

Abstract
AIM: Inactivation of p53 gene is one of the most frequent genetic alterations in carcinogenesis. The mutation status of p53 gene was analyzed, in order to understand the effect of p53 mutation on chemical hepatocarcinogenesis of rats.
METHODS: During hepatocarcinogenesis of rats induced by 3’-methyl-4- dimethylaminoazobenzene (3’-Me-DAB), prehepatocarcinoma and hepatocarcinoma foci were collected by laser capture microdissection (LCM), and quantitatively analyzed for levels of p53 mRNA by LightCyclerTM real-time RT-PCR and for mutations in p53 gene exons 5-8 by direct sequencing.
RESULTS: Samples consisting of 44 precancerous foci and 24 cancerous foci were collected by LCM. A quantitative analysis of p53 mRNA showed that p53 mRNA peaked at an early stage (week 6) in the prehepatocarcinoma lesion, more than ten times that of adjacent normal tissue, and gradually decreased from week 6 to week 24. The expression of p53 mRNA in adjacent normal tissue was significantly lower than that in prehepatocarcinoma. Similar to prehepatocarcinoma, p53 mRNA in cancer was markedly higher than that in adjacent normal tissue at week 12, and was closer to normal at week 24. Direct p53 gene sequencing showed that 35.3% (24/68) (9 precancer, 15 cancer) LCM samples exhibited point mutations, 20.5% of prehepatocarcinoma LCM samples presented missense mutations at exon 6/7 or/and 8, and was markedly lower than 62.5% of hepatocarcinoma ones (P < 0.01). Mutation of p53 gene formed the mutant hot spots at 5 codons. Positive immunostaining for p53 protein could be seen in prehepatocarcinoma and hepatocarcinoma foci at 24 weeks.
CONCLUSION: p53 gene mutation is present in initial chemical hepatocarcinogenesis, and the mutation of p53 gene induced by 3’-Me-DAB is an important factor of hepatocarcinogenesis.
INTRODUCTION
Abnormalities of some tumor suppressor genes and oncogenes play important roles in the development and progression of hepatoma[1,2]. Gene abnormalities in precancerous liver lesions (adenomatous hyperplasia and atypical adenomatous hyperplasia) and early hepatocellular carcinoma have been reported[3-5]. p53 tumor suppressor protein plays an important role in preventing malignant development, and p53 function is lost or compromised in most human cancers[6-8]. One of the principal functions of p53 is to inhibit cell growth, and p53 shows a strong cell cycle arrest and apoptotic activities[9,10]. As a result, cell proliferation is suppressed and/or programmed cell death is induced[11,12]. In cells with DNA injury, p53 can stop the cell cycle through p21 protein and then promote DNA repair. When DNA is seriously damaged, p53 can induce the cell to undergo programmed cell death to maintain the stability of genome and cells. Loss of p53 function activates oncogenes and inactivates cancer suppressor genes, playing an essential role in multistage carcinogenesis[13,14]. Some study has shown that mutation and loss of p53 gene are closely related to the conversion of adenoma to early colorectal cancer, and so are those in liver[15]. When cancer cell differentiation is low and the tumor becomes large, p53 gene mutation frequently arises in hepatocarcinoma, making p53 gene most closely concerned with the progress of hepatocarcinoma[16]. It is not yet clear which of these gene alterations is responsible for hepatocarcinogenesis, especially in a prehepatocarcinoma lesion.
3’-methyl-4-dimethylaminoazobenzene (3’-Me-DAB) could produce prehepato- carcinomatous lesions (altered focus and neoplastic nodules) in rats. Our study showed that mutation of p53 gene, in precancerous and cancerous foci of the F344 rat liver induced by 3’-Me-DAB, was successfully detected by integrating LCM, LightCycler RT-PCR with direct sequencing.
MATERIALS AND METHODS
Animals
Thirty-nine male F344 rats at 10 weeks of age were kept in a room with a 12h light and dark cycle and maintained at 22 °C. They were provided with a diet containing 0.06% 3’-Me-DAB[17] and tap water ad libitum for 6 weeks, 12 weeks, and 24 weeks, respectively. After the last day of each experimental period, the animals were anesthetized with ether and hepatectomized. The cavum thoracis and abdominal cavity were opened immediately with a sterilized scalpel, and part of the liver was quickly dissected out, placed in a cryomold, covered with Tissue-Tek O. C. T compound before being frozen in liquid nitrogen, and preserved at -80 °C until use. The remaining liver was perfused with 30% PBS-buffered sucrose before being removed and fixed in 10% neutral formalin, and then embedded in paraffin.
LCM of sample
The liver preserved at -80 °C was sequentially sliced into twenty 10 μ m thick sections, in a cryostat, which were mounted on clean microscope slides. The sections were stored at -80 °C. Of the 10 successive slide sections, three were stained chemically with H&E and immunohistochemically with glutathione S transferase placenta (GST-P) and AFP polyclonal antibody using Elite ABC kit (Funakoshi Co. Ltd., Tokyo, Japan). Prehepatocarcinoma and hepatocarcinoma foci were diagnosed by Pathologists and their positions were identified on the slides. Other slide sections underwent quick H&E staining based on the LCM manufacturer’s protocols, and were then cleaned in xylene for over 1 min. Once air-dried, slides were ready for LCM. Based on the position of GST-P (+) or AFP (+) foci from immunostained slide sections and the cancer pathology on the H&E section, a LCM cap was placed over the target area of the slide section[18,19]. Target foci in the same position as GST-P (+) or AFP (+) foci were then microdissected.
