

Summary
The innate immune system is an ancient surveillance system able to sense microbial invaders as well as aberrations in normal cell function. No longer viewed as a static and non‐specific part of immunity, the innate immune system employs a plethora of specialized pattern recognition sensors to monitor and achieve homeostasis; these include the Toll‐like receptors, the retinoic acid‐inducible gene‐like receptors, the nucleotide‐binding oligomerization domain receptors (NLRs), the C‐type lectins and the complement system. In order to increase specificity and diversity, innate immunity uses homotypic and heterotypic associations among these different components. Multi‐molecular assemblies are formed both on the cell surface and in the cytosol to respond to pathogen and danger signals. Diverse, but tailored, responses to a changing environment are orchestrated depending on the the nature of the challenge and the repertoire of interacting receptors and components available in the sensing cell. It is now emerging that innate immunity operates a system of ‘checks and balances’ where interaction among the sensors is key in maintaining normal cell function. Complement sits at the heart of this alarm system and it is becoming apparent that it is capable of interacting with all the other pathways to effect a tailored immune response. In this review, we will focus on complement interactions with NLRs, the so‐called ‘inflammasomes’, describing the molecular mechanisms that have been revealed so far and discussing the circumstantial evidence that exists for these interactions in disease states.
Keywords: cell surface molecules, complement, inflammasome, inflammation, innate lymphoid cells
Introduction
The complement system, named for its ability to ‘complement’ the humoral immune response, was the first part of the innate immune system to be discovered over a century ago. Since then, this complex fluid‐phase and membrane‐bound system of proteins has been considered to be the first line of our immune defence against infection. Thought to be a static and non‐specific apparatus, it was seen as an undifferentiated system, non‐specifically tagging invaders for phagocyte engulfment and digestion, in contrast to the sophisticated framework of T and B cells that have elaborate clonal mechanisms to form a plethora of highly specific receptors and antibodies by DNA rearrangement.
This view of the innate immune system changed dramatically with the discovery of Toll‐like receptors (TLRs) in the late 1990s and the verification that the innate immune system is actually highly specific, relying on germline‐encoded pattern recognition receptors (PRRs) that, in addition to TLRs, include retinoic acid‐inducible gene‐I‐like receptors (RLRs) and the nucleotide‐binding oligomerization domain receptors (NLRs) that have evolved to detect components of foreign pathogens referred to as pathogen‐associated molecular patterns (PAMPs). This system of sensors can detect not only PAMPs but also host‐derived damage‐associated molecular patterns (DAMPs), triggering infectious and sterile inflammation.
With the discovery of the TLRs, the complement system was momentarily overshadowed; however, it soon became apparent that all parts of the innate immune system followed the dogma of ‘pattern recognition’ with complement being the archetypal PRR – recognizing motifs and signatures from pathogens (carbohydrate structures) as well as host proteins (antibodies) so as to trigger activation. Hence, it has emerged that the innate immune system has virtues that are equally specific and just as elaborate as the features of adaptive immunity and that there are various levels of crosstalk among the different sensors shaping the inflammatory response. These sensors are not independent pathways, but rather act in concert in trigger‐dependent patterns and via several initiation and regulatory mechanisms to produce an anticipated result in immune surveillance. Complement sits at the heart of this surveillance system: TLR–complement interactions have been described that have both synergistic and antagonist consequences;1, 2, 3 there are TLR–NLR interactions, most notably forming a molecular scaffold complex, termed the inflammasome, which requires input from both the TLR and NLR pathways and leads to the activation of the pro‐inflammatory cytokines interleukin‐1β (IL‐1β) and IL‐18; triggering of TLRs and RLRs seems to act as a priming step for the inflammasome, and we now have data demonstrating direct interaction between complement and the inflammasome. The current review aims to summarize these interactions and describe complement‐induced inflammasome activation; we propose a model of innate immune activation where the terminal complement components, including complement‐induced pore formation on the plasma membrane, trigger inflammasome activation.
The complement system
The discovery of the complement system dates back to the late nineteenth century. In recent years attention has been shifted away from complement by the discoveries of the TLRs, RLRs and NLRs; indeed, immunologists have overlooked the fact that complement was the first PRR to be discovered and that the whole complement system follows the same dogma of activation – ‘pattern recognition’.
Complement is a large collection of plasma proteins that can be activated in a cascade‐like fashion leading to opsonization of pathogens for phagocytosis and the assembly and deposition of the membrane attack complex (MAC) that kills bacteria or infected/damaged cells through disruption of their membrane integrity. Hence, its job is to recognize PAMPs or DAMPs, to tag the cells of interest and to destroy them. Complement can therefore be viewed as another ‘sensor’ of the pattern recognition system of activation that the innate immune system employs. It recognizes motifs and signatures that trigger the cascade of activation.
Activation occurs via three pathways: (i) the classical pathway (CP) with C1q as its main pattern recognition sensor, (ii) the lectin pathway (LP) with mannose‐binding lectin (MBL) and ficolins as the recognition molecules, and finally (iii) the alternative pathway (AP) with a thioester moiety of C3b as well as properdin as the recognition molecules. Once activated all three pathways converge at C3.
The CP employs C1q as a PRR and it is able to bind host proteins, such as IgM and IgG clusters, apoptotic cells and structures on microbial cells. The C1 complex contains the proteases C1r and C1s, which become activated once there is C1q surface binding.4 C4 is subsequently cleaved by C1s into C4a and C4b, followed by C2, creating the C4bC2a enzyme (C3 convertase).
The LP employs MBL and ficolins as PRRs to recognize and bind carbohydrates. Both MBL and ficolins bind MBL‐associated serine proteases (MASPs) in structures similar to the C1 complex of the classical pathway – MBL/ficolins resembling C1q, whereas MASPs resemble C1r and C1s. In this pathway, MASP2 cleaves C4 and C2 to generate the C3 convertase.5
The AP has C3 as its central molecule. C3b generated by cleavage of C3 by CP/LP convertases or C3(H2O) formed spontaneously forms the nidus for binding of factor B (fB), which is then cleaved by the serum protease factor D (fD) to form the AP C3 convertase C3bBb (or C3(H2O)Bb). Cleavage of C3 exposes a thioester moiety in C3b that is able to recognize and bind surfaces,6 leading to complement tagging and opsonization. In addition, properdin, a stabilizer of the AP C3 convertase, is able to recognize several PAMPs and DAMPs, initiating a complement response.7 Therefore the AP has its own PRRs so as to be able to recognize host and pathogen signatures and trigger activation.
Membrane attack complex
Irrespective of how the complement cascade has been initiated, whether in response to host immunoglobulins, apoptotic bodies, or specific PAMPS or DAMPS, all activation cascades converge to the terminal pathway, a pathway that begins with the cleavage of C5 and ends with the deposition of an oligomeric structure of complement components embedded in the cell membrane, creating a pore. This oligomeric structure, the MAC, comprises five complement components, C5b‐9 and its sole purpose was thought to be the lysis of bacteria, and infected or ‘damaged’ cells.
However, in addition to lysis, MAC can trigger diverse effects in the target cells, such as inflammatory mediator release, cell proliferation and apoptosis.8, 9 Nucleated cells are resistant to MAC killing because of the presence of ion pumps and mechanisms that shed MAC. The fate of a nucleated cell attacked by complement seems to be dependent mainly on calcium. A large influx of calcium is the first event following MAC attack on a cell and increasing concentrations of calcium contribute to cell death.10 Increasing concentrations of calcium lead to loss of membrane potential of the mitochondria, resulting in an energy crisis within the cell. The situation becomes even worse with the loss of ATP through the MAC pore, rendering the cell incapable of sustaining its metabolic processes.11 Although we have known for over 25 years that these events occur following MAC attack on nucleated cells, it was not until recently that it was revealed that these events occur in cooperation with NLRs and the oligomeric structure termed the inflammasome.
The inflammasome
The term inflammasome was first used in 2002 by Martinon et al.,12 to describe a novel structure that was similar to the apoptosome and had the ability to cause inflammation. The inflammasomes are a group of multimeric protein complexes that assemble in the cytosol; all consist of an activated NLR molecule, apoptosis‐associated speck‐like protein containing CARD (ASC) and pro‐caspase‐1. The formation of the inflammasome complex is triggered by a variety of substances derived from infections, tissue damage, or metabolic dysfunctions.13 Ultimately, the arrangement of the inflammasome complex triggers the activation of pro‐caspase‐1, which proteolytically activates the pro‐inflammatory cytokines IL‐1β and IL‐18.
