered, termed the RP-MDM2-p53 pathway, which acts as a stress response to ribosome biogenesis disruption (Deisenroth and Zhang, 2010, Warner and McIntosh, 2009) (Fig. 5B). Enablers of this pathway will bind to MDM2 inhibiting p53 ubiquitination thus leading to cell cycle arrest, apoptosis, or senescence (Vogelstein et al., 2000). These effects are dependent on p53 because deletion of p53 prevents activation (McGowan et al., 2008, Terzian and Box, 2013). p53 ties into Pol I transcription through inhibition of SL1 binding to UBF (Ruggero and Pandolfi, 2003). Thus, disruption of ribosome biogenesis can impair rDNA transcription, ribosome generation, and mRNA translation while activating the tumor suppressor p53 to cause cell cycle arrest and apoptosis (Fig. 5B). This suggests that therapies to disrupt ribosome biogenesis could inhibit cancer growth.
Figure 5. Targeting ribosome biogenesis.

(A) Under normal conditions cells produce 60S and 40S ribosomal subunits through ribosome biogenesis in order to translate mRNA into protein. In addition, p53 is targeted for degradation by the E3 ubiquitin ligase MDM2. (B) Disruption of ribosome biogenesis, whether from ribosomal protein (RP) knockdown or from RNA Polymerase I (Pol I) disruption, causes cells to slow or stop the production of fully functional ribosomal subunits, which impairs mRNA translation. Furthermore, disruption of ribosomal protein production causes enabler RPs to bind to MDM2 inhibiting p53 ubiquitination. This stabilizes p53 leading to cell cycle arrest, apoptosis, or senescence.
Many RPs have been identified as enablers, the most prominent being RPL5 and RPL11 (Dai and Lu, 2004, Zhang et al., 2003) (Fig. 5B). However, others include RPL23 (Dai et al., 2004), RPL26 (Zhang et al., 2010), RPS7 (Zhu et al., 2009), RPL37 (Daftuar et al., 2013), RPS3 (Yadavilli et al., 2009), RPS15 (Daftuar et al., 2013), RPS20 (Daftuar et al., 2013), RPS27 (Xiong et al., 2011), and RPS27A (Sun et al., 2011). Large subunit ribosomal proteins have been grouped into separate categories based on effect on melanoma cell viability (Kardos et al., 2014). It was shown that disruption of RPs that are not enablers of the RP-MDM2-p53 pathway have the potential to not only impair protein synthesis, but also stabilize p53 (Fig. 5B). In fact, siRNA knockdown of RPs, such as RPL13, that are not enablers of this pathway resulted in p53 stabilization and inhibition of protein synthesis (Kardos et al., 2014). This led to a greater effect on melanoma cell viability than normal cells and decreased melanoma tumor growth (Kardos et al., 2014). Others have shown in prostate cancer, glioma, and gastric cancer that knockdown of RPL19, RPS9, and RPL6, respectively, can disrupt cancer growth (Bee et al., 2011, Lindstrom and Nister, 2010, Wu et al., 2011). Thus, targeting RPs may be a viable option for melanoma treatment, and likely other cancer types (Fig. 5B).
Disruption of ribosome biogenesis by targeting certain RPs appears to exert its effect through inhibition of global protein synthesis, specific protein synthesis, and extra-ribosomal functions (Terzian and Box, 2013). Disruption of ribosome biogenesis can impair the formation of fully functional ribosomes, thus global protein synthesis will be impaired due to the depletion of functioning ribosomes. Depending on the particular RP targeted, the synthesis of specific mRNAs may be differentially affected. This is an area that is currently poorly understood. However, recent studies on RPL38 have shown that this RP is important for the translation of a specific subset of Hox mRNA (Kondrashov et al., 2011) suggesting that certain RPs assist in the translation of particular mRNAs. Furthermore, some RPs have been implicated in extra-ribosomal functions such as mRNA translational regulation (off the ribosome) (Mazumder et al., 2003) and modulation of regulatory proteins (Wan et al., 2007). Disruption of these RPs might affect mRNA translation on multiple levels, some of which remain to be fully dissected. Much is still unknown about the nuances of ribosomal proteins and how they may contribute to the translational requirements of melanoma cells. Targeting the protein synthetic machinery in melanoma requires that the ribosome itself be considered as a viable therapeutic target.
CONCLUSION
Targeting the protein synthetic machinery in melanoma cells is often overlooked as a therapeutic strategy. Because major signaling pathways in melanoma cells regulate protein synthesis at multiple levels, a greater focus on known and unknown regulatory mechanisms of protein synthesis in melanoma is needed. A better understanding of mRNA translation regulatory factors, amino acid biosynthesis, and ribosomal proteins will enable novel therapeutic approaches to be evaluated. It is just now being discovered how ribosomal proteins and ribosome biogenesis tie into cell signaling and tumor development. RPs are not simply structural components of the ribosome but have complex functions and identifying the role played in translation and cell signaling can open new therapeutic approaches. Disrupting amino acid biosynthesis can inhibit protein synthesis while also affecting certain metabolic processes required for melanoma cell survival. Thus, agents could be developed to simultaneously target increased metabolic and protein synthesis demands required by melanoma cells for the long-term treatment of melanoma patients. It has not been firmly established which cancer types would benefit most from targeting mRNA translation, thus many approaches warrant further study not only in melanoma but also in other cancer types to determine potential efficacy. A better comparison of the role of the protein synthetic machinery between different cancer types will be essential to determine which cancer types would benefit most from therapies targeting translational apparatuses and provide an explanation as to why certain treatments might not be universally effective.
Despite all the advances in our understanding of protein synthesis and melanoma, some important questions remain. First, to better understand the contribution of protein synthesis to melanoma development, the connections between the MAPK and PI3K pathway and protein synthesis need to be more thoroughly dissected. Second, mechanisms leading to the reactivation of the protein synthetic machinery during resistance to V600EBRAF inhibition should be dissected. Third, if the protein synthetic machinery is one of the central foci of mutant BRAF melanoma viability, it is equally important to dissect its role in wildtype BRAF melanomas. Finally, unraveling the variable responses of inhibitors of the protein synthetic pathway in preclinical models and in clinical trials needs explanation. The answer to this important question may be because the target is not essential to tumors, overlapping compensating pathways, or because resistance mechanisms exist. In conclusion, more effective melanoma treatment will require a better understanding of the deregulation of the protein synthetic machinery, and how amino acid biosynthesis as well as ribosome biogenesis contribute to this process in melanoma cells.