Extraction of DNA of LCM samples
After microdissection, the LCM cap was inserted into an Eppendorf tube containing 50 μL of digestion buffer of 0.04% proteinase K, 10 mM Tris-HCl pH 8.0, 1 mM EDTA, and 1% Tween-20. The tube was then placed upside down overnight at 37 °C. Following ethanol precipitation, DNA was extracted by the phenol/chloroform/isopropanol method and used directly as a template for PCR.
Extraction of total RNA of LCM samples
After laser transfer, the LCM cap was gently placed on the Eppendorf tube containing 200 μL of reaction mixture. The tube was inverted back and shaken several times for over two minutes to digest the tissue on the cap. The digestive solution was removed from the Eppendorf tube and placed into a 1.5 mL sturdy tube, extraction and purification of total RNA were conducted according to conventional methods. The pellet of total RNA was then resuspended in H2O, and stored at -80 °C.
Primers for RT-PCR, direct sequencing of p53
We designed upstream PCR primers P1, P2, and P3 in the region corresponding to introns 4, 5, and 7 (Figure 1 and Table 1). They were used in combination with downstream primers P4, P5, and P6 within the region corresponding to introns 5, 7 and 8 of p53 gene (Figure 1 and Table 1), to specifically amplify exons 5, 6/7, and 8 of the functional p53 gene without p53 pseudo-gene. The primers Pmu and Pmd for RT-PCR were located on exons 7 and 8 (Figure 1 and Table 1).
Figure 1.
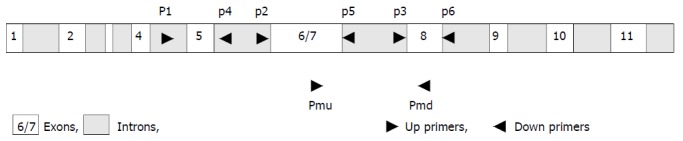
Strategy of primers. To avoid p53 pseudogene co-amplification, the upstream PCR primers P1, P2, and P3 in the region corresponding to introns 4, 5, and 7 were used in combination with downstream primers P4, P5, and P6 within the region corre-sponding to introns 5, 7, and 8 of p53 gene. Primers Pmu and Pmd for RT-PCR located in exons 7 and 8, respectively.
Table 1.
Primer sequences and PCR conditions used in sequencing DNA of p53 gene and RT-PCR
| Exons | Primer | Primer sequences of p53 | Conditions |
| 5 | P1 | GACCTTTGATTCTTTCTCCTCTCC | 94 °C , 3 min (94 °C , 30 sec; 56 °C , 30 sec; 72 °C , 30 sec) × 30 |
| P4 | GGGAGACCCTGGACAACCAG | ||
| 6/7 | P2 | GCCTCTGACTTATTCTTGCTC | 94 °C , 3 min (94 °C , 30 sec; 56 °C , 30 sec; 72 °C , 30 sec) × 30 |
| P5 | CCCAACCTGGCACACAGCTTC | ||
| 8 | P3 | CTGTGCCTCCTCTTGTCCCG | 94 °C , 3 min (94 °C , 30 sec; 56 °C , 30 sec; 72 °C , 30 sec) × 30 |
| P6 | CCACCTTCTTTGTCCTGCCTG | ||
| P53 | Pu | GTCGGCTCCGACTATACCACTATC | 95 °C , 2 min (95 °C , 0 sec; 56 °C , 5 sec; 72 °C , 11 sec) × 30 |
| mRNA | Pd | CTCTCTTTGCACTCCCTGGGGG | |
| GST-P | Pu | ATCGTCCACGCAGCTTTGA | 95 °C , 2 min (95 °C , 0 sec; 57 °C , 5 sec; 72 °C , 13 sec) × 30 |
| mRNA | Pd | AGCCTCCTTCTGGTCTTTC | |
| β-actin | Pu | ACCACCATGTACCCAGGCAT | 95 °C , 2 min (95 °C , 0 sec; 56 °C , 5 sec; 72 °C , 10 sec) × 30 |
| mRNA | Pd | CCGGACTCATCGTACTCCTG |
Quantitative analysis of mRNA
RNAs extracted from prehepatocarcinoma and hepatocarcinoma foci captured by LCM were reverse-transcripted into first strand cDNA at 42 °C for 50 min and at 99 °C for 5 min using Olig-dT-adaptor-primer of an RNA PCR kit (Takara, Co. Ltd., Japan) as the primer. A 1 μL aliquot of first-strand cDNA or H2O (as a negative control) was put into LightCycler capillary, together with 1 μL of 20 pM primer (Table 1) for target DNA and 18 μL of mixture of LightCycler-DNA Master SYBR Green I mixture. Various concentrations of standard sample cDNA were also used to construct a standard curve. After instantaneous centrifugation, capillaries were loaded onto a LightCycler instrument. Quantitative analysis of mRNA was conducted under PCR conditions in Table 1. The standard curve was shown as a straight line of linear regression with cycle number versus log-concentration of standard samples. This standard curve, in turn, was used to estimate the concentration of each sample. Since the expression of β -actin mRNA is constant in all types of cells, it was used to calibrate the original concentration of mRNA, i.e., the concentration unit of mRNA in tissue was defined as the ratio of target mRNA copies versus β-actin mRNA copies[20].
