

Abstract
We describe high resolution MAS solid-state NMR experiments that utilize 1H detection with 60 kHz magic angle spinning; simultaneous cross-polarization from 1H to 15N and 13C nuclei; bidirectional cross-polarization between 13C and 15N nuclei; detection of both amide nitrogen and aliphatic carbon 1H; and measurement of both 13C and 15N chemical shifts through multi-dimensional correlation experiments. Three-dimensional experiments correlate amide 1H and alpha 1H selectively with 13C or 15N nuclei in a polypeptide chain. Two separate three-dimensional spectra correlating 1Hα/13Cα/1HN and 1HN/15N/1Hα are recorded simultaneously in a single experiment, demonstrating that a two-fold savings in experimental time is potentially achievable. Spectral editing using bidirectional coherence transfer pathways enables simultaneous magnetization transfers between 15N, 13Cα(i) and 13C′(i−1), facilitating intra- and inter- residue correlations for sequential resonance assignment. Non-uniform sampling is integrated into the experiments, further reducing the length of experimental time.
Keywords: magic angle spinning, dual observation, peptides, proteins, triple-resonance
Introduction
High-resolution magic angle spinning (MAS) solid-state NMR spectroscopy is in the process of becoming a powerful tool for studying crystalline peptides [1] and proteins [2] as well as proteins immobilized in biological supramolecular assemblies such as membranes [3], fibrils [4], and virus particles [5]. Recent advances include high speed spinning, 1H-detection [6], and non-uniform sampling (NUS) [7–9]. Rapid spinning at the magic angle attenuates the severe line-broadening resulting from the dense 1H/1H homonuclear dipole-dipole coupling network present in peptides and proteins. Due to its high gyromagnetic ratio and natural abundance, 1H-detection has been the optimal choice for solution NMR for quite some time, and now with the ability to narrow 1H resonances with fast MAS, it is finding increasing applications in solid-state NMR. Moreover, NUS can be used to reduce the amount of time required for the experiments.
In addition to detecting and resolving individual resonances, assignment to specific sites is an essential aspect of protein NMR spectroscopy. A number of 1H-detection experiments in solid state have been reported that yield sequence-specific resonance assignments [10, 11] similar to the solution NMR experiment [12]. Current assignment procedures utilize double- and triple- resonance experiments to correlate 13C and 15N chemical shift frequencies with those of amide hydrogens (1HN). In contrast to solution NMR experiments, where backbone and side chain 1H, 13C and 15N resonances are assigned using established two- and three- dimensional triple-resonance experiments, in order to measure the chemical shift frequencies of non-amide hydrogens in solid state NMR, four-dimensional experiments or multiple three-dimensional experiments are typically required. For example a four-dimensional experiment correlating 1HXX1H, where X stands for 13C or 15N, has been shown to fully resolve the spectrum of a protein [7]. However, four-dimensional experiments can be extremely time consuming. It is possible for two separate three-dimensional experiments with either 13C or 15N editing to replace four-dimensional experiments, however, this approach can also require long measurement times.
The long times required for the higher dimensional NMR experiments can be addressed by incorporating simultaneous cross-polarization (SIM-CP) from 1H to 13C and 15N sites. Multiple two- and three- dimensional spectra can be obtained from a single experiment in this way [13–16]. These experiments were originally developed for 13C-detection at moderate spinning frequencies. Notably, 13C/13C homonuclear correlation under proton assisted recoupling (PAR) and 13C/15N heteronuclear correlation under proton-assisted insensitive nuclei (PAIN) can be performed using simultaneous spin-lock pulses on three channels under second-order recoupling schemes. In addition, recently, it has been shown that bidirectional cross-polarization between 13C and 15N nuclei can be used to transfer coherence without significant losses in signal intensities [15, 17]. This has enabled triple-resonance experiments to be executed with multiple acquisitions using a single receiver within a single experiment. Further savings of experimental time can be achieved with application of NUS [7, 18–21].
Here we demonstrate 1H-detection experiments that utilize SIM-CP and bidirectional coherence transfer to obtain two separate two- or three- dimensional spectra in a single experiment. These experiments utilize cross-polarization from 1H to both 13C and 15N sites without a substantial loss in sensitivity compared to individual 1H/13C or 1H/15N cross-polarization. Experiments are optimized for bidirectional coherence transfer at 60 kHz MAS in order to transfer magnetization simultaneously and selectively from the amide 15N to the 13Cα of the same residue and the 13C′ of the preceding residue. The experiments also enable sequential homo- and hetero- nuclear correlations between amide 1H and alpha 1H sites to 13C and 15N nuclei in order to improve resolution and to facilitate resonance assignment.
Experimental
Sample preparation
Uniformly 13C and 15N labeled fd bacteriophage with its major coat protein containing the Y21M mutation was obtained using our previously published protocol [5]. Bacteriophage particles from a single batch were concentrated to ~100 mg/ml by ultracentrifugation at 60,000 rpm for 2 hours. The pH of the sample was adjusted to 8.0 using 5 mM sodium borate buffer containing 0.1 mM sodium azide prior to ultracentrifugation. ~30 μL of fully hydrated phage particles at pH 8.0 were transferred to a 1.3 mm zirconium rotor. The sample in the rotor contained ~1 mg of protein.
Uniformly 13C and 15N labeled Met-Leu-Phe tripeptide was purchased from Cortecnet (www.cortecnet.com). ~3.5 mg of microcrystalline peptide was packed in a 1.3 mm MAS rotor.
