

Abstract
Cellular response to genotoxic stress is a very complex process, and it usually starts with the “sensing” or “detection” of the DNA damage, followed by a series of events that include signal transduction and activation of transcription factors. The activated transcription factors induce expressions of many genes which are involved in cellular functions such as DNA repair, cell cycle arrest, and cell death. There have been extensive studies from multiple disciplines exploring the mechanisms of cellular genotoxic responses, which have resulted in the identification of many cellular components involved in this process, including the mitogen-activated protein kinases (MAPKs) cascade. Although the initial activation of protein kinase cascade is not fully understood, human protein kinases ATM (ataxia-telangiectasia, mutated) and ATR (ATM and Rad3-related) are emerging as potential sensors of DNA damage. Current progresses in ATM/ATR research and related signaling pathways are discussed in this review, in an effort to facilitate a better understanding of genotoxic stress response.
INTRODUCTION
Cellular response to genotoxic stress is a very complex process. However, it can be “simply” envisioned as a signal transduction cascade in which DNA lesions act as the initial signal that is detected by sensors and passed down through transducers. Eventually the effectors receive the signal and execute various cellular functions (Figure 1). Much knowledge has been gained over the years concerning the signal transducers, and a large group of serine-threonine protein kinases, namely the mitogen-activated protein kinases (MAPKs), along with their upstream kinases, have been shown to play prominent roles in cellular genotoxic responses[1]. Three major classes of MAPKs, i.e., extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK), and p38 (also known as SAPK2, RK, CSBP, or Mxi2), could all be activated by various genotoxic stresses[1-7]. Although the precise mechanism has not been fully understood, it is known that damage to cellular DNA somehow leads to the activation of a group of serine-threonine kinases called MAPK kinase kinases (MAPKKK, or MEKK, MEK kinase), which phosphorylate the downstream dual-specificity kinases called MAPK kinases (MAPKK, or MEK, MAPK/ERK kinase). These MAPKKs then phosphorylate the threonine and tyrosine residues in MAPKs. This three-component module could be assembled together by scaffold proteins that ensure the efficiency and specificity of each individual MAPK pathway[8-10]. The activated MAPKs then translocate to the nucleus and phosphorylate scores of target proteins, including many transcription factors. Among these transcription factors is the tumor suppressor p53 protein, which plays such an important role in the genotoxic stress response that has earned the reputation as the “universal sensor for genotoxic stress”[2,11,12].
Figure 1.
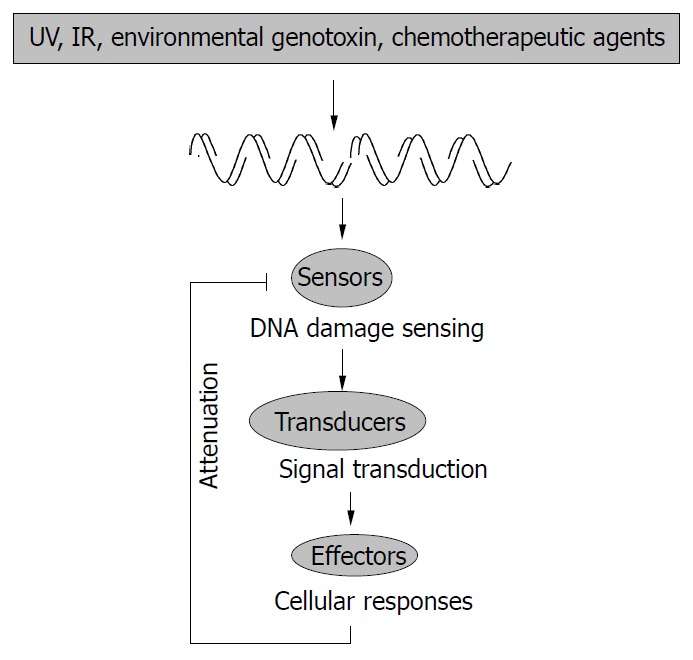
A general schematic representation of cellular re-sponses to genotoxic stress. Ultraviolet (UV), ionizing radia-tion (IR), and various chemicals can induce DNA damage, such as double strand breaks (DSBs), which can be detected by “sensors”. This generates some signal that can be transduced by the transducers to effector molecules. Finally, there is the presence of an attenuation mechanism to control the cellular response to genotoxic stress.
Although studies on MAPKs have provided a lot useful information about signal transducers, the initial sensors for DNA damage remain to be identified. Recently, it has been proposed that some multi-protein complexes that are involved in DNA maintenance or repair, such as the Rad family member Rad1, 9, 17, 26, and Hus1, might function as DNA damage sensors[13-18]. Members of the phosphatidylinositol 3-kinase (PI-3) superfamily, which are activated at the very early stages of DNA damage response, could also serve as sensors, as well as initiators, of the ensuing cellular genotoxic stress response, including ATM and ATR in humans[19]. Although these proteins share the PI-3-like kinase domain, they could not function as lipid kinases, but rather as serine-threonine protein kinases[14,20-25].
ATM AND ATR
Biochemistry of ATM and ATR
One distinguishing characteristic of the PI-3 family members is their unusually large size, which ranges from around 300 kDa to over 500 kDa. ATM is a 3 056 amino acid (aa) protein while ATR is a 2 644 aa protein, and both have a C-terminal catalytic domain (-300 aa) which is flanked by two loosely conserved domains. Although it has not been known how exactly these two kinases sense the DNA damage, it is clear that both kinases can be activated by DNA damage. However, it has been found that ATM responds primarily to double-strand breaks induced by ionizing irradiation (IR), while ATR also reacts to UV or stalled replication forks[13,14,26-29].