Mutation screening of p53 exons 5, 6/7, and 8
DNAs from the same LCM samples were amplified by PCR under the conditions in Table 1. PCR products were refined by a Microcon-100 kit (Takara, Co. Ltd., Japan). The sense strand of PCR products was sequenced using a cycler sequencing ready reaction kit (ABI, Perkin-Elmer Corp., USA) and analyzed on an ABI PrismTM 310 genetic analyzer (ABI, Perkin-Elmer Corp., USA).
Immunohistochemical staining
Immunohistochemical staining was conducted using the avidin-biotin-peroxidase complex technique (Vectastain Elite ABC Kit, Funakoshi Co. Ltd., Tokyo, Japan)[21]. Paraffin-embedded or frozen rat liver specimens were sectioned at 5 μm and placed on a precleaned glass microscope slide. After deparaffination and blocking of endogenous biotin activity, the sections were incubated with primary antibodies (anti-GST-P IgG diluted at 1:500, anti-AFP IgG diluted at 1:200 (polyclonal antibody) and anti-p53 monoclonal antibody Ab1 (Oncogene Science, Inc., USA) diluted at 1:100) for 90 min at 30 °C, then incubated with biotinylated anti-rabbit (for polyclonal antibody) or anti-mouse (for monoclonal antibody) secondary antibody for 30 min at 30 °C. The slides were incubated for 30 min with avidin-peroxidase conjugates. Finally, the sections were reacted with 3’3-diaminobenzidine tetrahydrochloride and hydrogen peroxide for 3 min followed by counter-staining with hematoxylin. For a negative control, pre-immune serum instead of primary antibody was used.
Statistical methods
Student-t test was used to identify the differences of mRNA concentration in normal tissue, precancerous and cancerous foci.
RESULTS
Hepatocarcinogenesis
Prehepatocarcinoma foci could be found in all (13) the livers of rats treated with 3’-Me-DAB for 12 weeks, which were intensely stained by GST-P (Figure 2, B). By H&E staining, the size of prehepatocarcinoma cells was similar to that of normal hepatocytes, but the cytoplasm of precancer cells was clearer. Under low power magnification, the edge of prehepatocarcinoma focus was distinct and bright (Figure 2, A). Hepatocarcinoma foci were seen in 6 liver sections. By H&E staining, the sizes of nuclei were different, the cytoplasm was a little basophilic and a lot of fat vescles were present in cytoplasm of hepatocarcinoma cells (Figure 2, C). Immunohistochemically staining for AFP was intensive in hepatocarcinoma foci. At week 24 (Figure 2, D), all (13) the livers exhibited hepatocarcinoma foci and some showed prehepatocarcinoma foci simultaneously. The diameter of precancerous foci was 0.5 mm-1.0 mm at week 6, 1.2 mm-1.5 mm at week 12 and > 2.5 mm at week 23. Forty-four precancerous and 24 cancerous samples were obtained for DNA sequencing analysis and quantitative analysis of p53 mRNA.
Figure 2.
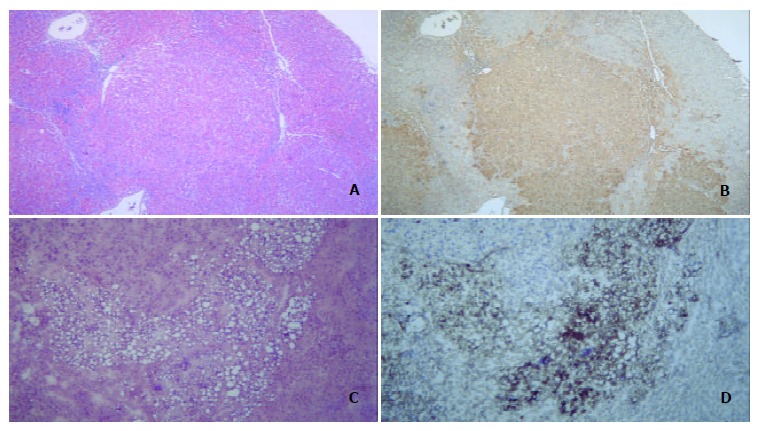
Hepatocarcinogenesis. A: Prehepatocarcinoma foci were stained by H&E, morphology of prehepatocarcinoma cells was similar to that of normal hepatocytes, and the edge of prehepatocarcinoma focus was distinct and bright. B: Prehepatocarcinoma focus was as intensely stained by GST-P as A. C: Hepatocarcinoma foci by H&E staining showed a lot of foam cells. D: Immuno-histochemical staining for AFP was intense in hepatocarcinoma foci.
Expression of p53 and GST-P mRNA
The time course of p53 gene expression showed that the mRNA levels peaked at week 6 in prehepatocarcinoma foci and at week 12 in hepatocarcinoma foci, although they declined significantly by week 24 (P < 0.01, Figure 3). The relative concentration of p53 mRNA in adjacent normal tissue remained significantly lower than in prehepatocarcinoma and hepatocarcinoma foci. In hepatocarcinoma foci, the relative concentration of p53 mRNA was 10 odd times higher than in adjacent normal tissue at week 6 of the experiment (P = 5.6 × 10-9) and 4 times as high at week 12 (P = 4.8 × 10-9). p53 mRNA concentration was also significantly higher in cancer than in adjacent normal tissue at week 12 (P = 0.028). However, at week 24, p53 mRNA of both prehepatocarcinoma and hepatocarcinoma foci was obviously elevated. Relative p53 mRNA concentration was the highest in prehepatocarcinoma foci and the lowest in adjacent normal tissue among the three tissues examined (Figure 3).