References
- AJANI JA, DIMERY I, CHAWLA SP, PINNAMANENI K, BENJAMIN RS, LEGHA SS, KRAKOFF IH. Phase II studies of homoharringtonine in patients with advanced malignant melanoma; sarcoma; and head and neck, breast, and colorectal carcinomas. Cancer Treat Rep. 1986;70:375–9. [PubMed] [Google Scholar]
- ANDERSON CM, BUZAID AC, LEGHA SS. Systemic treatments for advanced cutaneous melanoma. Oncology (Williston Park) 1995;9:1149–58. discussion 1163-4, 1167-8. [PubMed] [Google Scholar]
- ARSENEAU JC, SCHOENFELD DA, BORDEN EC. A phase II study of dihydroxyanthracenedione (DHAD, mitoxantrone, NSC 301739) in advanced malignant melanoma. Invest New Drugs. 1986;4:53–6. doi: 10.1007/BF00172017. [DOI] [PubMed] [Google Scholar]
- ASCIERTO PA, SCALA S, CASTELLO G, DAPONTE A, SIMEONE E, OTTAIANO A, BENEDUCE G, DE ROSA V, IZZO F, MELUCCI MT, ENSOR CM, PRESTAYKO AW, HOLTSBERG FW, BOMALASKI JS, CLARK MA, SAVARAJ N, FEUN LG, LOGAN TF. Pegylated arginine deiminase treatment of patients with metastatic melanoma: results from phase I and II studies. J Clin Oncol. 2005;23:7660–8. doi: 10.1200/JCO.2005.02.0933. [DOI] [PubMed] [Google Scholar]
- AZIZ SA, JILAVEANU LB, ZITO C, CAMP RL, RIMM DL, CONRAD P, KLUGER HM. Vertical targeting of the phosphatidylinositol-3 kinase pathway as a strategy for treating melanoma. Clin Cancer Res. 2010;16:6029–39. doi: 10.1158/1078-0432.CCR-10-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARZILAI A, GOLDBERG I, YULASH M, PAVLOTSKY F, ZUCKERMAN A, TRAU H, AZIZI E, KOPOLOVIC J. Silver-stained nucleolar organizer regions (AgNORs) as a prognostic value in malignant melanoma. Am J Dermatopathol. 1998;20:473–7. doi: 10.1097/00000372-199810000-00008. [DOI] [PubMed] [Google Scholar]
- BASU A, KRISHNAMURTHY S. Cellular responses to Cisplatin-induced DNA damage. J Nucleic Acids. 20102010 doi: 10.4061/2010/201367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAXTER GC, STANNERS CP. The effect of protein degradation on cellular growth characteristics. J Cell Physiol. 1978;96:139–45. doi: 10.1002/jcp.1040960202. [DOI] [PubMed] [Google Scholar]
- BEE A, BREWER D, BEESLEY C, DODSON A, FOROOTAN S, DICKINSON T, GERARD P, LANE B, YAO S, COOPER CS, DJAMGOZ MB, GOSDEN CM, KE Y, FOSTER CS. siRNA knockdown of ribosomal protein gene RPL19 abrogates the aggressive phenotype of human prostate cancer. PLoS One. 2011;6:e22672. doi: 10.1371/journal.pone.0022672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEN-SHEM A, GARREAU DE LOUBRESSE N, MELNIKOV S, JENNER L, YUSUPOVA G, YUSUPOV M. The structure of the eukaryotic ribosome at 3.0 A resolution. Science. 2011;334:1524–9. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- BERLANGA JJ, SANTOYO J, DE HARO C. Characterization of a mammalian homolog of the GCN2 eukaryotic initiation factor 2alpha kinase. Eur J Biochem. 1999;265:754–62. doi: 10.1046/j.1432-1327.1999.00780.x. [DOI] [PubMed] [Google Scholar]
- BLAGDEN SP, WILLIS AE. The biological and therapeutic relevance of mRNA translation in cancer. Nat Rev Clin Oncol. 2011;8:280–91. doi: 10.1038/nrclinonc.2011.16. [DOI] [PubMed] [Google Scholar]
- BORDELEAU ME, MATTHEWS J, WOJNAR JM, LINDQVIST L, NOVAC O, JANKOWSKY E, SONENBERG N, NORTHCOTE P, TEESDALE-SPITTLE P, PELLETIER J. Stimulation of mammalian translation initiation factor eIF4A activity by a small molecule inhibitor of eukaryotic translation. Proc Natl Acad Sci U S A. 2005;102:10460–5. doi: 10.1073/pnas.0504249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BORDELEAU ME, MORI A, OBERER M, LINDQVIST L, CHARD LS, HIGA T, BELSHAM GJ, WAGNER G, TANAKA J, PELLETIER J. Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat Chem Biol. 2006;2:213–20. doi: 10.1038/nchembio776. [DOI] [PubMed] [Google Scholar]
- BORDELEAU ME, ROBERT F, GERARD B, LINDQVIST L, CHEN SM, WENDEL HG, BREM B, GREGER H, LOWE SW, PORCO JA, JR., PELLETIER J. Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J Clin Invest. 2008;118:2651–60. doi: 10.1172/JCI34753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOUSSEMART L, MALKA-MAHIEU H, GIRAULT I, ALLARD D, HEMMINGSSON O, TOMASIC G, THOMAS M, BASMADJIAN C, RIBEIRO N, THUAUD F, MATEUS C, ROUTIER E, KAMSU-KOM N, AGOUSSI S, EGGERMONT AM, DESAUBRY L, ROBERT C, VAGNER S. eIF4F is a nexus of resistance to anti-BRAF and anti-MEK cancer therapies. Nature. 2014 doi: 10.1038/nature13572. [DOI] [PubMed] [Google Scholar]
- BRUNN GJ, HUDSON CC, SEKULIC A, WILLIAMS JM, HOSOI H, HOUGHTON PJ, LAWRENCE JC, JR., ABRAHAM RT. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science. 1997;277:99–101. doi: 10.1126/science.277.5322.99. [DOI] [PubMed] [Google Scholar]
- BURGER K, MUHL B, HARASIM T, ROHRMOSER M, MALAMOUSSI A, ORBAN M, KELLNER M, GRUBER-EBER A, KREMMER E, HOLZEL M, EICK D. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J Biol Chem. 2010;285:12416–25. doi: 10.1074/jbc.M109.074211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUXADE M, PARRA-PALAU JL, PROUD CG. The Mnks: MAP kinase-interacting kinases (MAP kinase signal-integrating kinases) Front Biosci. 2008;13:5359–73. doi: 10.2741/3086. [DOI] [PubMed] [Google Scholar]
- BYWATER MJ, PEARSON RB, MCARTHUR GA, HANNAN RD. Dysregulation of the basal RNA polymerase transcription apparatus in cancer. Nat Rev Cancer. 2013;13:299–314. doi: 10.1038/nrc3496. [DOI] [PubMed] [Google Scholar]
- BYWATER MJ, POORTINGA G, SANIJ E, HEIN N, PECK A, CULLINANE C, WALL M, CLUSE L, DRYGIN D, ANDERES K, HUSER N, PROFFITT C, BLIESATH J, HADDACH M, SCHWAEBE MK, RYCKMAN DM, RICE WG, SCHMITT C, LOWE SW, JOHNSTONE RW, PEARSON RB, MCARTHUR GA, HANNAN RD. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012;22:51–65. doi: 10.1016/j.ccr.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARRACEDO A, MA L, TERUYA-FELDSTEIN J, ROJO F, SALMENA L, ALIMONTI A, EGIA A, SASAKI AT, THOMAS G, KOZMA SC, PAPA A, NARDELLA C, CANTLEY LC, BASELGA J, PANDOLFI PP. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–74. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CENCIC R, DESFORGES M, HALL DR, KOZAKOV D, DU Y, MIN J, DINGLEDINE R, FU H, VAJDA S, TALBOT PJ, PELLETIER J. Blocking eIF4E-eIF4G interaction as a strategy to impair coronavirus replication. J Virol. 2011a;85:6381–9. doi: 10.1128/JVI.00078-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CENCIC R, HALL DR, ROBERT F, DU Y, MIN J, LI L, QUI M, LEWIS I, KURTKAYA S, DINGLEDINE R, FU H, KOZAKOV D, VAJDA S, PELLETIER J. Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc Natl Acad Sci U S A. 2011b;108:1046–51. doi: 10.1073/pnas.1011477108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAN JC, HANNAN KM, RIDDELL K, NG PY, PECK A, LEE RS, HUNG S, ASTLE MV, BYWATER M, WALL M, POORTINGA G, JASTRZEBSKI K, SHEPPARD KE, HEMMINGS BA, HALL MN, JOHNSTONE RW, MCARTHUR GA, HANNAN RD, PEARSON RB. AKT promotes rRNA synthesis and cooperates with c-MYC to stimulate ribosome biogenesis in cancer. Sci Signal. 2011;4:ra56. doi: 10.1126/scisignal.2001754. [DOI] [PubMed] [Google Scholar]
- CHANEY SG, CAMPBELL SL, BASSETT E, WU Y. Recognition and processing of cisplatin- and oxaliplatin-DNA adducts. Crit Rev Oncol Hematol. 2005;53:3–11. doi: 10.1016/j.critrevonc.2004.08.008. [DOI] [PubMed] [Google Scholar]
- CHEN JJ. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: relevance to anemias. Blood. 2007;109:2693–9. doi: 10.1182/blood-2006-08-041830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN JJ, PERRINE S. Stressing HbF synthesis: role of translation? Blood. 2013;122:467–8. doi: 10.1182/blood-2013-06-506139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN L, AKTAS BH, WANG Y, HE X, SAHOO R, ZHANG N, DENOYELLE S, KABHA E, YANG H, FREEDMAN RY, SUPKO JG, CHOREV M, WAGNER G, HALPERIN JA. Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget. 2012;3:869–81. doi: 10.18632/oncotarget.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN T, OZEL D, QIAO Y, HARBINSKI F, CHEN L, DENOYELLE S, HE X, ZVEREVA N, SUPKO JG, CHOREV M, HALPERIN JA, AKTAS BH. Chemical genetics identify eIF2alpha kinase heme-regulated inhibitor as an anticancer target. Nat Chem Biol. 2011;7:610–6. doi: 10.1038/nchembio.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN W, SWANSON S. Silvestrol induces autophagy and cell death in human melanoma cells. Planta Medica. 80:PG1. [Google Scholar]
