

Summary
The physiological role of autophagic flux within the vascular endothelial layer remains poorly understood. Here, we show that in primary endothelial cells, oxidized and native LDL stimulates autophagosome formation. Moreover, by both confocal and electron microscopy, excess native or modified LDL appears to be engulfed within autophagic structures. Transient knockdown of the essential autophagy gene ATG7 resulted in higher levels of intracellular 125I‐LDL and oxidized LDL (OxLDL) accumulation, suggesting that in endothelial cells, autophagy may represent an important mechanism to regulate excess, exogenous lipids. The physiological importance of these observations was assessed using mice containing a conditional deletion of ATG7 within the endothelium. Following acute intravenous infusion of fluorescently labeled OxLDL, mice lacking endothelial expression of ATG7 demonstrated prolonged retention of OxLDL within the retinal pigment epithelium (RPE) and choroidal endothelium of the eye. In a chronic model of lipid excess, we analyzed atherosclerotic burden in ApoE−/−mice with or without endothelial autophagic flux. The absence of endothelial autophagy markedly increased atherosclerotic burden. Thus, in both an acute and chronic in vivo model, endothelial autophagy appears critically important in limiting lipid accumulation within the vessel wall. As such, strategies that stimulate autophagy, or prevent the age‐dependent decline in autophagic flux, might be particularly beneficial in treating atherosclerotic vascular disease.
Keywords: autophagy, lipids, atherosclerosis, mouse
Cardiovascular disease, driven in part by the accumulation of modified lipids within the vessel wall, represents the leading cause of death in developed nations (Go et al., 2013). Considerable attention and study has been directed at the molecular consequences following the subendothelial deposition of lipids. These consequences include the recruitment of inflammatory cells and the subsequent engulfment of this deposited subendothelial lipid by resident macrophages (Chinetti‐Gbaguidi et al., 2014; Randolph, 2014). In contrast, relatively little is known regarding the steps proximal to lipid deposition and what regulatory role, if any, the endothelium might play in this process.
While evidence is limited for the case of the endothelium, in other cell types it appears that autophagy may play a critical role in maintaining overall lipid homeostasis. For instance, mice bearing ATG5‐deficient macrophages appear to exhibit increased plaque formation when bred with pro‐atherogenic mouse strains (Liao et al., 2012; Razani et al., 2012). While the basis for this increased plaque burden is undoubtedly complex, evidence suggests that macrophage‐specific ATG5 deletion results in impaired lipophagy (Sergin & Razani, 2014). Lipophagy refers to the specific degradation of lipids by the autophagic machinery. This concept was perhaps first described in the liver where genetic disruption of macroautophagy led to the accumulation of lipid droplets (Singh et al., 2009). These initial observations have been extended and lipophagy now appears to be important in maintaining lipid homeostasis in a diverse set of cell types from neurons to fibroblasts (Singh et al., 2009; Liu & Czaja, 2013; Settembre & Ballabio, 2014). Here, we demonstrate an important role for endothelial autophagy in maintaining vascular lipid homeostasis.
It has been previously observed that when endothelial cells in culture are exposed to OxLDL they respond with an increase in autophagosomes (Nowicki et al., 2007; Zhang et al., 2010; Muller et al., 2011; Menghini et al., 2014). In general, autophagic flux can be assessed by simultaneously analyzing the levels of LC3‐II, which labels autophagosome membranes, and measuring the levels of p62, a protein cleared via autophagic degradation. The combination of increased LC3‐II along with decreased p62 levels is consistent with an overall increase in autophagic flux. Consistent with past observations, following exposure of human umbilical vein endothelial cells (HUVECs) to OxLDL, the level of LC3‐I to LC3‐II conversion was increased (Fig. 1A,B). As we have previously described (Torisu et al., 2013), siRNA ‐mediated knockdown of ATG7 was effective in reducing ATG7 expression in HUVECs (Fig. S1A,B, Supporting information), and endothelial cells with reduced ATG7 expression had markedly impaired capacity to generate LC3‐II before or after OxLDL exposure (Fig. 1A,B). The level of LC3‐II following OxLDL exposure was further enhanced by the presence of the lysosomal inhibitor chloroquine (Fig. 1C). In contrast, levels of p62 were not as markedly affected after OxLDL exposure (Fig. 1A and Fig. S1C) raising the possibility, as previously noted, of alteration in lysosomal function after OxLDL exposure (Jerome, 2010). When primary endothelial cells were electroporated with a plasmid encoding GFP‐LC3 and subsequently exposed to fluorescently labeled OxLDL, we noted an increase in LC3 dot formation, again consistent with an increase in autophagosomes (Fig. S1D,E). Interestingly, the OxLDL within endothelial cells was often surrounded by GFP‐LC3 positive structures whose appearance was consistent with autophagosomes (Fig. 1D). These observations suggested that following uptake of OxLDL, lipids may be directly or indirectly trafficked into autophagosomes. Moreover, given the growing evidence that intracellular lipids can be degraded by lysosomal lipases (Singh & Cuervo, 2012; Liu & Czaja, 2013; Settembre & Ballabio, 2014), intact endothelial autophagy might modulate levels of OxLDL accumulation. Consistent with this hypothesis, OxLDL accumulated at higher levels in endothelial cells lacking an intact autophagy machinery (Fig. 1E).