Figure 3.
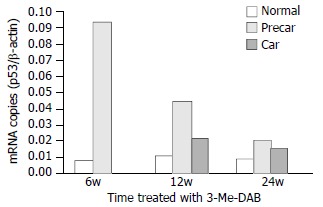
Quantitative analysis of p53 mRNA. After rats were treated with 3’-Me-DAB for 6, 12 and 24 weeks, the expressions of p53 mRNA were markedly higher in prehepatocarcinoma than in hepatocarcinoma and adjacent normal tissue, P < 0.001. The time course of p53 mRNA showed that mRNA levels peaked at week 6, and gradually decreased to minimum at week 24. Normal = adjacent normal tissue, Precar = prehepatocarcinoma, Car = hepatocarcinoma.
Moreover, the expression of GST-P mRNA was low in adjacent normal tissue from week 6 to week 24, and was significantly higher in prehepatocarcinoma foci than in adjacent normal tissue and hepatocarcinoma foci (P < 0.001), and higher in hepatocarcinoma foci than in adjacent normal tissue (Figure 4).
Figure 4.
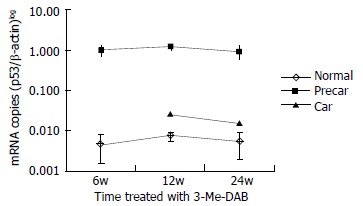
Quantitative analysis of GST-P mRNA. After rats were treated with 3’-Me-DAB for 6, 12 and 24 weeks, the expressions of GST-P mRNA were markedly higher in prehepatocarcinoma than that in hepatocarcinoma and adjacent normal tissue, P < 0.001, and also higher in hepatocarcinoma than that in adjacent normal tissue, P < 0.001. GST-P mRNA levels were expressed on a logarithmic scale. Normal = a djacent normal tissue, Precar = prehepatocarcinoma, Car = hepatocarcinoma.
Direct sequencing analysis of p53 mutation
Direct sequencing analysis of genomic DNA from 24 LCM samples showed 39 mutations at 18 codons of exons 6/7 and 8 in p53 gene (Figure 5, A, C, E). Of these 39 mutations, 10 transition mutations (1 A:T→G:C, 9 G:C→A:T) (Figure 5, A,C) and 29 transversion mutations (9 G:C→C:G, 9 G:C→T:A , 5 C:G→A:T, 2 C:G→G:C, 2 A:T→C:G, 1 T:A→A:T and 1 A:T→T:A) (Figure 5, E) were identified. A base inserting mutation following the first base substitution, G→TA, in codon 283 of exon 7 resulted in a putative substitution “end” for glutamic acid in p53 protein. Interestingly, four LCM samples had triple mutations and eight LCM samples had double mutations in exons 6/7 or/and 8 simultaneously. One LCM sample with triple mutations and 3 with double mutations were prehepatocarcinoma foci, and 3 other samples with triple mutations and 5 with double mutations were hepatocarcinoma foci.
Figure 5.
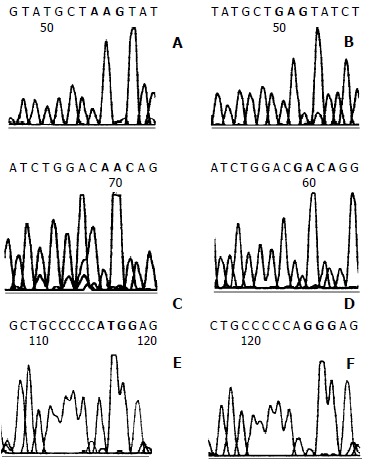
Direct sequencing of p53 exons 6/7 and 8. A: GAG to AAG transition mutation (red box) at codon 202 of exon 6/7 was seen in prehepatocarcinoma foci at 6 weeks. C: Four hepatocarcinoma foci with GAC to AAC transition mutation (red box) at codon 206 formed one of the mutation hot spots. E: GGG to TGG transversion mutation (red box) at codon 300 of exon 8 in hepatocarcinoma foci. B, D and F were normal sequence.
In brief, 24/68 (35.3%) of LCM samples had mutations of p53 gene. No mutation in exon 5 was found by direct sequencing.
Incidence of p53 gene mutation in hepatocarcinogenesis
20.5% precancerous foci samples expressed point mutation or inserting mutation in exons 6/7 and 8, and the incidence of mutation was markedly higher than that of cancerous samples (χ2 = 12.02, P < 0.01). At 12 weeks, the incidence of mutation was significantly higher in hepatocarcinoma samples than in prehepatocarcinoma samples, P = 0.034 by Fisher’s exact probabilities (Figure 6). At 24 weeks, the incidence of mutation was not different between prehepatocarcinoma and hepatocarcinoma foci samples.
Figure 6.
