

Abstract
Resistant hypertension (RHTN), defined as an uncontrolled blood pressure despite the use of multiple antihypertensive medications, is an increasing clinical problem associated with increased cardiovascular (CV) risk, including stroke and target organ damage. Genetic variability in blood pressure (BP)-regulating genes and pathways may, in part, account for the variability in BP response to antihypertensive agents, when taken alone or in combination, and may contribute to the RHTN phenotype. Pharmacogenomics focuses on the identification of genetic factors responsible for inter-individual variability in drug response. Expanding pharmacogenomics research to include patients with RHTN taking multiple BP-lowering medications may identify genetic markers associated with RHTN. To date, the available evidence surrounding pharmacogenomics in RHTN is limited and primarily focused on candidate genes. In this review, we summarize the most current data in RHTN pharmacogenomics and offer some recommendations on how to advance the field.
Keywords: Pharmacogenomics, Resistant hypertension, Blood pressure, Aldosterone
Introduction
Hypertension (HTN) is the most significant and common chronic modifiable condition, affecting about 1 billion adults worldwide, and about 80 million adults in the US [1–3]. Elevated blood pressure (BP) is a major contributor to cardiovascular (CV) morbidity and mortality, including heart failure, stroke, myocardial infarction, and renal failure. These HTN-related CV complications are largely preventable if BP is treated and successfully reduced. However, the majority of patients with HTN worldwide do not achieve adequate BP reduction, despite the use of multiple antihypertensive medications, in many cases. Uncontrolled BP despite treatment with multiple antihypertensive medications is referred to as resistant HTN (RHTN).
There is no uniform definition of RHTN; however, the American Heart Association in its 2008 position statement [4] defines RHTN as a BP that remains ≥140/90 mmHg despite the use of ≥3 antihypertensive medications, including a thiazide diuretic ideally; or BP<140/90 achieved with use of ≥4 medications. RHTN is a complex phenotype with a multifactorial etiology that is driven by the interplay between a number of pathophysiological mechanisms. It is usually not possible to attribute RHTN to a single cause (Fig. 1). Primary hyperaldosteronism is more prevalent in HTN than generally recognized and is estimated to be present in 20 % of patients with RHTN [5]. Even in the absence of primary hyperaldosteronism, patients with RHTN demonstrate higher levels of aldosterone and volume overload than individuals without RHTN [6]. The role of sympathetic hyperactivity in RHTN development is evident and well described [7, 8], and is likely interrelated with volume overload in RHTN. Activation of the renin-angiotensin aldosterone system (RAAS) may constitute the mechanistic link between elevated aldosterone levels and the sympathetic hyperactivity [9]. Finally, excessive vascular inflammation, and endothelial dysfunction a net result of the heightened sympathetic system and aldosterone may be implicated in the development of RHTN [10–12].
Fig. 1.
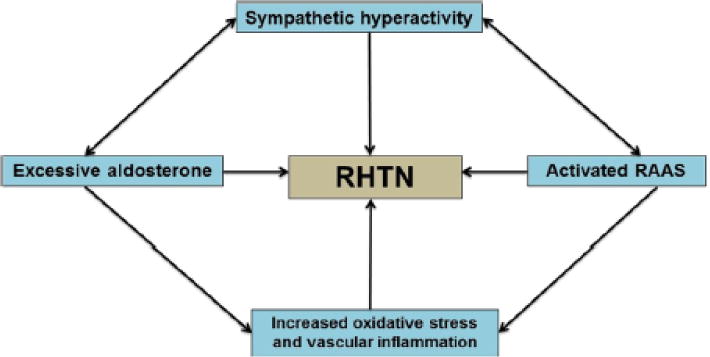
Multiple interrelated pathways involved in the pathogenesis of RHTN. Increased activity of sympathetic nervous system may activate the renal sympathetic efferents with a subsequent activation of RAAS. Excessive aldosterone promotes vascular inflammation and remodeling and may also trigger the activation of sympathetic nervous system. Vascular inflammation derived by excessive aldosterone or activated RAAS may contribute to endothelial dysfunction and arterial stiffness leading to RHTN. The double-headed arrows highlight the interplay of several pathogenic mechanisms in RHTN
Inter-individual variation in drug response, or BP reduction, a more specific surrogate, is hypothesized to result from differences in individual genetic architecture. Pharmacogenomics focuses on studying genetic variants underlying the variability in response to drugs [13, 14], with the goal of personalizing pharmacological treatment to an individual patient. The most frequently occurring genetic variation within the genome is called a single nucleotide polymorphism or SNP, which constitutes a single base substitution. It is estimated that there are approximately 10 million SNPs across the human genome. These SNPs account for the variability in disease risk as well as differences in drug response. Additionally, it is hypothesized that individuals who respond variably to drugs may present different subtypes of the disease. For example, patients who demonstrate a good BP reduction with one drug may have a different genetic and biological basis of their HTN than patients who demonstrate a better BP reduction with another drug. Ultimately, the study of the genetic variation associated with differences in BP reduction may help elucidate some of the genetic and mechanistic basis of HTN.