Due to the large number of ligands that have been identified as triggering the inflammasome, it has been suggested that the inflammasome does not directly bind the ligands but rather is activated indirectly by a two‐step activation mechanism consisting of a first priming step involving pro‐IL‐1β synthesis and a second step in which caspase‐1 activates and cleaves IL‐1β to produce the active cytokine.14
The priming step is believed to be triggered by PRR recognition of their ligands and the subsequent activation of nuclear factor‐κB. What occurs during the second step is still unclear; it is suggested that conformational change allows the NLR to bind ASC, which is the adaptor for all inflammasomes. This interaction triggers ASC to bring monomers of pro‐caspase‐1 into close proximity, initiating the activation of caspase‐1 self‐cleavage and the formation of its active form, which in turn mediates the conversion of pro‐IL‐1β into its bioactive cytokine IL‐1β.15, 16, 17 As there is no direct ligand binding, four mechanisms leading to ‘Signal 2’ of inflammasome activation have been proposed: (i) extracellular ATP triggers K+ efflux,18 (ii) release of lysosomal contents caused by lysosome disruption,19, 20 (iii) production of reactive oxygen species (ROS),21 and (iv) Ca2+ mobilization.22, 23 Generally it seems that the NLR senses ion fluxes or aberrations in homeostasis within the cytosol that lead to its activation.
NLR pyrin domain‐3 (NLRP3) inflammasome
The NLRP3 inflammasome is the best characterized inflammasome. Unlike NLRP1, the NLRP3 has a typical tripartite structure organization: an N‐terminal pyrin domain, a central nucleotide binding and oligomerization (NACHT) domain and a C‐terminal Leurine rich repeat (LRR) domain. The NACHT domain has ATPase activity and upon stimulation causes the receptor to oligomerize and recruit ASC via its pyrin domain. NLRP3 lacks a CARD domain so it can only recruit pro‐caspase‐1 through the adaptor molecule ASC.15
There are a plethora of triggers that activate NLRP3, including crystalline material,20 fibrillar amyloid‐β, 19 peptide aggregates, viral and bacterial PAMPs, and other DAMPs. NLRP3 has been shown to be activated by whole pathogens such as the fungi Candida albicans and Saccharomyces cerevisiae, and the viruses Sendai virus, adenovirus, influenza virus, respiratory syncytial virus and rhinovirus.24, 25, 26 NLRP3 has also been found to be activated by host‐derived molecules such as extracellular ATP that are sensed by the purinoceptor 7 (P2X7) receptor.27
‘Complementing’ the innate immune response
When the host is challenged by a variety of microorganisms as well as danger signals, it needs to focus on particular signatures and motifs to be able to effectively recognize the challenge and deploy a fast immune response – hence, pattern recognition is a key feature and central focus of the innate immune system. Over the years researchers have been thinking of the different PRRs of the innate immune system as receptors acting in isolation, but in vivo the single‐receptor model of activation is an oversimplified one. Realistically, cells will be challenged by and respond to multiple stimuli simultaneously and so several PRRs will be engaged deploying an inflammatory response as a result of the activation of several signalling cascades.
In such circumstances the location of each PRR would be key in triggering a co‐ordinated immune response. The complement system represents the extracellular surveillance system, with several soluble factors looking for microbes in the extracellular space and surface receptors detecting activation. The TLRs guard the ‘toll‐gates’ of the cells both on the cell surface and in endosomes along the internalization route of microbes, whereas the RLRs and NLRs (inflammasome) are the keepers of the cytosol. Upon concomitant detection of pathogens, the different PRRs of the innate immune system would need to coordinate responses to combat the infection. A system of checks and balances should be in place to control this network of immune sensors capable of commanding the inflammatory response, and complement sits at the centre of it. Since the discovery of the TLRs, it has been shown that there is co‐operation between the TLRs and the complement system.28 Complement acts synergistically with TLRs to amplify the inflammatory response through its membrane‐bound receptors, C3aR and C5aR, while antagonist crosstalk has also been observed.
Complement can also negatively regulate RLRs. Viral infection mediates the translocation of the receptor for the globular heads of C1q (gC1qR) to the mitochondria, promoting its associations with mitochondrial antiviral signalling protein (MAVS), the adaptor molecule for RLRs.29 The interaction of gC1qR with MAVS disrupts MAVS interaction with the RLRs (retinoic acid‐inducible gene‐I and MDA5) inhibiting RLR‐mediated signalling and anti‐viral responses. MBL, the LP PRR, has also been shown to be able to control anti‐viral responses; MBL binds dsRNA and modifies TLR3‐induced signalling.30
Complement and inflammasome activation
Most recently it has been shown that complement is also able to coordinate inflammasome activation and IL‐1β production. Inflammasomes are cytosolic oligomeric structures of NLRs and ASC molecules that regulate the secretion of IL‐1β and IL‐18. We and others have recently shown that sublytic MAC can trigger NLRP3 inflammasome activation.31 Deposition of sublytic MAC on the cell surface led to increased intracellular Ca2+ concentrations, which in turn accumulated in the mitochondrial matrix leading to loss of mitochondrial transmembrane potential and triggering of the NLRP3 inflammasome. This study has been corroborated by Laudisi et al.,32 where they have confirmed in mouse dendritic cells that sublytic MAC drives the assembly of the NLRP3 inflammasome and caspase‐1 activation.
The mechanism of activation seems to be dependent on Ca2+ ions, a consequence of pore formation on the cell membrane, leading us to believe that MAC must be acting in a manner similar to viroporins26 or bacterial pore‐forming toxins that have also been shown to activate the inflammasome. Viroporins, such as influenza M2,33 and respiratory syncytial virus SH25 deliver Signal 1 of inflammasome activation via a triggering PRR (i.e. TLRs) whereas Signal 2 is triggered by perforating the cell membrane. In the case of some bacterial toxins, as well as the Rhinovirus 2B viroporin,34 the toxins seem able to trigger both Signal 1 and Signal 2 of inflammasome activation by disrupting the membrane. The question that remains is how does complement membrane attack trigger this activation. The evidence clearly shows that MAC is delivering Signal 2 by changes in the Ca2+ concentration, but does it trigger Signal 1 – and if so, how? Is it possible that MAC provides both signals of activation, or are complement components upstream of MAC responsible for the priming signal?
Anaphylatoxins in inflammasome activation: providing Signal 1?
Although recent studies have implicated MAC, the end product of the complement cascade, as the trigger for inflammasome activation, the question that arises is whether the complement cascade upstream of MAC is able to also contribute to inflammasome activation. Anaphylatoxins, C3a and C5a, bind specific 7‐TM G‐protein‐coupled receptors (GPCR) on cells and could be triggering Signal 1 of inflammasome activation upstream of MAC.
C3a and C5a are small, highly cationic polypeptides (~70 amino acids) that are extremely potent inflammatory mediators.35 They regulate a spectrum of immune and non‐immune functions, such as vasodilatation,36 pro‐inflammatory cytokine production, histamine release, chemoattraction,37 and tissue regeneration.38 Control mechanisms have been evolved that regulate their activity, especially their pro‐inflammatory activity. Carboxypeptidases rapidly cleave off a C‐terminal arginine residue,39, 40 resulting in C5adesArg and C3adesArg [also known as acylation stimulating protein (ASP)], the former retaining minimal inflammatory activity (1–10%) by still being able to bind to C5a receptor (C5aR) whereas the latter is unable to bind to C3a receptor (C3aR) and lacks any pro‐inflammatory activity.41, 42 Interestingly, although C3adesArg has lost the pro‐inflammatory activity of its predecessor C3a, it still possesses metabolic activity, controlling triglyceride synthesis in the adipose tissue,43 an example of the many ways that the complement system can perform non‐immune functions.
C3a as Signal 1?
The anaphylatoxins exert their activities by binding to GPCR, the C3aR, the C5aR and C5a receptor‐like 2 (C5L2; this latter is not G‐protein coupled).44 C3aR binds C3a, but does not bind C3adesArg or C5a.45 Once bound to its ligand, C3aR triggers signalling via heterotrimeric G‐proteins, in particular via pertussis‐toxin sensitive G proteins (Gi), mobilizing intracellular calcium.46 In addition to Gi, C3aR can also use the pertussis‐toxin‐insensitive G proteins G12, G1347 and G1645 for signal transduction, triggering downstream signalling events such as activation of protein kinase C, phospholipase C, the mitogen‐activated protein kinases, extracellular signal‐regulated kinase 1 (ERK1) and ERK2,41, 48 as well as phosphatidylinositide 3‐kinase (PI3K) followed by Akt‐phosphorylation.49 Therefore, C3a is more than capable of delivering Signal 1 of inflammasome activation leading to generation of pro‐IL‐1β by triggering signalling cascades in both myeloid and non‐myeloid cells types where C3aR is expressed.
C3a has recently been shown to modulate IL‐1β production in human monocytes. Although C3a was implicated in IL‐1β production in an earlier study,50 the results were compromised by possible lipopolysaccharide contamination of the C3a preparations. In the most recent study, it was shown that C3aR augments the release of TLR4‐mediated cytokines, leading to enhanced T helper type 17 responses.51 The authors demonstrated that C3a binding of C3aR drives IL‐1β production by triggering ERK1/2 activation, followed by increased ATP efflux. This increase in extracellular ATP leads to the activation of the P2X7 which in turn triggers NLRP3 activation. This study is a perfect example of how complement, TLRs and the inflammasome co‐ordinate in response to danger.
C5a detecting crystalline material as Signal 1?