- CHEN YJ, SU JH, TSAO CY, HUNG CT, CHAO HH, LIN JJ, LIAO MH, YANG ZY, HUANG HH, TSAI FJ, WENG SH, WU YJ. Sinulariolide induced hepatocellular carcinoma apoptosis through activation of mitochondrial-related apoptotic and PERK/eIF2alpha/ATF4/CHOP pathway. Molecules. 2013;18:10146–61. doi: 10.3390/molecules180910146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOO AY, YOON SO, KIM SG, ROUX PP, BLENIS J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–9. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLIS L, PELTONEN K, SIRAJUDDIN P, LIU H, SANDERS S, ERNST G, BARROW JC, LAIHO M. DNA intercalator BMH-21 inhibits RNA polymerase I independent of DNA damage response. Oncotarget. 2014;5:4361–9. doi: 10.18632/oncotarget.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CROFT A, TAY KH, BOYD SC, GUO ST, JIANG CC, LAI F, TSENG HY, JIN L, RIZOS H, HERSEY P, ZHANG XD. Oncogenic activation of MEK/ERK primes melanoma cells for adaptation to endoplasmic reticulum stress. J Invest Dermatol. 2014;134:488–97. doi: 10.1038/jid.2013.325. [DOI] [PubMed] [Google Scholar]
- DAFTUAR L, ZHU Y, JACQ X, PRIVES C. Ribosomal Proteins RPL37, RPS15 and RPS20 Regulate the Mdm2-p53-MdmX Network. PLoS One. 2013;8:e68667. doi: 10.1371/journal.pone.0068667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAI MS, LU H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem. 2004;279:44475–82. doi: 10.1074/jbc.M403722200. [DOI] [PubMed] [Google Scholar]
- DAI MS, ZENG SX, JIN Y, SUN XX, DAVID L, LU H. Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol. 2004;24:7654–68. doi: 10.1128/MCB.24.17.7654-7668.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE INGENIIS J, RATNIKOV B, RICHARDSON AD, SCOTT DA, AZA-BLANC P, DE SK, KAZANOV M, PELLECCHIA M, RONAI Z, OSTERMAN AL, SMITH JW. Functional specialization in proline biosynthesis of melanoma. PLoS One. 2012;7:e45190. doi: 10.1371/journal.pone.0045190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEISENROTH C, ZHANG Y. Ribosome biogenesis surveillance: probing the ribosomal protein-Mdm2-p53 pathway. Oncogene. 2010;29:4253–60. doi: 10.1038/onc.2010.189. [DOI] [PubMed] [Google Scholar]
- DELAGE B, FENNELL DA, NICHOLSON L, MCNEISH I, LEMOINE NR, CROOK T, SZLOSAREK PW. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int J Cancer. 2010;126:2762–72. doi: 10.1002/ijc.25202. [DOI] [PubMed] [Google Scholar]
- DERENZINI M, MONTANARO L, TRERE D. What the nucleolus says to a tumour pathologist. Histopathology. 2009;54:753–62. doi: 10.1111/j.1365-2559.2008.03168.x. [DOI] [PubMed] [Google Scholar]
- DERENZINI M, TRERE D, PESSION A, MONTANARO L, SIRRI V, OCHS RL. Nucleolar function and size in cancer cells. Am J Pathol. 1998;152:1291–7. [PMC free article] [PubMed] [Google Scholar]
- DEVER TE, GREEN R. The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb Perspect Biol. 2012;4:a013706. doi: 10.1101/cshperspect.a013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DHILLON AS, HAGAN S, RATH O, KOLCH W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–90. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- DOLDAN A, CHANDRAMOULI A, SHANAS R, BHATTACHARYYA A, LEONG SP, NELSON MA, SHI J. Loss of the eukaryotic initiation factor 3f in melanoma. Mol Carcinog. 2008;47:806–13. doi: 10.1002/mc.20436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONALD SP, SUN XY, HU CA, YU J, MEI JM, VALLE D, PHANG JM. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res. 2001;61:1810–5. [PubMed] [Google Scholar]
- DONG J, QIU H, GARCIA-BARRIO M, ANDERSON J, HINNEBUSCH AG. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol Cell. 2000;6:269–79. doi: 10.1016/s1097-2765(00)00028-9. [DOI] [PubMed] [Google Scholar]
- DONZE O, JAGUS R, KOROMILAS AE, HERSHEY JW, SONENBERG N. Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. Embo j. 1995;14:3828–34. doi: 10.1002/j.1460-2075.1995.tb00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DORRELLO NV, PESCHIAROLI A, GUARDAVACCARO D, COLBURN NH, SHERMAN NE, PAGANO M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–71. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- DRONCA RS, ALLRED JB, PEREZ DG, NEVALA WK, LIESER EA, THOMPSON M, MAPLES WJ, CREAGAN ET, POCKAJ BA, KAUR JS, MOORE TD, MARCHELLO BT, MARKOVIC SN. Phase II Study of Temozolomide (TMZ) and Everolimus (RAD001) Therapy for Metastatic Melanoma: A North Central Cancer Treatment Group Study, N0675. Am J Clin Oncol. 2014;37:369–76. doi: 10.1097/COC.0b013e31827b45d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DRYGIN D, LIN A, BLIESATH J, HO CB, O'BRIEN SE, PROFFITT C, OMORI M, HADDACH M, SCHWAEBE MK, SIDDIQUI-JAIN A, STREINER N, QUIN JE, SANIJ E, BYWATER MJ, HANNAN RD, RYCKMAN D, ANDERES K, RICE WG. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011;71:1418–30. doi: 10.1158/0008-5472.CAN-10-1728. [DOI] [PubMed] [Google Scholar]
- DRYGIN D, SIDDIQUI-JAIN A, O'BRIEN S, SCHWAEBE M, LIN A, BLIESATH J, HO CB, PROFFITT C, TRENT K, WHITTEN JP, LIM JK, VON HOFF D, ANDERES K, RICE WG. Anticancer activity of CX-3543: a direct inhibitor of rRNA biogenesis. Cancer Res. 2009;69:7653–61. doi: 10.1158/0008-5472.CAN-09-1304. [DOI] [PubMed] [Google Scholar]
- DUNCAN R, MILBURN SC, HERSHEY JW. Regulated phosphorylation and low abundance of HeLa cell initiation factor eIF-4F suggest a role in translational control. Heat shock effects on eIF-4F. J Biol Chem. 1987;262:380–8. [PubMed] [Google Scholar]
- EBERLE J, KRASAGAKIS K, ORFANOS CE. Translation initiation factor eIF-4A1 mRNA is consistently overexpressed in human melanoma cells in vitro. Int J Cancer. 1997;71:396–401. doi: 10.1002/(sici)1097-0215(19970502)71:3<396::aid-ijc16>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- ELLERHORST JA, BEDIKIAN A, RING S, BUZAID AC, ETON O, LEGHA SS. Phase II trial of doxil for patients with metastatic melanoma refractory to frontline therapy. Oncol Rep. 1999;6:1097–9. doi: 10.3892/or.6.5.1097. [DOI] [PubMed] [Google Scholar]
- EMMANUEL R, WEINSTEIN S, LANDESMAN-MILO D, PEER D. eIF3c: a potential therapeutic target for cancer. Cancer Lett. 2013;336:158–66. doi: 10.1016/j.canlet.2013.04.026. [DOI] [PubMed] [Google Scholar]
- ENSOR CM, HOLTSBERG FW, BOMALASKI JS, CLARK MA. Pegylated arginine deiminase (ADI-SS PEG20,000 mw) inhibits human melanomas and hepatocellular carcinomas in vitro and in vivo. Cancer Res. 2002;62:5443–50. [PubMed] [Google Scholar]
- FALCHOOK GS, LONG GV, KURZROCK R, KIM KB, ARKENAU TH, BROWN MP, HAMID O, INFANTE JR, MILLWARD M, PAVLICK AC, O'DAY SJ, BLACKMAN SC, CURTIS CM, LEBOWITZ P, MA B, OUELLET D, KEFFORD RF. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379:1893–901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAYE MD, HOLCIK M. The role of IRES trans-acting factors in carcinogenesis. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbagrm.2014.09.012. [DOI] [PubMed] [Google Scholar]
- FLAHERTY KT, PUZANOV I, KIM KB, RIBAS A, MCARTHUR GA, SOSMAN JA, O'DWYER PJ, LEE RJ, GRIPPO JF, NOLOP K, CHAPMAN PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–19. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLAHERTY KT, ROBERT C, HERSEY P, NATHAN P, GARBE C, MILHEM M, DEMIDOV LV, HASSEL JC, RUTKOWSKI P, MOHR P, DUMMER R, TREFZER U, LARKIN JM, UTIKAL J, DRENO B, NYAKAS M, MIDDLETON MR, BECKER JC, CASEY M, SHERMAN LJ, WU FS, OUELLET D, MARTIN AM, PATEL K, SCHADENDORF D. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–14. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- FOUCHIER F, FORGET P, BELLAN C, MARVALDI J, CHAMPION S, PICHON J. The effects of ribavirin on the GTP level and the VIP receptor dynamic of human IGR39 cells. J Recept Signal Transduct Res. 1996;16:39–58. doi: 10.3109/10799899609039940. [DOI] [PubMed] [Google Scholar]