Activation of ATM and ATR
Several mechanisms have been proposed for the activation of ATM and ATR by DNA damage: a) direct activation through interaction with damaged DNA, b) indirect activation through interaction with DNA repair or maintenance proteins, or c) a combination of both[30]. Existing experimental data support the third mechanism, that they are activated both through interactions with DNA and members of the repair complexes. For example, ATM could bind directly to DNA. Furthermore, pre-treatment of DNA-cellulose matrix with IR or restriction enzymes could stimulate ATM binding, suggesting that ATM binds to DNA ends[31,32]. ATR could also bind to DNA, with a higher affinity to UV-damaged than undamaged DNA. In addition, damaged DNA could stimulate the kinase activity of ATR to a significantly higher level than undamaged DNA[33,34]. ATM and ATR also interact with many proteins that co-localize at the site of DNA damage. For example, ATM as a part of a super protein complex called BRCA1-associated genome surveillance complex (BASC), is involved in the recognition and repair of aberrant DNA structures. It has been found this complex contains several other proteins such as breast cancer gene 1 (BRCA1), mismatch-repair protein hRad50, and BLM helicase[35]. ATM could bind to histone deacetylase HDAC1 both in vitro and in vivo, and the extent of this association was increased after exposure of MRC5CV1 human fibroblasts to IR[36]. ATR was also able to bind to Rad17[37] and BRCA1[38], and associated with components of the nucleosome remodeling and deacetylating (NRD) complex such as chromodomain-helicase-DNA-binding protein 4 (CHD4) and histone-deacetylase-2 (HDAC2)[39]. All these data support the model that multiple checkpoint protein complexes localize at the sites of DNA damage independently and interact to trigger the checkpoint-signaling cascade.
Interaction with c-Abl
c-Abl, a non-receptor tyrosine kinase that is ubiquitously expressed and localized in both nucleus and cytoplasm, could be up-regulated following exposure to IR or genotoxic chemicals such as cisplatin, methyl methane sulfonate (MMS), mitomycin-C, hydrogen peroxide, but not UV[3,40-42]. IR-induced activation of c-Abl has been shown to require the involvement of ATM in some cases, with ATM phosphorylating serine residue 465 located within the kinase domain of c-Abl[43-45]. However, other studies found that c-Abl was not essential for ATM function in chromosomal maintenance, suggesting that c-Abl and ATM are at least partially independent[46].
An important effect which has been found following the activation of c-Abl, is the induction of cell cycle arrest in a p53-dependent manner, with the possible involvement of Rb, but not P21Cip[47,48]. c-Abl could directly interact with and phosphorylate p53, and regulate the level of p53 by preventing its nuclear export and ubiquitination-dependent degradation[49,50]. It could also induce apoptosis in response to DNA damage[51,52], although this activity involved collaboration with p73 more than p53[53-55]. c-Abl binds to p73 through its Src-homology (SH3) domain to phosphorylate p73 at tyrosine residues, which in turn activates p73-dependent apoptosis pathway.
Regulation of the tumor suppressor p53 protein
Since p53 is such an important mediator in cellular response to genotoxic stress, it is no wonder that ATM/ATR can regulate p53 activity at multiple levels (Figure 2). The most straightforward way to manage p53 is through direct interaction,e.g. , phosphorylation of p53. Both ATM and ATR have been shown to phosphorylate p53 protein at serine 15 to enhance its transactivating activity[56-59]. ATM is also required for dephosphorylation of Ser 376, which can create a binding site for 14-3-3 protein. The association of p53 and 14-3-3 could increase the affinity of p53 for its specific DNA sequence, therefore enhancing its transcriptional activity[60]. Other sites that could be phosphorylated by ATM on p53 include Ser 6, 9, 46, and Thr 18, which may be important for the apoptotic activity of p53 (Ser 46) or may enhance the acetylation of p53 (Ser 6, 9, Thr18)[61]. In addition, ATM/ATR could regulate p53 through the action of other kinases. For example, ATM-activated c-Abl could phosphorylate p53 at Ser 20, which is important for the stabilization of p53 since this modification interferes with the binding between p53 and its regulator murine double minute 2 (Mdm2)[49,62]. ATM-activated Chk2 could also phosphorylate p53 protein at Ser 20 and possibly at other sites, leading to the activation of p53[63-65]. Furthermore, ATM has been found able to bind and phosphorylate Mdm2 and HDM2 (the human homologue of Mdm2), thus inhibiting p53 degradation and promoting its accumulation in cells[66-68].
Figure 2.
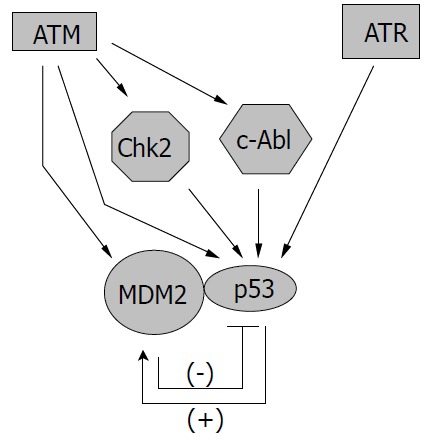
Regulation of p53 protein by ATM and ATR. ATM and ATR can influence the activity of p53 directly through phos-phorylation or indirectly through the action of other kinases. Furthermore, ATM can regulate p53 through phosphorylation of Mdm2 molecule, the negative regulator of p53, which can be up-regulated by p53.
