

Abstract
Ginseng (Panax ginseng C.A. Mey.) is commonly used in traditional oriental medicine for its wide spectrum of medicinal properties, including anti-inflammatory, antitumorigenic, adaptogenic and anti-aging properties. 20(S)-Protopanaxadiol (PPD), the main intestinal metabolite of ginsenosides, is one of the active ingredients in ginseng. In this study, we aimed to investigate the neuroprotective effects of PPD on PC12 cells; however, the underlying mechanisms remain elusive. We examined cell viability by MTT assay and the morphological changes of PC12 cells following glutamate-induced cell damage and evaluated the anti-apoptotic effects of PPD using Hoechst 33258 staining, western blot analysis and Muse™ Cell Analyzer and the antioxidant effects of PPD using FACS analysis and immunofluorescence. Furthermore, PPD exerted protective effects on PC12 cells via the inhibition of mitochondrial damage against glutamate-induced excitotoxicity using immunofluorescence, electron microscopy and FACS analysis. We demonstrate that treatment with PPD suppresses apoptosis, which contributes to the neuroprotective effects of PPD against glutamate-induced excitotoxicity in PC12 cells. Treatment with PPD inhibited nuclear condensation and decreased the number of Annexin V-positive cells. In addition, PPD increased antioxidant activity and mitochondrial homeostasis in the glutamate-exposed cells. These antioxidant effects were responsible for the neuroprotection and enhanced mitochondrial function following treatment with PPD. Furthermore, PD inhibited the glutamate-induced morphological changes in the mitochondria and scavenged the mitochondrial and cytosolic reactive oxygen species (ROS) induced by glutamate. In addition, mitochondrial function was significantly improved in terms of mitochondrial membrane potential (MMP) and enhanced mitochondrial mass compared with the cells exposed to glutamate and not treated with PPD. Taken together, the findings of our study indicate that the antioxidant effects and the enhanced mitochondrial function triggered by PPD contribute to the inhibition of apoptosis, thus leading to a neuroprotective response, as a novel survival mechanism.
Keywords: 20(S)-protopanaxadiol, mitochondrial membrane potential, mitochondrial mass, reactive oxygen species
Introduction
Ginsenosides, or ginseng saponins, are the major components of ginseng (Panax ginseng C.A. Mey.) and are believed to be responsible for the majority of the beneficial effects of ginseng (1). The pharmacologically active ginsenosides reportedly have diverse beneficial effects on the circulatory, endocrine, immune and central nervous systems (2). For example, the ginsenosides Rb1 and Rg1 have been shown to improve the function of neurotransmitters, such as acetylcholine (3,4) as well as to exert neuroprotective and neurite outgrowth-promoting effects in cultured neurons (5). In addition, cultured neurons treated with ginsenoside Rd demonstrated a higher survival rate against excitotoxicity and oxidative stress-induced injury (6–8). Ginsenoside Rg3 has been shown to attenuate cell damage induced by neurotoxicity and to inhibit the overproduction of nitric oxide and malondialdehyde (MDA) formation induced by glutamate (9,10). Of note, it has been demonstrated that ginsenosides prevent neuronal cell death during ischemia and glutamate-induced excitotoxicity (11), and enhance neurological function recovery while reducing the infarct size (12). In animals, the application of ginsenosides has been shown to rescue neurons in the forebrain from cell death and to prevent myocardial infarction. A recent study suggested that the purified ginsenoside, 20(S)-protopanaxadiol (PPD), exerted protective effects against human lung cancer cells and permanent focal cerebral ischemic injury in rats (13,50). The molecular composition of PPD (Fig. 1) has been suggested to play a role in regulating Ca2+ levels, as well as the activity of superoxide dismutase (SOD) and nitric oxide synthase (NOS) (14–16). However, the precise mechanisms responsible for the neuroprotective effects of PPD remain poorly understood.
Figure 1.
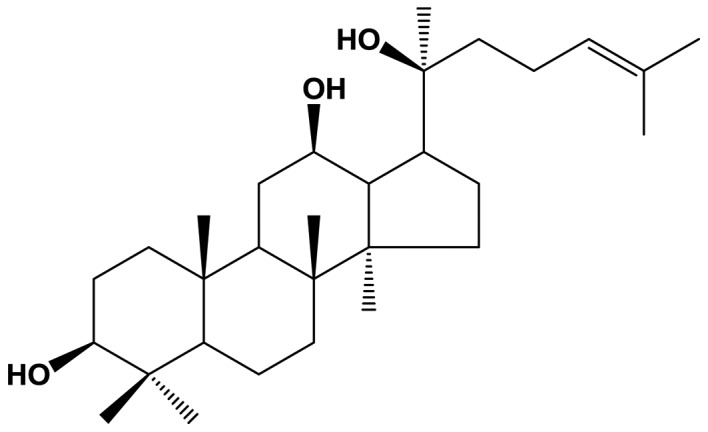
Chemical structure of 20(S)-protopanaxadiol (PPD).
The mitochondria play a critical role in maintaining cellular function (17). Studies have shown that ginsenosides exert preventive effects against mitochondrial dysfunction (18), and that they enhance cell longevity (19,20), possibly by improving mitochondrial quality control (21). The regulation of reactive oxygen species (ROS) production by mitochondria plays a critical role in neurons by affecting the homeostasis of mitochondrial morphology and function. In addition, ginsenoside Rg3 has been shown to exert protective effects on mitochondrial function and energy status during ischemia/reperfusion in the rat brain (22). Ischemia/reperfusion injury is related to ROS production and mitochondrial damage, which ultimately lead to cellular damage (23).