While pharmacogenomics research in HTN has witnessed advancement in recent years and many genetic variants for different antihypertensive drug classes have been discovered [15–19], there has been slow progress in the application of pharmacogenomics research to the RHTN phenotype. Nevertheless, there are a handful of genetic studies that included patients with RHTN, the majority of them focused on revealing the genetic basis of RHTN. A few additional studies focused on a specific genotype-drug treatment response phenotype in patients with RHTN. The purpose of this review is to highlight the most recent RHTN genetic and pharmacogenetic literature (Table 1), with a focus on identifying studies that could potentially guide treatment selection in RHTN and others that could potentially reveal a genetic basis for RHTN. Additionally, the potential for RHTN pharmacogenomics research beyond the candidate gene approach will be discussed, which would likely identify additional genetic markers and advance the field of RHTN pharmacogenomics in the future.
Table 1.
Summary of the most recent RHTN studies
| Study | Subjects | Gene/candidate SNP | Findings/conclusion |
|---|---|---|---|
| Pharmacogenomic studies related to aldosterone and aldosterone pathways | |||
| Jones ES et al.[32] | 1468 HTN, 471 controls from three different ethnic groups | ENaC (R563Q variant) | R563Q variant may be involved in HTN pathogenesis: patients with RHTN, and the variant may benefit from amiloride treatment |
| Fontana et al.[35] | 62 RHTN from Brazil | CYP11B2 (−344C/T) | Spironolactone may not be the preferred treatment for the −344 TT genotype |
| Laffer et al.[38] | 83 African Americans with volume-dependent RHTN randomized to spironolactone, amiloride, combination of spironolactone and amiloride, or placebo | CYP4A11 (rs3890011) | rs3890011 may be associated with increased ENAC activity Spironolactone may not be effective for rs3890011 CC genotype |
| Genetic studies related to vascular inflammation | |||
| Oliveira-Paula et al.[59] | 113 normotensives, 115 controlled HTN, 70 RHTN from Brazil | iNOS variants: g.2087G>A, microsatellite (CCTTT)n, g-1026C>A | AA+GA variant of g 2087 was associated with HTN |
| iNOS haplotype: (CCTTT)n, g1026C>A, g2087G>A | SCA haplotype was associated with protective effect against RHTN | ||
| Genetic studies in unrelated genes | |||
| Yugar-Toledo et al.[54] | 70 normotensives, 80 controlled HTN, 82 RHTN from Brazil | AGT (M235T), ACE (ACEI/D), NO3 (Glu298Asp) | The T allele of M235 was associated with RHTN in patients older than 50 |
| Lynch et al.[77] | 2203 RHTN, 2354 controlled HTN from ALLHAT trial | 78 candidate SNPs | The Met allele and G allele of rs699 and rs5051 associated with RHTN in whites |
| Fontana et al.[81•] | 1225 controlled HTN, 516 RHTN from INVEST | ~50,000 SNPs in 2000 genes from IBC chip | ATP2B1 was associated with RHTN |
Recent RHTN Pharmacogenomics Studies
Excessive aldosterone plays an important role in the pathophysiology of RHTN [6]. Aldosterone exerts its sodium retaining effects via the kidney’s distal collecting tubules, by binding to the mineralocorticoid receptor (MR). This leads to subsequent translocation of the receptor in the nucleus and transcription of genes, ultimately leading to activation of the epithelial sodium channel (ENaC) [20, 21]. In addition to these effects that are mediated through the kidney, aldosterone exerts adverse effects on the vasculature including inflammation, oxidative stress, vascular remodeling, and endothelial dysfunction [22]. ENaC plays an important role