C5a triggers signalling via its C5aR a GPCR linked to a heterotrimeric G‐protein complex including the pertussis‐toxin‐sensitive α units Gi252 and the pertussis‐toxin‐insensitive G16.53, 54 Ligation of C5aR causes calcium mobilization from the extracellular space as well as from intracellular stores. Activation leads to phosphorylation of the C‐terminus of the receptors by GPCR kinases, which bind to arrestins targeting C5aR for internalization via clathrin‐coated pits,55 as well as interact with intracellular signalling components such as PI3K, mitogen‐activated protein/extracellular signal‐regulated kinases and Akt.56 Different signalling cascades have been linked to C5aR activation, including phospholipase C,57 phospholipase D,58 PI3K,59, 60 Raf‐1‐mediated activation of MEK‐1,61 demonstrating that C5aR is a signalling PRR that is able to deliver Signal 1 of inflammasome activation.
Recent studies have demonstrated that crystalline material along with C5a are able to trigger inflammasome activation.62, 63 The complement system as well as the inflammasomes have been individually linked to the innate recognition of crystalline material. Crystalline substances have been previously shown to be able to activate the NLRP3 inflammasome and lead to the production of IL‐1β and 1L‐18. In particular, cholesterol crystals (CC),64 monosodium urate (MSU) and calcium phosphate crystals have all been shown to be readily phagocytosed by macrophages and lead to NLRP3 activation through lysosomal damage and ROS production.65 In parallel, such crystalline material has been previously shown to activate the complement system. CC,66, 67, 68 and MSU crystals,69 have been shown to activate the complement cascade via the AP. In an MSU‐crystal‐induced rabbit synovitis model, complement triggered pro‐inflammatory responses as well as chemotaxis of neutrophils into the synovium,70 suggesting that complement is central in the innate immune response against MSU crystals.
A recent study by Samstad et al.62 demonstrated that CC induce complement‐dependent inflammasome activation via C5a. Both CP and AP were activated in response to CC, confirming earlier studies that had suggested that CC activated the AP.66, 67, 68 The amounts of C3bBb and properdin in plasma were increased in response to CC, whereas C5a enhanced CC‐induced cytokine release, including tumour necrosis factor‐α and IL‐1β. The study suggested that C5a in combination with tumour necrosis factor‐α might act as a priming signal (Signal 1) of inflammasome activation. Most crystalline structures trigger a short‐lived ROS production as Signal 2 of inflammasome activation and their data in a whole blood assay demonstrated complement‐dependent ROS production in response to CC. Therefore, complement not only provides Signal 1 (via CC and C5a) but also Signal 2 of inflammasome activation by the production of ROS. Furthermore, they identified complement receptor 3 (CR3) as the receptor responsible for the internalization of CC, implicating complement as a key orchestrator in CC‐induced inflammation.
In a study by An et al.,63 the authors investigated MSU‐crystal‐induced pro‐inflammatory responses in whole blood, and demonstrated regulation by C5a via the C5a–C5aR axis. They showed that C5a alone is able to deliver Signal 1 of inflammasome activation, by inducing pro‐IL‐1β in human monocytes, and potentiates IL‐1β production. This activation was caspase‐1‐dependent and required intracellular Ca2+, K+ efflux and Cathepsin B activity. This study demonstrated that C5a can act as an endogenous priming signal for the initiation of inflammasome activation in response to crystalline material.
C1q is an inhibitor of inflammasome activation
Although so far we have described evidence that complement enhances inflammasome activation, a recent study has revealed the possibility that complement components act as inhibitors of the inflammasome. The role of C1q as a PRR molecule of the complement system is well established. C1q binds apoptotic cells via its globular heads,71, 72 and acts to clear cellular debris by binding to phagocytic receptors via its collagen tail.73 C1q has been found to be synthesized in tissue macrophages and dendritic cells lacking C1r and C1s,74, 75 its local interaction with macrophages and dendritic cells results in down‐regulation of pro‐inflammatory cytokine production.76, 77 It is hypothesized that C1q in this form provides a system of rapid opsonization of dying cells in tissue that down‐regulates local inflammation in response to DAMPs.
A recent study by Benoit et al.78 using a unique system of primary autologous lymphocytes and monocyte‐derived macrophages, demonstrated that C1q directly suppressed caspase‐1‐dependent IL‐1β production, suggesting that C1q is a down‐regulator of inflammasome activation. They showed that C1q bound to apoptotic lymphocytes triggered Janus kinase–signal transducer and activator of transcription signalling and increased the expression of immunoregulatory cytokines, such as IL‐10, IL‐27 and IL‐33, as well as inhibiting NLRP3 activation by reducing the cleavage of caspase‐1. The authors proposed that increased expression of NLRP12, a negative regulator of the inflammasome, was the mechanism for C1q‐induced NLRP3 inhibition, although this remains to be confirmed.78
Inflammatory diseases: circumstantial evidence for complement–inflammasome interactions
The complement system is emerging as one of the key regulators of the innate immune response, able to co‐operate with TLRs and RLRs as well as NLRs. The NLRs are guardians of normal cell function, triggered in response to aberrations of normal homeostasis. Recurrent themes linking NLR triggers are loss of membrane integrity (i.e. through pore‐forming toxins, viroporins, lysosomal rupture or MAC), changes in cellular homeostasis (i.e. ROS production, Ca2+ efflux, K+, Na+ ions) and loss of mitochondrial membrane potential.79 Complement controls the NLR ‘cellular emergency alarm system’: it can provide Signal 1 for inflammasome activation by triggering pro‐IL1β production via the C5a–C5aR axis,62, 63 it can trigger Signal 2 either via C3a‐induced ATP release51 or via MAC insertion into the membrane and Ca2+ fluxes.31, 32 Finally it can ‘switch off’ the alarm using C1q and inhibiting cleavage of caspase‐178 (Fig. 1).
Figure 1.
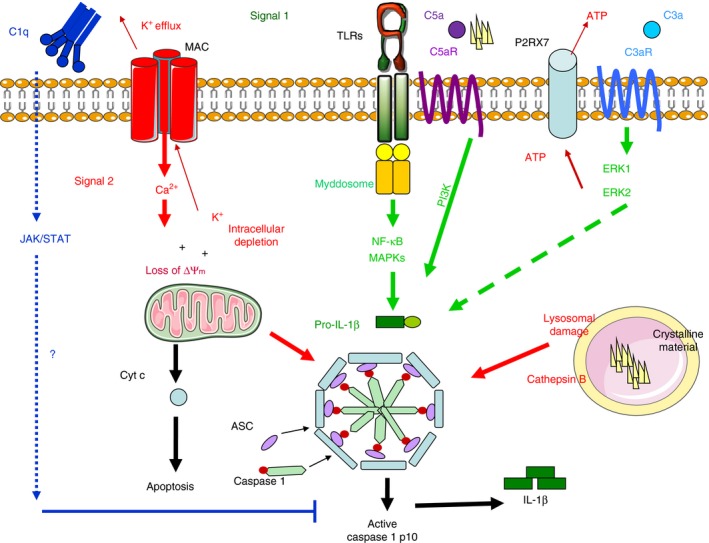
Complement‐dependent inflammasome activation. Complement controls the nucleotide‐binding oligomerization domain receptor (NLR) ‘cellular emergency alarm system’: it provides Signal 1 (green arrows) for inflammasome activation by triggering pro‐interleukin‐1β IL‐1β) production via the C5a–C5aR axis, it triggers signal 2 (red arrows) either via C3a‐induced ATP release or via membrane attack complex (MAC) insertion into the membrane and Ca2+ fluxes. Finally, it can ‘switch off’ the alarm through C1q inhibition of caspase‐1 cleavage.
Although these innate immune pathways are designed to co‐operate so as to protect the host against invading microorganisms, this interaction is a double‐edged sword in the sense that it may induce undesirable inflammation if activated improperly or in an uncontrolled manner. Numerous studies have shown that triggering of complement and NLRs can contribute to various immune, inflammatory, neurodegenerative, ischaemic and age‐related diseases, but none has investigated the co‐operation of these two pathways in inflammatory diseases. Understanding this intricate network of pathways is crucial in controlling several conditions of unmet clinical need such as sepsis, ischaemia–reperfusion injury, capillary leak syndrome, neuroinflammatory disease, nephritis, transplant rejection, age‐related macular degeneration, rheumatoid arthritis, osteoarthritis and even atherosclerosis just to name a few. In the section below, we will present the evidence that currently exists, hinting at an involvement of complement–inflammasome interactions in disease states (Fig. 2).
Figure 2.
