

Abstract
As average life expectancy rises throughout the world, neurodegenerative diseases have emerged as one of the greatest global public heath challenges in modern times. Substantial efforts have been made in researching neurodegenerative diseases over the last few decades, yet their predominantly sporadic nature has made uncovering their etiologies challenging. Mounting evidence has suggested that factors like damage-associated molecular patterns (DAMPs) released by stressed and dying neurons are likely involved in disease pathology and in stimulating chronic activation of microglia that contributes to neuronal oxidative stress and degeneration. This review focuses on how the microglial integrin receptor Mac1 and its downstream effector NADPH oxidase (NOX2) contribute to maintaining chronic neuroinflammation and are crucial in inflammation-driven neurotoxicity in neurodegenerative diseases. Our hope is to provide new insights on novel targets and therapies that could slow or even halt neurodegeneration.
Introduction
Neurodegenerative diseases are characterized by a progressive, yet selective regional neuronal loss, whereby chronic neuroinflammation has been verified to contribute to the degenerative process [1–4]. Microglia are widely considered the predominant resident effector cells of the immune system in the central nervous system (CNS) and become activated in response to stimuli such as chemicals and toxins detected in their microenvironment. Microglia typically are activated until they, and other infiltrating leukocytes that may have been recruited, have sufficiently cleared the insult or source of deleterious stimuli. Yet in condition where the deleterious stimuli persist, this results in unresolved neuroinflammation that can damage proximal neurons. Collateral damage to neurons releases endogenous proteins and cell membrane fragments that can re-stimulate the activation of microglia forming a chronic state of activation known as reactive microgliosis. Evidence suggests that this low-grade chronic neuroinflammation contributes to progressive neurodegeneration in diseases such as Parkinson’s and Alzheimer’s diseases. Though the receptors that detect microbial pathogens are shared among all innate immune cells including microglia, these cells are also attuned to detect endogenous cellular components released by cells during stress, injury and death through the same receptors [5]. Though the structure of these intrinsic and extrinsic stimuli are highly variable, they are thought to activate microglia through receptor-mediated signaling of pattern recognition receptors such as Toll-like receptors (TLRs), Nod-like receptors (NLRs), and RIG-1 like receptors (RLRs) or through scavenger, integrin, or RAGE receptors that have similarly promiscuous binding capabilities [1,6–11].
Traditionally, neuroinflammation was also described through the immunological activation of astrocytes, yet today the functional role of astrocytes during neuroinflammation is less clear. The expression of the above-mentioned receptors by astrocytes has been inconclusive and many of their immune functions are still controversial since they were historically confirmed in vitro using isolation methods that sill resulted in significant microglial contamination that could alter the outcome of the studies. A recent study from our group demonstrated that, contrary to much of the reported literature, astrocytes may not possess the ability to directly recognize innate immune stimuli such as the bacterial endotoxin LPS. In fact, they rather depend on crosstalk with activated microglia to elicit their activation and promote the release of neurotrophic factors as a counterbalance that supports neuronal survival from the collateral damage generated by activated microglia during neuroinflammation [12]. For this reason, we have chosen to focus on microglia with this review. Furthermore, even though circulating monocytes are known infiltrate the CNS and differentiate into microglial-like macrophages during neuroinflammation [13,14], we will also not be addressing the role of these infiltrating leukocytes since they are only thought to play a pivotal role in disease with high-grade neuroinflammation such as traumatic brain injury and multiple sclerosis and are not thought to be as important in conditions of low-grade neuroinflammation.
Neuroinflammation is a self-defense reaction to combat pathogen infection or clear and restore injuries in the CNS; this reaction is initially carried out by microglia and shaped by reactive astrocytes and other infiltrating leukocytes. Neuroinflammatory responses are typically transient and help restore CNS homeostasis, however in pathological conditions neuroinflammation may continue unresolved becoming persistent [15]. Yet the mechanism by which acute neuroinflammation turns to chronic in neurodegenerative diseases remains largely unknown. Here we have reviewed the literature on reactive microgliosis and hypothesize that persistent Mac1 signaling on microglia is required for continuous activation of NADPH oxidase (NOX2), the main catalytic enzyme responsible for generating extracellular superoxide required to maintain self-propelling reactive microgliosis found in chronic neuroinflammation.