Figure 1.
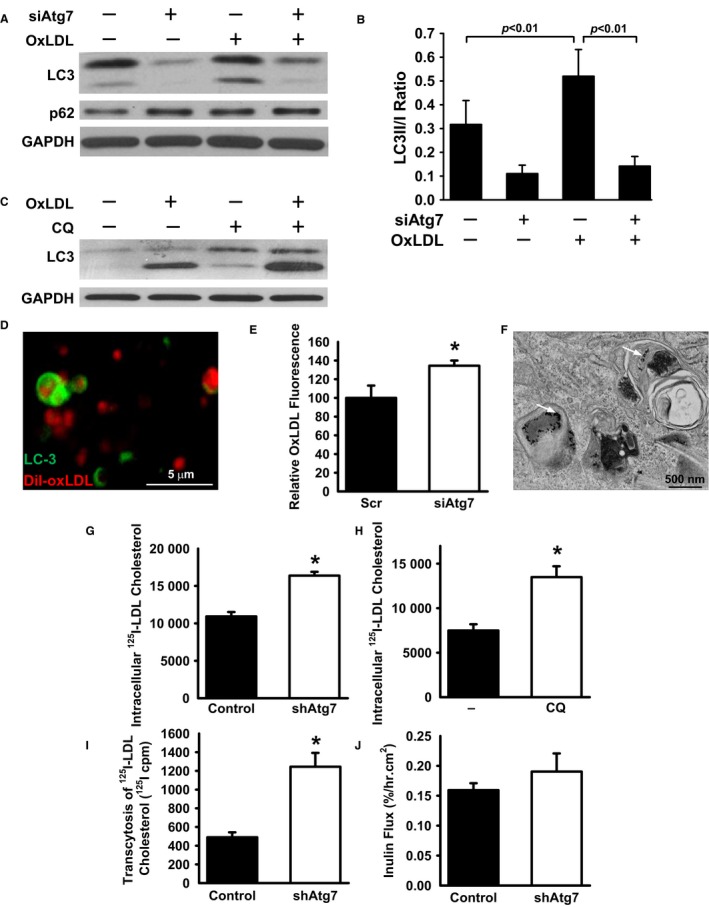
Role of autophagy in endothelial lipid homeostasis. (A) Western blot analysis of HUVECs exposed to OxLDL (50 μg mL−1 for 24 h) that were knocked down with either a scrambled RNAi (−) or an RNAi targeting the essential autophagy gene ATG7 (siAtg7). Shown are levels of LC3, where the top band represents LC3‐I and the bottom band LC3‐II, p62, and GAPDH as a loading control. (B) Quantification of the LC3‐II/LC3‐I ratio as a marker of autophagosome formation (n = 4–6 blots per group; one‐way ANOVA followed by Bonferroni post hoc test). (C) Levels of LC3‐I and LC3‐II in HUVECs in the presence or absence of OxLDL and the lysosomal inhibitor chloroquine (CQ). (D) Confocal image of HUVECs transfected with a GFP‐LC3 plasmid (green) and exposed to fluorescently labeled OxLDL (red). The lipid, in some cases, appears to be surrounded by circular LC‐3‐coated structures, consistent with an autophagosome. (E) Accumulation of OxLDL in endothelial cells transfected with a scrambled siRNA or one targeting ATG7 (n = 5 per condition, *P < 0.05). (F) Electron micrographs of HUVECs incubated with native LDL that had been coupled to gold beads. Micrographs demonstrate gold beads within autophagosomes (white arrows). (G) Intracellular‐labeled 125I‐LDL accumulation in control knocked down cells or in cells knocked down for ATG7. (n = 3, *P < 0.01 by two‐tailed unpaired t‐test with Welch's correction, representative of four similar independent experiments). (H) Intracellular 125I‐LDL accumulation in un‐infected HUVECs following treatment with chloroquine (CQ; n = 4, *P < 0.01 by two‐tailed unpaired t‐test with Welch's correction, representative of three similar independent experiments). (I) Level of in vitro transcytosis of labeled 125I‐LDL across HUVEC monolayer cultures following control or ATG7 knockdown. (n = 3, *P < 0.01 by two‐tailed unpaired t‐test with Welch's correction, representative of four similar independent experiments. (J) Levels of fluorescently tagged inulin flux through HUVEC monolayer cultures. (n = 3, P = NS).