NMR Spectroscopy
The experiments were performed on a spectrometer with a 1H resonance frequency of 900 MHz. The spectrometer was equipped with a Bruker Avance III HD console and a Bruker 1.3 mm 1H/13C/15N triple-resonance MAS probe (www.bruker.com). The spinning rate was controlled at 60.0 kHz. The temperature of the nitrogen gas was maintained at −18° C throughout the experiments. The 1H resonance frequency of water was used to monitor the temperature of the protein-containing sample, and served as an internal chemical shift reference frequency at 4.8 ppm at 20 °C. The chemical shift frequencies of the polycrystalline sample were referenced externally to solid samples with the methylene 13C resonance of adamantane at 38.48 ppm and the 15N resonance of ammonium sulfate at 26.8 ppm [22].
The experimental data were acquired using the pulse sequences diagrammed in Figure 1. Heteronuclear decoupling was accomplished using XiX for 1H [23] and GARP [24] for 15N and 13C nuclei. 30% ramped radio frequency (RF) irradiation on the 1H channel was used for initial cross-polarization (CP) from 1H to 13C, 1H to 15N, and back to 1H during reverse CP. Spin-exchange between 15N and 13C was accomplished using spectrally induced filtering in combination with cross-polarization (SPECIFIC-CP) [25] with 10% ramped amplitude irradiation on the 13C channel. All RF irradiations on the 1H, 13C and 15N channels were matched to the (n-1) Hartmann-Hahn resonance condition for optimal magnetization transfer. The amplitude of the ramped 1H irradiation ranged from 120 kHz to 60 kHz. The 13C and 15N RF amplitudes were adjusted to be near 30 kHz to meet the Hartmann-Hahn condition. Mixing intervals of 1 ms and 0.4 ms were used for initial and final steps in all experiments. A 6 ms mixing time was used to exchange magnetization between 15N and 13C. Phase alternated RF pulses were used to suppress the water signals [26], with 15 kHz RF irradiation for 300 ms. For the fully hydrated biological sample water suppression was improved by increasing the amplitude of the 1H irradiation to ~40 kHz and implementing supercycled phase alternated pulses. All spectra were processed with Bruker TOPSPIN and represented with SPARKY (University of California, San Francisco) programs.
Figure 1.
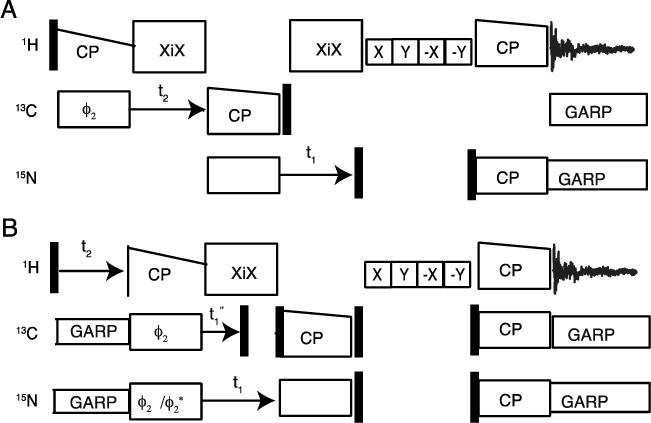
Three channel pulse sequence diagrams for 1H detection in solid state NMR with 60 kHz magic angle spinning. A. and B. Three-dimensional experiments for simultaneous 13C and 15N edited heteronuclear correlation. A. Experiment for inter and intra residue 13C/15N/1HN three-dimensional correlation. B. Experiment for three-dimensional correlation spectroscopy for simultaneous observations of 1Hn/N(Cα)/1Hα and 1Hα/13Cα (N)/1HN resonances. Phase cycling for A ; 13C CP (0 0 0 0 2 2 2 2), 13C and 15N DCP (0), 15N reverse CP (0 0 2 2), 1H reverse CP (0 0 0 0 1 1 1 1 2 2 2 2 3 3 3 3), receiver (1 3 3 1 3 1 1 3 2 0 0 2 0 2 2 0). Phase cycling for B; 13C and 15N CP (0), 13C DCP (0 0 0 0), 15N DCP (0 0 2 2), 1H, 13C and 15N reverse CP (0), receiver (1 3 3 1).
Results
The pulse sequences utilized in this study are diagrammed in Figure 1. All of the experiments were performed with 60 kHz MAS. The sequence for a three-dimensional 1H detection experiment is shown in Figure 1A. One and two-dimensional experiments with 1H detection and simultaneous correlation to 13C and 15N nuclei were adapted from previous publications using simultaneous cross polarization (CP) [14, 15, 17] and concurrent cross polarization pulse schemes [27]. 1H magnetization is transferred to 13C and 15N simultaneously using Hartmann-Hahn CP. Following CP and chemical shift evolution with 1H decoupling, the 13C and 15N magnetizations are stored as Zeeman order using 90° pulses. Water signals and other unwanted 1H signals are suppressed using RF irradiation applied in the XY plane [26]. Then the 13C and 15N magnetizations are flipped back to the transverse direction using 90° pulses. By applying matched RF irradiation on the 1H, 13C, and 15N channels, polarization is transferred back to the 1H nuclei, and subsequently detected under 13C and 15N heteronuclear decoupling.