Role of Toll-like receptors in the pathogenesis of acute and chronic neuroinflammation
Acute inflammation
Acute neuroinflammation is typically initiated by detecting the pathogen-associated molecular patterns (PAMPs) from microorganisms, or the damage-associated molecular patterns (DAMPs) molecules released from injured or dying neurons. At this stage, microglia are recruited to clear these signals by preventing infection or restoring injuries in the brain [16]. Toll-like receptors are the most extensively studied receptors stimulated by both PAMPs and DAMPs and are readily expressed on microglia [17]. TLRs signal through various adaptor molecules to stimulate the activation of nuclear factor-κB (NF-κB) and the mitogen-activated protein kinases (MAPKs) like extracellular signal-regulated kinase ½ (Erk1/2), p38, and JNK to induce the production of proinflammatory cytokines and chemokines [18]. DAMPs associated with the pathology of several neurodegenerative diseases act on TLRs [19] such as α-synuclein that acts on TLR2 [20] and both β-amyloid [21–23] and HMGB-1 that act on TLR2, TLR4 and TLR9 [24,25]. Furthermore, brains from patients with neurodegenerative diseases or that have undergone normal aging had far greater TLR gene expression and expression of pro-inflammatory genes associated with TLRs signal transduction [26,27] likely due to increased DAMPs during neurodegeneration. These results put forward the hypothesis that an aberrant TLR activation may contribute to the process of aging and neurodegenerative diseases.
Chronic inflammation
It is widely accepted that chronic low-grade neuroinflammation plays a key role in the pathogenesis of neurodegeneration. However, the molecular mechanism mediating chronic neuroinflammation is less clear. To avoid the excessive damage of host cells by harmful and inappropriate inflammatory responses during the acute phase of inflammation, TLR signals are often promptly dampened or terminated through multiple mechanisms, such as dissociation of adaptor complexes, degradation of signaling proteins, and transcriptional regulation (also known TLR tolerance) [28]. Although TLRs have been widely implicated in neuroinflammation, their role in maintaining reactive microgliosis and chronic neuroinflammaiton has not been well studied. For this reason, we suspect that although TLRs are crucial in the initiation of immune responses during the early phases of neurodegenerative diseases, the maintenance of chronic neuroinflammation is likely mediated by another receptor that detects neuronal DAMPs.
Microglial Mac1 is essential in maintaining chronic neuroinflammation
Our group and others have demonstrated in rodents that the integrin receptor Mac1 (also known as CD11b/CD18, complement receptor 3 [CR3], or αMβ2) is essential in maintaining chronic reactive microgliosis and in driving inflammation-mediated neurodegeneration [29–31]. Historically, Mac1 has been recognized as an adhesion molecule that participates in cell signaling during cell-to-cell contact. Yet, during inflammation, Mac1 is required for chemotaxis and phagocytosis in activated neutrophils and macrophages [32,33]. Additionally, the expression of Mac1 is elevated in brains from patients with Alzheimer’s disease [34] and in MPTP animal models of Parkinson’s disease [35]. Most importantly, microglial Mac1 is capable of binding neuronal DAMPs including α-synuclein [36], β-amyloid [8], HMGB-1 [9], and myelin [37] that are typically associated with neurodegeneration to induce signaling and their subsequent activation. Furthermore, in vivo and in vitro studies have shown that the genetic ablation of Mac1 results in decreased neurodegeneration when stimulated with the neurotoxicant MPTP [31] or the administration of the exogenous DAMP peptides α-synuclein [36] and β-amyloid [8]. Based on these findings we suspect Mac1 on microglia is the DAMP receptor responsible for maintaining the self-perpetuating reactive microgliosis required in chronic neuroinflammation (Figure 1; [9]).
Figure 1.
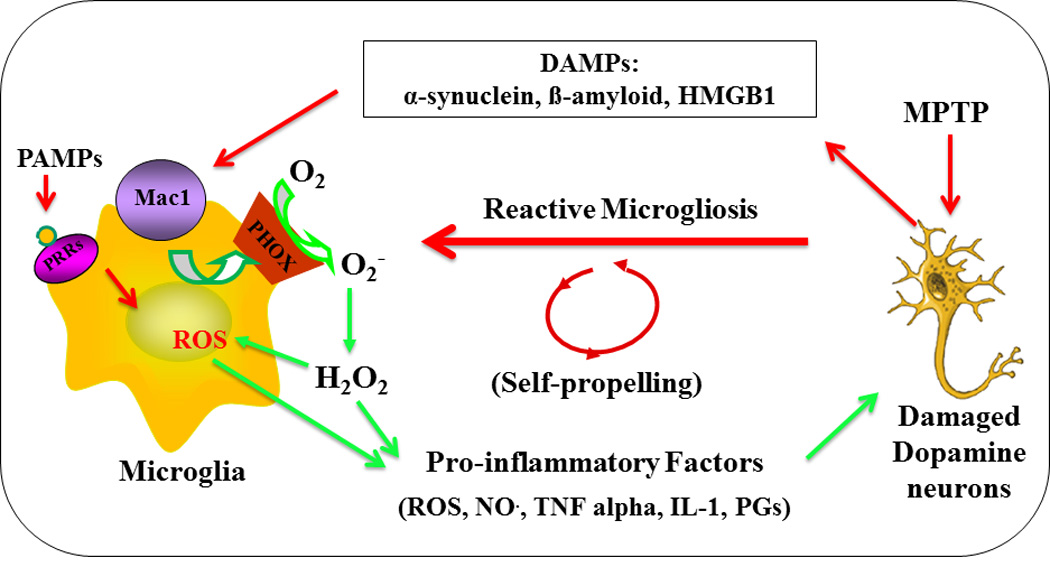
Reactive microgliosis drives chronic and progressive neurotoxicity. Microglia can initiate neurotoxicity by recognizing pro-inflammatory stimuli, such as cytokines, pathogen associated molecular patterns (PAMPs) from microbial pathogens to become activated and producing cytotoxic factors to damage neurons. Damage-associated molecular patterns (DAMPs) released from damaged/dead neurons can sustain microglia activation (reactive microgliosis), which cause further neuronal damage/death. Microglia Mac1 could recognize DAMPs and activate downstream NADPH oxidase (NOX2) to produce superoxide anions and its associated reactive oxygen species (ROS), such as hydrogen peroxide, which play a critical role in reactive microgliosis and driving the chronic neurodegeneration.