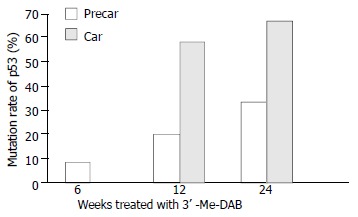
Mutation ratio of p53 exons 6/7 and 8 in prehepatoca-rcinoma (precar) and hepatocarcinoma foci (car).
Hot spot of mutation
Among the 34 mutation codons of p53 exons 6/7 and 8, 9 were at codon 233, 5 at codons 278 and 279, respectively, 4 at codon 206 (Figure 5,C), and 3 mutations were at codon 212. These mutational codons formed the mutational hot spots of precancerous and cancerous foci in this study, and were located in highly conservative DNA binding domain of p53 protein.
Expression of p53 protein in cells
As shown in Figure 7, paraffin-embedded or frozen liver sections from rats treated with 3’-Me-DAB for 24 weeks exhibited intensive immunostaining of p53 protein in at least 30% cells of prehepatocarcinoma (Figure 7B) and hepatocarcinoma foci (Figure 7, C, D). Staining was predominantly limited to enlarged nuclei with intensive staining for the anti-p53 antibody. In contrast, no liver sections from rats treated with 3’-Me-DAB for 6 or 12 weeks demonstrated immunostaining for p53 protein (Figure 7, A).
Figure 7.
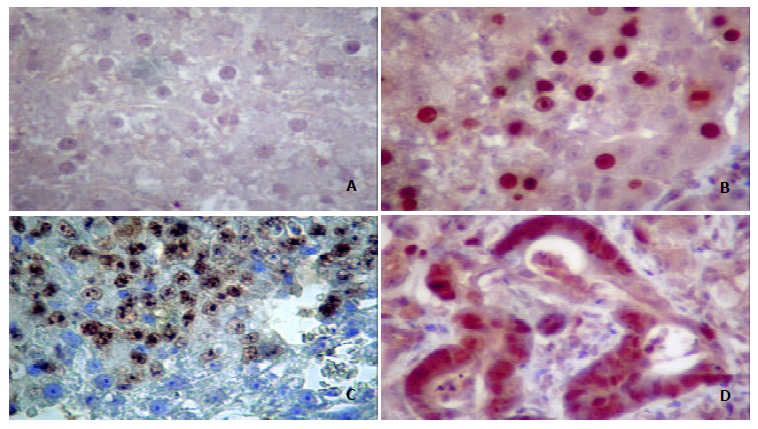
Mutant p53 protein expressions in prehepatocancer and hepatocancer. A: Normal tissue. B: Nuclei of prehepatocarcinoma cells immunostained by anti-p53 Ab1. C: Nuclei of hepatocellular carcinoma cells stained by anti-p53 Ab1. D: Nuclei of adenohepatoma cells stained by anti-p53 Ab1. Enlarged nuclei intensely stained for Ab1 were seen in cells. Original magnification 400 × .
DISCUSSION
Researchers are used to studying human tumors by obtaining human hepatocarcinoma tissue after hepatectomy, but it is difficult to obtain pre-hepatocarcinoma foci in cases without clinical symptoms. We and other investigators were therefore constrained to using rats as the animal model for assessing the effects of p53 gene mutations on hepatocarcinogenesis[22,23]. It has been found that rat p53 gene differs from human p53 gene, in that it processes pseudogenes in its genome[24,25]. Pitfalls associated with p53 pseudogene co-amplification from genomic DNA could be avoided, however, by designing PCR primers based on the intron sequence[24,26].
Carcinogenesis is a complex process characterized by the cumulative activation of various oncogenes and the inactivation of suppressor genes. About 30%-40% of human hepatocarcinomas and 20%-60% of rat experimental tumors demonstrated mutations of p53 gene[2,27-30]. Chemically induced rat liver cancer proceeds through multiple, distinct initiation-promotion-progression stages and mutation of the suppressor p53 gene has been found in relatively early preneoplastic lesions in the rat liver.
We used quantification of p53 mRNA expression to detect genetic alterations at RNA level in the present study. The results showed that in precancerous foci of the rat liver, p53 mRNA rose quickly and peaked at week 6 of 3’-Me-DAB treatment. It then gradually decreased from week 12 and fell to a minimum at week 24 of the experiment. Similar results were also seen in cancer foci. The time course of mRNA expression differed from that of p53 protein accumulation in nuclei. GST-P is one of the detoxification enzymes involved in the metabolism of 3’-Me-DAB as well as other carcinogens and plays a protective role during chemical hepatocarcinogenesis. It is considered as an early marker of preneoplastic lesion. When GST-P played a protective role, mRNA transcript increased coincidentally with positive GST-P immunostaining in prehepatocancerous foci of the rats at all experimental time points tested (data not shown). GST-P overexpression was not consistent with an aberration in p53 protein expression[31].
In this paper, 20.5% of precancerous foci occurring after week 6, week 12, and week 24 exhibited missense mutations of p53 exons 6/7 or/and 8, with double or triple codon mutations found in the same sample by direct sequencing. Of the hepatocancerous foci, 58.3% and 66.7%had mutations in p53 gene at week 12 and week 24 of 3’-Me-DAB treatment, respectively. Mutations were distributed widely throughout exons 6/7and 8, but not in exon 5. p53 mutation rate increased from week 6 to week 24, suggesting that p53 gene mutation is closely associated with hepatocarcinoma development and progression. It is possible that p53 mutation ran through the initiative, intermediate, and late stages of hepatocarcinogenesis, and was not an event occurring only at the advanced stage of liver cancer. Therefore, mutant p53 molecules have been thought to have some unique properties that are important in carcinogenesis in rats[32].