Native LDL is thought to be taken up by receptor‐mediated endocytosis and delivered to lysosomes through the endosomal pathway. It has been appreciated for some time that genetic deficiencies in lysosomal lipase impairs cholesterol hydrolysis leading to intracellular lipid accumulation (Goldstein et al., 1975). Increasing evidence also suggests that endosomal and autophagosomal pathways are closely connected and share common machinery (Rusten et al., 2012; Hyttinen et al., 2013). Electron micrographs of HUVECs incubated with gold‐labeled native LDL identified gold‐labeled particles within double membrane autophagosomal structures (Fig. 1F). Similar to what we observed following OxLDL treatment, native LDL also appeared to increase the number of autophagosomes (Fig. S1F,G) and to be in close contact with GFP‐LC3 positive structures (Fig. S1H). As we have previously described, we next used lentivirus‐mediated shRNAs to stably knockdown ATG7 expression (Torisu et al., 2013). We noted that 125I‐LDL cholesterol accumulated to a higher level in cells with reduced Atg7 (Fig. 1G). Increased 125I‐LDL cholesterol accumulation was also observed following treatment of endothelial cells with the autophagy and lysosomal inhibitor chloroquine (Fig. 1H). This increase was not due to any apparent alterations in levels of the LDL receptor of surface binding of 125I‐LDL cholesterol (Fig. S1I,J).
The deposition of native or modified lipids in the sub‐endothelial space is thought to contribute to vascular disease. To begin to model this phenomenon in vitro, we analyzed the transcytosis of 125I‐LDL cholesterol in confluent monolayers of HUVECs following control or ATG7 knockdown. As noted in Fig. 1I, following a 24‐h incubation, there was evidence for increased 125I‐LDL transcytosis across ATG7‐deficient endothelial monolayers. In contrast, Atg7 knockdown did not affect the permeability to inulin (Fig. 1J). The differences in transcytosis rates may simply reflect higher rates of accumulation of 125I‐LDL cholesterol in ATG7 knockdown cells (Fig. 1G) or perhaps may reflect recent observations directly linking autophagy to barrier function (Nighot et al., 2015).
While a number of sophisticated in vitro assays have been developed for assessing endothelial barrier function (Wegener & Seebach, 2014), we sought to assess a potentially more physiologically relevant model by analyzing mice with a conditional endothelial deletion of ATG7 (Torisu et al., 2013). We took advantage of the rich vascular plexus in the eye and the previously described transport of lipids from the choroidal blood vessels to the retinal pigment epithelium (RPE) (Picard et al., 2010). To assess whether the acute handling of modified lipids depends on intact endothelial autophagy, we injected control mice (WT/WT; VE‐Cadherin Cre) or Atg7endo mice (fl/fl;VE‐Cadherin Cre) mice with fluorescently labeled OxLDL (dil‐OxLDL).We next analyzed the retinas of mice 24 and 48 h after OxLDL injection by confocal microscopy. Deposits of dil‐OxLDL were evident in the mouse retinal pigment epithelium (RPE) and the choroidal endothelium beneath the RPE. Twenty‐four hours post injection, dil‐OxLDL uptake in the mouse eye was similar in Atg7 endo and control mice (data not shown). However, 48 h after dil‐OxLDL administration, the fluorescent tagged dil‐OxLDL was retained at higher levels within the RPE and choroidal endothelium of the Atg7 endo mice than in wild‐type tissue (Fig. 2A,B). Quantification of dil‐OxLDL fluorescence confirmed the increased persistence of dil‐OxLDL in Atg7 endo mice (Fig. 2C, n = 6 eyes per group, P < 0.0001 by two‐tailed t‐test). Thus, in this acute model of modified LDL exposure, autophagy‐deficient mice demonstrate increased and persistent lipid accumulation.