Mac1–NOX2 signaling bridges chronic neuroinflammation and progressive neurodegeneration
Recent studies have indicated microglial NOX2 activation as an important downstream effector of Mac1 signaling [9,29,38]. NOX2 is composed of cytosolic subunits (p47phox, p67phox, p40phox, and the small Rho GTPase, Rac1 or Rac2) and membrane-bound subunits p22phox and gp91phox [39]. Upon stimulation, cytosolic subunits of NOX2 translocate and bind to the membrane subunits to assemble the catalytically active form of NOX2 that produces extracellular superoxide [40]. When HMGB-1 binds to Mac1 it induces the expressions of several proinflammatory factors including TNF-α, IL-1β and NO through the activation of NF-κB signal pathway and the production of superoxide through the activation of NOX2 [9]. Further interrogation into the mechanism that links DAMPs binding to Mac1to NOX2 activation using β-amyloid shows that the conformational changes of Mac1 upon binding increases the level of PI3K, phosphorylating p47phox and PIP3 to trigger their translocation from the cytosol to membrane-bound NOX2 to generate superoxide production [8]. Interestingly, when examining the chemotactic role of Mac1 towards aggregated α-synuclein, NOX2 activation was required to produce superoxide that rapidly transmutes into H2O2 that activates tyrosine protein kinase Lyn to phosphorylate the F-actin–associated protein cortactin—mediating actin rearrangement required for directional microglia migration [36].
During chronic neuroinflammation, microglia maintain chronic low-grade neuroinflammation through continuous transcription of mRNA of pro-inflammatory factors such as TNF-α, IL-1β, COX2 as well as NOX2 [41]. The importance of NOX2-generated superoxide in the progression of inflammation-driven degeneration in both in vitro and in vivo models has been established by using microglia derived from NOX2 knockout mice and using NOX2 inhibitors diphenyleneiodonium (DPI) or apocynin [42–44]. Interestingly, the genetic ablation of CD11b, the alpha subunit of the Mac1 receptor in mice, showed a similar ability to prevent superoxide release and attenuated neurodegeneration during neuroinflammation [31,38]. This attenuation occurs by preventing reactive microgliosis along the Mac1-NOX2 axis, whereby microglial Mac1 re-stimulated microglial activation in response to neuronal-derived DAMPs to induce receptor-mediated signal transduction that activates NOX2 to generate superoxide [31]. This pathway, beyond that of TLRs, is crucial for maintaining chronic neuroinflammation and driving inflammation-mediated oxidative stress that leads to neurodegeneration.
Neuronal NOX2 increases neuronal oxidative stress in aging and inflammatory conditions
How microglia-produced proinflammatory factors cause neuronal damage or death is a critical question in neurodegeneration that has not been clarified yet. Evidence suggests that ROS produced from the Mac1-NOX2 signaling pathway plays a crucial role in neuroinflammation-mediated oxidative stress in neurons that results in their degeneration [40]. Neurons are highly sensitive to oxidative stress. During inflammatory conditions, continuous bombardment of pro-inflammatory factors released from microglia gradually increase intracellular ROS (i.e. hydrogen peroxide and peroxynitrite produced by a reaction between superoxide and nitric oxide) within neighboring neurons. ROS-related oxidative stress impairs neuronal mitochondrial functions by the reduction of membrane potential, inhibition of ATP production, and greater production of mitochondria-derived ROS [41,45]. Moreover, upregulation of neuronal NOX expression results from the increased level of ROS and further enhances the production of ROS inside neurons. This feed forward chain reaction likely drives another vicious cycle within neurons generating even more ROS as mitochondria begin to fail until ROS over-production and oxidative stress begin to form protein aggregates, lysosomal malfunction and impaired clearance of dysfunctional mitochondria overall driving neuron death (Figure 2) [40].