For heteronuclear correlation, 15N and 13C chemical shift frequencies evolve in the indirect (t1) dimension under 1H irradiation for decoupling. The chemical shift frequencies are recorded in the phase sensitive mode by incrementing the RF phase by 90° during the initial CP. In one experiment, both 13C and 15N signals are acquired with the same phase. In a second experiment, out of phase signals are acquired by alternating the phase of either the 13C or 15N irradiation by 180°. We have also observed relatively inverted signals (i.e., amide 1H and alpha 1H) by altering the 13C and 15N RF phases during the reverse CP transfer to the 1H nuclei.
Figure 1A illustrates a pulse sequence for a 13C to amide 1H and 15N correlation experiment. The experiment transfers magnetization from 1H to 13C followed by chemical shift evolution in the indirect (t2) dimension under 1H decoupling. The magnetization from 13Cα and 13C′ are transferred selectively to 15N using Hartmann-Hahn CP followed by 15N chemical shift evolution in the indirect (t1) dimension under 1Hdecoupling. Water resonance suppression pulses are applied before the reverse CP from 15N for 1H-detection.
Three-dimensional correlation experiments with simultaneous 13C and 15N editing were carried out using the pulse scheme shown in Figure 1B. The pulse sequence correlates alpha 1H and amide 1H with either 13Cα or 15N amide nuclei in a peptide plane. The pulse sequence is similar to that in Figure 1A, except that a spectral editing double cross-polarization between 13C and 15N nuclei is included between the initial and final CP. Both 13C and 15N chemical shift evolutions occur after initial CP. The signals are encoded in the indirect dimensions, and a correlation spectrum of 1HN/15N(13Cα)/1Hα and 1Hα/13Cα (15N)/1HN results.
The spectra shown in Figures 2 and 3 were obtained from a polycrystalline sample of uniformly 13C and 15N labeled Met-Leu-Phe (MLF) peptide utilizing the pulse sequences diagrammed in Figure 1. One-dimensional 1H NMR spectra are shown in Figure 2A–C. Simultaneous observation of amide and aliphatic 1H signals in Figure 2A resulted from using both 13C and 15N editing. The 15N edited amide 1H NMR spectrum is shown in Figure 2B. Similarly the 13C edited aliphatic 1H NMR spectrum in Figure 2C. Notably, all of the magnetization was recovered with simultaneous cross-polarization. Following the one-dimensional experiments, heteronuclear correlation of 1H with 13C and 15N nuclei was obtained by evolving both nuclei simultaneously in the indirect dimension. This is demonstrated with the two-dimensional spectrum shown in Figure 2D, which contains cross-peaks for both 13C and 15N nuclei. The spectrum was obtained with the 13C and 15N magnetization locked with the same phase during Hartmann-Hahn CP. In order to distinguish the chemical shifts by nucleus, two separate data sets were collected. The first experiment was performed using the spin-lock pulses with the same phase. The second data set was acquired with 180° phase shifted 15N spin lock pulses. This resulted in a spectrum where the 13C and 15N cross-peaks are opposite in phase. By adding and subtracting the two data sets, two separate 1H/15N and 1H/13C heteronuclear correlation spectra are obtained, as shown in Figure 2E and F, respectively. The spectral width/dwell time was kept constant for chemical shift evolution in the indirect dimensions for both 13C and 15N nuclei. However, sensitivity was found to be independent of the dwell times used for each nucleus. A three-dimensional spectrum obtained using different dwell times is shown in Figure 2G. The 13C and 15N spin-lock pulses differed by 180° in phase and the 15N dwell time was set to twice that used for the detection of 13C signals. The cube shows a total of six cross-peaks correlating amide 1H, with alpha 1H, and with 15N amide or 13Cα chemical shift frequencies. The color codes indicate the signals acquired with opposite phases. The blue and green colors indicate 1HN/15N/1Hα and 1Hα/13Cα/1HN chemical shift correlation and were generated using 1HN (t2) > 15N (t1) > 13Cα> 1Hα(t3) and 1Hα(t2) > 13Cα(t1) > N > 1HN (t3) coherence transfer pathways, respectively. Two separate three-dimensional experiments were performed in a similar fashion to that used for the two-dimensional experiments described above. By addition and subtraction of the three-dimensional spectra, separate chemical shift correlation spectra are obtained. The 180° phase shifted spectra are obtained by changing the initial CP irradiation phases followed by chemical shift evolution in the indirect dimension. It is also possible to obtain the phase shifted 1H spectrum with direct detection by changing the phases of the final CP irradiations by 180°.
Figure 2.