in BP regulation via sodium and volume handling [23, 24]. Inappropriate activation of ENaC due to genetic variation can lead to increased sodium and water reabsorption accompanied by reduced renin secretion, and therefore a low-renin subtype of RHTN [20, 25, 26]. Low-renin RHTN is commonly present in patients of African origin who harbor genetic variants in the ENAC, as an adaptive survival advantage in the sub-Saharan desert [27]. Mineralocorticoid receptor antagonists (MRAs) (e.g., spironolactone and eplerenone) are highly efficacious medications in RHTN, and their effects on BP lowering and vascular remodeling can occur even at low to moderate doses [28–30]. However, MRAs may not be effective when RHTN is driven by the over-activation of ENaC. In the ENAC over-activation scenario, amiloride, a diuretic that directly blocks ENaC, is more effective for BP lowering than an MRA [31]. Below, we review studies that evaluated antihypertensive drug-associated BP response according to a specific genotype.
Genetic Variation in Sodium and Water Handling Pathways
Epithelial Sodium Channel
The R563Q SNP, rs80311498, is a genetic variant in ENAC discovered in South African blacks and is associated with low-renin HTN [25]. Recently, the association of this variant with HTN and with response to amiloride in those with RHTN, among patients from Southern Africa, was investigated [32]. The R563Q variant was found to be associated with HTN in the urban areas, but not in Namibian or Northern Cape San, perhaps due to low dietary sodium consumption in these regions. Moreover, 22 patients with RHTN who carried one risk allele for the R563Q variant were treated with amiloride in addition to their antihypertensive regimen. BP was reduced by an average of 36/17 mmHg from an average of 172/99 mmHg prior to amiloride treatment (p<0.0001). This is an example of the application of pharmacogenomics in RHTN where the selection of a specific pharmacologic agent may be guided by genotype of the patients. However, this association needs to be confirmed among other populations before this could be universally implemented.
Aldosterone Synthase (Cytochrome P45011B2, CYP11B2)
Aldosterone synthase (Cytochrome P45011B2, CYP11B2) is the rate limiting step of aldosterone synthesis in humans [33]. Based on the critical role of aldosterone synthase in aldosterone biosynthesis and previous associations with hypertension [34], Brazilian investigators assessed the effect of the −344 C/T polymorphism (rs1799998) in CYP11B2 on plasma aldosterone levels in 62 patients with documented RHTN [35]. After adjusting for gender, body mass index, and ambulatory BP, patients who were homozygote for the CYP11B2 variant (TT) had higher plasma aldosterone than those who were carriers of the wild-type C allele. Additionally, patients with TT genotype who were treated with an MRA demonstrated an increased level of aldosterone compared to those treated with an MRA with the CT or CC genotype. Further, patients not treated with an MRAwho were TT homozygotes had a higher levels of aldosterone compared with those not treated with an MRA who were carriers of a C allele. The authors concluded that TT homozygote patients may be more susceptible to the aldosterone breakthrough phenomena [36], which is characterized by an increase in aldosterone following MRA exposure. If this finding is confirmed in additional studies, it may be possible to conclude that treatment with an MRA is not preferred in patients with RHTN who are homozygote for the CYP11B2 variant (TT) due to aldosterone breakthrough.