Activation of MAPKs
Accumulative data support the notion that the activation of MAPKs in response to genotoxic stress is ATM/ATR dependent. For example, DNA damaging stimuli, including etoposide (ETOP), adriamycin (ADR), IR, and UV could activate ERK1/2 in primary (MEF and IMR90), immortalized (NIH3T3) and transformed (MCF-7) cells. It has further been shown that ERK activation in response to ETOP could be abolished in ATM-/- fibroblasts (GM05823) independenty of p53[69]. UVA (320-400 nm) triggered ATM-dependent p53 phosphorylation and JNK activation that resulted in apoptosis, while ATR was required for UVC (200-290 nm)-mediated p53 phosphorylation and JNK activation[70]. In addition, activation of ATM by gamma irradiation could lead to the activation of MKK6 and p38γ isoform, and that activation of both MKK6 and p38γ was essential for the proper regulation of G2 checkpoint in mammalian cells[71].
Although the link between ATM/ATR and MAPKs has been established, it is still not clear how ATM/ATR activates MAPKs. In general, MAPK pathways are activated by extracellular signals or signals generated in the cytoplasm, and then the activated MAPKs transduce the specific “messages” to the nucleus. However, in response to genotoxic stress, the signal seems to flow from the nucleus to the cytoplasm to activate MAPKs. In this case, c-Abl kinase may provide an explanation. It has been found that c-Abl can activate p38 through MKK6[72-74], and JNK by translocating from nucleus to cytoplasm to phosphorylate hematopoietic progenitor kinase (HPK1), an upstream kinase of JNK[75]. Therefore, c-Abl may fulfill a role as the message carrier to transduce signals between subcellular locations. This may further explain why in response to genotoxic stress the activation of p38 was rather late (- 1 h) and prolonged[71], while the cytokine activation of p38 was rapid and transient (maximum around 30-60 min)[76].
In addition to their ability to activate MAPKs, ATM/ATR may also regulate these kinases through their negative regulators, the dual specificity of phosphatase MAPK and phosphatase family (MKP). One member of the MKP family, MKP-5, is known to dephosphorylate and inactivate the stress-activated JNK and p38. Thephosphorylation - dephosphorylation cycle of JNK and p38 stimulated by radiomimetic chemical neocarzinostatin (NCS), which can induce double strand breaks (DSBs), could be attenuated in A-T cells[77], further emphasizing the role of ATM as a master regulator in the cellular response to genotoxic stress.
Mutations in ATM in association with cancer
Homozygous mutations in the ATM gene can cause human genetic disorder ataxia-telangiectasia (A-T), which is characterized by cerebellar degeneration, immunodeficiency, cancer predisposition, and acute sensitivity to IR. The affected individual has been found to be prone to develop T cell pro-lymphocytic leukemia, B cell chronic lymphocytic leukemia, as well as sporadic colon cancer with microsatellite instability[78]. Atm-deficient mice also showed a striking predisposition to lymphoid malignancies, particularly thymic lymphomas, to which they succumbed before the age of 1 year. However, much of the literature on ATM mutations and cancer was not about A-T patients, but was, instead, on heterozygous carriers of A-T mutations. For example, recent studies have found an unusually high occurrence of breast cancer in the relatives of A-T patients, and that loss of heterozygosity of ATM occurred frequently during the early stages of breast cancer development[79]. Furthermore, heterozygous mice were more sensitive to radiation-induced cataracts than their wild-type counterparts[80]. Spring et al[81] established a knock-in mouse mutant in which an inframe deletion was previously found to cause A-T in humans was induced. Mice homozygous for this mutation could produce small amounts of inactive ATM and usually showed the hallmarks of the Atm-knockout phenotype. Notably, mice heterozygous for this mutation were predisposed to various cancers, unlike the animals that carry a single knockout allele that does not produce any protein. Therefore, ATM heterozygotes in human population might also be more radiosensitive, and have a higher risk for cancer[82](Figure 3).
Figure 3.

The relationship between ATM and carcinogenesis.
No human disease has been found to link to defects in ATR, although it was found that defects in ATR led to embryonic lethality in mice, suggesting that ATR is essential for of development of ATR[83,84]. Nonetheless, it is known that over-expressing the inactive form of ATR had a dominant negative effect, causing increased sensitivity to DNA damaging stimuli and failure to activate cell cycle checkpoints in response to IR[28,85]. Finally, over-expressing active ATR could restore S phase checkpoint defect in A-T cells, suggesting that ATM and ATR may complement each other in the cellular genotoxic stress response[85].
Down-regulation of ATM and ATR
Once the sensors detect DNA damage and initiate the signaling pathway, and the biological consequences (including DNA repair, cell cycle arrest, and apoptosis) take effect, the signals need to be inactivated or attenuated. The regulation of some downstream components in the cellular genotoxic stress response has been rather clearly defined, and usually involves a negative feedback mechanism. One such example is the p53-Mdm2 regulation loop. In this loop p53 could activate the expression of Mdm2, and Mdm2 could mediate the rapid degradation of p53 through the ubiquitin pathway[62]. MAPKs have a similar feedback regulation mechanism with MKPs. MAPKs could induce the expression of MKPs, and MKPs then could interact with specific MAPKs to deactivate them through dephosphorylation[1]. On the other hand, the mechanisms for the regulation of ATM and ATR, remain obscure, although some recent studies have significantly advanced our understanding.