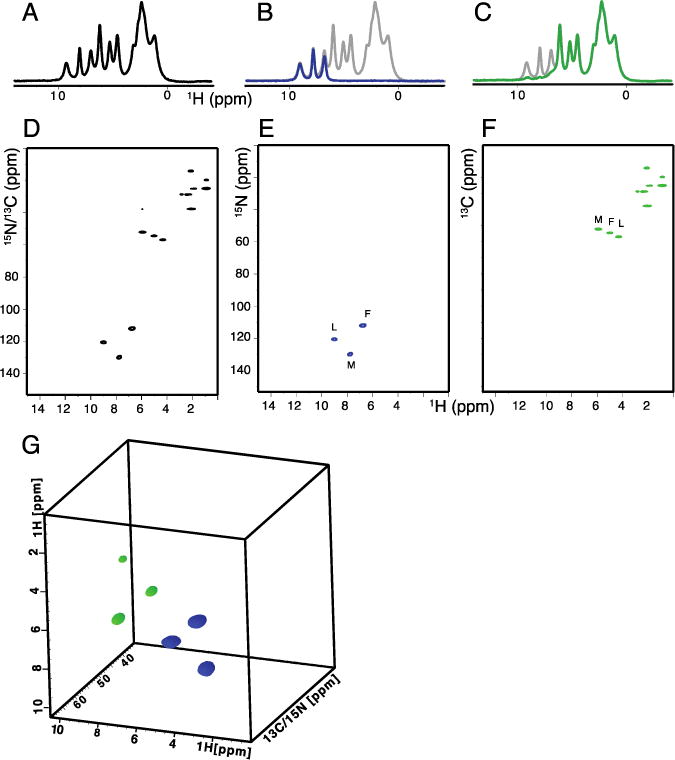
One-, two-, and three- dimensional spectra of uniformly 13C and 15N labeled polycrystalline Met-Leu-Phe tripeptide obtained with 60 kHz MAS. A.-C. One-dimensional 1H spectra obtained using simultaneous CP. Four scans were co-added for each spectrum. A. 13C and 15N-edited 1H spectrum. B. 15N -edited spectrum. C. 13C edited spectrum. For visual comparison, the one dimensional spectrum from A is plotted in grey in the background. D and E, two-dimensional heteronuclear correlation spectra obtained for a total number of 4 scans. G. Three-dimensional correlation spectrum obtained using the pulse scheme shown in Figure 1C. The color codes are blue for positive and green for negative cross-peaks for 1Hα/13Cα (N)/1HN and 1Hn/N(Cα)/1Hα correlations, respectively. The three-dimensional data was acquired with a total number of 16 scans; 4 second recycle delay; and evolution periods of 11 ms (t3) 1H, 1.5 ms (t2) 13C, and 2.5 ms (t1) 15N. An equal number of data points were linear predicted during data processing.
Figure 3.
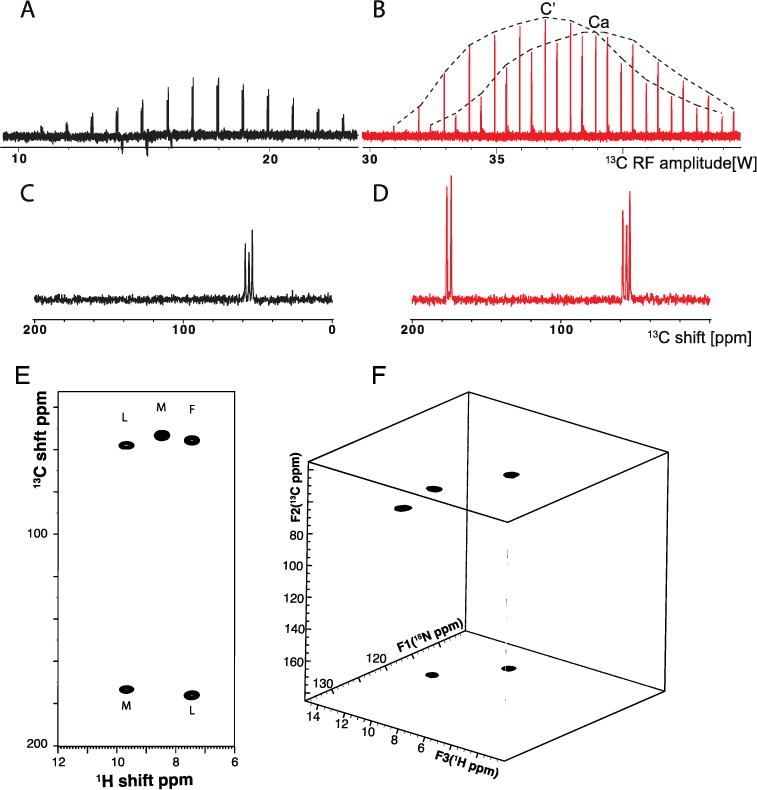
13C- and 1H- detected spectra of MLF acquired with 60 kHz MAS. A. Variable amplitude 13C spectra of SPECIFIC-CP between 15N and 13Cα at fixed 25 kHz 15N amplitude. 13C and 15N are irradiated at 55 ppm and 120 ppm respectively. B. Same as A, but 13C irradiated at 115 ppm and 15N amplitude at 15 kHz (2.8 watts). Envelopes on top are drawn to visually guide the magnetization transfer to 13Cα and 13C′. C. Optimal 1D spectra obtained from A. D. Same as C, but for 13C amplitude at 38 watts. E. 15N-edited and 1H-detected 1HN/13C two-dimensional correlation spectrum. F. 1H-detected 13C/15N/1HN three-dimensional correlation spectrum. Both A. and B. were obtained with 16 scans, and 4 second recycle delays. The three-dimensional spectrum was reconstructed from 10% sampling density of a 400 (t2)/64 (t1) complex data matrix. The sampling schedule was generated in TOPSPIN 3.2 by incorporating T2 and J coupling values for 13C nuclei. The spectrum was reconstructed using the compressed sensing package in TOPSPIN.