Figure 2.
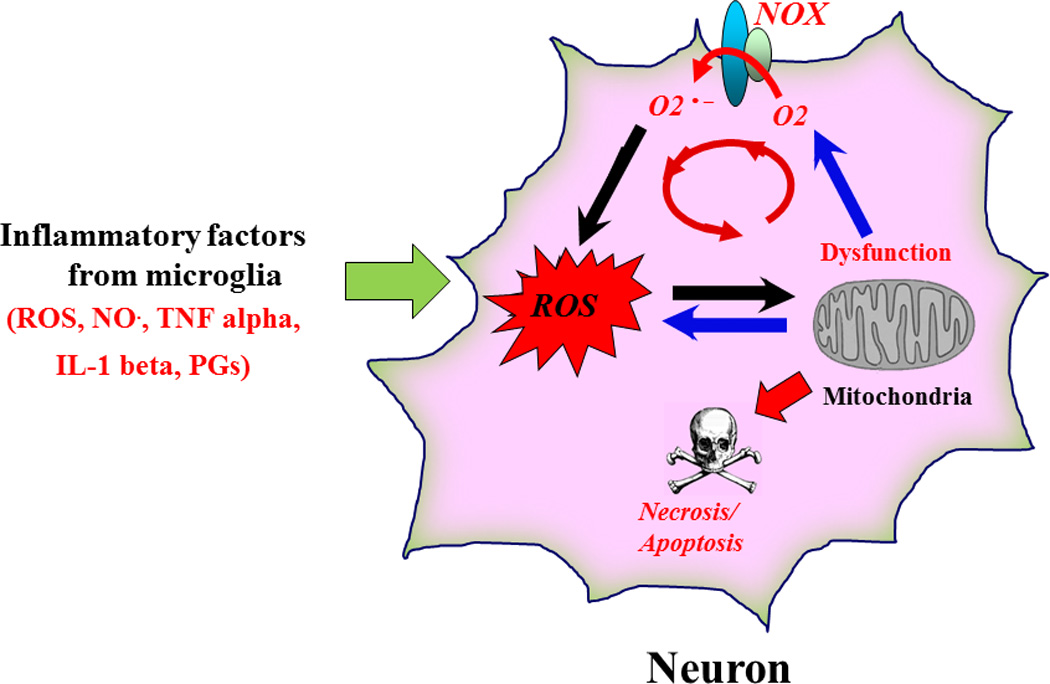
Inflammation-derived oxidative stress leads to a vicious cycle inside the damaged neurons and causes neuronal death. Sustained release of neurotoxic factors from activated microglia continually bombards neurons and increases neuronal oxidative stress during chronic neuroinflammation. The oxidative stress causes mitochondria dysfunction, which could upregulate the expression of neuronal NOX, produce more ROS and lead to progressive neurodegeneration.
Novel anti-inflammatory therapeutic strategies for neurodegenerative diseases by targeting NOX2
Oxidative stress and neuroinflammation are among the most common features shared in all neurodegenerative diseases. Antioxidant therapy has been considered as a strategy to treat neurodegenerative diseases, however the administration of antioxidants (e.g., vitamin C, vitamin E and co-enzyme Q10) showed marginal symptomatic improvements and was unable to halt disease progression in clinical trials for Alzheimer’s and Parkinson’s disease [46–49]. Since anti-oxidants are designed to neutralize unpaired electrons of free radicals, they are ineffective against hydrogen peroxide and peroxynitrite, the two species thought to be most effective at driving oxidative stress in neurons. Furthermore, targeting inflammation as a method of neutralizing inflammation-mediated oxidative stress in neurodegenerative diseases has also been investigated using nonsteroidal anti-inflammatory drugs (NSAIDs) in clinical trials with similar ineffective results. This is partly because anti-inflammatory therapies target the production of specific cytokines or prostaglandins rather than the mechanism underlying chronic neuroinflammation generation (i.e., the Mac1-NOX2 axis). For this reason, therapies that disrupt the Mac1-NOX2 axis could be more effective strategies in extinguishing chronic neuroinflammation, limiting the neuronal oxidative stress that contributes to neurodegeneration. There are two known anti-inflammatory compounds that reduce Mac1 expression, the natural flavonoid baicalin derived from the roots and leaves of the