

Abstract
AIM: To evaluate the effects of 3,3’-diethyl-9-methylthia-carbocyanine iodide (DMTCCI) on DNA primase activity and on apoptosis of human hepatocellular carcinoma BEL-7402 cells.
METHODS: DNA primase assay was used to investigate DNA primase activity. MTT assay was applied to determine cell proliferation. Flow cytometric analysis, transmission electron microscopy, DNA fragmentation assay were performed to detect DMTCCI-induced apoptosis. Expression levels of p53, Bcl-2, Bcl-xL, Bad, Bax, survivin, Caspase-3 and poly (ADP-ribose) polymerase (PARP) were evaluated by immunoblot analysis. Caspase-3 activity was assessed with ApoAlert Caspase-3 colorimetric assay kit.
RESULTS: DMTCCI had inhibitory effects on eukaryotic DNA primase activity with IC50 value of 162.2 nmol/L. It also inhibited proliferation of human hepatocellular carcinoma BEL-7402 cells with IC50 value of 2.09 μmol/L. Furthermore, DMTCCI-induced BEL-7402 cell apoptosis was confirmed by DNA fragmentation (DNA ladders and sub-G1 formation) and transmission electron microscopy (apoptotic bodies formation). During the induction of apoptosis, expression of Bcl-2, Bcl-xL and survivin was decreased, and that of p53, Bad and Bax was increased. Caspase-3 was activated and poly (ADP-ribose) polymerase (PARP) was cleaved in BEL-7402 cells treated with DMTCCI.
CONCLUSION: The present data suggest that DMTCCI has inhibitory effects on eukaryotic DNA primase and can induce apoptosis of BEL-7402 cells. The modulation of expression of p53 and Bcl-2 family proteins, and activation of Caspase-3 might be involved in the induction of apoptosis.
INTRODUCTION
In eukaryotes, DNA primase is the only enzyme catalyzing RNA primer synthesis and initiating DNA replication as well. DNA primase combines tightly with DNA polymerase α. The complex, consisting of four subunits with a molecular weight of 180 kD, 68 kD, 58 kD and 49 kD respectively, is formed. DNA primase consists of p58 and p49 subunit, and the latter is the catalytic core[1]. PRIM1, the gene encoding DNA primase, is located on 12q13. It was found that 12q13-15 was amplified in many kinds of tumors, such as bladder cancer, glioblastoma, neuroblastoma, chronic lymphocytic leukemia, follicular central lymphoma, malignant fibrous histiocytoma, testicular germ cell tumor, breast cancer, sarcoma, primary alveolar rhabdomyosarcoma, Ewing’s sarcoma and osteosarcoma. It was found that DNA primase activity in replicating cells was 5 to 6 times that in static cells. DNA primase could be regarded as a candidate target for cancer chemotherapy. In recent years, a few DNA primase inhibitors have been discovered, including fludarabine and its analogues, sphingosine, suramin, actinomycin, L-ATP, nucleotide analogues, Evans blue and aurintricarboxylic acid[2-4]. However, there have been no anticancer drugs targeting DNA primase mainly in clinical use.
Hepatocellular carcinoma (HCC) is one of the most lethal malignancies in the world and there has been no effective preventive measure for this highly malignant disease up to date. In China, HCC has ranked the second of cancer mortality since 1990’s[5,6]. In this study, we found 3,3’-diethyl-9-methylthiacarbocyanine iodide (DMTCCI) could inhibit DNA primase activity. To explore the therapeutic potential of DMTCCI, we treated hepatocellular carcinoma BEL-7402 cells with DMTCCI and observed the proliferation inhibition and apoptosis induction effects. It was suggested that the modulation of expression of p53 and Bcl-2 family proteins, and activation of Caspase-3 might be involved in the induction of apoptosis.
MATERIALS AND METHODS
Drugs and reagents
DMTCCI (Figure 1), poly(dT) and MTT were purchased from Sigma-Aldrich (USA). RPMI-1640 medium was purchased from GIBCO. DE-52 was purchased from Serva. Phosphocellulose–11 was obtained from Whatman. [α-32P]ATP was purchased from Amersham Biosciences. Anti-Caspase-3 antibody was obtained from Cell Signal Technology. Other antibodies were purchased from Santa Cruz Biotechnology. ApoAlert Caspase-3 colorimetric assay kit was purchased from Clontech. Ehrlich’s ascites carcinoma (EAC) cells and hepatocellular carcinoma cell line BEL-7402 were obtained from the Cancer Center, Sun Yat-sen University, Guangzhou, China.
Figure 1.
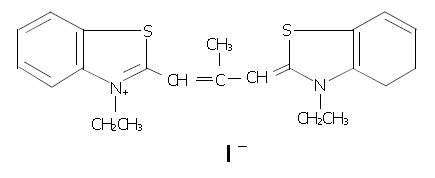
Chemical structure of 3, 3’-diethyl-9-methylthia-carbocyanine iodide (DMTCCI).