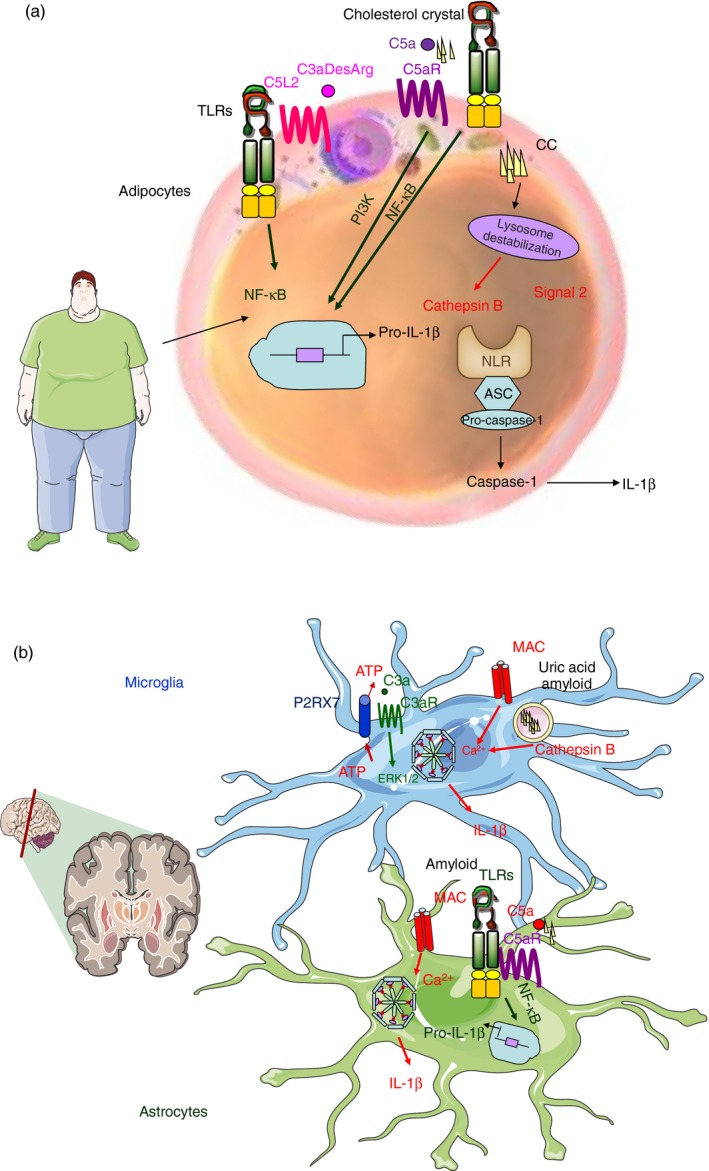
Complement–inflammasome interactions in chronic inflammatory conditions. Proposed mechanisms of complement‐dependent inflammasome activation in metabolic diseases (a) and neurodegenerative conditions (b). (a) In metabolic diseases, in adipocytes Toll‐like receptors (TLRs) might cooperate with (i) C5aR in response to cholesterol crystals or (ii) C5L2 in response to C3adesArg in order to provide Signal 1. Internalization of cholesterol crystals could lead to lysosomal destabilization and release of cathepsin B, providing the second signal for inflammasome activation. (b) In neurodegenerative conditions, in microglia and astrocytes: (i) TLRs might cooperate with C5aR for the recognition of amyloid or other crystalline material triggering Signal 1 of inflammasome activation or (ii) C3aR might trigger signalling and release ATP and activate the inflammasome via the P2X7 receptor. Signal 2 of inflammasome activation could be triggered by elevated cytoplasmic Ca2+ caused by membrane attack complex (MAC), or cathepsin B release resulting from lysosomal damage.
Complement–inflammasome interactions in sepsis
Sepsis, the syndrome that results from overproduction of cytokines, is an often fatal response of the immune system against microbial pathogens. It is now widely accepted that the over‐reaction in the host occurs at the level of the innate immune system and is directly linked to the recognition of bacterial cell wall components, such as lipopolysaccharide. The TLR family and in particular TLR4 have been identified as the main signalling receptors for triggering the response,80 but the involvement of complement has also been suggested.81 Originally it was thought that C5a was the main complement‐derived contributor in sepsis, it was later shown that C5a‐triggered inflammation was due not only to C5aR ligation but also to C5L2, only their combined inhibition was found to be protective in sepsis.81 Cross‐talk of TLR4 and C5aR signalling in response to lipopolysaccharide82 indicated complex interactions of the complement system and other danger‐sensing systems in sepsis; these include the NLRs. NLRP3 deficiency provides partial protection against bacterial challenge,83 whereas deficiency of NLRP3 inflammasome components, such as caspase‐184 and ASC85 has been shown to be protective against the lethal effects of endotoxin. Therefore, it seems that all three main innate immune pathways, the TLRs, NLRs and complement, are contributing in sepsis, the question that remains is what are the exact molecular events that control the crosstalk among these pathways in sepsis and how can we target them therapeutically?
Although much is known about the individual role of these innate immune sensors in sepsis, our understanding of their interactions in the context of sepsis is limited. For both septic acute kidney injury and acute lung injury the NLRP3 inflammasome86, 87 and the complement system88, 89 have been implicated; however, the role that possible interactions between the two systems might play in the pathogenesis of both remains undetermined. The combined data suggest that NLRs, TLRs and complement collaborate to deliver signals for inflammasome activation in the context of sepsis; it would be interesting to explore interactions between these pathways in both septic acute kidney injury and acute lung injury.
Complement–inflammasome interactions in metabolic diseases
Many modern metabolic diseases, ranging from obesity and atherosclerosis to type 2 diabetes mellitus have been associated with a chronic inflammatory state. Both complement and the inflammasome have been individually linked to such conditions, but no one so far has looked at the interactions of these two pathways in the context of metabolic disease.
In the case of obesity, chronic low‐grade inflammation (metabolic inflammation) has been a feature of chronic nutritional surplus.79 One of the first links between obesity‐associated inflammation and type 2 diabetes mellitus was discovered from studying the levels of pro‐inflammatory cytokines; tumour necrosis factor‐α, IL‐6 and IL‐1β were found to be increased in the bloodstream as well as in the adipose tissue of obese and diabetic patients.79, 90, 91, 92 Subsequent studies in mice demonstrated the existence of activated macrophages in adipose tissue and showed that these were the main source of the pro‐inflammatory cytokines. In particular, they showed that adipose tissue macrophages from obese individuals were mainly of the pro‐inflammatory M1 phenotype rather than the anti‐inflammatory M2 phenotype. Their presence was directly correlated with ectopic lipid accumulation and insulin resistance in mice.93
In the past decade, members of the TLR family as well as inflammasome have been shown to be expressed on these adipose tissue macrophages sensing metabolic disturbances. TLR2 and TLR4, which are signalling PRRs that can provide Signal 1 of inflammasome activation, as well as components of the NLRP3 inflammasome (NLRP3, ASC, caspase‐1) have been shown to be increased in adipose tissue of obese/diabetic individuals.94, 95 It is believed that the mechanism of inflammasome activation involves defective autophagy and ROS.95 Interestingly, adipocyte precursors from caspase‐1−/− mice, differentiate more efficiently into adipocytes and have an increased oxidation rate,96 leading us to believe that there might be adipocyte‐specific proteins that can serve as caspase‐1 substrates and cause changes observed in adipocytes in obese mice. The identity of such sensor proteins remains to be elucidated. In light of the recent findings of complement control of inflammasome activation, we could suggest that complement components or receptors might play such a role. C5L2 is the cellular receptor for C3adesArg,97 playing key roles in lipid metabolism in adipocytes and fibroblasts;98 it is possible that the C5L2–C3adesArg axis is also responsible for inflammasome activation in adipocytes. C5L2 was originally believed to be an orphan decoy receptor, but is now known to be a positive modulator of responses to C5a and C3a.99 It is not clear whether C5L2 is a signalling receptor itself, or a modulator of the signalling pathways used by other receptors capable of delivering Signal 1, such as TLR4.100 The fact that C5a has recently been shown to contribute to obese adipose tissue inflammation and insulin resistance101 lends more support to the hypothesis that complement products, such as C3adesArg and C5a might contribute to inflammasome activation.
In the case of atherosclerosis, complement activation products of the terminal pathway (C5a and MAC) are abundant in atherosclerotic plaques and have been suggested to play critical roles in atherogenesis, through mechanisms that remain unclear.102, 103, 104 On the other hand, NLRP3 has also been implicated in CC‐induced inflammasome activation.64 The fact that CCs induce complement (C5a)‐dependent inflammasome activation,62 further demonstrates the link between metabolic disease and complement–inflammasome interactions.
Complement–inflammasome interactions in neuroinflammatory conditions
Neurodegenerative diseases are becoming a major issue in an ever aging population. Alzheimer's disease (AD) is a neurodegenerative disorder and the most common cause of dementia in the elderly. Patients present with a gradual memory loss and other cognitive deficits. Accumulation of amyloid β (Aβ) in senile plaques is a principal event in the pathogenesis of AD,105, 106 but the cellular events leading to plaque‐induced neuronal dysfunction are still unclear. Genetic factors have been suggested to either cause or predispose to AD, including mutation in β‐amyloid precursor protein, which is the protein that generates Aβ by proteolytic processing, mutation in presenilins 1 and 2 as well as polymorphism to apolipoprotein A and Complement receptor 1. Inflammatory and immunological processes have also been implicated in the initiation and progression of AD.105
Neuroinflammation is a key feature in the progression of the disease and is concentrated at sites of Aβ plaques along with increased levels of pro‐inflammatory cytokines, complement components and proteases.107, 108 TLRs109 and the complement system (both the CP and AP) are directly activated by Aβ in an antibody‐independent fashion,110 and the NLRP3 inflammasome19 has been linked to AD, but currently, there is little evidence for cooperation between these three systems in AD. Preliminary data from our group suggest that TLRs, NLRs and complement co‐operate so as to trigger inflammatory responses against Aβ (Triantafilou & Morgan unpublished data), suggesting that future therapies should focus on targeting all three cascades. A recent study by Fonseca et al.111 demonstrated that treatment with a C5aR antagonist decreased pathology and enhanced behavioural performance in murine models of AD, suggesting that C5aR is a novel therapeutic target for AD.