13C-detected experiments optimized to quantify the magnetization transfer between 13C and 15N at 60 kHz MAS are illustrated in Figure 3. The experiment was carried out using a tailored double CP (DCP) pulse sequence where 1H magnetization is transferred to 15N and subsequently transferred to 13C for detection. During the DCP, no 1H RF irradiation is applied, and the13C spectrum is irradiated at 115 ppm, 55 ppm, or 175 ppm. For specific transfer of 15N magnetization to either 13Cα or 13C′, we optimized the experimental conditions in a similar manner as described in an earlier publication [10]. We fixed the 15N and 13C RF amplitudes at 25 kHz and 35 kHz and performed the DCP with the 13C resonance frequency at 55 ppm for 13Cα and 175 ppm for 13C′. The RF amplitude applied to 13C was varied to find the maximum sensitivity. Magnetization transfer for 13Cα only is shown in Figure 3A. For simultaneous magnetization transfer to 13Cα and 13C′, we optimized the experiment with fixed RF amplitudes of 15 kHz for 15N and 75 kHz for 13C irradiated at 115 ppm. This is demonstrated in Figure 3B.
One-dimensional results for the MLF tripeptide powder sample, demonstrating the optimal polarization transfer to either carbon or simultaneous selective transfer of magnetization to 13Cα and 13C′, are shown in Figure 3C and D, respectively. For comparison, a spectrum obtained by 1H to 13C direct CP (with a 1 ms mix time) was also obtained (data not shown). All experiments were carried out under similar conditions and the data were processed in the same manner. 15N-edited two-dimensional 13C/1HN heteronuclear correlation is shown in Figure 3E. The experiment was carried out using the pulse sequence shown in Figure 1A, except that no 15N chemical shift evolution was incorporated. The peaks are marked for resonances assigned to 13Cα and 13C′ chemical shift frequencies of the three amino acid residues.
A three-dimensional experiment correlating the 13C/15N/1HN chemical shift frequencies is shown in Figure 3F. The spectrum was obtained using the pulse sequence in Figure 1A and incorporating a 10% non-uniform sampling schedule generated in TOPSPIN and processed using compressed sensing provided in the same software package. The spectrum was reconstructed for a total evolution period of 11 ms 1H (t3), 6 ms 13C (t2) and 12 ms 15N (t1).
The experiments were also applied to a protein sample to demonstrate the breadth of their utility. For this purpose, we used the structural form of the coat protein in fd bacteriophage. NMR signals were obtained from a concentrated solution of uniformly 13C and 15N labeled virus particles. The virus particles are ~90% by weight coat protein subunits, thus the individual signals come from the symmetrically arranged coat proteins. Like wild type fd bacteriophage proteins [28], the mutant Y21M fd coat protein used in these experiments is a 50 amino acid long polypeptide chain with an alpha helical conformation [5]. At 60 kHz MAS, simultaneous CP and 1H detection experiments showed essentially complete polarization transfer similar to that observed for the polycrystalline tripeptide sample. However, due to spectral overlap, only ~30 individual cross-peaks could be identified in the two-dimensional 1H/15N heteronuclear single quantum correlation spectrum shown in Figure 4A. Similarly only ~20 cross-peaks could be resolved in the 1Hα/13Cαcorrelation spectrum in Figure 4D. Both spectra were acquired using simultaneous-CP with the pulse sequence shown in Figure 1B. Nearly all protein signals are resolved in the three-dimensional spectra. Two separate three-dimensional correlation spectra such as 1HN/15N/1Hα and 1Hα/13Cα/1HN were obtained simultaneously using the pulse sequence diagrammed in Figure 1C. Two-dimensional slices correlating 1H/13C and 1H/15N shifts at various 1H chemical shifts are shown in Figure 4. 15N/1HN chemical shift correlation in Figure 4B and C were extracted at 4.2 ppm and 3.8 ppm 1H chemical shift frequencies, respectively. Similarly 1Hα/13Cα two-dimensional planes were obtained at 8.5 ppm and 7.6 ppm 1HN chemical shift frequencies. Simultaneous observation of NCO and NCA correlation with 1HN detection was carried out using the pulse sequence depicted in Figure 1B. The experiment was carried out in a similar manner to that used for MLF in Figure 3. The two-dimensional plane correlating 1HN with 13C shifts in Figure 4G was obtained from the three-dimensional data at 109.7ppm 15N chemical shift.
Figure 4.
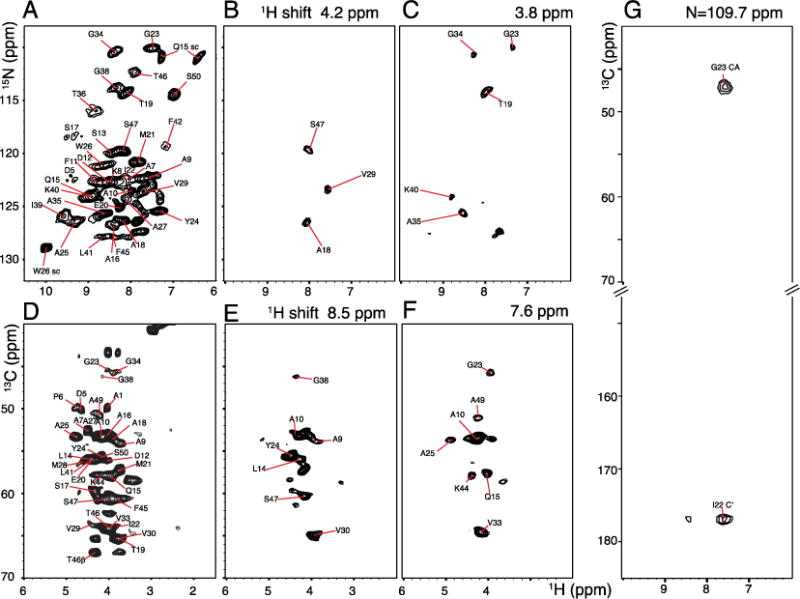
Two- and three-dimensional spectra of uniformly 13C and 15N labeled fd coat protein in bacteriophage particles obtained with 60 kHz MAS. A. Two-dimensional 1H/15N heteronuclear correlation spectrum. B. and C. 1H/15N two-dimensional slices obtained at 4.2 and 3.8 ppm 1H chemical shifts, respectively. D. Two-dimensional 1H/13C heteronuclear correlation spectrum. E. and F. Two-dimensional slices obtained at 8.5 and 7.6 ppm 1H chemical shifts. A. and D. were obtained using the pulse sequence shown in Figure 1B without incorporating DCP editing pulses. Three-dimensional experiments were carried out using the pulse scheme shown in Figure 1B with a total number of 16 scans, 4 s recycle delay; total evolution periods of 10 ms (t3) 1H; 3 ms (t2) 1H; 7.2 ms (t1) 15N, and 3.6 ms (t1′) 13C. Equal numbers of data points were linear predicted during data processing.