DNA primase assay
DNA primase-polymerase complex was purified from mice EAC cells by chromatography (with DE-52 and complex. Appropriate amounts of DMTCCI stock suspended in 0.1% DMSO were incubated with the standard reaction mixture for 30 min at 37 °C. Then, the reaction products were spotted onto GF/C filters (Whatman), which were then washed three times in 5% trichoroacetic acid and twice in 95% ethanol. Filters were dried and the acid-precipitable radioactivity was determined with a liquid scintillation counter (Beckman). DNA primase reaction was measured by the formation of RNA oligomers. In order to explore the mode of inhibition of DMTCCI on DNA primase activity with respect to templet poly[dT], the concentrations of poly[dT] were established at 67, 100, 133, 167 and 200 mg/L with 0, 25 and 50 nmol/L of DMTCCI, respectively. The CPM counts were determined. In terms of Dixon plots, the mode of inhibition of DMTCCI on DNA primase activity was shown. Ki values of DMTCCI on DNA primase activity were calculated in accordance with Michaelis-Menton equation.
Cell culture
Human hepatocellular carcinoma cell line BEL-7402 was cultured in RPMI1640 medium supplemented with 10% fetal bovine serum, penicillin (100 IU/mL) and streptomycin (100 μg/mL) at 37 °C in a 5% CO2 humidified atmosphere.
MTT assay
The logarithmically growing BEL-7402 cells were plated at a density of 1 × 104 cells/well into a 96-well plate. The stock of DMTCCI was diluted with medium, then added to the wells for the desired final concentrations. After 3 days’ exposure to DMTCCI, 10 μL MTT (5 mg/mL) was added to each well and incubated for an additional 4 h, and then the liquid in the wells was evaporated. To dissolve the formazam, 200 μL of DMSO was added. Control wells were treated with 0.1% DMSO alone. The absorbance was detected in the microplate reader 550 model at 570 nm wavelength. Growth inhibition was equal to (1-absorbance of the treated wells)/(absorbance of the control wells) × 100%. IC50 value was determined using a POMS software.
Flow cytometric analysis
Cells were harvested by trypsinization, washed twice with ice-cold PBS, resuspended in ice-cold PBS, and fixed with 70% ethanol. When ready to stain with propidium iodide (PI), the cells were centrifuged. After the ethanol was removed, the cells were washed once in PBS. The cell pellets were then resuspended in 1 mL of PI/Triton X-100 staining solution (0.1% Triton X-100 in PBS, 0.2 mg/mL RNase A, and 10 μg/mL propidium iodide) and incubated for at least 30 min at room temperature. The stained cells were analyzed using a FACScan flow cytometer in combination with BD Lysis II Software (Becton Dickinson).
Transmission electron microscopy
BEL-7402 cells treated with DMTCCI (8 μmol/L) for 48 h were collected and washed once in PBS. Cells were fixed in half-strength Karnofsky’s fixative, postfixed in 1% collidine-buffered osmium tetroxide, dehydrated in a graded series of ethanol and propylene oxide, and embedded in LR white resin. Sections were cut and stained with both uranyl acetate and Reynold’s lead stain. Electron micrographs were photographed with a JEM-1200 electron microscope.
DNA fragmentation assay
The integrity of DNA was assessed by agarose gel electrophoresis. Cells (1 × 106) were centrifuged for 5 min at 3000 g, washed once in PBS, and cell pellets were solubilized in 100 μL of lysis buffer (50 mmol/L Tris-HCl, pH8.0, 10 mmol/L tetraacetic acid, 0.4% SDS, 0.5 g/L proteinase K) and incubated for 8 h at 50 °C, then 10 μL of 0.5 g/L RNase A was added. The samples were incubated for 1 h at 50 °C and heated to 70 °C for 5 min, then 100 μL of phenol:chloroform:isopropanol (25:24:1) was added. After centrifugation, the supernatants were transferred to new tubes, and twice volume ethanol (ice cold) was added. After centrifugation, the pellets were suspended in TE buffer (10 mmol/L Tris-HCl, 1 mmol/L tetraacetic acid) and loaded on 1.8% agarose gel for electrophoresis. The gel was stained with ethidium bromide, and photographed with UV illumination.
Immunoblot analysis
After treatment, the cells were washed in phosphate-buffered saline (PBS), and lysed in 100 μL of lysis buffer (10 mmol/L Tris-HCl, pH7.4, 5 mmol/L MgCl2, 1 mmol/L EDTA, 25 mmol/L NaF, 100 μmol/L fresh Na3VO4 and l mmol/L dithiothreitol). Cell lysates were centrifuged for 10 min at 14000 g. Protein concentration in the supernatant was determined by Bradford protein assay. Equal amounts of protein (40 μg) were resolved on 10% SDS-polyacrylamide gel, and transferred electrophoretically to PVDF membrane. The membranes were blocked with fat free dry milk (5%) in TBST (10 mmol/L Tris-HCl, pH7.4, 150 mmol/L NaCl, 0.1% Tween-20), and then incubated with primary antibodies for 1 h at 4 °C, washed three times in TBST for 30 min, and then incubated with secondary antibody (horseradish peroxidase-conjugated) for 2 h at room temperature. After the secondary antibody was washed, the bound antibody complex was detected using an ECL chemiluminescence reagent. All Western blots were performed three times in independent experiments.
Caspase-3 activity assay
To analyze Caspase-3 activity, DMTCCI-treated cells (1 × 106 cells) were washed twice in PBS and resuspended in cell lysis buffer (10 mmol/L HEPES, pH7.4, 2 mmol/L EDTA, 0.1% CHAPS, 5 mmol/L DTT, 1 mmol/L PMSF, 10 μg/mL pepstatin A, 10 μg/mL aprotinin, and 20 μg/mL leupeptin). The rest of the protocol followed the manufacturer’s instructions (Bio-Rad). A specific inhibitor of Caspase-3, DEVD-fmk, was used to confirm the specificity, as suggested by the manufacturer. Absorbance was read with the microplate reader 550 model at 410 nm and the activity units were determined according to the instructions provided with the kit.