In the present study, we demonstrate that PPD exerts anti-apoptotic effects through its antioxidant activity, and that PPD prevents glutamate-induced excitotoxicity through mitochondrial homeostasis in PC12 cells. Our data suggest that PPD may prove to be a novel therapeutic agent which may provide neuroprotection and prevent mitochondrial dysfunction.
Materials and methods
Materials
PPD (chemical structure shown in Fig. 1) was HPLC grade and purchased from the Ambo Institute (Seoul, Korea). All chemicals and solvents used were of the highest analytical grade available. Cell culture supplies and media, fetal bovine serum (FBS), phosphate-buffered saline (PBS) and penicillin-streptomycin were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Anti-cleaved capase-3 antibody (#9664) was purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and Hoechst 33258 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Annexin V-FITC was purchased from BD Biosciences (Flanklin Lakes, NJ, USA). Dihydroethidium (DHE) was purchased from Calbiochem (EMD Millipore; Billerica, MA, USA). Tetramethylrhodamine ethyl ester (TMRE), MitoSOX™ Red and MitoTracker Green FM were purchased from Invitrogen (Waltham, MA, USA).
Cell culture
The PC12 cell line was purchased from the American Tissue Culture Collection (ATCC; Rockville, MD, USA). The cells were cultured under sterile conditions at 37°C in a humid environment with 5% of CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (both from Thermo Fisher Scientific), 4 mM glutamine, 100 U/ml penicillin and 100 mg/ml streptomycin. The cultures were regularly checked and trypsinized when the cell confluence reached 85%.
MTT assay
To determine the half maximal inhibitory concentration (IC50 value) of glutamate on the PC12 cells, 5×103 cells in single cell suspensions were seeded in individual wells of 96-well plates and incubated for 24 h at 37°C prior to exposure to glutamate at the indicated concentrations (1, 5 and 10 mM) for 24 h. After determining that the glutamate IC50 value was 5 mM, the cells were exposed to 5 mM glutamate for 24 h, 10 μM PPD, or a combination of both (5 mM glutamate plus 10 μM PPD). MTT solution was added to each well followed by incubation for 4 h at 37°C prior to removing the culture medium. DMSO was then added and mixed for 30 min at room temperature. Cell viability was determined by measuring the absorbance at 562 nm. The cell viability for each group was calculated as a percentage of that of the control group.
Morphological observation by microscopy
The cells were seeded at a density of 1×105 cells/well into 24-well plates and were left to grow exponentially at 37°C in a 5% CO2 atmosphere. The PC12 cells were then treated with the different drugs [untreated control (U), glutamine (Glut), PPD or Glut + PPD], and incubated for 12 h. Subsequently, the cells were fixed with 4% paraformaldehyde, washed with PBS 3 times and observed under an inverted microscope (Olympus CKX41; Olympus Corp., Tokyo, Japan).
Hoechst 33258 staining
To evaluate cell apoptosis, 1×106 cells/well were plated on a coverslip in a 12-well plate in triplicate. The cells were treated with the different drugs [U, Glut, PPD or Glut + PPD] and kept in a CO2 incubator at 37°C for 24 h. Following incubation and the addition of fresh medium, the cells were incubated with 5 μl/well of Hoechst 33258 (Sigma-Aldrich) solution at 37°C for 10 min, followed by observation under a laser confocal microscope (LSM-700; Carl Zeiss, Oberkochen, Germany). Strong fluorescence with shrunken nuclei was observed in the apoptotic cells, whereas weak fluorescence with normal-sized nuclei was observed in the non-apoptotic cells. The quantification of apoptotic cells was performed by capturing images in random fields and counting at least 200 cells in 4 random fields in each well.
Western blot analysis
Briefly, 30–50 μg of protein were resolved by 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then electro-blotted onto polyvinylidene difluoride membranes for western blot analysis. The blots were probed with 1:1,000-diluted primary antibodies [anti-cleaved caspase-3 (#9664; Cell Signaling Technology, Inc.), actin (sc-7210; Santa Cruz Biotechnology, Dallas, TX, USA)] overnight at 4°C, followed by horseradish peroxidase-conjugated secondary antibodies (anti-rabbit IgG, HRP-linked antibody, #7074; Cell Signaling Technology, Inc.). The proteins were then visualized using enhanced chemiluminescent reagent (ECL; Millipore Corp., Billerica, MA, USA) and exposure to X-ray film.
Flow cytometric analysis
The cells were analyzed by flow cytometry using a BD FACSCanto II flow cytometer as indicated by the manufacturer (BD Biosciences). Following 2 washes with PBS, the cells were fixed in 4% paraformaldehyde for 10 min at 37°C and permeabilized with 0.25% Triton X-100 in PBS for 10 min. The cells were stained with individual dye for 1 to 2 h at 4°C and the secondary antibodies (1:100) for 1 h on ice. Following 2 washes with PBS, the cells were fixed in 4% para-formaldehyde and assayed immediately. Flow cytometry data were collected using 10,000–30,000 cells and were analyzed using FlowJo software (Tree Star, Inc., Ashland, OR, USA).