Discussion
60 kHz MAS enables pulse sequences for simultaneous observation of 13C and 15N signals with 1H detection to be performed. Several pulse sequences were developed with protein structure determination in mind. In particular, in order to measure 1H chemical shifts in a sequential manner, multiple coherence pathways are used in combination.
Simultaneous cross-polarization transfer enables measurement of the chemical shifts of hydrogens bonded to amide nitrogens and aliphatic carbons. Three resolved amide nitrogen 1H and three alpha carbon 1H resonances are observed in spectra obtained from one-dimensional experiments performed on a polycrystalline sample of the tripeptide MLF, as shown in Figure 2 A–C. Significantly, efficient polarization transfer was achieved using SIM-CP by matching the 13C and 15N RF fields to the (n-1) Hartmann-Hahn resonance condition with 1H.
The two-dimensional correlation spectra in Figure 2D show one-bond correlations of 15N/1HN and 13C/1H resonances. The residue specific chemical shift assignments were obtained from previously reported 13C-detected experiments [1]. The pulse sequence in Figure 1C enables two separate three-dimensional spectra to be obtained from a single experiment. In particular, these experiments enable the simultaneous observation of 13C and 15N chemical shifts correlated to amide 1H and alpha 1H, in particular 1Hn/N(Cα)/1Hα and 1Hα/13Cα (N)/1HN. The advantage of using bidirectional polarization transfer is that amide 1HN are correlated selectively with the 13C and/or 15N, and 1Hα within a residue. This is a prerequisite to assign backbone 1H resonances before they can be correlated to other 1H for assignments and distance measurements.
The experiments simultaneously measure the amide 1H and aliphatic 1H resonance frequencies and have the advantage over conventional methods of using amide 1H-detection only to assign back bone resonances and promote distance measurements through homonuclear spin exchange [14]. This requires multiple step coherence transfers and may cause sensitivity losses for samples that undergo fast relaxation. Secondly, simultaneous 13C and 15N editing experiments may be useful for lifting the ambiguity of resonance assignments for heavily crowded spectra, such as the case shown for fd coat protein.
Additionally, the bidirectional polarization transfer between 13C and 15N has the advantage of correlating amide 15N and 13Cα chemical shifts within a residue with the 13C′ chemical shift of the preceding residue. This step of spectral editing/selective magnetization transfer allows the experiment to be used for sequential resonance assignments in proteins. As observed, 13C-detected experiments show nearly 67% and 33% magnetization transfer from 15N to 13C′ and to 13Cα, respectively. We also observed that the magnetization transfer for the current optimization surprisingly showed 30–50% higher sensitivity compared to the previous optimization method by Barbet-Masin et al. [10] and under similar experimental conditions. Inter- and intra- residue correlation spectra are shown in Figure 3. It is clear from the well-resolved two- and three-dimensional spectra of MLF, which show the amide nitrogen 1H of leucine correlated with the Leu 13Cα and Met 13C′ chemical shifts. In the same spectra, the Phe 1HN resonance is correlated with its 13Cα and the Leu 13C′ chemical shifts. The cross-peaks are marked in Figure 3E by residue type. The three-dimensional data correlating amide 15N and 13C resonances were acquired using a 10% non-uniform sampling schedule in order to reduce the amount of time needed to perform the experiment. The successfully reconstructed spectrum is shown in Figure 3F. The spectrum showed increase in sensitivity and resolution when compared to the linear method of data acquisition. This was achieved due to the long evolution periods that enabled the line narrowing and improved signal-to-noise ratios. We also performed homonuclear 13C/13C mixing prior to heteronuclear 13C/15N mixing to simultaneously obtain intra- and inter- residue correlations such as 1HN/NCACO and 1HN/NCOCA in a single experiment (data not shown). We note that similar results performed at moderate spinning rate have been recently reported [11].