RESULTS
Inhibition of DNA primase activity by DMTCCI
DNA primase is responsible for the synthesis of RNA primers on both leading and lagging strand DNA during semi-conservative DNA replication. When DNA primase activity was assayed in the presence of various concentrations of DMTCCI, our data showed that at the concentrations of 18.5, 37.0, 111.0, 333.3 and 1 000 nmol/L of DMTCCI, the inhibitory rates were (24.9 ± 2.0)%, (31.4 ± 5.0)%, (38.5 ± 3.8)%, (52.5 ± 6.4)% and (80.4 ± 2.9)%, respectively, with IC50 value of (162.2 ± 7.8) nmol/L (Figure 2). The Dixon plot analysis demonstrated that DMTCCI inhibited mammalian DNA primase by a non-competitive mechanism (Ki = 60.5 nmol/L) (Figure 3). The kinetics of the blockade indicated an allosteric modulation by DMTCCI of DNA primase activity.
Figure 2.
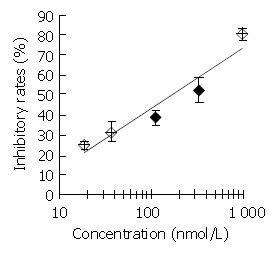
Inhibitory effect of DMTCCI on DNA primase activity. Appropriate amounts of DMTCCI were incubated with the standard reaction mixture containing DNA primase-poly-merase complex purified from mice Ehrilich’s ascites carci-noma cells for 30 min at 37 °C. DNA primase reaction was mea-sured by the formation of RNA oligomers. Results were ex-pressed as mean ± SD for three independent experiments.
Figure 3.
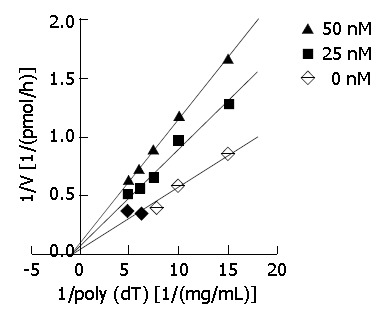
Kinetic analysis. Primase activity was measured in the absence or presence of DMTCCI using the indicated concentrations of DNA template poly[dT]. A Dixon plot was made on the basis of the data. Basically similar results were obtained in three separate repetitions of the experiment.
Growth inhibition of BEL-7402 cells by DMTCCI
After 3 days’ exposure to DMTCCI, cell survival was analyzed by MTT assay. The results of MTT assay showed that DMTCCI inhibited cell proliferation of BEL-7402. At the concentrations of 0.3125, 0.625, 1.25, 2.5, 5, 10 and 20 μmol/L, the inhibitory rates were 16.9%, 27.7%, 38.5%, 49.2%, 67.7%, 81.5% and 87.7% respectively. IC50 value was 2.09 μmol/L (Figure 4).
Figure 4.
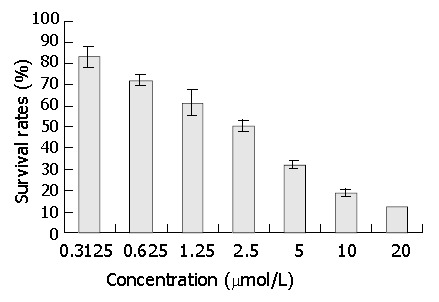
Effect of DNA primase inhibitor DMTCCI on proliferation of BEL-7402 cells. BEL-7402 cells were treated with various concentrations of DMTCCI as indicated for 72 h. Cell viability was determined with MTT assay and shown as survival rate.
Detection of apoptosis by flow cytometry
To examine whether BEL-7402 cells underwent apoptosis when they were treated with DMTCCI, the cells were stained with propidium iodide, followed by examination of the appearance of sub-G1 population using flow cytometry. After treatment with 2 μmol/L, 4 μmol/L, 8 μmol/L of DMTCCI for 24 h, cell death became apparent. As evidenced by the appearance of sub-G1 population, the apoptotic indexes in BEL-7402 cells were 10.8%, 16.5% and 24.3%, respectively. At 36 h, the apoptotic indexes were 14.0%, 26.2% and 33.1%, respectively. At 48 h, the apoptotic indexes were 21.8%, 28.6% and 41.0%, respectively (Figure 5).
Figure 5.
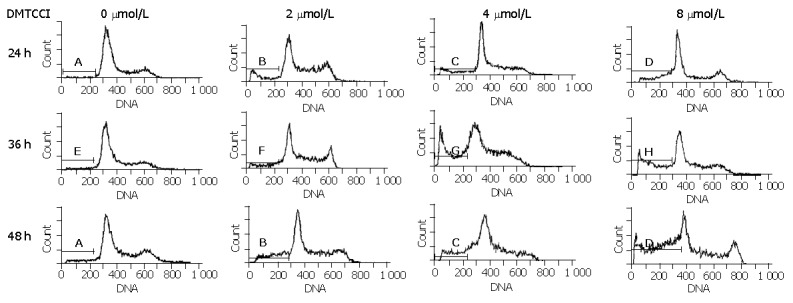
Flow cytometric analysis of BEL-7402 cells treated with DMTCCI. Untreated cells (control) or cells treated with DMTCCI were stained with propidium iodide, their DNA content was measured by flow cytometry. The data shown are representative of three experiments performed.