In contrast to the vast volume of reports about the activation of ATM under genotoxic stress, very few studies have been conducted to evaluate how ATM was inactivated. The results from these studies so far all pointed toward inactivation of ATM through Caspase-mediated cleavage during apoptosis[86-88]. This same mechanism has also been shown to regulate many other proteins involved in apoptosis, including serine/threonine protein kinase Cδ (PKCδ), Mdm2, PARP, replication factor C, 70 kDa U1snRNP, fodrin and lamins[87]. It was reported that during apoptosis induced by c-Myc or DNA-damaging agents (such as etoposide or IR), ATM but not ATR, was specifically cleaved by members of the Caspase family. Detailed studies revealed that the Caspase responsible for this cleavage was either Caspase-3 or -7, but not Caspase-6. This cleavage abrogated the kinase activity of ATM to phosphorylate p53, although the resulting two fragments retained their DNA binding ability and interacted with each other. This finding led to the hypothesis that cleaved ATM protein, without its kinase activity, might act in a trans-dominant-negative fashion to compete with the intact ATM, thus preventing DNA repair and DNA damage signaling through its binding to DNA[86-88].
Even less information is available regarding the inactivation of ATR. However, the recent identification of an ATR-interacting protein (ATRIP) might provide a lead for future studies[89]. ATRIP is an 86-kDa protein with a coiled-coil domain near its N-terminal and its expression is regulated by ATR. The deletion of ATR mediated by Cre recombinase could cause the loss of both ATR and ATRIP expression, along with the loss of DNA damage checkpoint responses and cell death. ATRIP could be phosphorylated by ATR and co-localized at intranuclear foci with ATR after DNA damage caused by hydroxyurea (HU), IR, or UV, or inhibition of DNA replication. Conversely, ATRIP could also regulate the expression of ATR, as inhibition of ATRIP expression with small interference RNA (siRNA) would result in decreased ATR protein expression, while ATR mRNA levels would not be affected. Interference with ATRIP function could cause the same loss of G2-M response to DNA damage as that seen in the case of ATR deletion, suggesting that these two proteins work as mutually dependent partners in cell cycle checkpoint pathways[89].
CONCLUSION
Human cancer is a major health issue for society, causing millions of deaths each year and huge economical losses. Since most of human carcinogens are genotoxins[90,91], considerable resources have been and are being expended in efforts to understand the mechansm of genotoxin - induced carcinogenesis, thus leading to a better prevention or even the treatment of cancer. Since the sensing of DNA damage is one of the earliest steps in the cellular response to gentoxic stress, identification of these “sensors” is the most prominent challenge. As discussed in this review, ATM and ATR are showing their promise as potential candidates. However, what we should keep in mind is that detection of DNA damage may not be such a simple process, and may require more than just one or two proteins to fulfill this role. Supporting this idea is the finding of “foci” at damaged DNA sites, where many proteins involved in DNA repair and maintenance aggregate. It is more likely that interactions of these proteins, combined with some unidentified factors might function as DNA damage sensors[92]. Further elucidation of these “foci” will be an exciting area for future research.
Footnotes
Supported by National Key Basic Research and Development Program No. 2002CB512901, China; National Natural Science Foundation No. 30300277, China; the Initial Funds for Returned Overseas Chinese Scholar from Zhejiang University and Ministry of Education, China
Edited by Zhu LH and Wang XL
References
- 1.Yang J, Yu Y, Duerksen-Hughes PJ. Protein kinases and their involvement in the cellular responses to genotoxic stress. Mutat Res. 2003;543:31–58. doi: 10.1016/s1383-5742(02)00069-8. [DOI] [PubMed] [Google Scholar]
- 2.Liu Y, Guyton KZ, Gorospe M, Xu Q, Lee JC, Holbrook NJ. Differential activation of ERK, JNK/SAPK and p38/CSBP/RK map kinase family members during the cellular response to arsenite. Free Radic Biol Med. 1996;21:771–781. doi: 10.1016/0891-5849(96)00176-1. [DOI] [PubMed] [Google Scholar]
- 3.Liu ZG, Baskaran R, Lea-Chou ET, Wood LD, Chen Y, Karin M, Wang JY. Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature. 1996;384:273–276. doi: 10.1038/384273a0. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez-Prieto R, Rojas JM, Taya Y, Gutkind JS. A role for the p38 mitogen-acitvated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res. 2000;60:2464–2472. [PubMed] [Google Scholar]