Scutellaria baicalensis plant [50] and the pharmaceutically synthesized leumedin NPC 15669 [51]. Unfortunately, NPC 15669 failed during Phase I clinical trials for undisclosed safety reasons. Interestingly, our group has shown that inhibition of NOX2 in the Mac1-NOX2 axis provides great efficacy at preventing inflammation-driven neurodegeneration in pre-clinical studies of Parkinson’s disease. Our laboratory has identified several compounds including morphinans (e.g. dextromethorphan, sinomenine, naloxone and naltrexone) [52–55], peptides (e.g. dynorphine and Gly-Gly-Phe) [56,57], and adrenergic receptor agonist (e.g. salmeterol) [58] that inhibit NOX2-generated superoxide to limit inflammation-driven oxidative stress and neurodegeneration (Fig. 3). Among the most recently published compounds, the established NOX inhibitor diphenyleneiodonium (DPI) was precluded from use in humans due to its high toxicity at micromolar concentrations. Interestingly, when DPI was administered in vivo at subpicomolar concentrations it not only had high specificity with long-term NOX2 inhibition but also resulted in no detectable acute cytotoxicity [42]. DPI was so effective, as it was shown to protect mice in multiple models of Parkinson’s disease even when administered post disease onset [42]. Though DPI had such great initial preclinical success, we are currently screening several novel blood-brain-barrier permeable NOX2 inhibitors with even greater efficacy and specificity that might become promising clinical therapeutics to extinguish chronic neuroinflammation in neurodegenerative diseases by targeting the Mac1-NOX2 axis.
Figure 3.
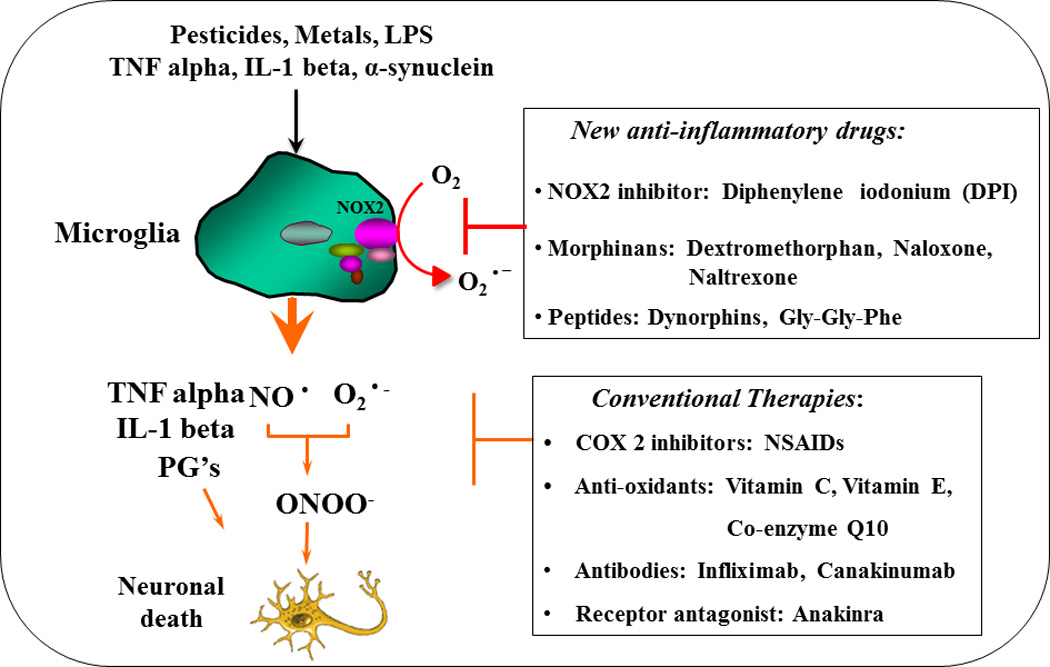
The novel promising anti-inflammatory therapies. The conventional therapies target a limited number of pro-inflammatory factors and failed to block disease progression. The novel anti-inflammatory therapy targets upstream neuro-inflammatory signaling by inhibiting microglial NOX2, which in turn reduces superoxide production and over-activation of microglia and thereby reducing the release of most pro-inflammatory and detrimental factors.