- FU YM, MEADOWS GG. Specific amino acid dependency regulates the cellular behavior of melanoma. J Nutr. 2007;137:1591S–1596S. doi: 10.1093/jn/137.6.1591S. discussion 1597S-1598S. [DOI] [PubMed] [Google Scholar]
- FUJIMOTO S, OGAWA M. Antitumor activity of mitoxantrone against murine experimental tumors: comparative analysis against various antitumor antibiotics. Cancer Chemother Pharmacol. 1982;8:157–62. doi: 10.1007/BF00255476. [DOI] [PubMed] [Google Scholar]
- FUKUMOTO S, HANAZONO K, FU DR, ENDO Y, KADOSAWA T, IWANO H, UCHIDE T. A new treatment for human malignant melanoma targeting L-type amino acid transporter 1 (LAT1): a pilot study in a canine model. Biochem Biophys Res Commun. 2013;439:103–8. doi: 10.1016/j.bbrc.2013.08.020. [DOI] [PubMed] [Google Scholar]
- GEBAUER F, HENTZE MW. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol. 2004;5:827–35. doi: 10.1038/nrm1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERSTBERGER S, HAFNER M, TUSCHL T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15:829–45. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GLISOVIC T, BACHORIK JL, YONG J, DREYFUSS G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–86. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRAFF JR, KONICEK BW, VINCENT TM, LYNCH RL, MONTEITH D, WEIR SN, SCHWIER P, CAPEN A, GOODE RL, DOWLESS MS, CHEN Y, ZHANG H, SISSONS S, COX K, MCNULTY AM, PARSONS SH, WANG T, SAMS L, GEEGANAGE S, DOUGLASS LE, NEUBAUER BL, DEAN NM, BLANCHARD K, SHOU J, STANCATO LF, CARTER JH, MARCUSSON EG. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J Clin Invest. 2007;117:2638–48. doi: 10.1172/JCI32044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GREGER JG, EASTMAN SD, ZHANG V, BLEAM MR, HUGHES AM, SMITHEMAN KN, DICKERSON SH, LAQUERRE SG, LIU L, GILMER TM. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther. 2012;11:909–20. doi: 10.1158/1535-7163.MCT-11-0989. [DOI] [PubMed] [Google Scholar]
- GRUMMT I. Life on a planet of its own: regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 2003;17:1691–702. doi: 10.1101/gad.1098503R. [DOI] [PubMed] [Google Scholar]
- GUERTIN DA, SABATINI DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- GUREL G, BLAHA G, MOORE PB, STEITZ TA. U2504 determines the species specificity of the A-site cleft antibiotics: the structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J Mol Biol. 2009;389:146–56. doi: 10.1016/j.jmb.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUTIERREZ E, SHIN BS, WOOLSTENHULME CJ, KIM JR, SAINI P, BUSKIRK AR, DEVER TE. eIF5A promotes translation of polyproline motifs. Mol Cell. 2013;51:35–45. doi: 10.1016/j.molcel.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAGEDORN CH, PHANG JM. Catalytic transfer of hydride ions from NADPH to oxygen by the interconversions of proline and delta 1-pyrroline-5-carboxylate. Arch Biochem Biophys. 1986;248:166–74. doi: 10.1016/0003-9861(86)90413-3. [DOI] [PubMed] [Google Scholar]
- HANNAN KM, BRANDENBURGER Y, JENKINS A, SHARKEY K, CAVANAUGH A, ROTHBLUM L, MOSS T, POORTINGA G, MCARTHUR GA, PEARSON RB, HANNAN RD. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol Cell Biol. 2003;23:8862–77. doi: 10.1128/MCB.23.23.8862-8877.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANNAN KM, SANIJ E, ROTHBLUM LI, HANNAN RD, PEARSON RB. Dysregulation of RNA polymerase I transcription during disease. Biochim Biophys Acta. 2013;1829:342–60. doi: 10.1016/j.bbagrm.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARRINGTON LS, FINDLAY GM, LAMB RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci. 2005;30:35–42. doi: 10.1016/j.tibs.2004.11.003. [DOI] [PubMed] [Google Scholar]
- HART LS, CUNNINGHAM JT, DATTA T, DEY S, TAMEIRE F, LEHMAN SL, QIU B, ZHANG H, CERNIGLIA G, BI M, LI Y, GAO Y, LIU H, LI C, MAITY A, THOMAS-TIKHONENKO A, PERL AE, KOONG A, FUCHS SY, DIEHL JA, MILLS IG, RUGGERO D, KOUMENIS C. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J Clin Invest. 2012;122:4621–34. doi: 10.1172/JCI62973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAUKE RJ, INFANTE JR, RUBIN MS, SHIH KC, ARROWSMITH ER, HAINSWORTH JD. Everolimus in combination with paclitaxel and carboplatin in patients with metastatic melanoma: a phase II trial of the Sarah Cannon Research Institute Oncology Research Consortium. Melanoma Res. 2013;23:468–73. doi: 10.1097/CMR.0000000000000014. [DOI] [PubMed] [Google Scholar]
- HAUSCHILD A, GROB JJ, DEMIDOV LV, JOUARY T, GUTZMER R, MILLWARD M, RUTKOWSKI P, BLANK CU, MILLER WH, JR., KAEMPGEN E, MARTIN-ALGARRA S, KARASZEWSKA B, MAUCH C, CHIARION-SILENI V, MARTIN AM, SWANN S, HANEY P, MIRAKHUR B, GUCKERT ME, GOODMAN V, CHAPMAN PB. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- HEIN N, HANNAN KM, GEORGE AJ, SANIJ E, HANNAN RD. The nucleolus: an emerging target for cancer therapy. Trends Mol Med. 2013;19:643–54. doi: 10.1016/j.molmed.2013.07.005. [DOI] [PubMed] [Google Scholar]
- HERNANDEZ-VERDUN D. The nucleolus: a model for the organization of nuclear functions. Histochem Cell Biol. 2006;126:135–48. doi: 10.1007/s00418-006-0212-3. [DOI] [PubMed] [Google Scholar]
- HESS G, HERBRECHT R, ROMAGUERA J, VERHOEF G, CRUMP M, GISSELBRECHT C, LAURELL A, OFFNER F, STRAHS A, BERKENBLIT A, HANUSHEVSKY O, CLANCY J, HEWES B, MOORE L, COIFFIER B. Phase III study to evaluate temsirolimus compared with investigator's choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2009;27:3822–9. doi: 10.1200/JCO.2008.20.7977. [DOI] [PubMed] [Google Scholar]
- HODI FS, O'DAY SJ, MCDERMOTT DF, WEBER RW, SOSMAN JA, HAANEN JB, GONZALEZ R, ROBERT C, SCHADENDORF D, HASSEL JC, AKERLEY W, VAN DEN EERTWEGH AJ, LUTZKY J, LORIGAN P, VAUBEL JM, LINETTE GP, HOGG D, OTTENSMEIER CH, LEBBE C, PESCHEL C, QUIRT I, CLARK JI, WOLCHOK JD, WEBER JS, TIAN J, YELLIN MJ, NICHOL GM, HOOS A, URBA WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HONG DS, KURZROCK R, OH Y, WHELER J, NAING A, BRAIL L, CALLIES S, ANDRE V, KADAM SK, NASIR A, HOLZER TR, MERIC-BERNSTAM F, FISHMAN M, SIMON G. A phase 1 dose escalation, pharmacokinetic, and pharmacodynamic evaluation of eIF-4E antisense oligonucleotide LY2275796 in patients with advanced cancer. Clin Cancer Res. 2011;17:6582–91. doi: 10.1158/1078-0432.CCR-11-0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOU J, LAM F, PROUD C, WANG S. Targeting Mnks for cancer therapy. Oncotarget. 2012;3:118–31. doi: 10.18632/oncotarget.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ILIC N, UTERMARK T, WIDLUND HR, ROBERTS TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proc Natl Acad Sci U S A. 2011;108:E699–708. doi: 10.1073/pnas.1108237108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INGOLIA NT, GHAEMMAGHAMI S, NEWMAN JR, WEISSMAN JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–23. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JAMIESON ER, LIPPARD SJ. Structure, Recognition, and Processing of Cisplatin-DNA Adducts. Chem Rev. 1999;99:2467–98. doi: 10.1021/cr980421n. [DOI] [PubMed] [Google Scholar]
- JASIULIONIS MG, LUCHESSI AD, MOREIRA AG, SOUZA PP, SUENAGA AP, CORREA M, COSTA CA, CURI R, COSTA-NETO CM. Inhibition of eukaryotic translation initiation factor 5A (eIF5A) hypusination impairs melanoma growth. Cell Biochem Funct. 2007;25:109–14. doi: 10.1002/cbf.1351. [DOI] [PubMed] [Google Scholar]
- JIANG CC, CROFT A, TSENG HY, GUO ST, JIN L, HERSEY P, ZHANG XD. Repression of microRNA-768-3p by MEK/ERK signalling contributes to enhanced mRNA translation in human melanoma. Oncogene. 2014;33:2577–88. doi: 10.1038/onc.2013.237. [DOI] [PubMed] [Google Scholar]