- 5.Kharbanda S, Saxena S, Yoshida K, Pandey P, Kaneki M, Wang Q, Cheng K, Chen YN, Campbell A, Sudha T, et al. Translocation of SAPK/JNK to mitochondria and interaction with Bcl-x(L) in response to DNA damage. J Biol Chem. 2000;275:322–327. doi: 10.1074/jbc.275.1.322. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Zhong S, Dong Z, Chen N, Bode AM, Ma W, Dong Z. UVA induces Ser381 phosphorylation of p90RSK/MAPKAP-K1 via ERK and JNK pathways. J Biol Chem. 2001;276:14572–14580. doi: 10.1074/jbc.M004615200. [DOI] [PubMed] [Google Scholar]
- 7.She QB, Chen N, Dong Z. ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J Biol Chem. 2000;275:20444–20449. doi: 10.1074/jbc.M001020200. [DOI] [PubMed] [Google Scholar]
- 8.Akechi M, Ito M, Uemura K, Takamatsu N, Yamashita S, Uchiyama K, Yoshioka K, Shiba T. Expression of JNK cascade scaffold protein JSAP1 in the mouse nervous system. Neurosci Res. 2001;39:391–400. doi: 10.1016/s0168-0102(01)00194-8. [DOI] [PubMed] [Google Scholar]
- 9.Tawadros T, Formenton A, Dudler J, Thompson N, Nicod P, Leisinger HJ, Waeber G, Haefliger JA. The scaffold protein IB1/JIP-1 controls the activation of JNK in rat stressed urothelium. J Cell Sci. 2002;115:385–393. doi: 10.1242/jcs.115.2.385. [DOI] [PubMed] [Google Scholar]
- 10.Ito M, Akechi M, Hirose R, Ichimura M, Takamatsu N, Xu P, Nakabeppu Y, Tadayoshi S, Yamamoto K, Yoshioka K. Isoforms of JSAP1 scaffold protein generated through alternative splicing. Gene. 2000;255:229–234. doi: 10.1016/s0378-1119(00)00335-8. [DOI] [PubMed] [Google Scholar]
- 11.Stenius U, Högberg J. Re: Yang, J. and Duerksen-Hughes, P. (1998) A new approach to identifying genotoxic carcinogens: p53 induction as an indicator of genotoxic damage. Carcinogenesis, 19, 1117-1125. Carcinogenesis. 1999;20:181–182. doi: 10.1093/carcin/20.1.181. [DOI] [PubMed] [Google Scholar]
- 12.Wahl GM, Linke SP, Paulson TG, Huang LC. Maintaining genetic stability through Tp53 mediated checkpoint control. Cancer Surv. 1997;29:183–219. [PubMed] [Google Scholar]
- 13.Lowndes NF, Murguia JR. Sensing and responding to DNA damage. Curr Opin Genet Dev. 2000;10:17–25. doi: 10.1016/s0959-437x(99)00050-7. [DOI] [PubMed] [Google Scholar]
- 14.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 15.Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297:547–551. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- 16.O'Connell MJ, Walworth NC, Carr AM. The G2-phase DNA-damage checkpoint. Trends Cell Biol. 2000;10:296–303. doi: 10.1016/s0962-8924(00)01773-6. [DOI] [PubMed] [Google Scholar]
- 17.Green CM, Erdjument-Bromage H, Tempst P, Lowndes NF. A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr Biol. 2000;10:39–42. doi: 10.1016/s0960-9822(99)00263-8. [DOI] [PubMed] [Google Scholar]
- 18.Roos-Mattjus P, Vroman BT, Burtelow MA, Rauen M, Eapen AK, Karnitz LM. Genotoxin-induced Rad9-Hus1-Rad1 (9-1-1) chromatin association is an early checkpoint signaling event. J Biol Chem. 2002;277:43809–43812. doi: 10.1074/jbc.M207272200. [DOI] [PubMed] [Google Scholar]
- 19.Durocher D, Jackson SP. DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme. Curr Opin Cell Biol. 2001;13:225–231. doi: 10.1016/s0955-0674(00)00201-5. [DOI] [PubMed] [Google Scholar]
- 20.Rotman G, Shiloh Y. ATM: a mediator of multiple responses to genotoxic stress. Oncogene. 1999;18:6135–6144. doi: 10.1038/sj.onc.1203124. [DOI] [PubMed] [Google Scholar]
- 21.Chan ED, Winston BW, Jarpe MB, Wynes MW, Riches DW. Preferential activation of the p46 isoform of JNK/SAPK in mouse macrophages by TNF alpha. Proc Natl Acad Sci USA. 1997;94:13169–13174. doi: 10.1073/pnas.94.24.13169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bao S, Tibbetts RS, Brumbaugh KM, Fang Y, Richardson DA, Ali A, Chen SM, Abraham RT, Wang XF. ATR/ATM-mediated phosphorylation of human Rad17 is required for genotoxic stress responses. Nature. 2001;411:969–974. doi: 10.1038/35082110. [DOI] [PubMed] [Google Scholar]
- 23.Plumb MA, Smith GC, Cunniffe SM, Jackson SP, O'Neill P. DNA-PK activation by ionizing radiation-induced DNA single-strand breaks. Int J Radiat Biol. 1999;75:553–561. doi: 10.1080/095530099140195. [DOI] [PubMed] [Google Scholar]
- 24.Jackson SP. DNA-dependent protein kinase. Int J Biochem Cell Biol. 1997;29:935–938. doi: 10.1016/s1357-2725(97)00006-x. [DOI] [PubMed] [Google Scholar]
- 25.Gately DP, Hittle JC, Chan GK, Yen TJ. Characterization of ATM expression, localization, and associated DNA-dependent protein kinase activity. Mol Biol Cell. 1998;9:2361–2374. doi: 10.1091/mbc.9.9.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pandita TK, Lieberman HB, Lim DS, Dhar S, Zheng W, Taya Y, Kastan MB. Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene. 2000;19:1386–1391. doi: 10.1038/sj.onc.1203444. [DOI] [PubMed] [Google Scholar]