Cytochrome P450, Family 4, Subfamily A, Polypeptide 11, CYP4A11
The CYP4A11 enzyme converts arachidonic acid to 20-hydroxyeicosatetraenoic acid (20-HETE), which is known to induce natriuresis through ENaC inhibition [37]. Genetic variants that reduce the activity of CYP4A11 may promote volume-dependent RHTN due to sodium and water retention as a result of ENaC activation. Laffer et al. sought to evaluate the effect of genetic variant rs3890011 in CYP4A11 on the response to drugs that act on the ENaC pathway, including spironolactone or amiloride [38•]. Eighty-three patients of African descent were included in the study. Plasma renin activity >2 ng/mL/h was an exclusion. Patients were randomized to spironolactone, amiloride, spironolactone, and amiloride, or placebo. Patients with the rs3890011 homozygous variant (CC) did not achieve a BP-lowering response to spironolactone (+6.8±7.9/+4.8±8.6 mmHg) while they demonstrated BP reduction with amiloride (−6.3±7.3/−3.2±4 mmHg, p<0.01/<0.05). On the other hand, patients with the GG and GC genotypes responded similarly to spironolactone and amiloride (−9.8±9.4/−6.3±6.5, −10.6±8.2/−5.9±6.4, respectively, p=0.41/0.43). BP was reduced in all three genotypes in the combination arm. These results suggest that patients with the homozygote variant genotype demonstrate increased activity of ENaC, and therefore respond to amiloride, and not to spironolactone.
Genetic Variation of Response to Dietary Salt Reduction and Diuretics
Patients with RHTN often have occult volume overload, and diuretics are considered the backbone of effective management [39]. Additionally, sodium intake is a significant contributor to RHTN making dietary salt restriction especially effective in patients with RHTN [40]. A study by Pimenta et al. observed that reduction of salt intake to 1.1g/day reduced 24-h ambulatory BP by 23/9 mmHg in patients with RHTN [41]. Variable response to both dietary salt reduction and diuretics exists suggesting the involvement of genetic variants in sodium-handling mechanisms, including renal sodium reabsorption; both of which may play an important role in the overall RHTN phenotype. These genetic variations may result in the inability to excrete sodium in the face of increased sodium-chloride load, leading to salt-sensitive type of HTN, which is usually responsive to diuretic [42]. This has encouraged the investigation of the effect of genetic variation on both response to dietary salt reduction and diuretics. We refer readers to a recent review by Armando et al. [43] that covers the genetic and pharmacogenomic variations involved in salt-sensitive HTN, which is beyond the scope of this review, and in the following text, we focus on some of the shared genetic variants between responses to dietary salt interventions and diuretics, particularly thiazide diuretics, which is a highly effective class in RHTN treatment.
α-Adducin
α-Adducins are cytoskeletal proteins directly involved in sodium handling through the effect on Na+/K+ ATPase, playing an active role in sodium reabsorption through renal tubules [44]. A non-synonymous SNP (Gly460Trp) in ADD1, the gene encoding adducins increases the activity of Na+/K+ ATPase, and thus sodium reabsorption. This SNP was associated with sodium sensitivity and patients with 460Trp carrier had a greater BP response (−15 mmHg) 2 months after treatment with HCTZ compared with the wild-type genotype (−7.4 mmHg, P=0.001) [45]. A rare intronic SNP (rs17833272) in ADD1 was associated with a higher SBP, DBP, mean arterial pressure in response to high dietary sodium (18 g/day), and a lower DBP in response to lower sodium intake (3 g/day) [46].
Guanine Nucleotide-Binding Protein β-Polypeptide 3
Similar to ADD1, guanine nucleotide-binding protein β-polypeptide 3 (GNB3) has been linked to salt-sensitive HTN and response to thiazide diuretics. The variant T allele of C825T was associated with lower plasma renin and pro-renin, higher aldosterone to renin ratio, and DBP [47]. Turner et al. tested the association of the SNP with response to HCTZ in a cohort of 197 African Americans and 190 whites from the Genetic Epidemiology of Responses to Antihypertensives (GERA) [48]. In the study, the variant allele was associated with responsiveness to HCTZ even after adjusting for clinical variables. The same association was observed in another study from the Netherlands where the T allele was also associated with a better BP reduction to HCTZ [49].