- JOHNSON GL, STUHLMILLER TJ, ANGUS SP, ZAWISTOWSKI JS, GRAVES LM. Molecular pathways: adaptive kinome reprogramming in response to targeted inhibition of the BRAF-MEK-ERK pathway in cancer. Clin Cancer Res. 2014;20:2516–22. doi: 10.1158/1078-0432.CCR-13-1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON SA, DUBEAU L, KAWALEK M, DERVAN A, SCHONTHAL AH, DANG CV, JOHNSON DL. Increased expression of TATA-binding protein, the central transcription factor, can contribute to oncogenesis. Mol Cell Biol. 2003;23:3043–51. doi: 10.1128/MCB.23.9.3043-3051.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON SA, MANDAVIA N, WANG HD, JOHNSON DL. Transcriptional regulation of the TATA-binding protein by Ras cellular signaling. Mol Cell Biol. 2000;20:5000–9. doi: 10.1128/mcb.20.14.5000-5009.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JORDAN P, CARMO-FONSECA M. Cisplatin inhibits synthesis of ribosomal RNA in vivo. Nucleic Acids Res. 1998;26:2831–6. doi: 10.1093/nar/26.12.2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JUAREZ P, MOHAMMAD KS, YIN JJ, FOURNIER PG, MCKENNA RC, DAVIS HW, PENG XH, NIEWOLNA M, JAVELAUD D, CHIRGWIN JM, MAUVIEL A, GUISE TA. Halofuginone inhibits the establishment and progression of melanoma bone metastases. Cancer Res. 2012;72:6247–56. doi: 10.1158/0008-5472.CAN-12-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KARDOS GR, DAI MS, ROBERTSON GP. Growth inhibitory effects of large subunit ribosomal proteins in melanoma. Pigment Cell Melanoma Res. 2014 doi: 10.1111/pcmr.12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KARDOS GR, WASTYK H, ROBERTSON GP. Disruption of Proline Biosynthesis in Melanoma Inhibits Protein Synthesis Mediated by the GCN2 Pathway. 2015 doi: 10.1158/1541-7786.MCR-15-0048. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAZEMI S, MOUNIR Z, BALTZIS D, RAVEN JF, WANG S, KRISHNAMOORTHY JL, PLUQUET O, PELLETIER J, KOROMILAS AE. A novel function of eIF2alpha kinases as inducers of the phosphoinositide-3 kinase signaling pathway. Mol Biol Cell. 2007;18:3635–44. doi: 10.1091/mbc.E07-01-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KELLER TL, ZOCCO D, SUNDRUD MS, HENDRICK M, EDENIUS M, YUM J, KIM YJ, LEE HK, CORTESE JF, WIRTH DF, DIGNAM JD, RAO A, YEO CY, MAZITSCHEK R, WHITMAN M. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol. 2012;8:311–7. doi: 10.1038/nchembio.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KHALILI JS, YU X, WANG J, HAYES BC, DAVIES MA, LIZEE G, ESMAELI B, WOODMAN SE. Combination small molecule MEK and PI3K inhibition enhances uveal melanoma cell death in a mutant GNAQ- and GNA11-dependent manner. Clin Cancer Res. 2012;18:4345–55. doi: 10.1158/1078-0432.CCR-11-3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KHOSRAVI S, WONG RP, ARDEKANI GS, ZHANG G, MARTINKA M, ONG CJ, LI G. Role of EIF5A2, a downstream target of Akt, in promoting melanoma cell invasion. Br J Cancer. 2014;110:399–408. doi: 10.1038/bjc.2013.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIMBALL SR. Eukaryotic initiation factor eIF2. Int J Biochem Cell Biol. 1999;31:25–9. doi: 10.1016/s1357-2725(98)00128-9. [DOI] [PubMed] [Google Scholar]
- KONDRASHOV N, PUSIC A, STUMPF CR, SHIMIZU K, HSIEH AC, XUE S, ISHIJIMA J, SHIROISHI T, BARNA M. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell. 2011;145:383–97. doi: 10.1016/j.cell.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KONICEK BW, STEPHENS JR, MCNULTY AM, ROBICHAUD N, PEERY RB, DUMSTORF CA, DOWLESS MS, IVERSEN PW, PARSONS S, ELLIS KE, MCCANN DJ, PELLETIER J, FURIC L, YINGLING JM, STANCATO LF, SONENBERG N, GRAFF JR. Therapeutic inhibition of MAP kinase interacting kinase blocks eukaryotic initiation factor 4E phosphorylation and suppresses outgrowth of experimental lung metastases. Cancer Res. 2011;71:1849–57. doi: 10.1158/0008-5472.CAN-10-3298. [DOI] [PubMed] [Google Scholar]
- KOROMILAS AE. Roles of the translation initiation factor eIF2alpha serine 51 phosphorylation in cancer formation and treatment. Biochim Biophys Acta. 2014 doi: 10.1016/j.bbagrm.2014.12.007. [DOI] [PubMed] [Google Scholar]
- KOROMILAS AE, ROY S, BARBER GN, KATZE MG, SONENBERG N. Malignant transformation by a mutant of the IFN-inducible dsRNA-dependent protein kinase. Science. 1992;257:1685–9. doi: 10.1126/science.1382315. [DOI] [PubMed] [Google Scholar]
- KRISHNAMOORTHY T, PAVITT GD, ZHANG F, DEVER TE, HINNEBUSCH AG. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol Cell Biol. 2001;21:5018–30. doi: 10.1128/MCB.21.15.5018-5030.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KROON HM, HUISMANS AM, KAM PC, THOMPSON JF. Isolated limb infusion with melphalan and actinomycin D for melanoma: a systematic review. J Surg Oncol. 2014;109:348–51. doi: 10.1002/jso.23553. [DOI] [PubMed] [Google Scholar]
- KUMAR SK, JETT J, MARKS R, RICHARDSON R, QUEVEDO F, MOYNIHAN T, CROGHAN G, MARKOVIC SN, BIBLE KC, QIN R, TAN A, MOLINA J, KAUFMANN SH, ERLICHMAN C, ADJEI AA. Phase 1 study of sorafenib in combination with bortezomib in patients with advanced malignancies. Invest New Drugs. 2013;31:1201–6. doi: 10.1007/s10637-013-0004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUZNETSOV G, XU Q, RUDOLPH-OWEN L, TENDYKE K, LIU J, TOWLE M, ZHAO N, MARSH J, AGOULNIK S, TWINE N, PARENT L, CHEN Z, SHIE JL, JIANG Y, ZHANG H, DU H, BOIVIN R, WANG Y, ROMO D, LITTLEFIELD BA. Potent in vitro and in vivo anticancer activities of des-methyl, des-amino pateamine A, a synthetic analogue of marine natural product pateamine A. Mol Cancer Ther. 2009;8:1250–60. doi: 10.1158/1535-7163.MCT-08-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KWITKOWSKI VE, PROWELL TM, IBRAHIM A, FARRELL AT, JUSTICE R, MITCHELL SS, SRIDHARA R, PAZDUR R. FDA approval summary: temsirolimus as treatment for advanced renal cell carcinoma. Oncologist. 2010;15:428–35. doi: 10.1634/theoncologist.2009-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LARKIN J, DEL VECCHIO M, ASCIERTO PA, KRAJSOVA I, SCHACHTER J, NEYNS B, ESPINOSA E, GARBE C, SILENI VC, GOGAS H, MILLER WH, JR., MANDALA M, HOSPERS GA, ARANCE A, QUEIROLO P, HAUSCHILD A, BROWN MP, MITCHELL L, VERONESE L, BLANK CU. Vemurafenib in patients with BRAF(V600) mutated metastatic melanoma: an open-label, multicentre, safety study. Lancet Oncol. 2014;15:436–44. doi: 10.1016/S1470-2045(14)70051-8. [DOI] [PubMed] [Google Scholar]
- LARSSON O, LI S, ISSAENKO OA, AVDULOV S, PETERSON M, SMITH K, BITTERMAN PB, POLUNOVSKY VA. Eukaryotic translation initiation factor 4E induced progression of primary human mammary epithelial cells along the cancer pathway is associated with targeted translational deregulation of oncogenic drivers and inhibitors. Cancer Res. 2007;67:6814–24. doi: 10.1158/0008-5472.CAN-07-0752. [DOI] [PubMed] [Google Scholar]
- LAZARIS-KARATZAS A, MONTINE KS, SONENBERG N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5' cap. Nature. 1990;345:544–7. doi: 10.1038/345544a0. [DOI] [PubMed] [Google Scholar]
- LEBWOHL D, ANAK O, SAHMOUD T, KLIMOVSKY J, ELMROTH I, HAAS T, POSLUSZNY J, SALETAN S, BERG W. Development of everolimus, a novel oral mTOR inhibitor, across a spectrum of diseases. Ann N Y Acad Sci. 2013;1291:14–32. doi: 10.1111/nyas.12122. [DOI] [PubMed] [Google Scholar]
- LEE SM, BETTICHER DC, THATCHER N. Melanoma: chemotherapy. Br Med Bull. 1995;51:609–30. doi: 10.1093/oxfordjournals.bmb.a072982. [DOI] [PubMed] [Google Scholar]
- LESLIE NR, DOWNES CP. PTEN function: how normal cells control it and tumour cells lose it. Biochem J. 2004;382:1–11. doi: 10.1042/BJ20040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI HH, SU JH, CHIU CC, LIN JJ, YANG ZY, HWANG WI, CHEN YK, LO YH, WU YJ. Proteomic investigation of the sinulariolide-treated melanoma cells A375: effects on the cell apoptosis through mitochondrial-related pathway and activation of caspase cascade. Mar Drugs. 2013;11:2625–42. doi: 10.3390/md11072625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI W, MELTON DW. Cisplatin regulates the MAPK kinase pathway to induce increased expression of DNA repair gene ERCC1 and increase melanoma chemoresistance. Oncogene. 2012;31:2412–22. doi: 10.1038/onc.2011.426. [DOI] [PubMed] [Google Scholar]