- 27.Andegeko Y, Moyal L, Mittelman L, Tsarfaty I, Shiloh Y, Rotman G. Nuclear retention of ATM at sites of DNA double strand breaks. J Biol Chem. 2001;276:38224–38230. doi: 10.1074/jbc.M102986200. [DOI] [PubMed] [Google Scholar]
- 28.Wright JA, Keegan KS, Herendeen DR, Bentley NJ, Carr AM, Hoekstra MF, Concannon P. Protein kinase mutants of human ATR increase sensitivity to UV and ionizing radiation and abrogate cell cycle checkpoint control. Proc Natl Acad Sci USA. 1998;95:7445–7450. doi: 10.1073/pnas.95.13.7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hekmat-Nejad M, You Z, Yee MC, Newport JW, Cimprich KA. Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr Biol. 2000;10:1565–1573. doi: 10.1016/s0960-9822(00)00855-1. [DOI] [PubMed] [Google Scholar]
- 30.Wahl GM, Carr AM. The evolution of diverse biological responses to DNA damage: insights from yeast and p53. Nat Cell Biol. 2001;3:E277–E286. doi: 10.1038/ncb1201-e277. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki K, Kodama S, Watanabe M. Recruitment of ATM protein to double strand DNA irradiated with ionizing radiation. J Biol Chem. 1999;274:25571–25575. doi: 10.1074/jbc.274.36.25571. [DOI] [PubMed] [Google Scholar]
- 32.Smith GC, Cary RB, Lakin ND, Hann BC, Teo SH, Chen DJ, Jackson SP. Purification and DNA binding properties of the ataxia-telangiectasia gene product ATM. Proc Natl Acad Sci USA. 1999;96:11134–11139. doi: 10.1073/pnas.96.20.11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo Z, Kumagai A, Wang SX, Dunphy WG. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000;14:2745–2756. doi: 10.1101/gad.842500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Unsal-Kaçmaz K, Makhov AM, Griffith JD, Sancar A. Preferential binding of ATR protein to UV-damaged DNA. Proc Natl Acad Sci USA. 2002;99:6673–6678. doi: 10.1073/pnas.102167799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the rec-ognition and repair of aberrant DNA structures. Genes Dev. 2000;14:927–939. [PMC free article] [PubMed] [Google Scholar]
- 36.Kim GD, Choi YH, Dimtchev A, Jeong SJ, Dritschilo A, Jung M. Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J Biol Chem. 1999;274:31127–31130. doi: 10.1074/jbc.274.44.31127. [DOI] [PubMed] [Google Scholar]
- 37.Zou L, Cortez D, Elledge SJ. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002;16:198–208. doi: 10.1101/gad.950302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tibbetts RS, Cortez D, Brumbaugh KM, Scully R, Livingston D, Elledge SJ, Abraham RT. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000;14:2989–3002. doi: 10.1101/gad.851000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmidt DR, Schreiber SL. Molecular association between ATR and two components of the nucleosome remodeling and deacetylating complex, HDAC2 and CHD4. Biochemistry. 1999;38:14711–14717. doi: 10.1021/bi991614n. [DOI] [PubMed] [Google Scholar]
- 40.Wang JY. Cellular responses to DNA damage. Curr Opin Cell Biol. 1998;10:240–247. doi: 10.1016/s0955-0674(98)80146-4. [DOI] [PubMed] [Google Scholar]
- 41.Wen ST, Van Etten RA. The PAG gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev. 1997;11:2456–2467. doi: 10.1101/gad.11.19.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kharbanda S, Ren R, Pandey P, Shafman TD, Feller SM, Weichselbaum RR, Kufe DW. Activation of the c-Abl tyrosine kinase in the stress response to DNA-damaging agents. Nature. 1995;376:785–788. doi: 10.1038/376785a0. [DOI] [PubMed] [Google Scholar]
- 43.Shafman T, Khanna KK, Kedar P, Spring K, Kozlov S, Yen T, Hobson K, Gatei M, Zhang N, Watters D, et al. Interaction between ATM protein and c-Abl in response to DNA damage. Nature. 1997;387:520–523. doi: 10.1038/387520a0. [DOI] [PubMed] [Google Scholar]
- 44.Baskaran R, Wood LD, Whitaker LL, Canman CE, Morgan SE, Xu Y, Barlow C, Baltimore D, Wynshaw-Boris A, Kastan MB, et al. Ataxia telangiectasia mutant protein activates c-Abl tyrosine kinase in response to ionizing radiation. Nature. 1997;387:516–519. doi: 10.1038/387516a0. [DOI] [PubMed] [Google Scholar]
- 45.Shangary S, Brown KD, Adamson AW, Edmonson S, Ng B, Pandita TK, Yalowich J, Taccioli GE, Baskaran R. Regulation of DNA-dependent protein kinase activity by ionizing radiation-activated abl kinase is an ATM-dependent process. J Biol Chem. 2000;275:30163–30168. doi: 10.1074/jbc.M004302200. [DOI] [PubMed] [Google Scholar]
- 46.Takao N, Mori R, Kato H, Shinohara A, Yamamoto Ki. c-Abl tyrosine kinase is not essential for ataxia telangiectasia mutated functions in chromosomal maintenance. J Biol Chem. 2000;275:725–728. doi: 10.1074/jbc.275.2.725. [DOI] [PubMed] [Google Scholar]
- 47.Yuan ZM, Huang Y, Whang Y, Sawyers C, Weichselbaum R, Kharbanda S, Kufe D. Role for c-Abl tyrosine kinase in growth arrest response to DNA damage. Nature. 1996;382:272–274. doi: 10.1038/382272a0. [DOI] [PubMed] [Google Scholar]