A tetramer of p53 molecules has been assembled through carboxyterminal oligomerization domains. This allows the central domains to interact directly with a consensus DNA element. As a consequence, aminoterminal transactivation domains could interact with basal transcription factors, resulting in increased gene expression[33,34]. Among the 39 mutation codons of p53 exons 6/7 and 8, 9 were at codon 233, 5 at codons 278 and 279, respectively, 4 at codon 206, and 3 mutations were at codon 212. These mutational codons formed mutational hot spots of precancerous and cancerous foci in this study, and were located in the highly conservative DNA binding domain of p53 protein[35].
Over 90% of mutations are missensed, causing the substitution of amino acids. It has been found that mutation hot spots were dispersed in three exons of p53 gene, and three hotspots fell within two evolutionarily highly conserved regions[35,36], suggesting no single spot is responsible for maintaining p53 tumor suppressor function. In fact, a certain oncogenic agent could act on only one or more spots of the p53 gene DNA sequence. The effect of the hotspots is yet uncertain, because a certain oncogenic agent has made different mutational hotspots of p53 gene in different species[37-40] and tissues[28,39,40].
Amino acid residues 278 and 279 of the p53 protein have been found to be located in helix H2 of loop-sheet-helix fitting in the major groove of DNA, allowing them to contact edges of the bases, and to play a central role in DNA recognition[41], so mutation occurring in this area may cause p53 to lose its growth regulation.
Positive immunostaining of p53 was only found in sections of LCM samples treated with 3’-Me-DAB for 24 weeks, not in those treated for 6 or 12 weeks, demonstrating that p53 protein accumulates relatively late during hepatocarcinogenesis. Positive immunostaining of p53 was consistent with the increased half-life of mutant p53 protein compared to wild-type p53[35,42,43]. As the half-life of wild-type p53 protein is short, it is usually too difficult to detect it in normal tissue with Western blotting or immunohistochemical staining. Mutant p53 protein could competitively inhibit the function of wild p53 protein, promoting hyperplasia of cells and leading to tumor development[44,45]. A few studies have reported that accumulation of p53 protein in nuclei in late chemical hepatocarcinogenesis was not synchronized with the increased expression of p53 mRNA[28,46,47]. The rise of p53 mRNA markedly preceded the rise of the expression quantity of p53 protein[19,48].
We showed that mutation of p53 gene occurred in early precancerous and cancerous foci of F344 rats treated with 3’-Me-DAB and mutation increased progressively from week 6 to week 24, but the expression of p53 mRNA decreased progressively from week 6 to week 24. The detection of p53 gene mutation may benefit the early diagnosis of tumors[49], and also may aid in understanding the mechanism behind hepatocarcinogenesis.
Footnotes
Supported by Jilin University Excellent Young Teacher Foundation, No. 2001033
Edited by Zhu LH and Wang XL
References
- 1.Teeguarden JG, Newton MA, Dragan YP, Pitot HC. Genome-wide loss of heterozygosity analysis of chemically induced rat hepatocellular carcinomas reveals elevated frequency of allelic imbalances on chromosomes 1, 6, 8, 11, 15, 17, and 20. Mol Carcinog. 2000;28:51–61. doi: 10.1002/(sici)1098-2744(200005)28:1<51::aid-mc7>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 2.De Miglio MR, Muroni MR, Simile MM, Virdis P, Asara G, Frau M, Calvisi DF, Seddaiu MA, Pascale RM, Feo F. Frequent loss of heterozygosity at the Hcr1 (hepatocarcinogenesis resistance) locus on chromosome 10 in primary hepatocellular carcinomas from LFF1 rat strain. Hepatology. 2001;33:1110–1117. doi: 10.1053/jhep.2001.23795. [DOI] [PubMed] [Google Scholar]
- 3.Kishimoto Y, Shiota G, Kamisaki Y, Wada K, Nakamoto K, Yamawaki M, Kotani M, Itoh T, Kawasaki H. Loss of the tumor suppressor p53 gene at the liver cirrhosis stage in Japanese patients with hepatocellular carcinoma. Oncology. 1997;54:304–310. doi: 10.1159/000227708. [DOI] [PubMed] [Google Scholar]
- 4.Ashida K, Kishimoto Y, Nakamoto K, Wada K, Shiota G, Hirooka Y, Kamisaki Y, Itoh T, Kawasaki H. Loss of heterozygosity of the retinoblastoma gene in liver cirrhosis accompanying hepatocellular carcinoma. J Cancer Res Clin Oncol. 1997;123:489–495. doi: 10.1007/BF01192203. [DOI] [PubMed] [Google Scholar]