Transmission electron microscopy
Transmission electron microscopy showed that morphological changes typical of apoptosis were observed in BEL-7402 cells treated with DMTCCI. The cytoplasm shrank and the chromatin of cells became condensed and marginated. Chromatin was fragmented into apoptotic bodies in which internal organelles were preserved, while most of control cells had centrally located round nuclei with prominent nucleoli and uniform morphology (Figure 6).
Figure 6.
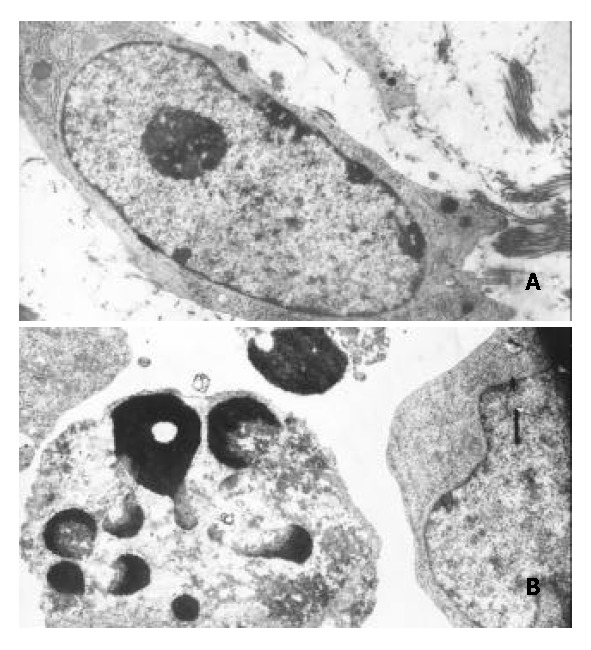
Morphological changes typical of apoptosis in BEL-7402 cells treated with 8 μmol/L of DMTCCI for 24 h. BEL-7402 cells showed apoptotic morphological changes of the chro-matin being condensed and marginated. A: Control. × 5000; B: Treated with DMTTCI 8 μmol/L. × 7000.
Internucleosomal DNA damage in DMTCCI-treated BEL-7402 cells
Genomic DNA fragmentation was assayed as a hallmark of apoptotic cell death. As shown in Figure 7, agarose gel electrophoresis of DNA extracted from cells treated with DMTCCI revealed ladders of DNA fragmentation, indicative of induced apoptosis in BEL-7402 cells.
Figure 7.
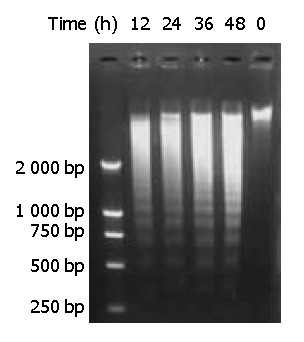
DNA fragmentation of BEL-7402 cells induced by DMTCCI. BEL-7402 cells were incubated in the absence or pres-ence of DMTCCI 8 μM for 12 h, 24 h, 36 h, 48 h. Cells were harvested. DNA was isolated and DNA fragmentation was visualized as oligonucleosome-sized fragmentation in ethidium bromide after DNA agarose gel electrophoresis. This figure is a representative of three experiments.
Effect of DMTCCI on p53 in BEL-7402 cells
DNA damage could activate tumor suppressor gene p53, thereby inducing cell growth arrest, which could allow time for DNA repair. To investigate whether p53 protein was associated with DMTCCI-induced apoptosis, we examined the change in p53 levels during the apoptotic process in BEL-7402 cells. Western blot analysis showed that the level of p53 protein increased following DMTCCI treatment in a time-dependent manner (Figure 8).
Figure 8.
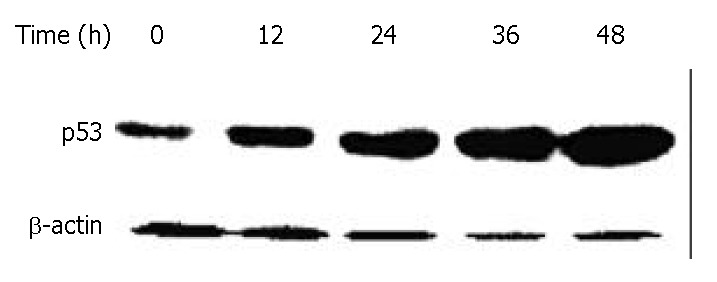
Down-regulation of p53 in BEL-7402 cells by DMTCCI. BEL-7402 cells were incubated with 8 μM DMTCCI for indicated time. Cells were harvested, lysed and resolved in 10% SDS-polyacrylamide gel. p53 protein levels were determined by immunoblot analysis. β-actin was used as a control.
Effect of DMTCCI on the expression of Bcl-2 family proteins
The Bcl-2 family proteins play a prominent role in the regulation of apoptosis. In this study, DMTCCI induced the expression of pro-apoptotic proteins, Bad and Bax, and downregulated the anti-apoptotic proteins, Bcl-2 and Bcl-xL in BEL-7402 cells. We deduced Bcl-2 family members might play a role in apoptosis induced by DMTCCI in BEL-7402 cells (Figure 9).
Figure 9.