- 48.Wen ST, Jackson PK, Van Etten RA. The cytostatic function of c-Abl is controlled by multiple nuclear localization signals and requires the p53 and Rb tumor suppressor gene products. EMBO J. 1996;15:1583–1595. [PMC free article] [PubMed] [Google Scholar]
- 49.Sionov RV, Moallem E, Berger M, Kazaz A, Gerlitz O, Ben-Neriah Y, Oren M, Haupt Y. c-Abl neutralizes the inhibitory effect of Mdm2 on p53. J Biol Chem. 1999;274:8371–8374. doi: 10.1074/jbc.274.13.8371. [DOI] [PubMed] [Google Scholar]
- 50.Sionov RV, Coen S, Goldberg Z, Berger M, Bercovich B, Ben-Neriah Y, Ciechanover A, Haupt Y. c-Abl regulates p53 levels under normal and stress conditions by preventing its nuclear export and ubiquitination. Mol Cell Biol. 2001;21:5869–5878. doi: 10.1128/MCB.21.17.5869-5878.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang Y, Yuan ZM, Ishiko T, Nakada S, Utsugisawa T, Kato T, Kharbanda S, Kufe DW. Pro-apoptotic effect of the c-Abl tyrosine kinase in the cellular response to 1-beta-D-arabinofuranosylcytosine. Oncogene. 1997;15:1947–1952. doi: 10.1038/sj.onc.1201376. [DOI] [PubMed] [Google Scholar]
- 52.Yuan ZM, Huang Y, Ishiko T, Kharbanda S, Weichselbaum R, Kufe D. Regulation of DNA damage-induced apoptosis by the c-Abl tyrosine kinase. Proc Natl Acad Sci USA. 1997;94:1437–1440. doi: 10.1073/pnas.94.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shaul Y. c-Abl: activation and nuclear targets. Cell Death Differ. 2000;7:10–16. doi: 10.1038/sj.cdd.4400626. [DOI] [PubMed] [Google Scholar]
- 54.Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG, Levrero M, Wang JY. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–809. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- 55.Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature. 1999;399:809–813. doi: 10.1038/21697. [DOI] [PubMed] [Google Scholar]
- 56.Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 57.Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 58.Nakagawa K, Taya Y, Tamai K, Yamaizumi M. Requirement of ATM in phosphorylation of the human p53 protein at serine 15 following DNA double-strand breaks. Mol Cell Biol. 1999;19:2828–2834. doi: 10.1128/mcb.19.4.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Waterman MJ, Stavridi ES, Waterman JL, Halazonetis TD. ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nat Genet. 1998;19:175–178. doi: 10.1038/542. [DOI] [PubMed] [Google Scholar]
- 61.Saito S, Goodarzi AA, Higashimoto Y, Noda Y, Lees-Miller SP, Appella E, Anderson CW. ATM mediates phosphorylation at multiple p53 sites, including Ser(46), in response to ionizing radiation. J Biol Chem. 2002;277:12491–12494. doi: 10.1074/jbc.C200093200. [DOI] [PubMed] [Google Scholar]
- 62.Alarcon-Vargas D, Ronai Z. p53-Mdm2--the affair that never ends. Carcinogenesis. 2002;23:541–547. doi: 10.1093/carcin/23.4.541. [DOI] [PubMed] [Google Scholar]
- 63.Hirao A, Kong YY, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ, Mak TW. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000;287:1824–1827. doi: 10.1126/science.287.5459.1824. [DOI] [PubMed] [Google Scholar]
- 64.Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 functions as a DNA damage checkpoint in G1 by stabilizing p53. Genes Dev. 2000;14:278–288. [PMC free article] [PubMed] [Google Scholar]
- 65.Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Natl Acad Sci USA. 1999;96:13777–13782. doi: 10.1073/pnas.96.24.13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khosravi R, Maya R, Gottlieb T, Oren M, Shiloh Y, Shkedy D. Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc Natl Acad Sci USA. 1999;96:14973–14977. doi: 10.1073/pnas.96.26.14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Toledo SM, Azzam EI, Dahlberg WK, Gooding TB, Little JB. ATM complexes with HDM2 and promotes its rapid phosphorylation in a p53-independent manner in normal and tumor human cells exposed to ionizing radiation. Oncogene. 2000;19:6185–6193. doi: 10.1038/sj.onc.1204020. [DOI] [PubMed] [Google Scholar]
- 68.Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O, Moas M, Buschmann T, Ronai Z, Shiloh Y, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15:1067–1077. doi: 10.1101/gad.886901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tang D, Wu D, Hirao A, Lahti JM, Liu L, Mazza B, Kidd VJ, Mak TW, Ingram AJ. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J Biol Chem. 2002;277:12710–12717. doi: 10.1074/jbc.M111598200. [DOI] [PubMed] [Google Scholar]
- 70.Zhang Y, Ma WY, Kaji A, Bode AM, Dong Z. Requirement of ATM in UVA-induced signaling and apoptosis. J Biol Chem. 2002;277:3124–3131. doi: 10.1074/jbc.M110245200. [DOI] [PubMed] [Google Scholar]