Conclusions
The role of low-grade, chronic neuroinflammation in the pathogenesis of neurodegenerative diseases continues to be strengthened. Yet our lack of understanding of the cellular and molecular mechanisms that shift inflammation from a tightly controlled acute event into a self-propelling cycle that causes collateral damage has prevented us from developing better targeted interventions to prevent chronic neuroinflammation. The most recent hypothesis based on data collected from healthy and diseased human brain tissue and animal models suggests that gradual increased release of neuronal DAMPs likely drive and maintain reactive microgliosis in neurodegenerative diseases. Although studies have shown that TLRs could be the central receptors for DAMPs during the progression of neurodegenerative disease, we present data that supports the Mac1-NOX2 signaling pathway is not only stimulated by DAMPs but also necessary for neuroinflammation to become sustained and pathological. Unlike TLRs, Mac1 can undergo multiple activations without tolerance-like modulations which more closely resemble the pattern of chronic neuroinflammation seen in neurodegenerative diseases. Thus, we believe designing therapies that disrupt the Mac1-NOX2 axis will likely show great promise in breaking the vicious cycle of uncontrolled neuroinflammation that drives oxidative stress and neurodegeneration in neurodegenerative diseases.
Highlights.
Neuroinflammation is a key risk factor in neurodegenerative diseases.
Mac1 signaling bridges chronic neuroinflammation and progressive neuronal loss.
NADPH oxidase could be a novel therapeutic target for neurodegenerative diseases.
Neuroinflammation is a key risk factor in neurodegenerative diseases.
Mac1 signaling bridges chronic neuroinflammation and progressive neuronal loss.
NADPH oxidase could be a novel therapeutic target for neurodegenerative diseases.
ACKNOWLEDGEMENTS
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interests
The authors have declared that no conflict of interest exists.
References
- 1.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 2.Gao HM, Hong JS. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 2008;29:357–365. doi: 10.1016/j.it.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener. 2009;4:47. doi: 10.1186/1750-1326-4-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 6.Hanamsagar R, Hanke ML, Kielian T. Toll-like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol. 2012;33:333–342. doi: 10.1016/j.it.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doens D, Fernandez PL. Microglia receptors and their implications in the response to amyloid beta for Alzheimer's disease pathogenesis. J Neuroinflammation. 2014;11:48. doi: 10.1186/1742-2094-11-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang D, Hu X, Qian L, Chen SH, Zhou H, Wilson B, Miller DS, Hong JS. Microglial MAC1 receptor and PI3K are essential in mediating beta-amyloid peptide-induced microglial activation and subsequent neurotoxicity. J Neuroinflammation. 2011;8:3. doi: 10.1186/1742-2094-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao HM, Zhou H, Zhang F, Wilson BC, Kam W, Hong JS. HMGB1 acts on microglia Mac1 to mediate chronic neuroinflammation that drives progressive neurodegeneration. J Neurosci. 2011;31:1081–1092. doi: 10.1523/JNEUROSCI.3732-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carty M, Bowie AG. Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem Pharmacol. 2011;81:825–837. doi: 10.1016/j.bcp.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 11.Walsh JG, Muruve DA, Power C. Inflammasomes in the CNS. Nat Rev Neurosci. 2014;15:84–97. doi: 10.1038/nrn3638. [DOI] [PubMed] [Google Scholar]
- 12.Chen SH, Oyarzabal EA, Sung YF, Chu CH, Wang Q, Chen SL, Lu RB, Hong JS. Microglial regulation of immunological and neuroprotective functions of astroglia. Glia. 2015;63:118–131. doi: 10.1002/glia.22738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Theriault P, ElAli A, Rivest S. The dynamics of monocytes and microglia in Alzheimer's disease. Alzheimers Res Ther. 2015;7:41. doi: 10.1186/s13195-015-0125-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15:300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- 15. Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–248. doi: 10.1016/j.neuron.2013.12.034. An excellent review outlining the role of reactive gliosis in neurological diseases.