Measurement of ROS production
The mitochondrial ROS levels were measured using the mitochondrion-specific fluorescent hydroethidine-derivative dye, MitoSOX Red (10 μM; Invitrogen), as previously described (24). The cells were exposed to glutamate with or without treatment with PPD and then incubated with MitoSOX Red for 30 min at 37°C in 5% CO2. The cells were washed twice with PBS, fixed with 4% paraformaldehyde, and analyzed under a confocal microscope. To measure the intracellular ROS levels, the cells were incubated for 30 min in Krebs-HEPES buffer containing DHE (Calbiochem) washed twice, and analyzed using a flow cytometer.
Immunofluorescence staining
The cells (1×106 cells/well) were prepared on sterilized glass coverslips (BD Biosciences) in triplicate. Following 24 h of treatment with the drugs (U, Glut, PPD or Glut + PPD), the cells were fixed in 4% paraformaldehyde in PBS for 10 min, permeabilized with 0.25% Triton X-100 in PBS for 10 min, and incubated with the primary antibody (Tomm20, ab56783; Cambridge, MA, USA) for 2 h at room temperature. The cells were washed to remove the excess primary antibody, and incubated with the appropriate fluorescently-labeled secondary antibodies for 1 h at room temperature. The nuclei were stained with DAPI and incubated for 5 min. After mounting, fluorescence images were captured using a confocal microscope (LSM 700; Carl Zeiss). To quantify the immunoreacted cells, the fluorescence intensity was measured in 10 randomly selected images.
Analysis of apoptosis
The determine cell apoptosis, monolayer cultures of PC12 cells treated with the different drugs [U, Glut, PPD and Glut + PPD] were supplemented with nutrient medium. The cultures were incubated for 24 h. The dead cells were analyzed prior to and subsequent to exposure to glutamate and treatment with PPD using the Muse™ Annexin V and Dead Cell Assay kit (Muse™Cell Analyzer; Millipore Corp.) according to the manufacturer's instructions.
Transmission electron microscopy (TEM)
For TEM analysis, the cells were collected and fixed with 4% paraformaldehyde and 2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) for 2 h, post-fixed with 1% osmium tetroxide for 1 h, washed, and stained for 1 h in 3% aqueous uranyl acetate. The samples were then washed again, dehydrated with graded alcohol, and embedded in Epon-Araldite resin (Canemco, Inc., Lakefield, QC, Canada). Ultrathin sections were cut on an ultramicrotome (Reichert-Jung, Inc., Cambridge, UK), counterstained with 0.3% lead citrate, and examined under a transmission electron microscope (HT7700; Hitachi, Ltd., Tokyo, Japan).
Measurement of mitochondrial membrane potential (MMP or ΔΨm)
The ΔΨm of intact cells was measured as previously described (25), with some modifications. Briefly, the cells were washed with PBS and trypsinized. The protein concentration of the cells was adjusted to 0.2 mg/ml in DMEM without phenol red (Invitrogen Life Technologies), FBS and antibiotics. TMRE (200 nM; T669; Invitrogen-Molecular Probes) was added to the cell suspension. The cells were incubated at 37°C for 30 min in the dark. ΔΨm was measured by flow cytometry, and the data were analyzed using FlowJo software. TMRE fluorescence was measured using the FL2 channel (582 nm).
Determination of mitochondrial mass and visualization of mitochondria in living cells
Mitochondrial mass was measured using fluorescence levels following staining with MitoTracker Green FM (100 nM; M7514; Invitrogen) at 37°C for 30 min. Subsequently, the cells were washed once in PBS and promptly used for flow cytometry. The percentage and mean fluorescence intensity level of the mass were calculated for each sample.
To visualize the mitochondria, the mitochondrial marker, Tomm20, was used, and was added with DMEM at 37°C for 30 min. The cover glasses were then washed twice with PBS and images were immediately captured using a confocal laser scanning microscope (LSM 700; Carl Zeiss). The images were evaluated as best-focus intensity projections.
Statistical analysis
Data obtained from independent experiments (means ± SD) were analyzed using a two-tailed Student's t-test. Differences were considered significant if P<0.05. 95% confidence intervals were computed, 1.96 x standard error in each direction. Statistical significance was evaluated using a log-rank (Mantel-Cox) test.
Results
Effect of PPD on cell viability
To determine the appropriate concentration of glutamate, the PC12 cells were exposed to various concentrations of glutamate for 24 h. Glutamate (1–10 mM) gradually reduced cell viability in a dose-dependent manner, and the cell viability (as shown by MTT assay) decreased by approximately 70% in the cells incubated with 5 mM glutamate for 24 h (Fig. 2A). Hence, the concentration of 5 mM glutamate was selected for use in the subsequent assays. The cells exposed to 5 mM glutamate were pre-treated with 5, 10 or 20 μM PPD (Fig. 2B). We wished to examine the possible beneficial effects of PPD on the viability of glutamate-exposed PC12 cells. The exposure of the neuronal PC12 cells to glutamate for 24 h resulted in cell damage, as shown by the changes in cell morphology, such as cell shrinkage (Fig. 2C). As shown by MTT assay, pre-treatment with PPD resulted in a higher cell viability compared with the glutamate-exposed cells not treated with PPD (Fig. 2D). Thus, treatment with PPD significantly improved the viability of the glutamate-exposed cells.
Figure 2.

20(S)-Protopanaxadiol (PPD) protects neuronal cells against glutamate-induced excitotoxicity. (A) PC12 cells were treated with various concentration of glutamate for 24 h and cell viability was determined by MTT assay. (B) PC12 cells were treated with 5, 10 and 20 μM PPD for 24 h. (C) PC12 cells were exposed to glutamate for 24 h in the presence or absence of PPD. At the end of treatment, cell morphology was observed under an inverted microscope. (D) Cell viability was determined by MTT assay. ***P<0.001 (two-tailed Student's t-test), compared with (A) the untreated control (U) or (C) exposure to glutamate alone (Glut).