Similarly, all three pathways seem to be involved in the chronic inflammatory response that is observed in multiple sclerosis (MS), which is a chronic autoimmune inflammatory and demyelinating disease of the central nervous system that currently affects over seven million people worldwide.112 Recent studies have implicated the NLRP3 inflammasome in the development of MS, by demonstrating increased levels of activated caspase‐1, IL‐1β, IL‐18 and activators of the inflammasome, such as ATP, uric acid and Cathepsin B, in patients with MS.113 In addition, caspase‐1, a key component of the NLRP3 inflammasome, has been found in high amounts in MS plaques.114, 115
The complement system has also been individually linked to the disease, because it has been demonstrated that there is C3 deposition in the brains of people with MS.116 The terminal complement components were also reported in MS within plaques and adjacent white matter,117 and more recently, we demonstrated the presence of complement proteins, activation products as well as regulators in MS brain, confirming that complement activation occurred in the plaque, peri‐plaque and adjacent white matter.118 The link between complement and the NLRP3 inflammasome in MS has yet to be studied, but it is clear that as their activation markers co‐exist in MS plaques and brain, there must be concomitant activation of both innate surveillance pathways.
Neuromyelitis optica (NMO) is a severe autoimmune demyelinating disease that affects the optic nerves and spinal cord. An IgG autoantibody (NMO‐IgG) against aquaporin 4 (AQP4) drives pathology and serves as a marker for NMO.119 Binding of the NMO‐IgG to AQ4 causes complement‐dependent cytotoxicity and inflammation,120 therefore the complement CP is central in the pathogenesis of the disease. More recently, NLRP3 activation has been found in lesions of NMO, suggesting that the inflammasome is activated mainly in reactive astrocytes and infiltrating macrophages involved in the active destruction of brain tissue.121 Aquaporins themselves have been shown to induce IL‐1β secretion, because they are able to disrupt water intake within the cell.122 As the inflammasome is able to sense aberrant homeostasis within the cell, water imbalances caused by binding of autoantibodies to AQP4 and subsequent destruction by complement could be triggering NLRP3 inflammasome activation. Future studies should focus on unravelling the molecular mechanisms involved.
Modulating complement–inflammasome interactions as a therapeutic intervention
Growing evidence suggests that the components of the innate immune response play an integral part in the pathogenesis of many acute and chronic inflammatory conditions. Therapeutic interventions that individually modulate these pathways are being developed. Drugs inhibiting IL‐1, P2X7R,123 and caspase‐1 [pralnacasan (VX‐740) and VX‐765]124 have been developed for inhibiting inflammasome activation. However, only IL‐1 inhibitors (anakinra and canakinumab)125 and colchicine, a drug frequently used for the treatment of autoinflammatory diseases such as gout and pseudogout,126 are already used in the clinic, the rest are currently in Phase 1 and Phase 2 clinical trials.
Interestingly, new ways to modulate NLRP3 activation are emerging that could be potential therapeutic interventions in the future. One of the most exciting discoveries is of the first small molecule inhibitor of the inflammasome, MCC950.127 MCC950 is a potent and selective inhibitor able to block the canonical and non‐canonical NLRP3 activation by blocking NLRP3‐induced ASC oligomerization;127 because of its selective nature it is an exciting potential therapeutic for NLRP3‐associated syndromes. In addition, ketone bodies128 as well as dopamine129 have been recently discovered as negative regulators of the inflammasome. Ketone bodies β‐hydroxybutyrate and acetoacetate that support mammalian survival during states of energy deficit, seem to reduce NLRP3‐induced IL‐1β and IL‐18 production as well as caspase‐1 activation in human monocytes as well as in mouse models of NLRP3‐associated diseases.128 Therefore suggesting that NLRP3‐associated diseases could be modulated by caloric restriction or ketogenic diets in the future. Interestingly, a recent study has demonstrated that dopamine, a neurotransmitter, inhibits NLRP3 inflammasome activation via dopamine D1 receptors.129 These findings suggest that there is an endogenous regulatory mechanism of inflammasome‐associated inflammation, and targeting receptors such as dopamine D1 receptors could be potential therapeutic targets for NLRP3‐driven inflammatory diseases.
In parallel, over the past decade, many small potent C5aR antagonists130 as well as compstatin,131 targeting C3, have been reported and used in various models as well as in humans. Eculizumab (Soliris), a humanized monoclonal antibody that is a first‐in‐class terminal complement inhibitor, has been used with great success in treating paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome,132 suggesting that targeting the complement terminal pathway might be beneficial for several inflammatory diseases. Recent pre‐clinical studies have investigated the use of MAC inhibitors with great success.133
Both pathways, the CP and the inflammasome, seem to co‐operate in many acute and chronic inflammatory conditions, so future pre‐clinical studies could focus on using combination treatments.
Concluding remarks and future direction
The innate immune response pathways are an archaic and essential system of host defence that is conserved throughout species and is fundamental for our survival. Originally defined as the ‘non‐specific’ part of our immune response, it is now clear that it is highly specialized. It has microbe‐sensing capabilities with several families of PRRs that have evolved to recognize microbial signatures that are indicative of the presence of bacteria, viruses, parasites or fungi – therefore, pattern recognition seems to be the key focus of the innate immune system. In addition to microbial signatures (PAMPs), PRRs can also sense host‐derived danger signals (DAMPs) that are indicative of tissue dysfunction. Sensing of either PAMPs or DAMPs leads to the production of pro‐inflammatory cytokines that orchestrate the switch from tissue homeostasis to a state of inflammation, the goal of which is to remove the ‘offending’ trigger and restore normal function.
Prototypic families of innate immune sensors include the TLRs, RLRs, C‐type lectins, NLRs and the complement system. To achieve the specificity and diversity that are required to be able to sense such a vast array of pathogen and host‐derived triggers, the sensors employ homotypic and heterotypic associations, forming oligomeric activation structures either on the cell membrane (such as MAC) or in the cytosol (such as the inflammasome). The assembly of these structures has evolved as a cellular alarm system. These structures not only deliver the cytokine responses required to remove the trigger, but also co‐operate among themselves in the fight against infection and in the maintenance of normal tissue function.
Evidence summarized above shows that the complement‐inflammasome axis plays an integral part in the pathogenesis of acute and chronic inflammatory conditions. Although for most of the diseases mentioned above, evidence of interactions of the inflammasome with complement is currently circumstantial, it is highly likely that these interactions will prove to be a cornerstone of the pathogenesis of these diseases. It is important that in the future we focus on understanding how these multi‐component surveillance systems collaborate to sense perturbations of cellular homeostasis in the context of disease; we need to understand at which stage in each disease these interactions impact on disease progression to be able to target the relevant pathways therapeutically. As these chronic inflammatory conditions engage multiple pathways of the innate immune system, a therapeutic intervention to be successful should be aimed at fixing the aberrant surveillance system as a whole.
Disclosures
There are no competing interests.