- 16.Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14:463–477. doi: 10.1038/nri3705. [DOI] [PubMed] [Google Scholar]
- 17.Lehnardt S. Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia. 2010;58:253–263. doi: 10.1002/glia.20928. [DOI] [PubMed] [Google Scholar]
- 18.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 19.Piccinini AM, Midwood KS. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm. 2010;2010 doi: 10.1155/2010/672395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562. doi: 10.1038/ncomms2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W, et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease. Cell Physiol Biochem. 2007;20:947–956. doi: 10.1159/000110455. [DOI] [PubMed] [Google Scholar]
- 22.Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, Rube CE, Walter J, Heneka MT, Hartmann T, et al. TLR2 is a primary receptor for Alzheimer's amyloid beta peptide to trigger neuroinflammatory activation. J Immunol. 2012;188:1098–1107. doi: 10.4049/jimmunol.1101121. [DOI] [PubMed] [Google Scholar]
- 23.Doi Y, Mizuno T, Maki Y, Jin S, Mizoguchi H, Ikeyama M, Doi M, Michikawa M, Takeuchi H, Suzumura A. Microglia activated with the toll-like receptor 9 ligand CpG attenuate oligomeric amyloid {beta} neurotoxicity in in vitro and in vivo models of Alzheimer's disease. Am J Pathol. 2009;175:2121–2132. doi: 10.2353/ajpath.2009.090418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 25.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 26.Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain research reviews. 2009;59:278–292. doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron. 2009;64:110–122. doi: 10.1016/j.neuron.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liew FY, Xu D, Brint EK, O'Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 29.Levesque S, Taetzsch T, Lull ME, Johnson JA, McGraw C, Block ML. The role of MAC1 in diesel exhaust particle-induced microglial activation and loss of dopaminergic neuron function. J Neurochem. 2013;125:756–765. doi: 10.1111/jnc.12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryu JK, Davalos D, Akassoglou K. Fibrinogen signal transduction in the nervous system. J Thromb Haemost. 2009;7(Suppl 1):151–154. doi: 10.1111/j.1538-7836.2009.03438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu X, Zhang D, Pang H, Caudle WM, Li Y, Gao H, Liu Y, Qian L, Wilson B, Di Monte DA, et al. Macrophage antigen complex-1 mediates reactive microgliosis and progressive dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. J Immunol. 2008;181:7194–7204. doi: 10.4049/jimmunol.181.10.7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson SI, Hotchin NA, Nash GB. Role of the cytoskeleton in rapid activation of CD11b/CD18 function and its subsequent downregulation in neutrophils. J Cell Sci. 2000;113(Pt 15):2737–2745. doi: 10.1242/jcs.113.15.2737. [DOI] [PubMed] [Google Scholar]
- 33.Podolnikova NP, Podolnikov AV, Haas TA, Lishko VK, Ugarova TP. Ligand recognition specificity of leukocyte integrin alphaMbeta2 (Mac-1, CD11b/CD18) and its functional consequences. Biochemistry. 2015;54:1408–1420. doi: 10.1021/bi5013782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akiyama H, McGeer PL. Brain microglia constitutively express beta-2 integrins. J Neuroimmunol. 1990;30:81–93. doi: 10.1016/0165-5728(90)90055-r. [DOI] [PubMed] [Google Scholar]
- 35.Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- 36. Wang S, Chu CH, Stewart T, Ginghina C, Wang Y, Nie H, Guo M, Wilson B, Hong JS, Zhang J. alpha-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc Natl Acad Sci U S A. 2015;112:E1926–E1935. doi: 10.1073/pnas.1417883112. A crucial paper illustrating how aggregates of α-synuclein activate and recruit microglia through Mac1.
- 37.Rotshenker S. Microglia and macrophage activation and the regulation of complement-receptor-3 (CR3/MAC-1)-mediated myelin phagocytosis in injury and disease. J Mol Neurosci. 2003;21:65–72. doi: 10.1385/JMN:21:1:65. [DOI] [PubMed] [Google Scholar]
- 38.Pei Z, Pang H, Qian L, Yang S, Wang T, Zhang W, Wu X, Dallas S, Wilson B, Reece JM, et al. MAC1 mediates LPS-induced production of superoxide by microglia: the role of pattern recognition receptors in dopaminergic neurotoxicity. Glia. 2007;55:1362–1373. doi: 10.1002/glia.20545. [DOI] [PubMed] [Google Scholar]
- 39.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 40. Nayernia Z, Jaquet V, Krause KH. New insights on NOX enzymes in the central nervous system. Antioxid Redox Signal. 2014;20:2815–2837. doi: 10.1089/ars.2013.5703. A comprehensive review explaining the source of reactive oxygen species, function and regulation of different NOX isoforms in the central nervous system, and their pathophysiology in neurological diseases.
- 41. Qin L, Liu Y, Hong JS, FT C. NADPH oxidase and aging drive microglial activation, oxidative stress and dopaminergic neurodegeneration following systemic LPS administration. Glia. 2013;61:855–868. doi: 10.1002/glia.22479. A crucial paper elucidating that chronic neuroinflammation significantly enhances oxidative stress in neurons during aging.
- 42. Wang Q, Qian L, Chen SH, Chu CH, Wilson B, Oyarzabal E, Ali S, Robinson B, Rao D, Hong JS. Post-treatment with an ultra-low dose of NADPH oxidase inhibitor diphenyleneiodonium attenuates disease progression in multiple Parkinson's disease models. Brain. 2015;138:1247–1262. doi: 10.1093/brain/awv034. An excellent paper demonstrating that low-dose DPI can specifically inhibit NOX2 activity and can elicit neuroperotective effect as post-treatment regimen.