We also applied the experiments to the 50-residue fd coat protein in intact bacteriophage particles. The heteronuclear correlation spectra in Figure 4A and D contain ~30 distinguishable 15N/1HN cross-peaks and ~2013C/1H cross-peaks, respectively. The relatively narrow spectral dispersion among the alpha hydrogen resonances prevents the two-dimensional from providing unambiguous resolution and assignments. In order to achieve adequate resolution, two separate three-dimensional spectra were acquired in a single experiment using the pulse sequence shown in Figure 1. Two two-dimensional slices correlating amide 1H chemical shifts with 15N chemical shifts obtained at 4.2 ppm and 3.8 ppm 1H chemical shifts are shown in Figure 4B and C, respectively. Similarly, two-dimensional slices correlating alpha carbon 1H resonances with 13C chemical shifts obtained at 8.5 ppm and 7.6 ppm are shown in Figure 4E and F, respectively. We also performed the intra- and inter- residue 13C correlation experiment for fd coat protein similar to the experiment performed on MLF shown in Figure 3D. A representative two-dimensional slice obtained at 109.7 ppm 15N chemical shift is shown in Figure 4G. The spectrum shows Gly23 1HN correlation with Gly23 13Cα and Ile22 13C′. This experiment is similar to the CANCO type correlation experiment routinely used for resonance assignment in proteins, but with the additional advantage of correlating 1HN chemical shifts. As seen from the representative slices, we were able to resolve nearly all resonance peaks and able to assign the proton, carbon and nitrogen chemical shifts for the fd coat protein back bone.
Here we demonstrate that pulse sequences utilizing simultaneous cross-polarization, bidirectional coherence transfer pathways and non-uniform sampling can be used to perform multi-dimensional experiments rapidly and efficiently. We have applied these experiments to fully protonated and uniformly 13C and 15N labeled polycrystalline tripeptide and hydrated bacteriophage samples. Multiple chemical shift correlations are observed using simultaneous CP and bidirectional coherence transfer techniques. Bidirectional CP between inter- and intra- residue 13C and 15N nuclei is efficient under fast MAS and can be exploited further in sequential resonance assignment experiments.
Highlights.
Bidirectional 13C-15N transfers at 60 kHz
Simultaneous inter- and intra- residue transfers with 1H irradiation
Simultaneous acquisition of two three-dimensional spectra with one receiver
1H assignments based on bidirectional 13C-15N transfers
Acknowledgments
The research was supported by grant P41EB002031, R01EB005161, R01GM099986, R01GM066978, and R01AI065361 from the National Institutes of Health. It utilized the Biotechnology Resource Center for NMR Molecular Imaging of Proteins at the University of California, San Diego.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rienstra CM, Tucker-Kellogg L, Jaroniec CP, Hohwy M, Reif B, McMahon MT, Tidor B, Lozano-Pérez T, Griffin RG. De novo determination of peptide structure with solid-state magic-angle spinning NMR spectroscopy. Proceedings of the National Academy of Sciences. 2002;99:10260–10265. doi: 10.1073/pnas.152346599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein H, Oschkinat H. Structure of a protein determined by solid-state magic angle spinning NMR spectroscopy. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- 3.Baker LA, Folkers GE, Sinnige T, Houben K, Kaplan M, van der Cruijsen EA, Baldus M. Magic-angle-spinning solid-state NMR of membrane proteins. Meth Enzymol. 2015;557:307–328. doi: 10.1016/bs.mie.2014.12.023. [DOI] [PubMed] [Google Scholar]
- 4.Tycko R, Wickner RB. Molecular structures of amyloid and prion fibrils: consensus versus controversy. Acc Chem Res. 2013;46:1487–1496. doi: 10.1021/ar300282r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeri AC, Mesleh MF, Nevzorov AA, Opella SJ. Structure of the coat protein in fd filamentous bacteriophage particles determined by solid-state NMR spectroscopy. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:6458. doi: 10.1073/pnas.1132059100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou DH, Shea JJ, Nieuwkoop AJ, Franks WT, Wylie BJ, Mullen C, Sandoz D, Rienstra CM. Solis-state protein-structure determination with proton-detected triple-resonance 3D magic-angle-spinning NMR spectroscopy. Angew Chemie Intl Ed. 2007;46:8380–8383. doi: 10.1002/anie.200702905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linser R, Bardiaux B, Andreas LB, Hyberts SG, Morris VK, Pintacuda G, Sunde M, Kwan AH, Wagner G. Solid-State NMR Structure Determination from Diagonal-Compensated, Sparsely Nonuniform-Sampled 4D Proton–Proton Restraints. Journal of the American Chemical Society. 2014;136:11002–11010. doi: 10.1021/ja504603g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holland DJ, Bostock MJ, Gladden LF, Nietlispach D. Fast Multidimensional NMR Spectroscopy Using Compressed Sensing. Angewandte Chemie International Edition. 2011;50:6548–6551. doi: 10.1002/anie.201100440. [DOI] [PubMed] [Google Scholar]
- 9.Kazimierczuk K, Orekhov VY. Accelerated NMR Spectroscopy by Using Compressed Sensing. Angewandte Chemie International Edition. 2011;50:5556–5559. doi: 10.1002/anie.201100370. [DOI] [PubMed] [Google Scholar]