References
- 1. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol 2010; 11:785–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hajishengallis G, Lambris JD. Microbial manipulation of receptor crosstalk in innate immunity. Nat Rev Immunol 2011; 11:187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang M, Krauss JL, Domon H, Hosur KB, Liang S, Magotti P et al Microbial hijacking of complement‐toll‐like receptor crosstalk. Sci Signal 2010; 3:ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gaboriaud C, Thielens N, Gregory L, Rossi V, Fontecilla‐Camps J, Arlaud G. Structure and activation of the C1 complex of complement: unraveling the puzzle. Trends Immunol 2004; 25:368–73. [DOI] [PubMed] [Google Scholar]
- 5. Chen C, Wallis R. Two mechanisms for mannose‐binding protein modulation of the activity of its associated serine proteases. J Biol Chem 2004; 279:26058–65. [DOI] [PubMed] [Google Scholar]
- 6. Sahu A, Kozel T, Pangburn M. Specificity of the thioester‐containing reactive site of human C3 and its significance to complement activation. Biochem J 1994; 302:429–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spilzer D, Mitchell L, Atkinson J, Hourcade D. Properdin can initiate complement activation by binding specific target surfaces and providing a platform for de novo convertase assembly. J Immunol 2007; 179:2600–8. [DOI] [PubMed] [Google Scholar]
- 8. Morgan BP. Complement membrane attack on nucleated cells: resistance, recovery and non‐lethal effects. Biochem J 1989; 264:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cole DS, Morgan BP. Beyond lysis: how complement influences cell fate. Clin Sci 2003; 104:455–66. [DOI] [PubMed] [Google Scholar]
- 10. Morgan BP, Luzio JP, Campbell AK. Intracellular Ca2+ and cell injury: a paradoxical role of Ca2+ in complement membrane attack. Cell Calcium 1986; 7:399–411. [DOI] [PubMed] [Google Scholar]
- 11. Papadimitriou JC, Phelps PC, Shin ML, Smith MW, Trump BF. Effects of Ca2+ deregulation on mitochondrial membrane potential and cell viability in nucleated cells following lytic complement attack. Cell Calcium 1994; 15:217–27. [DOI] [PubMed] [Google Scholar]
- 12. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL‐β . Mol Cell 2002; 10:417–26. [DOI] [PubMed] [Google Scholar]
- 13. Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol 2009; 27:229–65. [DOI] [PubMed] [Google Scholar]
- 14. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D et al Cutting edge: NF‐κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009; 183:787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 2011; 29:707–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Latz E, Xiao TZ, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol 2013; 13:397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stutz A, Golenbock DT, Latz E. Inflammasomes: too big to miss. J Clin Invest 2009; 119:3502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L et al Pannexin‐1‐mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll‐like receptor signaling. Immunity 2007; 26:433–43. [DOI] [PubMed] [Google Scholar]
- 19. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T et al The NALP3 inflammasome is involved in the innate immune response to amyloid‐β . Nat Immunol 2008; 9:857–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL et al Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008; 9:847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008; 320:674–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM et al Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A 2012; 109:11282–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chami M, Oules B, Paterlini‐Brechot P. Cytobiological consequences of calcium‐signaling alterations induced by human viral proteins. Biochim Biophys Acta 2006; 1763:1344–62. [DOI] [PubMed] [Google Scholar]
- 24. Schroder K, Tschopp J. The inflammasomes. Cell 2010; 140:821–32. [DOI] [PubMed] [Google Scholar]
- 25. Triantafilou K, Kar S, Vakakis E, Kotecha S, Triantafilou M. Human respiratory syncytial virus viroporin SH: a viral recognition pathway used by the host to signal inflammasome activation. Thorax 2012; 68:66–75. [DOI] [PubMed] [Google Scholar]
- 26. Triantafilou K, Triantafilou M. Ion flux in the lung: virus‐induced inflammasome activation. Trends Microbiol 2014; 22:580–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose‐Girma M et al Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006; 440:228–32. [DOI] [PubMed] [Google Scholar]
- 28. Hajishengallis G, Lambris JD. Crosstalk pathways between Toll‐like receptors and the complement system. Trends Immunol 2010; 31:154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu L, Xiao N, Liu F, Ren H, Gu J. Inhibition of RIG‐I and MDA5‐dependent antiviral response by gC1qR at mitochondria. Proc Natl Acad Sci U S A 2009; 106:1530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu H, Zhou J, Ma D, Lu X, Ming S, Shan G et al Mannan binding lectin attenuates double‐stranded RNA‐mediated TLR3 activation and innate immunity. FEBS Lett 2014; 588:866–72. [DOI] [PubMed] [Google Scholar]
- 31. Triantafilou K, Hughes TR, Triantafilou M, Morgan BP. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci 2013; 126(Pt 13):2903–13. [DOI] [PubMed] [Google Scholar]
- 32. Laudisi F, Spreafico R, Evrard M, Hughes TR, Mandriani B, Kandasamy M et al Cutting edge: the NLRP3 inflammasome links complement‐mediated inflammation and IL‐1b release. J Immunol 2013; 191:1006–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ichonohe T, Pang I, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol 2010; 11:404–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Triantafilou K, Kar S, van Kuppeveld FJ, Triantafilou M. Rhinovirus‐induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am J Respir Cell Mol Biol 2013; 49:923–34. [DOI] [PubMed] [Google Scholar]
- 35. Klos A, Tenner A, Johswich K, Ager R, Reis E, Kohl J. The role of the anaphylatoxins in health and disease. Mol Immunol 2009; 46:2753–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ember J, Jagels M, Hugli T. Characterisation of complement anaphylatoxins and their biological responses In: Volanakis J, Frank M, eds. The Human Complement System in Health and Disease. New York: Marcel Dekker Inc, 1998: 241–84. [Google Scholar]
- 37. Aksamit R, Falk W. Chemotaxis by mouse macrophage cell lines. J Immunol 1981; 126:2194–9. [PubMed] [Google Scholar]
- 38. Mastellos D, Papadimitriou J, Franchini S, Tsonis P, Lambris J. A novel role of complement: mice deficient in the fifth component of complement (C5) exhibit impaired liver regeneration. J Immunol 2001; 166:2479–86. [DOI] [PubMed] [Google Scholar]
- 39. Bokisch V, Muller‐Eberhard H. Anaphylatoxin inactivator of human plasma: its isolation and characterisation as a carboxypeptidase. J Clin Invest 1970; 49:2427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matthews K, Mueller‐Ortiz S, Wetsel R. Carboxypeptidase N: a pleiotropic regulator of inflammation. Mol Immunol 2004; 40:785–93. [DOI] [PubMed] [Google Scholar]
- 41. Sayah S, Jauneau A, Patte C, Tonon M, Vaudry H, Fontaine M. The two different transduction pathways are activated by C3a nad C5a anaphylatoxins on astrocytes. Brain Res Mol Brain Res 2003; 112:53–60. [DOI] [PubMed] [Google Scholar]
- 42. Kohl J. The anaphylatoxins and infectious and non‐infectious diseases. Mol Immunol 2001; 38:175–87. [DOI] [PubMed] [Google Scholar]
- 43. Cianflone K, Xia Z, Chen L. Critical review of acylation‐stimulating protein physiology in humans and rodents. Biochim Biophys Acta 2003; 1609:127–43. [DOI] [PubMed] [Google Scholar]
- 44. Lee D, George S, Cheng R, Nguyen T, Liu Y, Brown M et al Identification of four novel human G protein‐coupled receptors expressed in the brain. Brain Res Mol Brain Res 2001; 86:13–22. [DOI] [PubMed] [Google Scholar]
- 45. Crass T, Raffetseder U, Martin U, Grove M, Klos A, Kohl J et al Expression and cloning of the human C3a anaphylatoxin receptor (C3aR) from differentiated U‐937 cells. Eur J Immunol 1996; 26:1944–50. [DOI] [PubMed] [Google Scholar]
- 46. Norgauer J, Dobos G, Kownatzki E, Dahinden C, Burger R, Kupper R et al Complement fragment C3a stimulates Ca2+ influx in neutrophils via a pertussis‐toxin‐sensitive G protein. Eur J Immunol 1993; 217:289–94. [DOI] [PubMed] [Google Scholar]
- 47. Schraufstatter I, Trieu K, Sikora L, Sriramarao P, DiScipio R. Complement C3a and C5a induce different signal transduction cascades in endothelial cells. J Immunol 2002; 169:2102–10. [DOI] [PubMed] [Google Scholar]
- 48. Langkabel P, Zwirner J, Oppermann M. Ligand‐induced phosphorylation of anaphylatoxin receptors C3aR and C5aR is mediated by G‐protein‐coupled receptor kinases. Eur J Immunol 1999; 29:3035–46. [DOI] [PubMed] [Google Scholar]
- 49. Venkatesha R, Berla T, Zaidi A, Ali H. Distinct regulation of C3a‐induced MCP‐1/CCL2 and RANTES/CCL5 production in human mast cells by extracellular signal regulated kinase and PI3 kinase. Mol Immunol 2005; 42:581–7. [DOI] [PubMed] [Google Scholar]