- 5.Lu JP, Mao JQ, Li MS, Lu SL, Hu XQ, Zhu SN, Nomura S. In situ detection of TGF betas, TGF beta receptor II mRNA and telomerase activity in rat cholangiocarcinogenesis. World J Gastroenterol. 2003;9:590–594. doi: 10.3748/wjg.v9.i3.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol. 2001;13:332–337. doi: 10.1016/s0955-0674(00)00216-7. [DOI] [PubMed] [Google Scholar]
- 7.Miller DP, Liu G, De Vivo I, Lynch TJ, Wain JC, Su L, Christiani DC. Combinations of the variant genotypes of GSTP1, GSTM1, and p53 are associated with an increased lung cancer risk. Cancer Res. 2002;62:2819–2823. [PubMed] [Google Scholar]
- 8.Campling BG, El-Deiry WS. Clinical implication of p53 mutation in lung cancer. Mol Biotechnol. 2003;24:141–156. doi: 10.1385/MB:24:2:141. [DOI] [PubMed] [Google Scholar]
- 9.Vousden KH. p53: death star. Cell. 2000;103:691–694. doi: 10.1016/s0092-8674(00)00171-9. [DOI] [PubMed] [Google Scholar]
- 10.Wilson DR. Viral-mediated gene transfer for cancer treatment. Curr Pharm Biotechnol. 2002;3:151–164. doi: 10.2174/1389201023378445. [DOI] [PubMed] [Google Scholar]
- 11.Vousden KH. Switching from life to death: the Miz-ing link between Myc and p53. Cancer Cell. 2002;2:351–352. doi: 10.1016/s1535-6108(02)00186-1. [DOI] [PubMed] [Google Scholar]
- 12.Vousden KH. Activation of the p53 tumor suppressor protein. Biochim Biophys Acta. 2002;1602:47–59. doi: 10.1016/s0304-419x(02)00035-5. [DOI] [PubMed] [Google Scholar]
- 13.Harris CC. p53: at the crossroads of molecular carcinogenesis and risk assessment. Science. 1993;262:1980–1981. doi: 10.1126/science.8266092. [DOI] [PubMed] [Google Scholar]
- 14.Livingstone LR, White A, Sprouse J, Livanos E, Jacks T, Tlsty TD. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell. 1992;70:923–935. doi: 10.1016/0092-8674(92)90243-6. [DOI] [PubMed] [Google Scholar]
- 15.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 16.Oda T, Tsuda H, Scarpa A, Sakamoto M, Hirohashi S. p53 gene mutation spectrum in hepatocellular carcinoma. Cancer Res. 1992;52:6358–6364. [PubMed] [Google Scholar]
- 17.Sugioka Y, Fujii-Kuriyama Y, Kitagawa T, Muramatsu M. Changes in polypeptide pattern of rat liver cells during chemical hepatocarcinogenesis. Cancer Res. 1985;45:365–378. [PubMed] [Google Scholar]
- 18.Bonner RF, Emmert-Buck M, Cole K, Pohida T, Chuaqui R, Goldstein S, Liotta LA. Laser capture microdissection: molecular analysis of tissue. Science. 1997;278:1481,1483. doi: 10.1126/science.278.5342.1481. [DOI] [PubMed] [Google Scholar]
- 19.Fu Y, Deng WG, Li YL, Sugiyama T. [Quantitative analysis of p53 and related genes mRNA in rat hepatocarcinogenesis induced by 3'-Me-DAB] Ai Zheng. 2003;22:35–41. [PubMed] [Google Scholar]
- 20.Wittwer CT, Ririe KM, Andrew RV, David DA, Gundry RA, Balis UJ. The LightCycler: a microvolume multisample fluorimeter with rapid temperature control. Biotechniques. 1997;22:176–181. doi: 10.2144/97221pf02. [DOI] [PubMed] [Google Scholar]
- 21.Lehman TA, Haffty BG, Carbone CJ, Bishop LR, Gumbs AA, Krishnan S, Shields PG, Modali R, Turner BC. Elevated frequency and functional activity of a specific germ-line p53 intron mutation in familial breast cancer. Cancer Res. 2000;60:1062–1069. [PubMed] [Google Scholar]
- 22.Fukuda I, Ogawa K. Alternatively-spliced p53 mRNA in the FAA-HTC1 rat hepatoma cell line without the splice site mutations. Cell Struct Funct. 1992;17:427–432. doi: 10.1247/csf.17.427. [DOI] [PubMed] [Google Scholar]
- 23.Ohgaki H, Hard GC, Hirota N, Maekawa A, Takahashi M, Kleihues P. Selective mutation of codons 204 and 213 of the p53 gene in rat tumors induced by alkylating N-nitroso compounds. Cancer Res. 1992;52:2995–2998. [PubMed] [Google Scholar]
- 24.Weghorst CM, Buzard GS, Calvert RJ, Hulla JE, Rice JM. Cloning and sequence of a processed p53 pseudogene from rat: a potential source of false 'mutations' in PCR fragments of tumor DNA. Gene. 1995;166:317–322. doi: 10.1016/0378-1119(95)00629-x. [DOI] [PubMed] [Google Scholar]
- 25.Lin Y, Chan SH. Cloning and characterization of two processed p53 pseudogenes from the rat genome. Gene. 1995;156:183–189. doi: 10.1016/0378-1119(95)00015-x. [DOI] [PubMed] [Google Scholar]