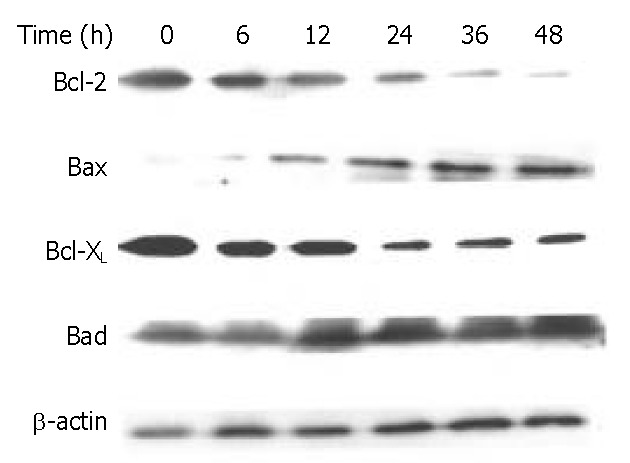
Effects of DMTCCI on levels of Bcl-2, Bax, Bcl-xL and Bad in BEL-7402 cells. BEL-7402 cells were treated with DMTCCI (8 μM) for indicated time, total protein was isolated by immunoblotting analysis. After electrophoresis on a 10% SDS-polyacrylamide gel, the protein was transferred to a PVDF membrane. The membrane was analyzed by immunoblotting with anti-Bcl-2, Bax, Bcl-xL and Bad antibodies, respectively. β-actin protein was used as a control.
Effect of DMTCCI on survivin in BEL-7402 cells
Survivin is a member of the inhibitors of apoptosis protein family that is believed to play a role in oncogenesis. BEL-7402 cells treated with 8 μM of DMTCCI for 6, 12, 24, 36 and 48 h were collected. Western blot analysis showed that the level of survivin protein decreased following DMTCCI treatment in a time-dependent manner (Figure 10).
Figure 10.
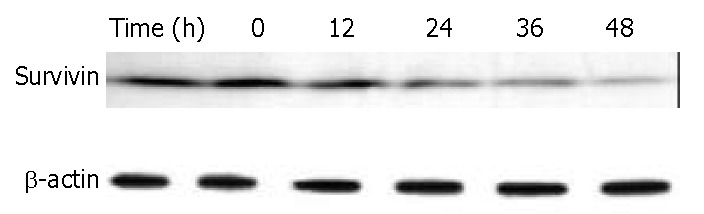
Down-regulation of survivin in BEL-7402 cells by DMTCCI. BEL-7402 cells were incubated with 8 μM DMTCCI for indicated time. Cells were harvested, lysed and resolved in 10% SDS-polyacrylamide gel. Survivin protein levels were determined by immunoblot analysis. β-actin was used as a control.
Caspase-3 activity in DMTCCI-treated BEL-7402 cells
The results shown above indicated that DMTCCI induced apoptosis in BEL-7402 cells. To probe into the mechanism of apoptosis, we detected Caspase-3 activity following treatment of BEL-7402 with DMTCCI. As shown in Figure 11, Caspase-3 activity was apparently increased at 6 h and reached a peak at 24 h in response to DMTCCI treatment in BEL-7402 cells.
Figure 11.
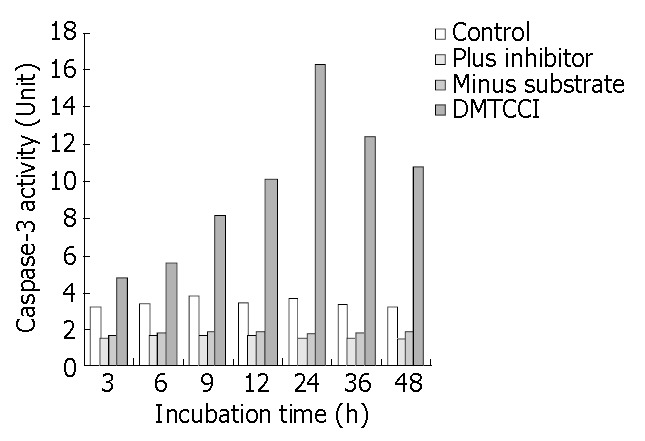
Inhibition of DMTCCI on activity of Caspase-3 in BEL-7402 cells. BEL-7402 cells were harvested after exposure to 8 μmol/L of DMTCCI for indicated time. Activity of Caspase-3 was measured by ApoAlert caspase-3 assay kits. Cell lysates were prepared for each sample and used in the Caspase-3 ac-tivity assay using the colorimetric substrate DEVD-pNA. Ab-sorbance was read at 410 nm and This figure is a representa-tive of three experiments.
Cleavage of Caspase-3 induced by DMTCCI in BEL-7402 cells
Caspase-3, a member of the Caspase family, was shown to play an essential role in apoptosis induced by a variety of stimuli[8-10]. We examined whether Caspase-3 was activated during DMTCCI-induced apoptosis in BEL-7402 cells. Activation of Caspase-3 was determined by monitoring proteolysis of the 35-kDa procaspase-3 to 17-kDa fragments. Figure 12 shows the appearance of the 17-kDa fragments of the active enzyme following exposure to 8 μmol/L DMTCCI for 24 h, indicating that DMTCCI efficiently activated Caspase-3 in BEL-7402 cells.
Figure 12.
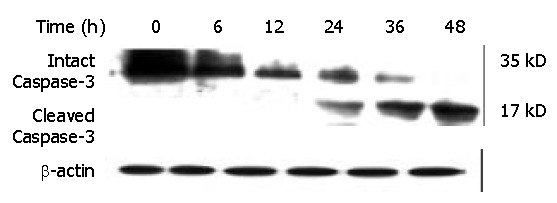
Effect of DMTCCI on levels of proteins of Caspase-3 in BEL-7402 cells. BEL-7402 cells were treated with 8 μM DMTCCI for the indicated time. Western blot analysis was performed with anti-Caspase-3 antibody. β-actin was used as a control.