- 50. Haeffner‐Cavaillon N, Cavaillon J, Laude M, Kazatchkine M. C3a (C3adesArg) induces production and release of interleukin‐1 by cultured human monocytes. J Immunol 1987; 139:794–9. [PubMed] [Google Scholar]
- 51. Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Kohl J et al C3a modulates IL‐1b secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood 2013; 122:3473–81. [DOI] [PubMed] [Google Scholar]
- 52. Skokowa J, Ali S, Felda O, Kumar V, Konrad S, Shushakova N et al Macrophages induce the inflammatory response in the pulmonary Arthus reaction through G α i2 activation that controls C5aR and Fc receptor cooperation. J Immunol 2005; 174:3050. [DOI] [PubMed] [Google Scholar]
- 53. Monk P, Partridge L. Characterisation of a complement‐fragement C5a stimulated calcium influx mechanism in U937 monocytic cells. Biochem J 1993; 295:679–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Amatruda T, Gerard N, Gerard C, Simon M. Specific interactions of chemoattractant factor receptors with G‐proteins. J Biol Chem 1993; 268:10139–44. [PubMed] [Google Scholar]
- 55. Braun L, Christophe T, Boulay F. Phosphorylation of key serine residues is required for internalization of the C5a anaphylatoxin receptor via a β‐arrestin, dynamin and clathrin‐dependent pathway. J Biol Chem 2002; 278:4277–85. [DOI] [PubMed] [Google Scholar]
- 56. Ribas C, Penela P, Murga C, Salcedo A, Garcia‐Hoz C, Jurado‐Pueyo M et al The G protein‐coupled receptor kinase (GRK) interactome: role of GRKs in GPCR regulation and signalling. Biochim Biophys Acta 2007; 1768:913–22. [DOI] [PubMed] [Google Scholar]
- 57. Jiang H, Kuang Y, Wu Y, Smrcka A, Simon MWD. Pertussis toxin‐sensitive activation of phospholipasse C by the C5a and fMet‐Leu‐Phe receptors. J Biol Chem 1996; 271:13430–4. [DOI] [PubMed] [Google Scholar]
- 58. Mullmann T, Siegel MER, Billah M. Complement C5a activation of phospholipase D in human neutrophils. A major route to the production of phosphatidates and diglycerides. J Immunol 1990; 144:1901–8. [PubMed] [Google Scholar]
- 59. Perianayagam M, Balakrishnan V, King A, Pereira B, Jaber B. C5a delays apoptosis of human neutrophils by a phosphatidylinositol 3 kinase signaling pathway. Kidney Int 2002; 61:456–63. [DOI] [PubMed] [Google Scholar]
- 60. la Sala A, Gadina M, Kelshall B. G(i)‐protein‐dependent inhibition of IL‐12 production is mediated by activation of the phosphatidylinositol 3 kinase‐protein 3 kinase B/akt pathway and JNK. J Immunol 2005; 175:2994–9. [DOI] [PubMed] [Google Scholar]
- 61. Buhl A, Avdi N, Worthen G, Johnson G. Mapping of the C5a receptor signal transduction network in human neutrophils. Proc Natl Acad Sci U S A 1994; 91:9190–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS et al Cholesterol crystals induce complement‐dependent inflammasome activation and cytokine release. J Immunol 2014; 192:2837–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. An LL, Mehta P, Xu L, Turman S, Reimer T, Naiman B et al Complement C5a potentiates uric acid crystal‐induced IL‐1b production. Eur J Immunol 2014; 44:3669–79. [DOI] [PubMed] [Google Scholar]
- 64. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG et al NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010; 464:1357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pazar B, Ea H, Narayan S, Kolly L, Bagnoud N et al Basic calcium phosphate crystals induce monocyte/macrophage IL‐1b secretion through the NLRP3 inflammasome in vitro . J Immunol 2011; 186:2495–502. [DOI] [PubMed] [Google Scholar]
- 66. Hammerschmidt DE, Greenberg CS, Yamada O, Craddock PR, Jacob HS. Cholesterol and atheroma lipis activate complement and stimulate granulocytes. A possible mechanism for amplification of ischemic injury in atherosclerotic states. J Lab Clin Med 1981; 98:68–77. [PubMed] [Google Scholar]
- 67. Seifert PS, Kazatchkline MD. Generation of complement anaphylatoxins and C5b‐9 by crystalline cholesterol oxidation derivatives depends on hydroxyl group number and position. Mol Immunol 1987; 24:1308. [DOI] [PubMed] [Google Scholar]
- 68. Vogt W, von Zabern I, Damerau B, Hesse D, Luhmann B, Nolte R. Mechanisms of complement activation by crystalline cholesterol. Mol Immunol 1985; 22:101–6. [DOI] [PubMed] [Google Scholar]
- 69. Doherty M, Whicher J, Dieppe P. Activation of the alternative pathway of complement by monosodium urate monohydrate crystals and other inflammatory particles. Ann Rheum Dis 1983; 42:285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tramontini N, Huber C, Liu‐Bryan R, Terkeltaub RA, Kilgore KS. Central role of complement membrane attack complex in monosodium urate crystal‐induced neutrophilic rabbit knee synovitis. Arthritis Rheum 2004; 50:2633–9. [DOI] [PubMed] [Google Scholar]
- 71. Korb L, Ahearn J. C1q binds directly and specifically to surface blebs of apoptotic human keartinocytes: complement deficiency and systemic lupus erythematosus revisited. J Immunol 1997; 158:4525–8. [PubMed] [Google Scholar]
- 72. Navratil J, Watkins S, Wisnieski J, Ahearn J. The globular heads of C1q specifically recognise surface blebs of apoptotic vascular endothelial cells. J Immunol 2001; 166:3231–9. [DOI] [PubMed] [Google Scholar]
- 73. Klickstein L, Barbashov S, Liu T, Jack R, Nicholson‐Weller A. Complement receptor type 1 (CR1, CD35) is a receptor for C1q. Immunity 1997; 7:345–55. [DOI] [PubMed] [Google Scholar]
- 74. Petry F, Reid B, Loos M. Gene expression of the A and B‐chain of mouse C1q in different tissues and the characterisation of the recombinant A‐chain. J Immunol 1991; 147:3988–93. [PubMed] [Google Scholar]
- 75. Castellano G, Trouw A, Fiore N, Daha M, Schena F, van Kooten C. Infiltrating dendritic cells contribute to local synthesis of C1q in murine and human lupus nephritis. Mol Immunol 2010; 47:2129–37. [DOI] [PubMed] [Google Scholar]
- 76. Fraser D, Bohlson S, Jasinskiene N, Rawal N, Palmarini G, Ruiz S et al C1q and MBL components of the innate immune system influence monocyte cytokine expression. J Leukoc Biol 2006; 80:107–16. [DOI] [PubMed] [Google Scholar]
- 77. Yamada M, Oritani K, Kaisho T, Ishikawa J, Yoshida H, Takahashi I et al Complement C1q regulates LPS‐induced cytokine production in bone marrow‐derived dendritic cells. Eur J Immunol 2004; 34:221–30. [DOI] [PubMed] [Google Scholar]
- 78. Benoit M, Clarke E, Morgado P, Fraser D, Tenner A. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J Immunol 2012; 188:5682–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Henao‐Mejia J, Elinav E, Thaiss C, Flavell RA. Inflammasomes and metabolic disease. Annu Rev Physiol 2014; 76:57–78. [DOI] [PubMed] [Google Scholar]
- 80. Poltorak A, He XL, Smirnova I, Liu MY, Van Huffel C, Du X et al Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998; 282:2085–8. [DOI] [PubMed] [Google Scholar]
- 81. Rittirsch D, Flierl MA, Nadeau BA, Day DE, Huber‐Lang M, Zetoune FS et al Functional roles for C5a receptors in sepsis. Nat Med 2008; 14:551–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang X, Kimura Y, Fang C, Zhou L, Sfyroera G, Lambris JD et al Regulation of Toll‐like receptor‐mediated inflammatory responses by complement in vivo . Blood 2007; 110:228–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sutterwala FS, Ogura Y, Szczepanik M, Lara‐Tejero M, Lichtenberger GS, Grant EP et al Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase‐1. Immunity 2006; 24:317–27. [DOI] [PubMed] [Google Scholar]
- 84. Sarkar A, Hall M, Exline M, Hart J, Knatz N, Gatson N et al Caspase‐1 regulates Escherichia coli sepsis and splenic B cell apoptosis independently of interleukin‐1β and interleukin 18. Am J Resp Crit Care Med 2006; 174:1003–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Mariathasan S, Newton K, Monack DVD, French D, Lee W, Roose‐Girma M et al Differential activation of the inflammasome by caspase‐1 adaptors ASC and Ipaf. Nature 2004; 430:213–8. [DOI] [PubMed] [Google Scholar]
- 86. Kim H, Lee D, Ravichandran K, Keys D, Akcay A, Nguyen Q et al NLRP3 inflammasome knockout mice are protected against ischemic but not cisplatin‐induced acute kidney injury. J Pharmacol Exp Ther 2013; 346:465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Jones H, Crother T, Gonzalez‐Villalobos R, Jupelli M, Chen S, Dagvadorj J et al The NLRP3 inflammasome is required for the development of hypoxemia in LPS/mechanical ventilation acute lung injury. Am J Respir Crit Care Med 2014; 50:270–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. McCullough J, Renner B, Thurman J. The role of the complement system in acute kidney injury. Semin Nephrol 2013; 33:543–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hu R, Chen Z, Yan J, Li Q, Huang Y, Xu H et al Complement C5a exacerbates acute lung injury induced through autophagy‐mediated alveolar macrophage apoptosis. Cell Death Dis 2014; 5:e1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bunout D, Munoz C, Lopez M, de la Maza M, Schlesinger L et al Interleukin 1 and tumour necrosis factor in obese alcoholics compared with normal weight patients. Am J Clin Nutr 1996; 63:373–6. [DOI] [PubMed] [Google Scholar]
- 91. Pickup J, Crook M. Is type II diabetes mellitus a disease of the innate immune system? Diabetologia 1998; 41:1241–8. [DOI] [PubMed] [Google Scholar]