Neural Precursor Cell Expressed Developmentally Down-Regulated 4-Like
Neural precursor cell expressed developmentally down-regulated 4-Like (NEDD4L) encodes for ubiquitin ligase that negatively regulates ENaC leading to increased sodium reabsorption [50]. The rs4149601 G>A polymorphism, a well-studied variant resulting in a cryptic splice site is associated with salt-sensitive HTN, where the G carriers of this SNP experience low plasma renin compared to non-carriers [51]. In the NORDIC Diltiazem (NORDIL) study, the G carriers, who were treated with either a thiazide diuretic or β-blocker had greater SBP and DBP reduction (−19.5 and −15.4 mmHg) than homozygous AA genotype (−15 and −14.1 mmHg); the genotype had no effect on BP response to diltiazem [52]. The association was confirmed in the Pharmacogenomic Evaluation of Antihypertensive Responses (PEAR) where increased reduction in BP response to HCTZ was observed with increasing copies of the rs4149601 G allele (SBP reduction for GG, GA, and AA: −12.4, −10.2, and −7.4; DBP reduction: −5.5, −5, and −2.2) [53].
Recent Genetic Studies in RHTN
The following section describes studies that investigate the effect of genetic variation on RHTN.
Genes Related to Vascular Function
The role of vascular inflammation and endothelial dysfunction in the development of RHTN [10] has encouraged the investigation of polymorphisms in genes involved in BP regulation and vascular function. Nitric oxide (NO) has been a focus of HTN research due to its role in BP regulation through the effect on the endothelium-derived relaxing factor activity [54]. Additionally, depletion of NO is implicated in the pathogenesis of hypertension [55, 56]. NO is produced by NO synthase (NOS), which has three isoforms: neuronal NOS (nNOS), inducible NOS (iNOS), and endothelial NOS (eNOS). While nNOS and eNOS are constitutively expressed, iNOS is induced by inflammation or cardiac damage [57]. During inflammation, NO produced by iNOS reacts with reactive oxygen species giving rise to peroxynitrite, which leads to vascular dysfunction [57, 58]. Oliveira-Paula et al. assessed the association between three known functional SNPs in iNOS and RHTN [59]. The SNPs evaluated included the following: a missense SNP (g.2087G>A) that changes an amino acid from serine to leucine and increases iNOS activity [60]; the microsatellite (CCTTT)n and g-1026C>A SNPs, which are located in the promotor region and increase expression of iNOS [61–63]. Moreover, the association between RHTN and the haplotype made by these three SNPs was evaluated. The analysis included 113 normotensive individuals, 115 with controlled HTN, and 82 with RHTN. The variant genotypes of g.2087G>A SNP (AA+GA) were more frequent in the HTN and RHTN groups compared with the normotensive group (OR=2.05, p=0.0016). Additionally, the haplotype made by the short microsatellite repeat, the variant allele of g.2087G>A, and the wild allele of g-1026C>A (SCA) was more frequent in the HTN group compared with the RHTN group (OR=0.14, p=0.012). The authors concluded that g.2087G>A variant may predispose to HTN and that the SCA haplotype is protective from RHTN. However, the effect of the genetic variants in relation to antihypertensive-associated BP response was not evaluated in the study. Additionally, the variants studied were more common in both HTN and RHTN, which makes it hard to draw a conclusion about the role of these variants in RHTN.