Cleavage of PARP induced by DMTCCI
Caspases are synthesized as inactive proenzymes. When activated, they could specifically cleave both relevant cellular substrates and zymogens to promote apoptosis[11,12]. One of the substrates for Caspase protease during apoptosis was PARP, an enzyme that appears to be involved in response to environment stress. Proteolytic cleavage of PARP could result in a characteristic shift of the protein upon electrophoresis from 116 to 89 kDa. PARP cleavage was used as an indicator of caspase activation in response to DMTCCI treatment. PARP could be cleaved 24 h after DMTCCI treatment (Figure 13).
Figure 13.
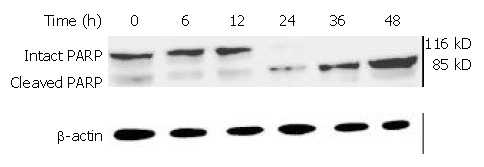
PARP cleavage in BEL-7402 cells induced by DMTCCI. After BEL-7402 cells were treated with 8 μM DMTCCI for the indicated time, total protein was isolated by immunoblotting analysis. After electrophoresis on a 10% SDS-polyacrylamide gel, the protein was transferred to a PVDF membrane . The membrane was analyzed by immunoblotting with antibody against PARP. β-actin protein was used as a control.
DISCUSSION
In this study, we found DMTCCI could inhibit proliferation of hepatocellular carcinoma BEL-7402 cells. DNA replication is a prime target of cancer chemotherapeutics. For example, DNA polymerase α inhibitors have been widely used in chemotherapy. DNA primase could combine tightly with DNA polymerase α as a complex. It could play a critical role in coupling replication to repair and in checkpoint control during S phase, as well as in telomere maintenance[1]. However, DNA primase has not been well studied as an anticancer target since its structure and function were not clarified until recently. DNA primase is considered as a key enzyme in DNA replication because it is the only enzyme that can initiate the synthesis of new DNA strands de novo. We believe new target helps improve existing treatment and overcome drug resistance. Nevertheless, our study showed DMTCCI could suppress cell proliferation at concentrations that were comparable to Ki values for its inhibition on DNA primase. The growth suppression of human hepatocellular carcinoma BEL-7402 cells by DMTCCI was well correlated with DNA primase inhibition.
Apoptosis plays important roles in the control of various biological systems, such as immune responses, hematopoiesis, embryonic development, and carcinogenesis. Apoptosis is an active cell death and could be triggered by a variety of pharmacological and physiological agents[13-15]. In this study, we reported firstly that DMTCCI induced apoptosis in BEL-7402 cells demonstrated by DNA fragmentation, flow cytometry analysis and transmission electron microscopy.
P53 protein has been implicated in DNA damage recognition, DNA repair, cell cycle regulation and particularly in triggering apoptosis after genetic injury[16-18]. To examine whether DMTCCI-induced apoptosis was associated with p53, we measured the level of p53 in BEL-7402 cells. Western blot analysis revealed that p53 protein level increased following DMTCCI treatment in a time-dependent manner, indicating that p53 might be involved in DMTCCI-mediated apoptosis.
It has been found that apoptotic process is controlled by several genes, including inducers and repressors, some of them belong to the Bcl-2 family[19]. Bcl-2 family could be divided into two classes: those such as Bcl-2 and Bcl-xL, are able to block apoptosis, and others, such as Bad and Bax, have proapoptotic effects[20-22]. The balance between the anti-apoptotic and pro-apoptotic Bcl-2 family members may be critical to determine if a cell undergoes apoptosis. The death promoting protein Bax counteracts the anti-apoptotic effects of Bcl-2 by forming a heterodimer with Bcl-2. The ratio of Bcl-2 to Bax, rather than the levels of individual proteins, was considered to be critical in determining the survival/death of cells[23-25]. Bad could compete for the binding to Bcl-xL and Bcl-2, thereby affecting the level of heterodimerization of these proteins with Bax[26]. Our data showed that the expression level of protein Bad and Bax was increased, and that of Bcl-2 and Bcl-xL was decreased in BEL-7402 cells treated with DMTCCI. Taken together, the significant increase of Bad and Bax, and decrease of Bcl-2 and Bcl-xL would favor Bad to compete for Bcl-xL, resulting in much more free Bax into homodimers, which might induce apoptotic response in BEL-7402 cells.
Survivin has recently been identified as a novel inhibitor of apoptosis (IAP). Acting as one of the substrate inhibitors of Caspases, survivin blocks the activation of different effector Caspases. Overexpression of survivin could lead to the suppression of apoptosis triggered by diverse proapoptotic signals[27-29]. In this study, DMTCCI decreased the level of survivin in BEL-7402 cells, indicating that survivin might be involved in DMTCCI-mediated apoptosis.