PPD prevents glutamate-induced apoptosis and cytotoxicity
We then wished to examine whether glutamate-induced cytotoxicity is mediated by apoptotic processes, using Hoechst 33342 staining, Annexin V staining and caspase-3 antibody. Hoechst 33342 staining revealed that treatment with PPD significantly decreased the amount of condensed chromatin compared with the glutamate-exposed cells not treated with PPD (Fig. 3A). Pre-treatment with PPD also significantly decreased the percentage of Annexin V-positive cells, compared the glutamate-exposed cells not treated with PPD (Fig. 3B). The results of western blot analysis also revealed that treatment with PPD prevented the glutamate-induced cleavage of caspase-3 (Fig. 3C), consistent with the results regarding the inhibition of apoptosis in the PPD pre-treated cells. Thus, PPD plays a crucial role in reducing the cytotoxic effects of glutamate by inhibiting apoptosis.
Figure 3.

20(S)-Protopanaxadiol (PPD) suppresses glutamate-induced apoptosis. PC12 cells were treated with or without PPD for 2 h and incubated with glutamate for 24 h. (A) Changes in nuclear morphology examined by Hoechst 33342 nuclear staining (left panel). Arrows indicate apoptotic nuclei. Bar graphs represent the percentage of apoptotic cells counted in each group (right panel). (B) Cells were stained with Annexin V-FITC and analyzed using a cell sorting system with Muse™. (C) Whole cell lysates were prepared and immunoblotted with antibodies against cleaved caspase-3. Actin was used as a loading control. **P<0.01 (two-tailed Student's t-test), compared to exposure to glutamate alone (Glut). U, untreated control.
Effects of PPD on cytosolic and mitochondrial ROS generation
Given that ginsenosides are able to reduce ROS generation, and that the glutamate-induced apoptotic pathway is linked to ROS of cytosolic and mitochondrial origin (26), in this study, we investigated the effects of PPD on the accumulation of ROS in glutamate-exposed PC12 cells. We measured cytosolic and mitochondrial ROS formation by flow cytometry and fluorescence microscopy using DHE and MitoSOX Red, a fluoroprobe that selectively detects ROS formation in live cells and mitochondria. The addition of PPD suppressed the glutamate-induced production of ROS, including the DHE-reactive superoxide anion (O2−) (Fig. 4A). Cytosolic ROS was measured by flow cytometric analysis of the DHE-stained cells. In additional experiments, mitochondrial ROS production was estimated using MitoSOX Red, a cell-permeable probe that is non-fluorescent when chemically reduced, but fluoresces following cellular oxidation and the removal of acetate groups by cellular esterases (Fig. 4B). The mitochondrial ROS levels were significantly decreased following treatment with PPD (Fig. 4C). PPD scavenged the mitochondrial ROS produced by glutamate-induced mitochondrial damage. These results suggest that PPD exerts major effects on cell survival through its antioxidant effects.
Figure 4.

Antioxidant effects of 20(S)-protopanaxadiol (PPD). (A) PC12 cells were pre-treated with PPD for 2 h and incubated with glutamate for 24 h. The cells were then incubated with dihydroethidium (DHE; 10 μM) for 30 min, washed thoroughly, and immediately analyzed by flow cytometry for cytosolic ROS production. (Left panel) Representative histogram. (Right panel) Data are presented as the average mean fluorescence intensity (MFI). (B and C) The cells were stained with MitoSOX Red for 30 min and analyzed for mitochondrial ROS by confocal microscopy and flow cytometry. Scale bar, 20 μm. ***P<0.001 (two-tailed Student's t-test), compared to exposure to glutamate alone (Glut). U, untreated control.
PPD protects the mitochondria against glutamate-induced damage
A recent study reported that glutamate is associated with mitochondrial damage (27). Thus, to determine whether glutamate induces mitochondrial damage, we first examined mitochondrial morphology. Mitochondrial morphology was examined using Tomm20 (mitochondrial outer membrane) staining following treatment with glutamate for 24 h. A significant change in mitochondrial morphology was observed at 24 h in the glutamate-exposed cells. Treatment of the glutamate-exposed cells to PPD led to a substantial increase in the proportion of mitochondria, compared to the cells exposed to glutamate and not treated with PPD (Fig. 5A). We also analyzed the changes in mitochondrial morphology using TEM (Fig. 5B). Ultrastructural imaging revealed that the rate of swollen mitochondria and broken mitochondrial matrix was increased in the glutamate-exposed cells, whereas treatment with PPD resulted in an apparent increase in the number normal mitochondria compared with the glutamate-exposed cells not treated with PPD. Thus, treatment with PPD may protect the mitochondria against glutamate-induced damage.
Figure 5.

Effects of 20(S)-protopanaxadiol (PPD) on mitochondria. (A) PC12 cells were stained with the mitochondrial marker, anti-Tomm20 (100 nM), and detected by confocal microscopy. A magnified view of corresponding boxed areas is shown in the lower panels. Scale bar, 5 μm. (B) Images of mitochondrial ultrastructure were obtained using transmission electron microscopy (TEM) in PC12 cells. Representative TEM images from 3 independent experiments are shown. Scale bar, 100 μm. U, untreated control; Glut, glutamate alone.