- 10.Barbet-Massin E, Pell AJ, Retel JS, Andreas LB, Jaudzems K, Franks WT, Nieuwkoop AJ, Hiller M, Higman V, Guerry P, Bertarello A, Knight MJ, Felletti M, Le Marchand T, Kotelovica S, Akopjana I, Tars K, Stoppini M, Bellotti V, Bolognesi M, Ricagno S, Chou JJ, Griffin RG, Oschkinat H, Lesage A, Emsley L, Herrmann T, Pintacuda G. Rapid Proton-Detected NMR Assignment for Proteins with Fast Magic Angle Spinning. Journal of the American Chemical Society. 2014;136:12489–12497. doi: 10.1021/ja507382j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herbst C, Bellstedt P, Görlach M, Ramachandran R. MAS solid state NMR of proteins: simultaneous 15N–13CA and 15N–13CO dipolar recoupling via low-power symmetry-based RF pulse schemes. Journal of Biomolecular NMR. 2015;62:7–15. doi: 10.1007/s10858-015-9910-2. [DOI] [PubMed] [Google Scholar]
- 12.Frueh D, Arthanari H, Wagner G. Unambiguous Assignment of NMR Protein Backbone Signals with a Time-shared Triple-resonance Experiment. Journal of Biomolecular NMR. 2005;33:187–196. doi: 10.1007/s10858-005-3204-z. [DOI] [PubMed] [Google Scholar]
- 13.Nielsen A, Székely K, Gath J, Ernst M, Nielsen N, Meier B. Simultaneous acquisition of PAR and PAIN spectra. Journal of Biomolecular NMR. 2012;52:283–288. doi: 10.1007/s10858-012-9616-7. [DOI] [PubMed] [Google Scholar]
- 14.Linser R, Bardiaux B, Higman V, Fink U, Reif B. Structure Calculation from Unambiguous Long-Range Amide and Methyl 1H−1H Distance Restraints for a Microcrystalline Protein with MAS Solid-State NMR Spectroscopy. Journal of the American Chemical Society. 2011;133:5905–5912. doi: 10.1021/ja110222h. [DOI] [PubMed] [Google Scholar]
- 15.Das BB, Opella SJ. Multiple acquisition/multiple observation separated local field/chemical shift correlation solid-state magic angle spinning NMR spectroscopy. Journal of Magnetic Resonance. 2014;245:98–104. doi: 10.1016/j.jmr.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herbst C, Herbst J, Leppert J, Ohlenschläger O, Görlach M, Ramachandran R. Chemical shift correlation at high MAS frequencies employing low-power symmetry-based mixing schemes. Journal of Biomolecular NMR. 2011;50:277–284. doi: 10.1007/s10858-011-9516-2. [DOI] [PubMed] [Google Scholar]
- 17.Gopinath T, Veglia G. Dual Acquisition Magic-Angle Spinning Solid-State NMR-Spectroscopy: Simultaneous Acquisition of Multidimensional Spectra of Biomacromolecules. Angewandte Chemie International Edition. 2012;51:2731–2735. doi: 10.1002/anie.201108132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyberts S, Robson S, Wagner G. Exploring signal-to-noise ratio and sensitivity in non-uniformly sampled multi-dimensional NMR spectra. Journal of Biomolecular NMR. 2013;55:167–178. doi: 10.1007/s10858-012-9698-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyberts SG, Takeuchi K, Wagner G. Poisson-gap sampling and forward maximum entropy reconstruction for enhancing the resolution and sensitivity of protein NMR data. Journal of the American Chemical Society. 2010;132:2145–2147. doi: 10.1021/ja908004w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palmer MR, Suiter CL, Henry GE, Rovnyak J, Hoch JC, Polenova T, Rovnyak D. Sensitivity of Nonuniform Sampling NMR. The Journal of Physical Chemistry B. 2015;119:6502–6515. doi: 10.1021/jp5126415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paramasivam S, Suiter CL, Hou G, Sun S, Palmer M, Hoch JC, Rovnyak D, Polenova T. Enhanced Sensitivity by Nonuniform Sampling Enables Multidimensional MAS NMR Spectroscopy of Protein Assemblies. The Journal of Physical Chemistry B. 2012;116:7416–7427. doi: 10.1021/jp3032786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morcombe CR, Zilm KW. Chemical shift referencing in MAS solid state NMR. Journal of Magnetic Resonance. 2003;162:479–486. doi: 10.1016/s1090-7807(03)00082-x. [DOI] [PubMed] [Google Scholar]
- 23.Detken A, Hardy EH, Ernst M, Meier BH. Simple and efficient decoupling in magic-angle spinning solid-state NMR: the XiX scheme. Chemical Physics Letters. 2002;356:298–304. [Google Scholar]
- 24.Shaka AJ, Barker PB, Freeman R. Computer-Optimized decoupling scheme for wideband applications and low-level operation. J Magn Reson. 1985;64:547–552. [Google Scholar]
- 25.Baldus M, Petkova AT, Herzfeld J, Griffin RG. Cross polarization in the tilted frame: assignment and spectral simplification in heteronuclear spin systems. Molecular Physics. 1998;95:1197–1207. [Google Scholar]
- 26.Zhou DH, Rienstra CM. High-performance solvent suppression for proton detected solid-state NMR. Journal of Magnetic Resonance. 2008;192:167–172. doi: 10.1016/j.jmr.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leskowitz GM, Ghaderi N, Olsen RA, Mueller LJ. Three-qubit nuclear magnetic resonance quantum information processing with a single-crystal solid. The Journal of Chemical Physics. 2003;119:1643–1649. [Google Scholar]
- 28.Morag O, Sgourakis NG, Baker D, Goldbourt A. The NMR–Rosetta capsid model of M13 bacteriophage reveals a quadrupled hydrophobic packing epitope. Proceedings of the National Academy of Sciences. 2015;112:971–976. doi: 10.1073/pnas.1415393112. [DOI] [PMC free article] [PubMed] [Google Scholar]