It has been found that apoptosis is coordinated by a family of cysteine proteases, the Caspases, which are activated upon receipt of divergent pro-apoptotic stimuli[30-32]. In this study, we found that Caspase-3 was activated during DMTCCI-induced apoptosis in BEL-7402 cells. Caspase-3 is believed to be the main executioner Caspase and its activation has been shown to be essential for DNA fragmentaion as well as chromatin condensation and proteolysis of certain Caspase substrates. During the process of DMTCCI-induced apoptosis, Caspase-3 activity was apparently increased at 6 h and reached a peak at 24 h in response to DMTCCI in BEL-7402 cells. One of the substrates for Caspase-3 during apoptosis was PARP, an enzyme that appears to be involved in response to environment stress. PARP cleavage was used as an indicator of Caspase-3 activation in response to DMTCCI treatment. PARP was cleaved 24 h after DMTCCI treatment. These results suggest that caspase-3 activation is involved in DMTCCI-mediated apoptosis in BEL-7402 cells.
In conclusion, DMTCCI could inhibit proliferation and induce apoptosis of human hepatocellular carcinoma BEL-7402 cells. In this study, the expression of Bcl-2, survivin and Bcl-xL was decreased, and that of Bad and Bax was increased in BEL-7402 cells treated with DMTCCI. Furthermore, we found that Caspase-3 was activated during apoptosis and the processing of Caspase-3 was accompanied by proteolytic cleavage of PARP. These finally led to apoptotic events including apoptotic bodies and DNA fragmentation. These results indicate that the induction of apoptosis by DMTCCI involves the modulation of expression of p53 and Bcl-2 family proteins and activation of Caspase-3. It can be a candidate for the development of new anti-tumor agents targeting DNA primase associated with human cancer, including human hepatocellular carcinoma.
Footnotes
Supported by the National High Technology Research and Development Program of China (863 Program), No. 2002AA2Z341C and the National Natural Science Foundation of China, No. 39870886
Edited by Zhu LH and Wang XL
References
- 1.Arezi B, Kuchta RD. Eukaryotic DNA primase. Trends Biochem Sci. 2000;25:572–576. doi: 10.1016/s0968-0004(00)01680-7. [DOI] [PubMed] [Google Scholar]
- 2.Cloutier S, Hamel H, Champagne M, Yotov WV. Mapping of the human DNA primase 1 (PRIM1) to chromosome 12q13. Genomics. 1997;43:398–401. doi: 10.1006/geno.1997.4833. [DOI] [PubMed] [Google Scholar]
- 3.Grosse F, Krauss G. The primase activity of DNA polymerase alpha from calf thymus. J Biol Chem. 1985;260:1881–1888. [PubMed] [Google Scholar]
- 4.Simbulan CM, Tamiya-Koizumi K, Suzuki M, Shoji M, Taki T, Yoshida S. Sphingosine inhibits the synthesis of RNA primers by primase in vitro. Biochemistry. 1994;33:9007–9012. doi: 10.1021/bi00196a019. [DOI] [PubMed] [Google Scholar]
- 5.Zhao WH, Ma ZM, Zhou XR, Feng YZ, Fang BS. Prediction of recurrence and prognosis in patients with hepatocellular carcinoma after resection by use of CLIP score. World J Gastroenterol. 2002;8:237–242. doi: 10.3748/wjg.v8.i2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts LR, LaRusso NF. Potential roles of tumor suppressor genes and microsatellite instability in hepatocellular carcinogenesis in southern African blacks. World J Gastroenterol. 2000;6:37–41. doi: 10.3748/wjg.v6.i1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tamiya-Koizumi K, Murate T, Suzuki M, Simbulan CM, Nakagawa M, Takemura M, Furuta K, Izuta S, Yoshida S. Inhibition of DNA primase by sphingosine and its analogues parallels with their growth suppression of cultured human leukemic cells. Biochem Mol Biol Int. 1997;41:1179–1189. doi: 10.1080/15216549700202271. [DOI] [PubMed] [Google Scholar]
- 8.Devarajan E, Sahin AA, Chen JS, Krishnamurthy RR, Aggarwal N, Brun AM, Sapino A, Zhang F, Sharma D, Yang XH, et al. Down-regulation of caspase 3 in breast cancer: a possible mechanism for chemoresistance. Oncogene. 2002;21:8843–8851. doi: 10.1038/sj.onc.1206044. [DOI] [PubMed] [Google Scholar]
- 9.Chen T, Yang I, Irby R, Shain KH, Wang HG, Quackenbush J, Coppola D, Cheng JQ, Yeatman TJ. Regulation of caspase expression and apoptosis by adenomatous polyposis coli. Cancer Res. 2003;63:4368–4374. [PubMed] [Google Scholar]
- 10.Ceruti S, Beltrami E, Matarrese P, Mazzola A, Cattabeni F, Malorni W, Abbracchio MP. A key role for caspase-2 and caspase-3 in the apoptosis induced by 2-chloro-2'-deoxy-adenosine (cladribine) and 2-chloro-adenosine in human astrocytoma cells. Mol Pharmacol. 2003;63:1437–1447. doi: 10.1124/mol.63.6.1437. [DOI] [PubMed] [Google Scholar]
- 11.Sharma K, Wang RX, Zhang LY, Yin DL, Luo XY, Solomon JC, Jiang RF, Markos K, Davidson W, Scott DW, et al. Death the Fas way: regulation and pathophysiology of CD95 and its ligand. Pharmacol Ther. 2000;88:333–347. doi: 10.1016/s0163-7258(00)00096-6. [DOI] [PubMed] [Google Scholar]