Effects of PPD on mitochondrial function
Treatment with ginsenosides has been shown to improve mitochondrial function in various cells (28,29). However, in neuronal cells, it is not known whether treatment with PPD is capable of controlling mitochondrial function, density and morphology, particularly following exposure to glutamate. Thus, in this study, to examine the protective role of PPD in maintaining mitochondrial function, we analyzed the MMP, as well as the changes in mitochondrial mass. The MMP was measured by flow cytometry using a potentiometric fluorescent probe (TRME) following exposure of the cells to glutamate (Fig. 6A). Significant depolarization of the mitochondria was observed at 24 h and sustained until 24 h. The MMP was significantly higher in the cells pre-treated with PPD than in the cells exposed to glutamate and not treated with PPD. PPD protected the cells against glutamate-induced mitochondrial damage by regulating the MMP.
Figure 6.

20(S)-Protopanaxadiol (PPD) protects against glutamate-induced mitochondrial dysfunction. (A) The mitochondrial membrane potential (MMP or ΔΨm) was measured by flow cytometric analysis following staining with potentiometric fluorescent probe (TMRE, 200 nM). (Left panel) Representative histogram. (Right panel) Data are presented as the average mean fluorescence intensity (MFI). (B) MitoTracker Green FM fluorescence signals were assessed by flow cytometric analysis. Bar graph indicating the mitochondrial mass as MFI. **P<0.01 and ***P<0.001 (two-tailed Student's t-test), compared with glutamate. U, untreated control; Glut, glutamate alone.
To further elucidate the possible mechanisms underlying the effects of PPD, we evaluated the mitochondrial mass. MitoTracker green staining indicated that treatment with PPD controlled mitochondrial mass following exposure to glutamate. The mitochondrial mass decreased by approximately 70% in cells exposed to glutamate and not treated with PPD (Fig. 6B). Thus, PPD improved mitochondrial function, as indicated by changes in MMP and mitochondrial mass, suggesting that PPD plays a role in maintaining mitochondrial functional.
Discussion
Potential mechanisms, such as oxidative stress, mitochondrial aberrations and inflammation, which lead to the degeneration and death of neurons, are considered important factors in the pathogenesis of a range of neurological disorders (30–32). Ameliorating therapeutic modulators of these universal mechanisms may provide new insight into therapeutic strategies for these fatal diseases by delaying the disease onset or progression. The interactions of glutamate with specific membrane receptors has been implicated in a number of neurological functions regulated by neuronal cells in the central nervous system (CNS), including synapse re-formation, perception and muscle movement (33–35). However, it has been suggested that glutamate participates in neuronal cell damage associated with several neurological deficits in the CNS (36). Glutamate, one of the major excitatory neurotransmitters in the brain (37), is the primary inducer of cell death during cerebral ischemia and has a direct neurotoxic effect by increasing ROS production (38). Glutamate from damaged cells is delivered to adjacent cells and results in excessive ion influx through the NMDA receptor, a specific glutamate receptor (39). Excessive glutamate stimulation sequentially disrupts cellular ion homeostasis and induces apoptosis. It has also been proposed that an overproduction of oxygen free radicals plays a pivotal role in hypoxic-ischemic injury (40,41).
Previous studies have demonstrated that extracts of ginseng (the root of Panax ginseng) have various biological effects, including anti-carcinogenic activities (42), as well as anticancer (43–45) and neuroprotective activities (46). It has been demonstrated that ginseng extracts enhance mitochondrial function and protect against oxidative stress (47). PPD is the most pharmacologically active ginsenoside that has been evaluated. PPD possesses various biological properties, including anti-inflammatory, antioxidant and antitumor properties (48,49). A recent study found that PPD protected against cerebral ischemia in a model of middle cerebral artery occlusion (MCAO) by reducing free radical formation, lipid peroxidation and calcium overload, and increasing the expression of anti-apoptotic proteins (50). In the present study, we demonstrated the ability of PPD to rescue PC12 cells from glutamate-induced excitotoxicity and examined the cellular events that underlie this effect.
The mitochondria play a potentially important homeostatic role in the molecular events surrounding cell death in pathological situations, such as ischemia and neurodegenerative diseases (51). Mitochondrial death signal results in the opening of the mitochondrial permeability transition pore and the collapse of ΔΨm, leading to the release of various substances, such as cytochrome c, which are associated with cell death. Previous studies have demonstrated that exposure to glutamate is accompanied by a loss of MMP (52), which may lead to ATP depletion (53) and energetic collapse (54–56), and ultimately contributes to cell death. The inhibition of mitochondrial membrane rupture, induced as a consequence of mitochondrial swelling caused by disturbed ionic homeostasis, has been postulated as a cell survival mechanism that follows the MMP (57). Thus, protection of the mitochondria may be involved in PPD-induced neuroprotection.
The present study found that the regulation of mitochondrial homeostasis leads to increased cell viability. In addition, it is speculated that the activation of mitochondrial function with PPD treatment affects overall cell survival. Taken together, the data from the current study strongly suggest that PPD prevents glutamate-induced excitotoxicity through multiple effects targeting mitochondria-dependent events, including the inhibition of ROS generation, the dissipation of MMP and a reduction in mitochondrial density. Our working hypothesis is that PPD functions as a neurotrophic agent to ameliorate the sensory deficit caused by glutamate-induced excitotoxicity through its antioxidant effects and enhances mitochondrial function (Fig. 7). Thus, PPD may have therapeutic value in the treatment of certain neurodegenerative diseases. However, further studies are warranted in order to fully elucidate the mechanisms responsible for its regulatory effects on the function of mitochondria.