Figure 2.
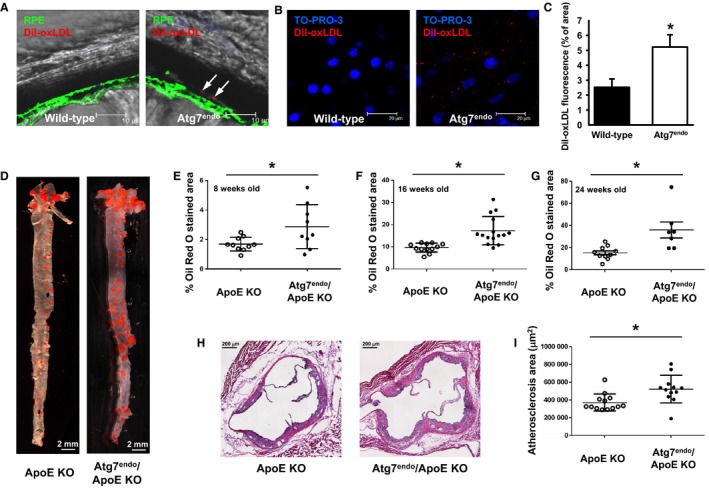
Autophagy regulates in vivo vascular lipid deposition. (A) Deposition of fluorescently labeled OxLDL in the retina of control (WT/WT; VE‐cadherin Cre) or Atg7endo mice 48 h after injection. Arrows represent retained diI‐OxLDL particles deposited sub‐RPE, at the level of the RPE basal membrane. (B) Representative whole mount images from Atg7endo mice demonstrating accumulation of fluorescent particles (presumptive vesicles) within binucleated RPE cells and in the adjacent extracellular matrix and endothelium of the choriocapillaris. (C) Quantification of labeled OxLDL in the retina of control or Atg7endo mice (n = 6 eyes per group) 48 h after infusion, *P < 0.05 by two‐tailed unpaired t‐test. (D) Representative Oil Red O stained aortas from control (WT/WT VE‐cadherin Cre/ApoE−/− abbreviated as ApoE KO) or Atg7endo/ApoE KO (fl/fl VE‐Cadherin Cre/ApoE−/−) mice. (E) Quantification of Oil Red O staining at 8 weeks (n = 10 ApoE KO and n = 9 Atg7endo/ApoE KO mice, *P < 0.05 two‐tailed unpaired t‐test with Welch's correction). (F) 16 weeks (n = 12 ApoE KO and n = 15 Atg7endoApoE KO mice, *P < 0.01 by two‐tailed unpaired t‐test with Welch's correction) and (G) 24 weeks of age (n = 10 ApoE KO and n = 7 Atg7endo/ApoE KO mice, *P < 0.03 by two‐tailed unpaired t‐test with Welch's correction). (H) Micrographs of the aortic root at 16 weeks of age. (I) Quantification of plaque area at 16 weeks between the two genotypes (n = 13 ApoE KO and n = 12 Atg7endo/ApoEKO mice, *P < 0.01 by two‐tailed unpaired t‐test with Welch's correction).