- 43.Qian L, Gao X, Pei Z, Wu X, Block M, Wilson B, Hong JS, Flood PM. NADPH oxidase inhibitor DPI is neuroprotective at femtomolar concentrations through inhibition of microglia over-activation. Parkinsonism Relat Disord. 2007;13(Suppl 3):S316–S320. doi: 10.1016/S1353-8020(08)70023-3. [DOI] [PubMed] [Google Scholar]
- 44.Simonyi A, Serfozo P, Lehmidi TM, Cui J, Gu Z, Lubahn DB, Sun AY, Sun GY. The neuroprotective effects of apocynin. Front Biosci (Elite Ed) 2012;4:2183–2193. doi: 10.2741/535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci. 2012;322:254–262. doi: 10.1016/j.jns.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 46.Gao HM, Zhou H, Hong JS. NADPH oxidases: novel therapeutic targets for neurodegenerative diseases. Trends Pharmacological Sci. 2012;33:295–303. doi: 10.1016/j.tips.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zandi PP, Anthony JC, Khachaturian AS, Stone SV, Gustafson D, Tschanz JT, Norton MC, Welsh-Bohmer KA, Breitner JC, Cache County Study G. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: the Cache County Study. Arch Neurol. 2004;61:82–88. doi: 10.1001/archneur.61.1.82. [DOI] [PubMed] [Google Scholar]
- 48.Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Aggarwal N, Wilson RS, Scherr PA. Dietary intake of antioxidant nutrients and the risk of incident Alzheimer disease in a biracial community study. JAMA. 2002;287:3230–3237. doi: 10.1001/jama.287.24.3230. [DOI] [PubMed] [Google Scholar]
- 49.Parkinson Study Group QEI. Beal MF, Oakes D, Shoulson I, Henchcliffe C, Galpern WR, Haas R, Juncos JL, Nutt JG, Voss TS, et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: no evidence of benefit. JAMA Neurol. 2014;71:543–552. doi: 10.1001/jamaneurol.2014.131. [DOI] [PubMed] [Google Scholar]
- 50.Shen YC, Chiou WF, Chou YC, Chen CF. Mechanisms in mediating the anti-inflammatory effects of baicalin and baicalein in human leukocytes. Eur J Pharmacol. 2003;465:171–181. doi: 10.1016/s0014-2999(03)01378-5. [DOI] [PubMed] [Google Scholar]
- 51.Bator JM, Weitzberg M, Burch RM. N-[9H-(2,7-dimethylfluorenyl-9-methoxy)carbonyl]-L-leucine, NPC 15669, prevents neutrophil adherence to endothelium and inhibits CD11b/CD18 upregulation. Immunopharmacology. 1992;23:139–149. doi: 10.1016/0162-3109(92)90038-e. [DOI] [PubMed] [Google Scholar]
- 52.Liu Y, Qin L, Li G, Zhang W, An L, Liu B, Hong JS. Dextromethorphan protects dopaminergic neurons against inflammation-mediated degeneration through inhibition of microglial activation. J Pharm Exp Ther. 2003;305:212–218. doi: 10.1124/jpet.102.043166. [DOI] [PubMed] [Google Scholar]
- 53.Qian L, Xu Z, Zhang W, Wilson B, Hong JS, Flood PM. Sinomenine, a natural dextrorotatory morphinan analog, is anti-inflammatory and neuroprotective through inhibition of microglial NADPH oxidase. J Neuroinflammation. 2007;4:23. doi: 10.1186/1742-2094-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu B, Du L, Hong JS. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharm Exp Ther. 2000;293:607–617. [PubMed] [Google Scholar]
- 55.Crain SM, Shen KF. Ultra-low concentrations of naloxone selectively antagonize excitatory effects of morphine on sensory neurons, thereby increasing its antinociceptive potency and attenuating tolerance/dependence during chronic cotreatment. Proc Natl Acad Sci U S A. 1995;92:10540–10544. doi: 10.1073/pnas.92.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Q, Shin EJ, Nguyen XK, Li Q, Bach JH, Bing G, Kim WK, Kim HC, Hong JS. Endogenous dynorphin protects against neurotoxin-elicited nigrostriatal dopaminergic neuron damage and motor deficits in mice. J Neuroinflammation. 2012;9:124. doi: 10.1186/1742-2094-9-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qin L, Liu Y, Qian X, Hong JS, Block ML. Microglial NADPH oxidase mediates leucine enkephalin dopaminergic neuroprotection. Ann N Y Acad Sci. 2005;1053:107–120. doi: 10.1196/annals.1344.009. [DOI] [PubMed] [Google Scholar]
- 58.Qian L, Wu HM, Chen SH, Zhang D, Ali SF, Peterson L, Wilson B, Lu RB, Hong JS, Flood PM. beta2-adrenergic receptor activation prevents rodent dopaminergic neurotoxicity by inhibiting microglia via a novel signaling pathway. J immunol. 2011;186:4443–4454. doi: 10.4049/jimmunol.1002449. [DOI] [PMC free article] [PubMed] [Google Scholar]