- 26.Hulla JE, Schneider RP. Structure of the rat p53 tumor suppressor gene. Nucleic Acids Res. 1993;21:713–717. doi: 10.1093/nar/21.3.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masui T, Nakanishi H, Inada K, Imai T, Mizoguchi Y, Yada H, Futakuchi M, Shirai T, Tatematsu M. Highly metastatic hepato-cellular carcinomas induced in male F344 rats treated with N-nitrosomorpholine in combination with other hepatocarcinogens show a high incidence of. p53 gene mutations along with altered mRNA expression of tumor-related genes. Cancer Lett. 1997;112:33–45. doi: 10.1016/s0304-3835(96)04543-0. [DOI] [PubMed] [Google Scholar]
- 28.Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature. 1991;350:429–431. doi: 10.1038/350429a0. [DOI] [PubMed] [Google Scholar]
- 29.Volkmann M, Hofmann WJ, Müller M, Räth U, Otto G, Zentgraf H, Galle PR. p53 overexpression is frequent in European hepatocellular carcinoma and largely independent of the codon 249 hot spot mutation. Oncogene. 1994;9:195–204. [PubMed] [Google Scholar]
- 30.Vancutsem PM, Lazarus P, Williams GM. Frequent and specific mutations of the rat p53 gene in hepatocarcinomas induced by tamoxifen. Cancer Res. 1994;54:3864–3867. [PubMed] [Google Scholar]
- 31.Liu YP, Lin Y, Ng ML. Immunochemical and genetic analysis of the p53 gene in liver preneoplastic nodules from aflatoxin-induced rats in one year. Ann Acad Med Singapore. 1996;25:31–36. [PubMed] [Google Scholar]
- 32.Haas MJ, Pitot HC. Characterization of rare p53 mutants from carcinogen-treated albumin-simian virus 40 T-antigen transgenic rats. Mol Carcinog. 1998;21:128–134. [PubMed] [Google Scholar]
- 33.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 34.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 35.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 36.Tam AS, Foley JF, Devereux TR, Maronpot RR, Massey TE. High frequency and heterogeneous distribution of p53 mutations in aflatoxin B1-induced mouse lung tumors. Cancer Res. 1999;59:3634–3640. [PubMed] [Google Scholar]
- 37.Denissenko MF, Koudriakova TB, Smith L, O'Connor TR, Riggs AD, Pfeifer GP. The p53 codon 249 mutational hotspot in hepatocellular carcinoma is not related to selective formation or persistence of aflatoxin B1 adducts. Oncogene. 1998;17:3007–3014. doi: 10.1038/sj.onc.1202214. [DOI] [PubMed] [Google Scholar]
- 38.Hulla JE, Chen ZY, Eaton DL. Aflatoxin B1-induced rat hepatic hyperplastic nodules do not exhibit a site-specific mutation within the p53 gene. Cancer Res. 1993;53:9–11. [PubMed] [Google Scholar]
- 39.Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature. 1991;350:427–428. doi: 10.1038/350427a0. [DOI] [PubMed] [Google Scholar]
- 40.Makino H, Ishizaka Y, Tsujimoto A, Nakamura T, Onda M, Sugimura T, Nagao M. Rat p53 gene mutations in primary Zymbal gland tumors induced by 2-amino-3-methylimidazo[4,5-f]quinoline, a food mutagen. Proc Natl Acad Sci USA. 1992;89:4850–4854. doi: 10.1073/pnas.89.11.4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 42.Oren M, Maltzman W, Levine AJ. Post-translational regulation of the 54K cellular tumor antigen in normal and transformed cells. Mol Cell Biol. 1981;1:101–110. doi: 10.1128/mcb.1.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stürzbecher HW, Chumakov P, Welch WJ, Jenkins JR. Mutant p53 proteins bind hsp 72/73 cellular heat shock-related proteins in SV40-transformed monkey cells. Oncogene. 1987;1:201–211. [PubMed] [Google Scholar]
- 44.Milner J, Medcalf EA. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell. 1991;65:765–774. doi: 10.1016/0092-8674(91)90384-b. [DOI] [PubMed] [Google Scholar]
- 45.Michalovitz D, Halevy O, Oren M. p53 mutations: gains or losses. J Cell Biochem. 1991;45:22–29. doi: 10.1002/jcb.240450108. [DOI] [PubMed] [Google Scholar]
- 46.Hsu HC, Tseng HJ, Lai PL, Lee PH, Peng SY. Expression of p53 gene in 184 unifocal hepatocellular carcinomas: association with tumor growth and invasiveness. Cancer Res. 1993;53:4691–4694. [PubMed] [Google Scholar]
- 47.Ng IO, Srivastava G, Chung LP, Tsang SW, Ng MM. Overexpression and point mutations of p53 tumor suppressor gene in hepatocellular carcinomas in Hong Kong Chinese people. Cancer. 1994;74:30–37. doi: 10.1002/1097-0142(19940701)74:1<30::aid-cncr2820740107>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 48.Mosner J, Mummenbrauer T, Bauer C, Sczakiel G, Grosse F, Deppert W. Negative feedback regulation of wild-type p53 biosynthesis. EMBO J. 1995;14:4442–4449. doi: 10.1002/j.1460-2075.1995.tb00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ono K, Tanaka T, Tsunoda T, Kitahara O, Kihara C, Okamoto A, Ochiai K, Takagi T, Nakamura Y. Identification by cDNA microarray of genes involved in ovarian carcinogenesis. Cancer Res. 2000;60:5007–5011. [PubMed] [Google Scholar]