We next sought to assess a more chronic model of lipid exposure by crossing our control and Atg7endo mice into an ApoE−/− background. The presence or absence of endothelial ATG7 did not significantly alter serum lipid or glucose levels, nor did it alter overall body weight or body composition (Table S1, Supporting information). In contrast, consistent with altered lipid handling, we observed increased en face Oil Red O‐positive staining in high‐fat‐fed ApoE−/−Atg7endo mice (Fig. 2D). These differences were maintained when mice were analyzed at 8 weeks of age (e.g., 4 weeks of high‐fat diet), 16 weeks of age (12 weeks of high‐fat diet) or 24 weeks of age (20 weeks of high‐fat diet; Fig. 2E–G). A similar analysis of cross‐sectional plaque area in the aortic root was also consistent with the observation that high‐fat‐fed ApoE−/−Atg7endo mice had increased atherosclerotic lesion size when compared with control animals (Fig. 2H,I). Preliminary analysis of the plaque composition revealed a trend for increased necrotic area in the ApoE−/−Atg7endo mice (Fig. S2, Supporting information).
Taken together, our results demonstrate an important role for endothelial autophagy in maintaining vascular lipid homeostasis. Further work is needed to better define how autophagy modulates the intracellular fate of engulfed lipids (Fig. S1K). Nonetheless, the ability of endothelial cells to target excessive native or modified lipids for autophagy‐dependent degradation appears to be an important mechanism to limit atherosclerotic plaque formation. Our in vitro results suggest that knockdown of ATG7 or treatment with chloroquine can increase endothelial lipid accumulation. Given that chloroquine works as an inhibitor of lysosomal processes, the most likely explanation is that similar to what has been described in other tissues; endothelial cells rely, at least in part, on autophagosomal‐mediated delivery of lipids to the lysosome for degradation. In this context, it may be relevant that the gene lysosomal acid lipase A (LIPA) has been recently identified in genome‐wide studies as a potential susceptibility locus for atherosclerotic disease (Wild et al., 2011; Vargas‐Alarcon et al., 2013). In addition, it is of potential interest that a number of genes associated with lipid metabolism have also been linked to age‐related retinal diseases (Black & Clark, 2015).
Given that autophagic flux is believed to decline as a function of age (Cuervo, 2008), the age‐dependent decline in endothelial autophagy might contribute to the sharp rise in age‐related cardiovascular disease. Interestingly, pharmacological efforts that augment autophagy appear to reverse some of the properties of mouse and human arterial aging (LaRocca et al., 2012). Previous studies have also suggested that the atherosclerotic plaque is enriched for autophagosomes (Martinet & De Meyer, 2009). In addition, recent mouse genetic studies have implicated the role of autophagy in macrophage foam cell formation (Muller et al., 2011; Ouimet et al., 2011; Le Guezennec et al., 2012; Razani et al., 2012; Sergin & Razani, 2014). Our results complement these observations and suggest that similar to what was observed in macrophages, endothelial autophagy is important in limiting atherosclerotic progression. It is tempting to speculate that enhancing autophagy may therefore be a beneficial strategy to reduce the rate of age‐dependent cardiovascular disease. Such speculation is supported by previous observations including that inhibition of mTOR (a kinase known to function as a negative regulator of autophagy) with pharmacological agents such as rapamaycin appears to inhibit atherosclerosis in a number of different animal models (Castro et al., 2004; Pakala et al., 2005; Mueller et al., 2008). Similarly, strategies such as calorie restriction, that are known to elevate autophagic flux, appear to reduce cardiovascular disease in both mice and humans (Guo et al., 2002; Fontana et al., 2004). Manipulation of vascular autophagy might therefore be an attractive therapeutic target to potentially limit atherosclerotic disease independent of serum lipid values.
Supporting information
Fig. S1. Characterization of the role of autophagy in endothelial lipid homeostasis.
Fig. S2. The role of autophagy in ApoE KO mouse atherosclerotic plaque formation.
Table S1. Serum chemistry and body weight.
Data S1. Experimental procedures.
Acknowledgments
We are grateful to Phillip W. Connelly and Graham F. Maguire (Keenan Research Centre for Biomedical Sciences, St. Michael's Hospital) for the measurement of mouse serum triglycerides, HDL, LDL and VLDL cholesterol. This work was supported by NIH Intramural Funds and the Leducq Foundation (T.F) and the Heart and Stroke Foundation of Canada and the Canadian Institutes of Health Research (S.V). T.T was a recipient of a Japan Society for the Promotion of Science Research Fellowship in Biomedical and Behavioral Research at the NIH.