Figure 7.

A schematic model of the mechanisms of 20(S)-protopanaxadiol (PPD)-induced neuroprotection. Glutamate induces reactive oxygen species (ROS) accumulation and mitochondrial depolarization causes mitochondrial damage, resulting apoptosis. PPD exerts antioxidant effect and enhances mitochondrial function, and thus exerts neuroprotective effects.
Acknowledgments
The present study was supported by a grant from the Agenda Program (PJ008568), Rural Development Administration, Republic of Korea.
References
- 1.Liu CX, Xiao PG. Recent advances on ginseng research in China. J Ethnopharmacol. 1992;36:27–38. doi: 10.1016/0378-8741(92)90057-X. [DOI] [PubMed] [Google Scholar]
- 2.Nah SY, Kim DH, Rhim H. Ginsenosides: are any of them candidates for drugs acting on the central nervous system? CNS Drug Rev. 2007;13:381–404. doi: 10.1111/j.1527-3458.2007.00023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benishin CG. Actions of ginsenoside Rb1 on choline uptake in central cholinergic nerve endings. Neurochem Int. 1992;21:1–5. doi: 10.1016/0197-0186(92)90061-U. [DOI] [PubMed] [Google Scholar]
- 4.Benishin CG, Lee R, Wang LC, Liu HJ. Effects of ginsenoside Rb1 on central cholinergic metabolism. Pharmacology. 1991;42:223–229. doi: 10.1159/000138801. [DOI] [PubMed] [Google Scholar]
- 5.Kim S, Ahn K, Oh TH, Nah SY, Rhim H. Inhibitory effect of ginsenosides on NMDA receptor-mediated signals in rat hippocampal neurons. Biochem Biophys Res Commun. 2002;296:247–254. doi: 10.1016/S0006-291X(02)00870-7. [DOI] [PubMed] [Google Scholar]
- 6.Zhang C, Du F, Shi M, Ye R, Cheng H, Han J, Ma L, Cao R, Rao Z, Zhao G. Ginsenoside Rd protects neurons against glutamate-induced excitotoxicity by inhibiting Ca(2+) influx. Cell Mol Neurobiol. 2012;32:121–128. doi: 10.1007/s10571-011-9742-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye R, Han J, Kong X, Zhao L, Cao R, Rao Z, Zhao G. Protective effects of ginsenoside Rd on PC12 cells against hydrogen peroxide. Biol Pharm Bull. 2008;31:1923–1927. doi: 10.1248/bpb.31.1923. [DOI] [PubMed] [Google Scholar]
- 8.Ye R, Li N, Han J, Kong X, Cao R, Rao Z, Zhao G. Neuroprotective effects of ginsenoside Rd against oxygen-glucose deprivation in cultured hippocampal neurons. Neurosci Res. 2009;64:306–310. doi: 10.1016/j.neures.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 9.Kim YC, Kim SR, Markelonis GJ, Oh TH. Ginsenosides Rb1 and Rg3 protect cultured rat cortical cells from glutamate-induced neurodegeneration. J Neurosci Res. 1998;53:426–432. doi: 10.1002/(SICI)1097-4547(19980815)53:4<426::AID-JNR4>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 10.Nah SY. Ginseng ginsenoside pharmacology in the nervous system: involvement in the regulation of ion channels and receptors. Front Physiol. 2014;5:98. doi: 10.3389/fphys.2014.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jang S, Ryu JH, Kim DH, Oh S. Changes of (3H)MK-801, (3H) muscimol and (3H)flunitrazepam binding in rat brain by the prolonged ventricular infusion of transformed ginsenosides. Neurochem Res. 2004;29:2257–2266. doi: 10.1007/s11064-004-7034-2. [DOI] [PubMed] [Google Scholar]
- 12.Zhang E, Shen J, So KF. Chinese traditional medicine and adult neurogenesis in the hippocampus. J Tradit Complement Med. 2014;4:77–81. doi: 10.4103/2225-4110.130372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang YL, Zhang R, Xu HL, Yu XF, Qu SC, Sui DY. 20(S)-protopanaxadiol triggers mitochondrial-mediated apoptosis in human lung adenocarcinoma A549 cells via inhibiting the PI3K/Akt signaling pathway. Am J Chin Med. 2013;41:1137–1152. doi: 10.1142/S0192415X13500778. [DOI] [PubMed] [Google Scholar]
- 14.Lee DC, Lau AS. Effects of Panax ginseng on tumor necrosis factor-α-mediated inflammation: a mini-review. Molecules. 2011;16:2802–2816. doi: 10.3390/molecules16042802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang MS, Lee SG, Rho HM. Transcriptional activation of Cu/Zn superoxide dismutase and catalase genes by panaxadiol ginsenosides extracted from Panax ginseng. Phytother Res. 1999;13:641–644. doi: 10.1002/(SICI)1099-1573(199912)13:8<641::AID-PTR527>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 16.Leung KW, Leung FP, Mak NK, Tombran-Tink J, Huang Y, Wong RN. Protopanaxadiol and protopanaxatriol bind to glucocorticoid and oestrogen receptors in endothelial cells. Br J Pharmacol. 2009;156:626–637. doi: 10.1111/j.1476-5381.2008.00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gutierrez J, Ballinger SW, Darley-Usmar VM, Landar A. Free radicals, mitochondria, and oxidized lipids: the emerging role in signal transduction in vascular cells. Circ Res. 2006;99:924–932. doi: 10.1161/01.RES.0000248212.86638.e9. [DOI] [PubMed] [Google Scholar]