- 92. Hotamisligil G, Shargill N, Spiegelman B. Adipose expression of tumour necrosis factor‐α: direct role in obesity‐linked insulin resistance. Science 1993; 259:87–91. [DOI] [PubMed] [Google Scholar]
- 93. Samuel V, Shulman G. Mechanisms of insulin resistance: common threads and missing links. Cell 2012; 148:852–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Konner A, Bruning J. Toll‐like receptors: linking inflammation and metabolism. Trends Endocrinol Metab 2011; 22:16–23. [DOI] [PubMed] [Google Scholar]
- 95. Vandanmagsar B, Youm Y, Ravussin A, Galgani J, Stadler K et al The NLRP3 inflammasome instigates obesity‐induced inflammation and insulin resistance. Nat Med 2011; 17:179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Stienstra R, Joosten L, Koenen T, van Tits B, van Diepen J et al The inflammasome‐mediated caspase‐1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab 2010; 12:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Kalant D, Cain S, Maslowska M, Sniderman A, Cianflone K, Monk P. The chemoattractant receptor‐like protein C5L2 binds the C3ades‐Arg77/Acylation‐stimulating protein. J Biol Chem 2003; 278:11123–9. [DOI] [PubMed] [Google Scholar]
- 98. Kalant D, MacLaren R, Cui W, Samanta R, Monk P, Laporte S et al C5L2 is a functional receptor for acylation‐stimulating protein. J Biol Chem 2005; 280:23936–44. [DOI] [PubMed] [Google Scholar]
- 99. Chen N, Mirtsos C, Suh D, Lu Y, Lin W, McKerlie C et al C5L2 is critical for the biological activities of the anaphylatoxins C5a and C3a. Nature 2007; 446:203–7. [DOI] [PubMed] [Google Scholar]
- 100. Raby AC, Holst B, Davies J, Colmont C, Laumonnier Y, Coles B et al TLR activation enhances C5a‐induced pro‐inflammatory responses by negatively modulating the second C5a receptor, C5L2. Eur J Immunol 2011; 41:2741–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Phieler J, Chung K, Chatzigeorgiou A, Klotzsche‐von Ameln A, Garcia‐Martin R, Sprott D et al The complement anaphylatoxin C5a receptor contributes to obese adipose tissue inflammation and insulin resistance. J Immunol 2013; 191:4367–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lewis RD, Jackson CL, Morgan BP, Hughes TR. The membrane attack complex of complement drives the progression of atherosclerosis in apolipoprotein E knockout mice. Mol Immunol 2010; 47:1098–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yun S, Leung WY, Botto M, Boyle JJ, Haskard DO. Accelerated atherosclerosis in low‐density lipoprotein receptor‐deficient mice lacking the membrane‐bound complement regulator CD59. Atherioscler Thromb Vasc Biol 2008; 28:1714–16. [DOI] [PubMed] [Google Scholar]
- 104. Wu G, Hu W, Shahsafaei A, Song W, Dobarro M, Sukhova GK et al Complement regulator CD59 protects against atherosclerosis by restricting the formation of complement membrane attack complex. Circ Res 2009; 104:550–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Weiner HL, Frenkel D. Immunology and immunotherapy of Alzheimer's disease. Nat Rev Immunol 2006; 6:404–16. [DOI] [PubMed] [Google Scholar]
- 106. Meyer‐Luehmann M, Spires‐Jones TL, Prada C, Garcia‐Alloza M, de Calignon A, Rozkalne A et al Rapid appearance and local toxicity of amyloid‐β plaques in a mouse model of Alzheimer's disease. Nature 2008; 451:720–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM et al Inflammation and Alzheimer's disease. Neurobiol Aging 2000; 21:383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. McGeer PL, Rogers J, McGeer EG. Inflammation, anti‐inflammatory agents and Alzheimer disease: the last 12 years. J Alzheimers Dis 2006; 9:271–6. [DOI] [PubMed] [Google Scholar]
- 109. Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W et al Role of the toll‐like receptor 4 in neuroinflammation in Alzheimer's disease. Cell Physiol Biochem 2007; 20:947–56. [DOI] [PubMed] [Google Scholar]
- 110. Daly J, Kotwal GJ. Pro‐inflammatory complement activation by the A β peptide of Alzheimer's disease is biologically significant and can be blocked by vaccinia virus complement control protein. Neurobiol Aging 1998; 19:619–27. [DOI] [PubMed] [Google Scholar]
- 111. Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM et al Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer's disease. J Immunol 2009; 183:1375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. World Health Organization . Atlas Multiple Sclerosis Resources in the World. Geneva, Switzerland: World Health Organization Press, 2008. [Google Scholar]
- 113. Furlan R, Filippi A, Bergami A, Rocca MA, Martinelli V, Poliani PL et al Peripheral levels of caspase‐1 mRNA correlate with disease activity in patients with multiple sclerosis: a preliminary study. J Neurol Neurosurg Psychiatr 1999; 67:785–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ming X, Li W, Maeda Y et al Caspase‐1 expression in multiple sclerosis plaques and cultured glial cells. J Neurol Sci 2002; 197:9–18. [DOI] [PubMed] [Google Scholar]
- 115. Huang WX, Huang P, Hillert J. Increased expression of caspase‐1 and interleukin 18 in peripheral blood mononuclear cells in patients with multiple sclerosis. Mult Scler 2004; 10:482–7. [DOI] [PubMed] [Google Scholar]
- 116. Lumsden CE. The immunogenesis of the multiple sclerosis plaque. Brain Res 1971; 28:365–90. [DOI] [PubMed] [Google Scholar]
- 117. Compston DA, Morgan BP, Campbell AK, Wilkins P, Cole G, Thomas ND et al Immunocytochemical localization of the terminal complement complex in multiple sclerosis. Neuropathol Appl Neurobiol 1989; 15:307–16. [DOI] [PubMed] [Google Scholar]
- 118. Ingram G, Loveless S, Howell O, Hakobyan S, Dancey B, Harris C et al Complement activation in multiple sclerosis plaques: an immunohistochemical analysis. Acta Neuropathol Commun 2014; 2:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lennon V, Wingerchuk D, Kryzer T, Pittock S, Lucchinetti C, Fujihara K et al A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004; 364:2106–12. [DOI] [PubMed] [Google Scholar]
- 120. Phuan P, Ratelade J, Rossi A, Tradtrantip L, Verkman A. Complement‐dependent cytotoxicity in neuromyelitis optica requires aquaporin‐4 protein assembly in orthogonal arrays. J Biol Chem 2015; 287:13829–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kawana N, Yamamoto Y, Ishida T, Saito Y, Konno H, Arima K et al Reactive astrocytes and perivascular macrophages express NLRP3 inflammasome in active demyelinating lesions of multiple sclerosis and necrotic lesions of neuromyelitis optica and cerebral infarction. Clin Exp Neuroimmunol 2013; 4:296–304. [Google Scholar]
- 122. Rabolli V, Walleme L, Lo Re S, Uwambayinema F, Palmai‐Pallag M, Thomassen L et al Critical role of aquaporins in interleukin 1β (IL‐1β)‐induced inflammation. J Biol Chem 2014; 289:13937–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Arulkumaran N, Unwin R, Tam F. A potential therapeutic role for P2X7 receptor (P2X7R) antagonists in the treatment of inflammatory diseases. Expert Opin Investig Drugs 2011; 20:897–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Cornelis S, Kersse K, Festjens N, Lamkanfi M, Vandenabeele P. Inflammatory caspases: targets for novel therapies. Curr Pharm Des 2007; 13:367–85. [DOI] [PubMed] [Google Scholar]
- 125. Nuki G, Bresnihan B, Bear M, McCabe D. Long‐term safety and maintenance of clinical improvement following treatment with anakinra (recombinant human interleukin 1 receptor antagonist) in patients with rheumatoid arthris: extension phase of a randomised, double‐blind, placebo‐controlled trial. Arthritis Rheum 2002; 46:2838–46. [DOI] [PubMed] [Google Scholar]
- 126. Molad Y. Update on colchicine and its mechanism of action. Curr Rheumatol Rep 2002; 4:252–6. [DOI] [PubMed] [Google Scholar]
- 127. Coll R, Robertson A, Chae J, Higgins S, Munoz‐Planillo R, Inserra M et al A small molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 2015; 21:248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Youm Y, Nguyen K, Grant R, Goldberg E, Bodogai M, Kim D et al The ketone metabolite β‐hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat Med 2015; 21:263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Yan Y, Jiang W, Wang X, Ding C, Tian Z, Zhou R. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015; 160:62–73. [DOI] [PubMed] [Google Scholar]
- 130. Qu H, Ricklin D, Lambris JD. Recent developments in low molecular weight complement inhibitors. Mol Immunol 2009; 47:185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Ricklin D, Lambris JD. Compstatin: a complement inhibitor on its way to clinical application. Adv Exp Med Biol 2008; 632:273–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Schrezenmeier H, Hochsmann B. Eculizumab opens a new era of treatment for paroxysmal nocturnal hemoglobinuria. Expert Rev Hematol 2009; 2:7–16. [DOI] [PubMed] [Google Scholar]
- 133. Lee M, Narayanan S, McGeer E, McGeer P. Aurin tricarboxylic acid protects against red blood cell hemolysis in patients with paroxysmal nocturnal hemoglobinemia. PLoS ONE 2014; 9:e87316. [DOI] [PMC free article] [PubMed] [Google Scholar]