- 71.Wang X, McGowan CH, Zhao M, He L, Downey JS, Fearns C, Wang Y, Huang S, Han J. Involvement of the MKK6-p38α cascade in α-radiation-induced cell cycle arrest. Mol Cell Biol. 2000;20:4543–4552. doi: 10.1128/mcb.20.13.4543-4552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sanchez-Prieto R, Sanchez-Arevalo VJ, Servitja JM, Gutkind JS. Regulation of p73 by c-Abl through the p38 MAP kinase pathway. Oncogene. 2002;21:974–979. doi: 10.1038/sj.onc.1205134. [DOI] [PubMed] [Google Scholar]
- 73.Pandey P, Raingeaud J, Kaneki M, Weichselbaum R, Davis RJ, Kufe D, Kharbanda S. Activation of p38 mitogen-activated protein kinase by c-Abl-dependent and -independent mechanisms. J Biol Chem. 1996;271:23775–23779. doi: 10.1074/jbc.271.39.23775. [DOI] [PubMed] [Google Scholar]
- 74.Cong F, Goff SP. c-Abl-induced apoptosis, but not cell cycle arrest, requires mitogen-activated protein kinase kinase 6 activation. Proc Natl Acad Sci USA. 1999;96:13819–13824. doi: 10.1073/pnas.96.24.13819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ito Y, Pandey P, Sathyanarayana P, Ling P, Rana A, Weichselbaum R, Tan TH, Kufe D, Kharbanda S. Interaction of hematopoietic progenitor kinase 1 and c-Abl tyrosine kinase in response to genotoxic stress. J Biol Chem. 2001;276:18130–18138. doi: 10.1074/jbc.M007294200. [DOI] [PubMed] [Google Scholar]
- 76.Liu RY, Fan C, Liu G, Olashaw NE, Zuckerman KS. Activation of p38 mitogen-activated protein kinase is required for tumor necrosis factor-α-supported proliferation of leukemia and lymphoma cell lines. J Biol Chem. 2000;275:21086–21093. doi: 10.1074/jbc.M001281200. [DOI] [PubMed] [Google Scholar]
- 77.Bar-Shira A, Rashi-Elkeles S, Zlochover L, Moyal L, Smorodinsky NI, Seger R, Shiloh Y. ATM-dependent activation of the gene encoding MAP kinase phosphatase 5 by radiomimetic DNA damage. Oncogene. 2002;21:849–855. doi: 10.1038/sj.onc.1205127. [DOI] [PubMed] [Google Scholar]
- 78.Ejima Y, Yang L, Sasaki MS. Aberrant splicing of the ATM gene associated with shortening of the intronic mononucleotide tract in human colon tumor cell lines: a novel mutation target of microsatellite instability. Int J Cancer. 2000;86:262–268. doi: 10.1002/(sici)1097-0215(20000415)86:2<262::aid-ijc17>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 79.Shen CY, Yu JC, Lo YL, Kuo CH, Yue CT, Jou YS, Huang CS, Lung JC, Wu CW. Genome-wide search for loss of heterozygosity using laser capture microdissected tissue of breast carcinoma: an implication for mutator phenotype and breast cancer pathogenesis. Cancer Res. 2000;60:3884–3892. [PubMed] [Google Scholar]
- 80.Worgul BV, Smilenov L, Brenner DJ, Junk A, Zhou W, Hall EJ. Atm heterozygous mice are more sensitive to radiation-induced cataracts than are their wild-type counterparts. Proc Natl Acad Sci USA. 2002;99:9836–9839. doi: 10.1073/pnas.162349699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Spring K, Ahangari F, Scott SP, Waring P, Purdie DM, Chen PC, Hourigan K, Ramsay J, McKinnon PJ, Swift M, et al. Mice heterozygous for mutation in Atm, the gene involved in ataxia-telangiectasia, have heightened susceptibility to cancer. Nat Genet. 2002;32:185–190. doi: 10.1038/ng958. [DOI] [PubMed] [Google Scholar]
- 82.Concannon P. ATM heterozygosity and cancer risk. Nat Genet. 2002;32:89–90. doi: 10.1038/ng0902-89. [DOI] [PubMed] [Google Scholar]
- 83.Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000;14:397–402. [PMC free article] [PubMed] [Google Scholar]
- 84.de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol. 2000;10:479–482. doi: 10.1016/s0960-9822(00)00447-4. [DOI] [PubMed] [Google Scholar]
- 85.Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998;17:159–169. doi: 10.1093/emboj/17.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Smith GC, d'Adda di Fagagna F, Lakin ND, Jackson SP. Cleavage and inactivation of ATM during apoptosis. Mol Cell Biol. 1999;19:6076–6084. doi: 10.1128/mcb.19.9.6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hotti A, Järvinen K, Siivola P, Hölttä E. Caspases and mitochondria in c-Myc-induced apoptosis: identification of ATM as a new target of caspases. Oncogene. 2000;19:2354–2362. doi: 10.1038/sj.onc.1203567. [DOI] [PubMed] [Google Scholar]
- 88.Tong X, Liu B, Dong Y, Sun Z. Cleavage of ATM during radiation-induced apoptosis: caspase-3-like apoptotic protease as a candidate. Int J Radiat Biol. 2000;76:1387–1395. doi: 10.1080/09553000050151664. [DOI] [PubMed] [Google Scholar]
- 89.Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- 90.Williams GM, Weisburger JH. Chemical Carcinogenesis. New York, NY, Pergamon Press. 1991. [Google Scholar]
- 91.Smart RC. Carcinogenesis. Norwalk, CT, Appleton Lange. 1994. [Google Scholar]
- 92.Yang J, Yu Y, Hamrick HE, Duerksen-Hughes PJ. ATM, ATR and DNA-PK: initiators of the cellular genotoxic stress responses. Carcinogenesis. 2003;24:1571–1580. doi: 10.1093/carcin/bgg137. [DOI] [PubMed] [Google Scholar]