- LINDSTROM MS, NISTER M. Silencing of ribosomal protein S9 elicits a multitude of cellular responses inhibiting the growth of cancer cells subsequent to p53 activation. PLoS One. 2010;5:e9578. doi: 10.1371/journal.pone.0009578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU W, LE A, HANCOCK C, LANE AN, DANG CV, FAN TW, PHANG JM. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc Natl Acad Sci U S A. 2012;109:8983–8. doi: 10.1073/pnas.1203244109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU Y, BORCHERT GL, SURAZYNSKI A, HU CA, PHANG JM. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: the role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene. 2006;25:5640–7. doi: 10.1038/sj.onc.1209564. [DOI] [PubMed] [Google Scholar]
- LIU Y, BORCHERT GL, SURAZYNSKI A, PHANG JM. Proline oxidase, a p53-induced gene, targets COX-2/PGE2 signaling to induce apoptosis and inhibit tumor growth in colorectal cancers. Oncogene. 2008;27:6729–37. doi: 10.1038/onc.2008.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIVINGSTONE M, ATAS E, MELLER A, SONENBERG N. Mechanisms governing the control of mRNA translation. Phys Biol. 2010;7:021001. doi: 10.1088/1478-3975/7/2/021001. [DOI] [PubMed] [Google Scholar]
- LOPEZ-FAUQUED M, GIL R, GRUESO J, HERNANDEZ-LOSA J, PUJOL A, MOLINE T, RECIO JA. The dual PI3K/mTOR inhibitor PI-103 promotes immunosuppression, in vivo tumor growth and increases survival of sorafenib-treated melanoma cells. Int J Cancer. 2010;126:1549–61. doi: 10.1002/ijc.24926. [DOI] [PubMed] [Google Scholar]
- LOW WK, DANG Y, SCHNEIDER-POETSCH T, SHI Z, CHOI NS, MERRICK WC, ROMO D, LIU JO. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol Cell. 2005;20:709–22. doi: 10.1016/j.molcel.2005.10.008. [DOI] [PubMed] [Google Scholar]
- LUTZKY J, NUNEZ Y, GRAHAM P. A phase II trial of oxaliplatin in patients with advanced melanoma. ASCO Annual Meeting Proceedings. 2006 [Google Scholar]
- LYNCH CJ. Role of leucine in the regulation of mTOR by amino acids: revelations from structure-activity studies. J Nutr. 2001;131:861s–865s. doi: 10.1093/jn/131.3.861S. [DOI] [PubMed] [Google Scholar]
- MA XH, PIAO SF, DEY S, MCAFEE Q, KARAKOUSIS G, VILLANUEVA J, HART LS, LEVI S, HU J, ZHANG G, LAZOVA R, KLUMP V, PAWELEK JM, XU X, XU W, SCHUCHTER LM, DAVIES MA, HERLYN M, WINKLER J, KOUMENIS C, AMARAVADI RK. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J Clin Invest. 2014;124:1406–17. doi: 10.1172/JCI70454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MA XM, BLENIS J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–18. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- MACCARTY WC. The value of the macronucleolus in the cancer problem. The American Journal of Cancer. 1936;26:529–532. [Google Scholar]
- MALINA A, CENCIC R, PELLETIER J. Targeting translation dependence in cancer. Oncotarget. 2011;2:76–88. doi: 10.18632/oncotarget.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MALINA A, MILLS JR, PELLETIER J. Emerging therapeutics targeting mRNA translation. Cold Spring Harb Perspect Biol. 2012;4:a012377. doi: 10.1101/cshperspect.a012377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARGOLIN K, LONGMATE J, BARATTA T, SYNOLD T, CHRISTENSEN S, WEBER J, GAJEWSKI T, QUIRT I, DOROSHOW JH. CCI-779 in metastatic melanoma: a phase II trial of the California Cancer Consortium. Cancer. 2005;104:1045–8. doi: 10.1002/cncr.21265. [DOI] [PubMed] [Google Scholar]
- MARGOLIN KA, MOON J, FLAHERTY LE, LAO CD, AKERLEY WL, 3RD, OTHUS M, SOSMAN JA, KIRKWOOD JM, SONDAK VK. Randomized phase II trial of sorafenib with temsirolimus or tipifarnib in untreated metastatic melanoma (S0438) Clin Cancer Res. 2012;18:1129–37. doi: 10.1158/1078-0432.CCR-11-2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAYER C, ZHAO J, YUAN X, GRUMMT I. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev. 2004;18:423–34. doi: 10.1101/gad.285504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAZUMDER B, SAMPATH P, SESHADRI V, MAITRA RK, DICORLETO PE, FOX PL. Regulated release of L13a from the 60S ribosomal subunit as a mechanism of transcript-specific translational control. Cell. 2003;115:187–98. doi: 10.1016/s0092-8674(03)00773-6. [DOI] [PubMed] [Google Scholar]
- MCGOWAN KA, LI JZ, PARK CY, BEAUDRY V, TABOR HK, SABNIS AJ, ZHANG W, FUCHS H, DE ANGELIS MH, MYERS RM, ATTARDI LD, BARSH GS. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat Genet. 2008;40:963–70. doi: 10.1038/ng.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIDDLETON MR, LORIGAN P, OWEN J, ASHCROFT L, LEE SM, HARPER P, THATCHER N. A randomized phase III study comparing dacarbazine, BCNU, cisplatin and tamoxifen with dacarbazine and interferon in advanced melanoma. Br J Cancer. 2000;82:1158–62. doi: 10.1054/bjoc.1999.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOERKE NJ, AKTAS H, CHEN H, CANTEL S, REIBARKH MY, FAHMY A, GROSS JD, DEGTEREV A, YUAN J, CHOREV M, HALPERIN JA, WAGNER G. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–67. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- MOUNIR Z, KRISHNAMOORTHY JL, ROBERTSON GP, SCHEUNER D, KAUFMAN RJ, GEORGESCU MM, KOROMILAS AE. Tumor suppression by PTEN requires the activation of the PKR-eIF2alpha phosphorylation pathway. Sci Signal. 2009;2:ra85. doi: 10.1126/scisignal.2000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MULLARKY E, MATTAINI KR, VANDER HEIDEN MG, CANTLEY LC, LOCASALE JW. PHGDH amplification and altered glucose metabolism in human melanoma. Pigment Cell Melanoma Res. 2011;24:1112–5. doi: 10.1111/j.1755-148X.2011.00919.x. [DOI] [PubMed] [Google Scholar]
- OTT PA, CARVAJAL RD, PANDIT-TASKAR N, JUNGBLUTH AA, HOFFMAN EW, WU BW, BOMALASKI JS, VENHAUS R, PAN L, OLD LJ, PAVLICK AC, WOLCHOK JD. Phase I/II study of pegylated arginine deiminase (ADI-PEG 20) in patients with advanced melanoma. Invest New Drugs. 2013;31:425–34. doi: 10.1007/s10637-012-9862-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAUSE A, BELSHAM GJ, GINGRAS AC, DONZE O, LIN TA, LAWRENCE JC, JR., SONENBERG N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5'-cap function. Nature. 1994;371:762–7. doi: 10.1038/371762a0. [DOI] [PubMed] [Google Scholar]
- PELTONEN K, COLIS L, LIU H, TRIVEDI R, MOUBAREK MS, MOORE HM, BAI B, RUDEK MA, BIEBERICH CJ, LAIHO M. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell. 2014;25:77–90. doi: 10.1016/j.ccr.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PERRY MD, GORE M, SEIGLER HF, JOHNSTON WW. Fine needle aspiration biopsy of metastatic melanoma. A morphologic analysis of 174 cases. Acta Cytol. 1986;30:385–96. [PubMed] [Google Scholar]
- PESTKA S. Inhibitors of protein synthesis. Academic Press; New York: 1977. [Google Scholar]
- PHANG JM, LIU W, HANCOCK C, CHRISTIAN KJ. The proline regulatory axis and cancer. Front Oncol. 2012;2:60. doi: 10.3389/fonc.2012.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PHILLIPS MM, SHEAFF MT, SZLOSAREK PW. Targeting arginine-dependent cancers with arginine-degrading enzymes: opportunities and challenges. Cancer Res Treat. 2013;45:251–62. doi: 10.4143/crt.2013.45.4.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PIANESE G. Beitrag zur Histologie und Aetiologie des Carcinoms. G. Fischer; 1896. [Google Scholar]
- POKLEPOVIC A, YOUSSEFIAN LE, WINNING M, BIRDSELL CA, CROSBY NA, RAMAKRISHNAN V, ERNSTOFF MS, ROBERTS JD. Phase I trial of bortezomib and dacarbazine in melanoma and soft tissue sarcoma. Invest New Drugs. 2013;31:937–42. doi: 10.1007/s10637-012-9913-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POLYAK K, XIA Y, ZWEIER JL, KINZLER KW, VOGELSTEIN B. A model for p53-induced apoptosis. Nature. 1997;389:300–5. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- POSSEMATO R, MARKS KM, SHAUL YD, PACOLD ME, KIM D, BIRSOY K, SETHUMADHAVAN S, WOO HK, JANG HG, JHA AK, CHEN WW, BARRETT FG, STRANSKY N, TSUN ZY, COWLEY GS, BARRETINA J, KALAANY NY, HSU PP, OTTINA K, CHAN AM, YUAN B, GARRAWAY LA, ROOT DE, MINO-KENUDSON M, BRACHTEL EF, DRIGGERS EM, SABATINI DM. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–50. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PYRONNET S, IMATAKA H, GINGRAS AC, FUKUNAGA R, HUNTER T, SONENBERG N. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. Embo j. 1999;18:270–9. doi: 10.1093/emboj/18.1.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAO R, WINDSCHITL H, ALLRED J, LOWE V, MAPLES W, GORNET M, SUMAN V, CREAGAN E, PITOT H, MARKOVIC S. Phase II trial of the mTOR inhibitor everolimus (RAD-001) in metastatic melanoma. J Clin Oncol. 2006;24:8043. [Google Scholar]