References
- Black JR, Clark SJ (2015) Age‐related macular degeneration: genome‐wide association studies to translation. Genet. Med. DOI 10.1038/gim.2015.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro C, Campistol JM, Sancho D, Sanchez‐Madrid F, Casals E, Andres V (2004) Rapamycin attenuates atherosclerosis induced by dietary cholesterol in apolipoprotein‐deficient mice through a p27 Kip1 ‐independent pathway. Atherosclerosis 172, 31–38. [DOI] [PubMed] [Google Scholar]
- Chinetti‐Gbaguidi G, Colin S, Staels B (2014) Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. DOI 10.1038/nrcardio.2014.173. [DOI] [PubMed] [Google Scholar]
- Cuervo AM (2008) Autophagy and aging: keeping that old broom working. Trends Genet. 24, 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Meyer TE, Klein S, Holloszy JO (2004) Long‐term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc. Natl Acad. Sci. USA 101, 6659–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB (2013) Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation 127, e6–e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS (1975) Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J. Biol. Chem. 250, 8487–8495. [PubMed] [Google Scholar]
- Guo Z, Mitchell‐Raymundo F, Yang H, Ikeno Y, Nelson J, Diaz V, Richardson A, Reddick R (2002) Dietary restriction reduces atherosclerosis and oxidative stress in the aorta of apolipoprotein E‐deficient mice. Mech. Ageing Dev. 123, 1121–1131. [DOI] [PubMed] [Google Scholar]
- Hyttinen JM, Niittykoski M, Salminen A, Kaarniranta K (2013) Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim. Biophys. Acta 1833, 503–510. [DOI] [PubMed] [Google Scholar]
- Jerome WG (2010) Lysosomes, cholesterol and atherosclerosis. Clin. Lipidol. 5, 853–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRocca TJ, Henson GD, Thorburn A, Sindler AL, Pierce GL, Seals DR (2012) Translational evidence that impaired autophagy contributes to arterial ageing. J. Physiol. 590, 3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guezennec X, Brichkina A, Huang YF, Kostromina E, Han W, Bulavin DV (2012) Wip1‐dependent regulation of autophagy, obesity, and atherosclerosis. Cell Metab. 16, 68–80. [DOI] [PubMed] [Google Scholar]
- Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I (2012) Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 15, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Czaja MJ (2013) Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 20, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinet W, De Meyer GR (2009) Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ. Res. 104, 304–317. [DOI] [PubMed] [Google Scholar]
- Menghini R, Casagrande V, Marino A, Marchetti V, Cardellini M, Stoehr R, Rizza S, Martelli E, Greco S, Mauriello A, Ippoliti A, Martelli F, Lauro R, Federici M (2014) MiR‐216a: a link between endothelial dysfunction and autophagy. Cell Death Dis. 5, e1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller MA, Beutner F, Teupser D, Ceglarek U, Thiery J (2008) Prevention of atherosclerosis by the mTOR inhibitor everolimus in LDLR−/− mice despite severe hypercholesterolemia. Atherosclerosis 198, 39–48. [DOI] [PubMed] [Google Scholar]
- Muller C, Salvayre R, Negre‐Salvayre A, Vindis C (2011) HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ. 18, 817–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nighot PK, Hu CA, Ma TY (2015) Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin‐2 protein degradation. J. Biol. Chem. 290, 7234–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicki M, Zabirnyk O, Duerrschmidt N, Borlak J, Spanel‐Borowski K (2007) No upregulation of lectin‐like oxidized low‐density lipoprotein receptor‐1 in serum‐deprived EA.hy926 endothelial cells under oxLDL exposure, but increase in autophagy. Eur. J. Cell Biol. 86, 605–616. [DOI] [PubMed] [Google Scholar]
- Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL (2011) Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 13, 655–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakala R, Stabile E, Jang GJ, Clavijo L, Waksman R (2005) Rapamycin attenuates atherosclerotic plaque progression in apolipoprotein E knockout mice: inhibitory effect on monocyte chemotaxis. J. Cardiovasc. Pharmacol. 46, 481–486. [DOI] [PubMed] [Google Scholar]