- 12.Nicholson DW. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999;6:1028–1042. doi: 10.1038/sj.cdd.4400598. [DOI] [PubMed] [Google Scholar]
- 13.Zimmermann KC, Bonzon C, Green DR. The machinery of programmed cell death. Pharmacol Ther. 2001;92:57–70. doi: 10.1016/s0163-7258(01)00159-0. [DOI] [PubMed] [Google Scholar]
- 14.Pallis M, Bradshaw TD, Westwell AD, Grundy M, Stevens MF, Russell N. Induction of apoptosis without redox catastrophe by thioredoxin-inhibitory compounds. Biochem Pharmacol. 2003;66:1695–1705. doi: 10.1016/s0006-2952(03)00471-4. [DOI] [PubMed] [Google Scholar]
- 15.Gil D, Garcia LF, Rojas M. Modulation of macrophage apoptosis by antimycobacterial therapy: physiological role of apoptosis in the control of Mycobacterium tuberculosis. Toxicol Appl Pharmacol. 2003;190:111–119. doi: 10.1016/s0041-008x(03)00162-5. [DOI] [PubMed] [Google Scholar]
- 16.Adimoolam S, Ford JM. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc Natl Acad Sci USA. 2002;99:12985–12990. doi: 10.1073/pnas.202485699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith ND, Rubenstein JN, Eggener SE, Kozlowski JM. The p53 tumor suppressor gene and nuclear protein: basic science review and relevance in the management of bladder cancer. J Urol. 2003;169:1219–1228. doi: 10.1097/01.ju.0000056085.58221.80. [DOI] [PubMed] [Google Scholar]
- 18.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 19.Konopleva M, Konoplev S, Hu W, Zaritskey AY, Afanasiev BV, Andreeff M. Stromal cells prevent apoptosis of AML cells by up-regulation of anti-apoptotic proteins. Leukemia. 2002;16:1713–1724. doi: 10.1038/sj.leu.2402608. [DOI] [PubMed] [Google Scholar]
- 20.Panaretakis T, Pokrovskaja K, Shoshan MC, Grandér D. Activation of Bak, Bax, and BH3-only proteins in the apoptotic response to doxorubicin. J Biol Chem. 2002;277:44317–44326. doi: 10.1074/jbc.M205273200. [DOI] [PubMed] [Google Scholar]
- 21.Bellosillo B, Villamor N, López-Guillermo A, Marcé S, Bosch F, Campo E, Montserrat E, Colomer D. Spontaneous and drug-induced apoptosis is mediated by conformational changes of Bax and Bak in B-cell chronic lymphocytic leukemia. Blood. 2002;100:1810–1816. doi: 10.1182/blood-2001-12-0327. [DOI] [PubMed] [Google Scholar]
- 22.Zhou HB, Zhu JR. Paclitaxel induces apoptosis in human gastric carcinoma cells. World J Gastroenterol. 2003;9:442–445. doi: 10.3748/wjg.v9.i3.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pettersson F, Dalgleish AG, Bissonnette RP, Colston KW. Retinoids cause apoptosis in pancreatic cancer cells via activation of RAR-gamma and altered expression of Bcl-2/Bax. Br J Cancer. 2002;87:555–561. doi: 10.1038/sj.bjc.6600496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou HB, Yan Y, Sun YN, Zhu JR. Resveratrol induces apoptosis in human esophageal carcinoma cells. World J Gastroenterol. 2003;9:408–411. doi: 10.3748/wjg.v9.i3.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li SM, Yao SK, Yamamura N, Nakamura T. Expression of Bcl-2 and Bax in extrahepatic biliary tract carcinoma and dysplasia. World J Gastroenterol. 2003;9:2579–2582. doi: 10.3748/wjg.v9.i11.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siervo-Sassi RR, Marrangoni AM, Feng X, Naoumova N, Winans M, Edwards RP, Lokshin A. Physiological and molecular effects of Apo2L/TRAIL and cisplatin in ovarian carcinoma cell lines. Cancer Lett. 2003;190:61–72. doi: 10.1016/s0304-3835(02)00579-7. [DOI] [PubMed] [Google Scholar]
- 27.Xu ZX, Zhao RX, Ding T, Tran TT, Zhang W, Pandolfi PP, Chang KS. Promyelocytic leukemia protein 4 induces apoptosis by inhibition of survivin expression. J Biol Chem. 2004;279:1838–1844. doi: 10.1074/jbc.M310987200. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Cao Z, Yan H, Wood WC. Coexistence of high levels of apoptotic signaling and inhibitor of apoptosis proteins in human tumor cells: implication for cancer specific therapy. Cancer Res. 2003;63:6815–6824. [PubMed] [Google Scholar]
- 29.Carter BZ, Wang RY, Schober WD, Milella M, Chism D, Andreeff M. Targeting Survivin expression induces cell proliferation defect and subsequent cell death involving mitochondrial pathway in myeloid leukemic cells. Cell Cycle. 2003;2:488–493. [PubMed] [Google Scholar]
- 30.Li HL, Chen DD, Li XH, Zhang HW, Lü JH, Ren XD, Wang CC. JTE-522-induced apoptosis in human gastric adenocarcinoma [correction of adenocarcinoma] cell line AGS cells by caspase activation accompanying cytochrome C release, membrane translocation of Bax and loss of mitochondrial membrane potential. World J Gastroenterol. 2002;8:217–223. doi: 10.3748/wjg.v8.i2.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolf BB, Green DR. Suicidal tendencies: apoptotic cell death by caspase family proteinases. J Biol Chem. 1999;274:20049–20052. doi: 10.1074/jbc.274.29.20049. [DOI] [PubMed] [Google Scholar]
- 32.Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]