- RAYMOND E, LAWRENCE R, IZBICKA E, FAIVRE S, VON HOFF DD. Activity of oxaliplatin against human tumor colony-forming units. Clin Cancer Res. 1998;4:1021–9. [PubMed] [Google Scholar]
- RAYNAUD FI, ECCLES S, CLARKE PA, HAYES A, NUTLEY B, ALIX S, HENLEY A, DI-STEFANO F, AHMAD Z, GUILLARD S, BJERKE LM, KELLAND L, VALENTI M, PATTERSON L, GOWAN S, DE HAVEN BRANDON A, HAYAKAWA M, KAIZAWA H, KOIZUMI T, OHISHI T, PATEL S, SAGHIR N, PARKER P, WATERFIELD M, WORKMAN P. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007;67:5840–50. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- REULAND SN, GOLDSTEIN NB, PARTYKA KA, SMITH S, LUO Y, FUJITA M, GONZALEZ R, LEWIS K, NORRIS DA, SHELLMAN YG. ABT-737 synergizes with Bortezomib to kill melanoma cells. Biol Open. 2012;1:92–100. doi: 10.1242/bio.2011035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERTS PJ, USARY JE, DARR DB, DILLON PM, PFEFFERLE AD, WHITTLE MC, DUNCAN JS, JOHNSON SM, COMBEST AJ, JIN J, ZAMBONI WC, JOHNSON GL, PEROU CM, SHARPLESS NE. Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clin Cancer Res. 2012;18:5290–303. doi: 10.1158/1078-0432.CCR-12-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RODRIGUEZ PC, QUICENO DG, OCHOA AC. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood. 2007;109:1568–73. doi: 10.1182/blood-2006-06-031856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROMO D, CHOI NS, LI S, BUCHLER I, SHI Z, LIU JO. Evidence for separate binding and scaffolding domains in the immunosuppressive and antitumor marine natural product, pateamine a: design, synthesis, and activity studies leading to a potent simplified derivative. J Am Chem Soc. 2004;126:10582–8. doi: 10.1021/ja040065s. [DOI] [PubMed] [Google Scholar]
- ROSENBERG B. Fundamental studies with cisplatin. Cancer. 1985;55:2303–l6. doi: 10.1002/1097-0142(19850515)55:10<2303::aid-cncr2820551002>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- ROSENWALD IB, WANG S, SAVAS L, WODA B, PULLMAN J. Expression of translation initiation factor eIF-2alpha is increased in benign and malignant melanocytic and colonic epithelial neoplasms. Cancer. 2003;98:1080–8. doi: 10.1002/cncr.11619. [DOI] [PubMed] [Google Scholar]
- RUDRA D, WARNER JR. What better measure than ribosome synthesis? Genes Dev. 2004;18:2431–6. doi: 10.1101/gad.1256704. [DOI] [PubMed] [Google Scholar]
- RUGGERO D, MONTANARO L, MA L, XU W, LONDEI P, CORDON-CARDO C, PANDOLFI PP. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10:484–6. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- RUGGERO D, PANDOLFI PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–92. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- SALAMA AK, KIM KB. Trametinib (GSK1120212) in the treatment of melanoma. Expert Opin Pharmacother. 2013;14:619–27. doi: 10.1517/14656566.2013.770475. [DOI] [PubMed] [Google Scholar]
- SAVARAJ N, YOU M, WU C, WANGPAICHITR M, KUO MT, FEUN LG. Arginine deprivation, autophagy, apoptosis (AAA) for the treatment of melanoma. Curr Mol Med. 2010;10:405–12. doi: 10.2174/156652410791316995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHNEIDER RJ, SONENBERG N. 15 Translational Control in Cancer Development and Progression. Cold Spring Harbor Monograph Archive. 2007;48:401–431. [Google Scholar]
- SCOTT L, LAMB J, SMITH S, WHEATLEY DN. Single amino acid (arginine) deprivation: rapid and selective death of cultured transformed and malignant cells. Br J Cancer. 2000;83:800–10. doi: 10.1054/bjoc.2000.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEARS R, LEONE G, DEGREGORI J, NEVINS JR. Ras enhances Myc protein stability. Mol Cell. 1999;3:169–79. doi: 10.1016/s1097-2765(00)80308-1. [DOI] [PubMed] [Google Scholar]
- SHAHSHAHAN MA, BECKLEY MN, JAZIREHI AR. Potential usage of proteasome inhibitor bortezomib (Velcade, PS-341) in the treatment of metastatic melanoma: basic and clinical aspects. Am J Cancer Res. 2011;1:913–24. [PMC free article] [PubMed] [Google Scholar]
- SHEEN JH, ZONCU R, KIM D, SABATINI DM. Defective regulation of autophagy upon leucine deprivation reveals a targetable liability of human melanoma cells in vitro and in vivo. Cancer Cell. 2011;19:613–28. doi: 10.1016/j.ccr.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEN LJ, SHEN WC. Drug evaluation: ADI-PEG-20--a PEGylated arginine deiminase for arginine-auxotrophic cancers. Curr Opin Mol Ther. 2006;8:240–8. [PubMed] [Google Scholar]
- SHI H, KONG X, RIBAS A, LO RS. Combinatorial treatments that overcome PDGFRbeta-driven resistance of melanoma cells to V600EB-RAF inhibition. Cancer Res. 2011;71:5067–74. doi: 10.1158/0008-5472.CAN-11-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHI J, KAHLE A, HERSHEY JW, HONCHAK BM, WARNEKE JA, LEONG SP, NELSON MA. Decreased expression of eukaryotic initiation factor 3f deregulates translation and apoptosis in tumor cells. Oncogene. 2006;25:4923–36. doi: 10.1038/sj.onc.1209495. [DOI] [PubMed] [Google Scholar]
- SILVERA D, FORMENTI SC, SCHNEIDER RJ. Translational control in cancer. Nat Rev Cancer. 2010;10:254–66. doi: 10.1038/nrc2824. [DOI] [PubMed] [Google Scholar]
- SIVAM GP, MARTIN PJ, REISFELD RA, MUELLER BM. Therapeutic efficacy of a doxorubicin immunoconjugate in a preclinical model of spontaneous metastatic human melanoma. Cancer Res. 1995;55:2352–6. [PubMed] [Google Scholar]
- SMALLEY KS, HERLYN M. Targeting intracellular signaling pathways as a novel strategy in melanoma therapeutics. Ann N Y Acad Sci. 2005;1059:16–25. doi: 10.1196/annals.1339.005. [DOI] [PubMed] [Google Scholar]
- SONENBERG N, HINNEBUSCH AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–45. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STAHL JM, CHEUNG M, SHARMA A, TRIVEDI NR, SHANMUGAM S, ROBERTSON GP. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003;63:2881–90. [PubMed] [Google Scholar]
- STEFANOVSKY VY, MOSS T. The splice variants of UBF differentially regulate RNA polymerase I transcription elongation in response to ERK phosphorylation. Nucleic Acids Res. 2008;36:5093–101. doi: 10.1093/nar/gkn484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEFANOVSKY VY, PELLETIER G, HANNAN R, GAGNON-KUGLER T, ROTHBLUM LI, MOSS T. An immediate response of ribosomal transcription to growth factor stimulation in mammals is mediated by ERK phosphorylation of UBF. Mol Cell. 2001;8:1063–73. doi: 10.1016/s1097-2765(01)00384-7. [DOI] [PubMed] [Google Scholar]
- SUGIMURA K, OHNO T, KUSUYAMA T, AZUMA I. High sensitivity of human melanoma cell lines to the growth inhibitory activity of mycoplasmal arginine deiminase in vitro. Melanoma Res. 1992;2:191–6. doi: 10.1097/00008390-199209000-00007. [DOI] [PubMed] [Google Scholar]
- SULLIVAN RJ, FLAHERTY KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer. 2013;49:1297–304. doi: 10.1016/j.ejca.2012.11.019. [DOI] [PubMed] [Google Scholar]
- SUN XX, DEVINE T, CHALLAGUNDLA KB, DAI MS. Interplay between ribosomal protein S27a and MDM2 protein in p53 activation in response to ribosomal stress. J Biol Chem. 2011;286:22730–41. doi: 10.1074/jbc.M111.223651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUZUKI A, IIZUKA A, KOMIYAMA M, TAKIKAWA M, KUME A, TAI S, OHSHITA C, KURUSU A, NAKAMURA Y, YAMAMOTO A, YAMAZAKI N, YOSHIKAWA S, KIYOHARA Y, AKIYAMA Y. Identification of melanoma antigens using a Serological Proteome Approach (SERPA) Cancer Genomics Proteomics. 2010;7:17–23. [PubMed] [Google Scholar]