Nitric oxide synthase 3 (eNOS) Variants and Phosphodiesterase Type 5 Inhibitors
As described above, NO plays a key role in maintaining vascular tone and vasodilation. NO exerts its vasodilation properties through stimulating guanyl cyclase, which converts GTP to cGMP leading to vascular smooth muscle relaxation and vasodilation [64]. cGMP is broken down by cGMP-specific phosphodiesterase type-5 (PDE5), which can be inhibited by PDE5 inhibitors leading to cGMP accumulation and smooth muscle relaxation. Agents like sildenafil and tadalafil, which are commonly used for erectile dysfunction treatment due to their vasodilatory effects, have been studied as potential antihypertensive agents [65]. Oliver et al. conducted a proof of concept study to evaluate the effect of combination of isosorbide mononitrate (ISMN) plus sildenafil on BP reduction in patients with RHTN. In this small study, six patients with RHTN were randomized in a double-blind four-way crossover to sildenafil (50 mg), ISMN (10 mg), a combination of sildenafil plus ISMN, or placebo [66]. The patients were maintained on their original antihypertensive medications during the study. While each of the agents resulted in a greater BP reduction than placebo, the combination of sildenafil and ISMN produced the largest decline in BP (SBP/DBP of 26/18 mmHg). This study suggests the effectiveness of PDE5 inhibitors, either alone or in combination, in the treatment of RHTN; however, the results have not been confirmed in larger studies with chronic administration.
Because polymorphisms in eNOS may affect the availability on NO availability, studies evaluated the effect of eNOS variants on response to PDE5 inhibitors in patients with ED. Several polymorphisms in eNOS were studied including T786C, variable number of tandem repeats (VNTR) in intron 4 and Glu298Asp [67–70]. Additionally, the (C-Glu-b) haplotype, made by the C allele of T786C, the Glu allele of Glu298Asp, and the 4b allele of tandem repeats, was associated with lower nitrate and nitrite, a surrogate for low endogenous NO, which may explain differences in response to drugs influencing NO activity [71–73]. The same haplotype was also associated with higher risk for HTN in adolescents and obese children [74]. Since the T786C polymorphism has been associated with lower transcriptional level [75], and thus lower levels of NO, investigators conducted exploratory evaluations of the SNP effect on hemodynamic and BP responses after acute use of sildenafil in RHTN patients [76]. Despite the improvement of hemodynamic properties after sildenafil administration including mean arterial pressure, total peripheral resistance (TPR), and diastolic dysfunction parameters, there was no significant change in nitrite levels or cGMP levels after treatment or by genotype. Also, the CC genotype was associated with a higher TPR than TT genotype [76]. If PDE5 inhibitors gain approval for use in RHTN management, a more thorough investigation of the effect of eNOS polymorphisms and haplotypes will be important to fully understand the impact on BP reduction in RHTN patients.
Genes in Unrelated Pathways
The multifactorial etiology of RHTN and the involvement of multiple pathways motivated Yugar-Toledo et al. to investigate the association of genetic variation in three genes: the M235T polymorphism (rs699) in the angiotensinogen gene (AGT), the insertion/deletion polymorphism (rs1799572) in the angiotensin I-converting enzyme gene (ACE), and the Glu298Asp polymorphism (rs1799983) in eNOS [54]. The individual effect of the three genetic variants, their interaction, and gene/environment interactions were tested among 70 patients with RHTN, 80 patients with controlled BP, and 70 normotensive individuals. No associations were observed for any of the studied genetic variants using logistic regression modeling after adjustment for age, gender, body mass index (BMI), low- and high-density lipoproteins, total cholesterol, and glomerular filtration rate. However, the T allele of the M235T polymorphism in AGT was associated with increased risk for RHTN, particularly in age >50.
Lynch et al. evaluated the association of 78 candidate gene polymorphisms in 2203 participants with RHTN and 2354 controls in the Genetics of Hypertension-Associated Treatment (GenHAT) [77•], the ancillary genetic study to the Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT) [78]. ALLHAT investigated the difference in fatal coronary heart disease and non-fatal myocardial infarction between the three classes of antihypertensive medications: calcium channel blockers (amlodipine), angiotensin-converting enzyme inhibitors (lisinopril), and a thiazide-like diuretic (chlorthalidone) [79]. After adjustment for clinical covariates and multiple comparisons, no significant genetic markers were identified in African Americans; however, two SNPs (rs699 and rs5051) in AGT were associated with RHTN in Caucasians. The wild-type allele of rs699 (Met allele) and the variant allele of rs5051 (G) were associated with increased risk for RHTN: OR=1.27 (1.12–1.44, P=0.0001) and OR=1.36 (1.20–1.53, P<0.0001), respectively. It was noted that these alleles were less common in African Americans. The interaction between the two SNPs, rs699 and rs5051, and race was evident when the data from Caucasians and African Americans were combined (P=0.0004 and 0.0001, respectively). It is interesting to note that the association found for the rs699 in AGT was opposite to that found in the previously described study by Yugar-Toledo [54] et al. Carriers of the T allele are at increased risk of RHTN. This may be explained by the different pattern of genetic structure of the populations investigated in the two studies; the Brazilian population had a higher degree of admixture including African, European, and Amerindian ancestry [80]. The effect of genotypes on BP response to antihypertensive drugs was not evaluated.