- 18.Liu D, Zhang H, Gu W, Liu Y, Zhang M. Neuroprotective effects of ginsenoside Rb1 on high glucose-induced neurotoxicity in primary cultured rat hippocampal neurons. PLoS One. 2013;8:e79399. doi: 10.1371/journal.pone.0079399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Attele AS, Wu JA, Yuan CS. Ginseng pharmacology: Multiple constituents and multiple actions. Biochem Pharmacol. 1999;58:1685–1693. doi: 10.1016/S0006-2952(99)00212-9. [DOI] [PubMed] [Google Scholar]
- 20.Liu WK, Xu SX, Che CT. Anti-proliferative effect of ginseng saponins on human prostate cancer cell line. Life Sci. 2000;67:1297–1306. doi: 10.1016/S0024-3205(00)00720-7. [DOI] [PubMed] [Google Scholar]
- 21.Sun M, Huang C, Wang C, Zheng J, Zhang P, Xu Y, Chen H, Shen W. Ginsenoside Rg3 improves cardiac mitochondrial population quality: mimetic exercise training. Biochem Biophys Res Commun. 2013;441:169–174. doi: 10.1016/j.bbrc.2013.10.039. [DOI] [PubMed] [Google Scholar]
- 22.Tian J, Zhang S, Li G, Liu Z, Xu B. 20(S)-ginsenoside Rg3, a neuroprotective agent, inhibits mitochondrial permeability transition pores in rat brain. Phytother Res. 2009;23:486–491. doi: 10.1002/ptr.2653. [DOI] [PubMed] [Google Scholar]
- 23.Perrelli MG, Pagliaro P, Penna C. Ischemia/reperfusion injury and cardioprotective mechanisms: role of mitochondria and reactive oxygen species. World J Cardiol. 2011;3:186–200. doi: 10.4330/wjc.v3.i6.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim JJ, Lee HM, Shin DM, Kim W, Yuk JM, Jin HS, Lee SH, Cha GH, Kim JM, Lee ZW, et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe. 2012;11:457–468. doi: 10.1016/j.chom.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Bauerfeld CP, Rastogi R, Pirockinaite G, Lee I, Hüttemann M, Monks B, Birnbaum MJ, Franchi L, Nuñez G, Samavati L. TLR4-mediated AKT activation is MyD88/TRIF dependent and critical for induction of oxidative phosphorylation and mitochondrial transcription factor A in murine macrophages. J Immunol. 2012;188:2847–2857. doi: 10.4049/jimmunol.1102157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–418. doi: 10.1023/A:1009616228304. [DOI] [PubMed] [Google Scholar]
- 27.Kumari S, Mehta SL, Li PA. Glutamate induces mitochondrial dynamic imbalance and autophagy activation: preventive effects of selenium. PLoS One. 2012;7:e39382. doi: 10.1371/journal.pone.0039382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Min JK, Kim JH, Cho YL, Maeng YS, Lee SJ, Pyun BJ, Kim YM, Park JH, Kwon YG. 20(S)-Ginsenoside Rg3 prevents endothelial cell apoptosis via inhibition of a mitochondrial caspase pathway. Biochem Biophys Res Commun. 2006;349:987–994. doi: 10.1016/j.bbrc.2006.08.129. [DOI] [PubMed] [Google Scholar]
- 29.Tamura T, Cui X, Sakaguchi N, Akashi M. Ginsenoside Rd prevents and rescues rat intestinal epithelial cells from irradiation-induced apoptosis. Food Chem Toxicol. 2008;46:3080–3089. doi: 10.1016/j.fct.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 30.Floyd RA. Antioxidants, oxidative stress, and degenerative neurological disorders. Proc Soc Exp Biol Med. 1999;222:236–245. doi: 10.1046/j.1525-1373.1999.d01-140.x. [DOI] [PubMed] [Google Scholar]
- 31.Tarnopolsky MA, Beal MF. Potential for creatine and other therapies targeting cellular energy dysfunction in neurological disorders. Ann Neurol. 2001;49:561–574. doi: 10.1002/ana.1028. [DOI] [PubMed] [Google Scholar]
- 32.Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C. The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci. 2002;202:13–23. doi: 10.1016/S0022-510X(02)00207-1. [DOI] [PubMed] [Google Scholar]
- 33.Headley PM, Grillner S. Excitatory amino acids and synaptic transmission: the evidence for a physiological function. Trends Pharmacol Sci. 1990;11:205–211. doi: 10.1016/0165-6147(90)90116-P. [DOI] [PubMed] [Google Scholar]
- 34.Zeng LH, Ouyang Y, Gazit V, Cirrito JR, Jansen LA, Ess KC, Yamada KA, Wozniak DF, Holtzman DM, Gutmann DH, et al. Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2007;28:184–196. doi: 10.1016/j.nbd.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Poul E, Boléa C, Girard F, Poli S, Charvin D, Campo B, Bortoli J, Bessif A, Luo B, Koser AJ, et al. A potent and selective metabotropic glutamate receptor 4 positive allosteric modulator improves movement in rodent models of Parkinson's disease. J Pharmacol Exp Ther. 2012;343:167–177. doi: 10.1124/jpet.112.196063. [DOI] [PubMed] [Google Scholar]