- TERZIAN T, BOX N. Genetics of ribosomal proteins: "curiouser and curiouser". PLoS Genet. 2013;9:e1003300. doi: 10.1371/journal.pgen.1003300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMAS G. An encore for ribosome biogenesis in the control of cell proliferation. Nat Cell Biol. 2000;2:E71–2. doi: 10.1038/35010581. [DOI] [PubMed] [Google Scholar]
- TOPALIAN SL, SZNOL M, MCDERMOTT DF, KLUGER HM, CARVAJAL RD, SHARFMAN WH, BRAHMER JR, LAWRENCE DP, ATKINS MB, POWDERLY JD, LEMING PD, LIPSON EJ, PUZANOV I, SMITH DC, TAUBE JM, WIGGINTON JM, KOLLIA GD, GUPTA A, PARDOLL DM, SOSMAN JA, HODI FS. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020–30. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOPISIROVIC I, RUIZ-GUTIERREZ M, BORDEN KL. Phosphorylation of the eukaryotic translation initiation factor eIF4E contributes to its transformation and mRNA transport activities. Cancer Res. 2004;64:8639–42. doi: 10.1158/0008-5472.CAN-04-2677. [DOI] [PubMed] [Google Scholar]
- VAN SLUIS M, MCSTAY B. Ribosome biogenesis: Achilles heel of cancer? Genes Cancer. 2014;5:152–3. doi: 10.18632/genesandcancer.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VAZQUEZ D. Inhibitors of protein biosynthesis. Mol Biol Biochem Biophys. 1979;30(i-x):1–312. doi: 10.1007/978-3-642-81309-2. [DOI] [PubMed] [Google Scholar]
- VILLANUEVA J, INFANTE JR, KREPLER C, REYES-URIBE P, SAMANTA M, CHEN HY, LI B, SWOBODA RK, WILSON M, VULTUR A, FUKUNABA-KALABIS M, WUBBENHORST B, CHEN TY, LIU Q, SPROESSER K, DEMARINI DJ, GILMER TM, MARTIN AM, MARMORSTEIN R, SCHULTZ DC, SPEICHER DW, KARAKOUSIS GC, XU W, AMARAVADI RK, XU X, SCHUCHTER LM, HERLYN M, NATHANSON KL. Concurrent MEK2 mutation and BRAF amplification confer resistance to BRAF and MEK inhibitors in melanoma. Cell Rep. 2013;4:1090–9. doi: 10.1016/j.celrep.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VOGELSTEIN B, LANE D, LEVINE AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- WAGLE N, VAN ALLEN EM, TREACY DJ, FREDERICK DT, COOPER ZA, TAYLOR-WEINER A, ROSENBERG M, GOETZ EM, SULLIVAN RJ, FARLOW DN, FRIEDRICH DC, ANDERKA K, PERRIN D, JOHANNESSEN CM, MCKENNA A, CIBULSKIS K, KRYUKOV G, HODIS E, LAWRENCE DP, FISHER S, GETZ G, GABRIEL SB, CARTER SL, FLAHERTY KT, WARGO JA, GARRAWAY LA. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4:61–8. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAN F, ANDERSON DE, BARNITZ RA, SNOW A, BIDERE N, ZHENG L, HEGDE V, LAM LT, STAUDT LM, LEVENS D, DEUTSCH WA, LENARDO MJ. Ribosomal protein S3: a KH domain subunit in NF-kappaB complexes that mediates selective gene regulation. Cell. 2007;131:927–39. doi: 10.1016/j.cell.2007.10.009. [DOI] [PubMed] [Google Scholar]
- WANG Q, BEAUMONT KA, OTTE NJ, FONT J, BAILEY CG, VAN GELDERMALSEN M, SHARP DM, TIFFEN JC, RYAN RM, JORMAKKA M, HAASS NK, RASKO JE, HOLST J. Targeting glutamine transport to suppress melanoma cell growth. Int J Cancer. 2014a;135:1060–71. doi: 10.1002/ijc.28749. [DOI] [PubMed] [Google Scholar]
- WANG R, DILLON CP, SHI LZ, MILASTA S, CARTER R, FINKELSTEIN D, MCCORMICK LL, FITZGERALD P, CHI H, MUNGER J, GREEN DR. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–82. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG X, REGUFE DA MOTA S, LIU R, MOORE CE, XIE J, LANUCARA F, AGARWALA U, PYR DIT RUYS S, VERTOMMEN D, RIDER MH, EYERS C, PROUD CG. Eukaryotic elongation factor 2 kinase activity is controlled by multiple regulatory inputs from oncogenic and anabolic pathways. Mol Cell Biol. 2014b doi: 10.1128/MCB.01035-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WARNER JR, MCINTOSH KB. How common are extraribosomal functions of ribosomal proteins? Mol Cell. 2009;34:3–11. doi: 10.1016/j.molcel.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WARNER JR, VILARDELL J, SOHN JH. Economics of ribosome biosynthesis. Cold Spring Harb Symp Quant Biol. 2001;66:567–74. doi: 10.1101/sqb.2001.66.567. [DOI] [PubMed] [Google Scholar]
- WASKIEWICZ AJ, FLYNN A, PROUD CG, COOPER JA. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. Embo j. 1997;16:1909–20. doi: 10.1093/emboj/16.8.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEBB BL, PROUD CG. Eukaryotic initiation factor 2B (eIF2B) Int J Biochem Cell Biol. 1997;29:1127–31. doi: 10.1016/s1357-2725(97)00039-3. [DOI] [PubMed] [Google Scholar]
- WEK SA, ZHU S, WEK RC. The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol Cell Biol. 1995;15:4497–506. doi: 10.1128/mcb.15.8.4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WENDEL HG, SILVA RL, MALINA A, MILLS JR, ZHU H, UEDA T, WATANABE-FUKUNAGA R, FUKUNAGA R, TERUYA-FELDSTEIN J, PELLETIER J, LOWE SW. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007;21:3232–7. doi: 10.1101/gad.1604407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITE RJ. RNA polymerases I and III, growth control and cancer. Nat Rev Mol Cell Biol. 2005;6:69–78. doi: 10.1038/nrm1551. [DOI] [PubMed] [Google Scholar]
- WU Q, GOU Y, WANG Q, JIN H, CUI L, ZHANG Y, HE L, WANG J, NIE Y, SHI Y, FAN D. Downregulation of RPL6 by siRNA inhibits proliferation and cell cycle progression of human gastric cancer cell lines. PLoS One. 2011;6:e26401. doi: 10.1371/journal.pone.0026401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XIONG X, ZHAO Y, HE H, SUN Y. Ribosomal protein S27-like and S27 interplay with p53-MDM2 axis as a target, a substrate and a regulator. Oncogene. 2011;30:1798–811. doi: 10.1038/onc.2010.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YADAVILLI S, MAYO LD, HIGGINS M, LAIN S, HEGDE V, DEUTSCH WA. Ribosomal protein S3: A multi-functional protein that interacts with both p53 and MDM2 through its KH domain. DNA Repair (Amst) 2009;8:1215–24. doi: 10.1016/j.dnarep.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YE J, KUMANOVA M, HART LS, SLOANE K, ZHANG H, DE PANIS DN, BOBROVNIKOVA-MARJON E, DIEHL JA, RON D, KOUMENIS C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J. 2010;29:2082–96. doi: 10.1038/emboj.2010.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YERLIKAYA A, DOKUDUR H. Investigation of the eIF2alpha phosphorylation mechanism in response to proteasome inhibition in melanoma and breast cancer cells. Mol Biol (Mosk) 2010;44:859–66. [PubMed] [Google Scholar]
- YOON KA, NAKAMURA Y, ARAKAWA H. Identification of ALDH4 as a p53-inducible gene and its protective role in cellular stresses. J Hum Genet. 2004;49:134–40. doi: 10.1007/s10038-003-0122-3. [DOI] [PubMed] [Google Scholar]
- YUAN TL, CANTLEY LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG C, COMAI L, JOHNSON DL. PTEN represses RNA Polymerase I transcription by disrupting the SL1 complex. Mol Cell Biol. 2005;25:6899–911. doi: 10.1128/MCB.25.16.6899-6911.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG P, MCGRATH BC, REINERT J, OLSEN DS, LEI L, GILL S, WEK SA, VATTEM KM, WEK RC, KIMBALL SR, JEFFERSON LS, CAVENER DR. The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol Cell Biol. 2002;22:6681–8. doi: 10.1128/MCB.22.19.6681-6688.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Y, WANG J, YUAN Y, ZHANG W, GUAN W, WU Z, JIN C, CHEN H, ZHANG L, YANG X, HE F. Negative regulation of HDM2 to attenuate p53 degradation by ribosomal protein L26. Nucleic Acids Res. 2010;38:6544–54. doi: 10.1093/nar/gkq536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Y, WOLF GW, BHAT K, JIN A, ALLIO T, BURKHART WA, XIONG Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23:8902–12. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO J, YUAN X, FRODIN M, GRUMMT I. ERK-dependent phosphorylation of the transcription initiation factor TIF-IA is required for RNA polymerase I transcription and cell growth. Mol Cell. 2003;11:405–13. doi: 10.1016/s1097-2765(03)00036-4. [DOI] [PubMed] [Google Scholar]
- ZHU Y, POYUROVSKY MV, LI Y, BIDERMAN L, STAHL J, JACQ X, PRIVES C. Ribosomal protein S7 is both a regulator and a substrate of MDM2. Mol Cell. 2009;35:316–26. doi: 10.1016/j.molcel.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]