- Picard E, Houssier M, Bujold K, Sapieha P, Lubell W, Dorfman A, Racine J, Hardy P, Febbraio M, Lachapelle P, Ong H, Sennlaub F, Chemtob S (2010) CD36 plays an important role in the clearance of oxLDL and associated age‐dependent sub‐retinal deposits. Aging (Albany NY) 2, 981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randolph GJ (2014) Mechanisms that regulate macrophage burden in atherosclerosis. Circ. Res. 114, 1757–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF (2012) Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 15, 534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusten TE, Vaccari T, Stenmark H (2012) Shaping development with ESCRTs. Nat. Cell Biol. 14, 38–45. [DOI] [PubMed] [Google Scholar]
- Sergin I, Razani B (2014) Self‐eating in the plaque: what macrophage autophagy reveals about atherosclerosis. Trends Endocrinol. Metab. 25, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Ballabio A (2014) Lysosome: regulator of lipid degradation pathways. Trends Cell Biol. DOI 10.1016/j.tcb.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Cuervo AM (2012) Lipophagy: connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012, 282041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ (2009) Autophagy regulates lipid metabolism. Nature 458, 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torisu T, Torisu K, Lee IH, Liu J, Malide D, Combs CA, Wu XS, Rovira II, Fergusson MM, Weigert R, Connelly PS, Daniels MP, Komatsu M, Cao L, Finkel T (2013) Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat. Med. 19, 1281–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas‐Alarcon G, Posadas‐Romero C, Villarreal‐Molina T, Alvarez‐Leon E, Angeles J, Vallejo M, Posadas‐Sanchez R, Cardoso G, Medina‐Urrutia A, Kimura‐Hayama E (2013) Single nucleotide polymorphisms within LIPA (Lysosomal Acid Lipase A) gene are associated with susceptibility to premature coronary artery disease. a replication in the genetic of atherosclerotic disease (GEA) Mexican study. PLoS ONE 8, e74703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener J, Seebach J (2014) Experimental tools to monitor the dynamics of endothelial barrier function: a survey of in vitro approaches. Cell Tissue Res. 355, 485–514. [DOI] [PubMed] [Google Scholar]
- Wild PS, Zeller T, Schillert A, Szymczak S, Sinning CR, Deiseroth A, Schnabel RB, Lubos E, Keller T, Eleftheriadis MS, Bickel C, Rupprecht HJ, Wilde S, Rossmann H, Diemert P, Cupples LA, Perret C, Erdmann J, Stark K, Kleber ME, Epstein SE, Voight BF, Kuulasmaa K, Li M, Schafer AS, Klopp N, Braund PS, Sager HB, Demissie S, Proust C, Konig IR, Wichmann HE, Reinhard W, Hoffmann MM, Virtamo J, Burnett MS, Siscovick D, Wiklund PG, Qu L, El Mokthari NE, Thompson JR, Peters A, Smith AV, Yon E, Baumert J, Hengstenberg C, Marz W, Amouyel P, Devaney J, Schwartz SM, Saarela O, Mehta NN, Rubin D, Silander K, Hall AS, Ferrieres J, Harris TB, Melander O, Kee F, Hakonarson H, Schrezenmeir J, Gudnason V, Elosua R, Arveiler D, Evans A, Rader DJ, Illig T, Schreiber S, Bis JC, Altshuler D, Kavousi M, Witteman JC, Uitterlinden AG, Hofman A, Folsom AR, Barbalic M, Boerwinkle E, Kathiresan S, Reilly MP, O'Donnell CJ, Samani NJ, Schunkert H, Cambien F, Lackner KJ, Tiret L, Salomaa V, Munzel T, Ziegler A, Blankenberg S (2011) A genome‐wide association study identifies LIPA as a susceptibility gene for coronary artery disease. Circ. Cardiovasc. Genet. 4, 403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YL, Cao YJ, Zhang X, Liu HH, Tong T, Xiao GD, Yang YP, Liu CF (2010) The autophagy‐lysosome pathway: a novel mechanism involved in the processing of oxidized LDL in human vascular endothelial cells. Biochem. Biophys. Res. Commun. 394, 377–382. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Characterization of the role of autophagy in endothelial lipid homeostasis.
Fig. S2. The role of autophagy in ApoE KO mouse atherosclerotic plaque formation.
Table S1. Serum chemistry and body weight.
Data S1. Experimental procedures.