- 36.Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van den Pol AN, Gao XB, Patrylo PR, Ghosh PK, Obrietan K. Glutamate inhibits GABA excitatory activity in developing neurons. J Neurosci. 1998;18:10749–10761. doi: 10.1523/JNEUROSCI.18-24-10749.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gliyazova NS, Huh EY, Ibeanu GC. A novel phenoxy thiophene sulphonamide molecule protects against glutamate evoked oxidative injury in a neuronal cell model. BMC Neurosci. 2013;14:93. doi: 10.1186/1471-2202-14-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salińska E, Danysz W, Łazarewicz JW. The role of excitotoxicity in neurodegeneration. Folia Neuropathol. 2005;43:322–339. [PubMed] [Google Scholar]
- 40.Baker JE. Oxidative stress and adaptation of the infant heart to hypoxia and ischemia. Antioxid Redox Signal. 2004;6:423–429. doi: 10.1089/152308604322899495. [DOI] [PubMed] [Google Scholar]
- 41.Peng YW, Buller CL, Charpie JR. Impact of N-acetylcysteine on neonatal cardiomyocyte ischemia-reperfusion injury. Pediatr Res. 2011;70:61–66. doi: 10.1203/PDR.0b013e31821b1a92. [DOI] [PubMed] [Google Scholar]
- 42.Bi X, Xia X, Mou T, Jiang B, Fan D, Wang P, Liu Y, Hou Y, Zhao Y. Anti-tumor activity of three ginsenoside derivatives in lung cancer is associated with Wnt/β-catenin signaling inhibition. Eur J Pharmacol. 2014;742:145–152. doi: 10.1016/j.ejphar.2014.08.032. [DOI] [PubMed] [Google Scholar]
- 43.Wang W, Rayburn ER, Hao M, Zhao Y, Hill DL, Zhang R, Wang H. Experimental therapy of prostate cancer with novel natural product anti-cancer ginsenosides. Prostate. 2008;68:809–819. doi: 10.1002/pros.20742. [DOI] [PubMed] [Google Scholar]
- 44.Lee JJ, Kwon HK, Jung IH, Cho YB, Kim KJ, Kim JL. Anticancer activities of ginseng extract fermented with Phellinus linteus. Mycobiology. 2009;37:21–27. doi: 10.4489/MYCO.2009.37.1.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Xu Y, Zhu Y, Li X. Anti-cancer effects of ginsenoside compound k on pediatric acute myeloid leukemia cells. Cancer Cell Int. 2013;13:24. doi: 10.1186/1475-2867-13-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim S, Lee Y, Cho J. Korean red ginseng extract exhibits neuroprotective effects through inhibition of apoptotic cell death. Biol Pharm Bull. 2014;37:938–946. doi: 10.1248/bpb.b13-00880. [DOI] [PubMed] [Google Scholar]
- 47.Dong GZ, Jang EJ, Kang SH, Cho IJ, Park SD, Kim SC, Kim YW. Red ginseng abrogates oxidative stress via mitochondria protection mediated by LKB1-AMPK pathway. BMC Complement Altern Med. 2013;13:64. doi: 10.1186/1472-6882-13-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lü JM, Yao Q, Chen C. Ginseng compounds: an update on their molecular mechanisms and medical applications. Curr Vasc Pharmacol. 2009;7:293–302. doi: 10.2174/157016109788340767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ren HC, Sun JG, Wang GJ, A JY, Xie HT, Zha WB, Yan B, Sun FZ, Hao HP, Gu SH. Sensitive determination of 20(S)-protopanaxadiol in rat plasma using HPLC-APCI-MS: application of pharmacokinetic study in rats. J Pharm Biomed Anal. 2008;48:1476–1480. doi: 10.1016/j.jpba.2008.09.045. [DOI] [PubMed] [Google Scholar]
- 50.Xu H, Yu X, Qu S, Chen Y, Wang Z, Sui D. Protective effect of 20(S)-protopanaxadiol saponins, isolated from Pana quinquefolium, on permanent focal cerebral ischemic injury in rats. Exp Ther Med. 2014;7:165–170. doi: 10.3892/etm.2013.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fiskum G, Murphy AN, Beal MF. Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. J Cereb Blood Flow Metab. 1999;19:351–369. doi: 10.1097/00004647-199904000-00001. [DOI] [PubMed] [Google Scholar]
- 52.Tirosh O, Sen CK, Roy S, Packer L. Cellular and mitochondrial changes in glutamate-induced HT4 neuronal cell death. Neuroscience. 2000;97:531–541. doi: 10.1016/S0306-4522(00)00028-2. [DOI] [PubMed] [Google Scholar]
- 53.Almeida A, Bolaños JP. A transient inhibition of mitochondrial ATP synthesis by nitric oxide synthase activation triggered apoptosis in primary cortical neurons. J Neurochem. 2001;77:676–690. doi: 10.1046/j.1471-4159.2001.00276.x. [DOI] [PubMed] [Google Scholar]
- 54.Khodorov B, Pinelis V, Vergun O, Storozhevykh T, Vinskaya N. Mitochondrial deenergization underlies neuronal calcium overload following a prolonged glutamate challenge. FEBS Lett. 1996;397:230–234. doi: 10.1016/S0014-5793(96)01139-8. [DOI] [PubMed] [Google Scholar]
- 55.Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- 56.Vergun O, Keelan J, Khodorov BI, Duchen MR. Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J Physiol. 1999;519:451–466. doi: 10.1111/j.1469-7793.1999.0451m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti F, et al. Mitochondria-ros crosstalk in the control of cell death and aging. J Signal Transduct. 2012;2012:329635. doi: 10.1155/2012/329635. [DOI] [PMC free article] [PubMed] [Google Scholar]