Fontana et al.[81•] used genetic data generated from the Human CVD Beadchip array, which contains approximately 50,000 SNPs in ~2000 genes involved in CV, inflammation and metabolic processes to assess the associations in 1225 controlled HTN and 526 RHTN participants from the genetic sub-study of the International Verapamil SR-Trandolapril Study (INVEST) [82]. INVEST randomized HTN patients with documented coronary artery disease to a verapamil-based or atenolol-based BP-lowering treatment strategy and followed patients for long-term CVoutcomes. For this analysis, logistic regression analysis was conducted separately in European Americans and Hispanics using an additive genetic model, adjusted for predictors of RHTN previously identified in INVEST, including age, gender, BMI, congestive heart failure, left ventricular hypertrophy, peripheral vascular disease, percutaneous coronary intervention, stroke, and randomized treatment assignment [83]. An intronic SNP (rs12817819) in the ATP2B1 gene was found to be associated with RHTN in both European Americans (OR=1.57 (1.17–2.01), P=2.44×10−3), and Hispanics (OR=1.76 (1.27–2.44), P=7.69×10−4). The ATP2B1 association was evaluated in the Women’s Ischemia Evaluation Syndrome (WISE), which aimed at improving ischemic heart disease diagnosis and recognition by enrolling women undergoing angiogram indicated for chest pain or myocardial ischemia [84]. Although the association of the ATP2B1 rs12817819 SNP was not statistically significant in WISE, the direction was consistent with INVEST. Additionally, the chip-wide significance level was achieved when INVEST and WISE were combined through a meta-analysis (OR=1.65 (1.36–1.95), meta-analysis p value=1.60×10−6). ATP2B1 has been associated with HTN in genome-wide [85] and gene-centric studies [86] and encodes a plasma membrane calcium/calmodulin-dependent ATPase, which is involved in intracellular calcium homeostasis and smooth muscle cell contraction.
The Need for Expansive Genetic Approaches in RHTN Pharmacogenomics
Therapeutic-related benefits are likely to be optimized if pharmacotherapy is customized to an individual’s clinical and genetic risk factors instead of the more commonly used “trial and error approach”. Despite the efforts to unravel the genetic determinants of RHTN, none of the findings from studies to date are ready for clinical implementation. This is partly due to lack of replication of findings and the small sample sizes. In addition, the focused scope of the studies evaluating only well-characterized genes and polymorphisms make it hard to gain new insights into RHTN, owing to the complex genetic architecture and the presence of multiple genetic variants interacting together to derive the phenotype. Therefore, it is of paramount importance to have a more comprehensive approach to survey the association of genetic variants across the whole genome and RHTN. The successful completion of the Human Genome Project, and the continuous efforts to catalog human genetic variations, including the International HapMap Project [87] and the 1000 Genomes [88] have provided the scientific community with a wealth of genetic information that can be used to reveal the genetic basis of diseases and phenotypes. Additionally, the availability of more advanced genetic platforms at more affordable cost makes it possible to employ the genome-wide association study (GWAS) approach to reveal genetic associations with RHTN.
GWAS analysis of pharmacogenomic phenotypes (for example, drug response) is based on the principle of identifying common genetic variants that are statistically different between cases and controls, where the cases are those experiencing the phenotype of interest (in this case, RHTN) and the controls are those without the phenotype of interest. For a specific drug response phenotype, cases and controls are selected from the extremes of the drug response distribution, which maximizes the power to detect significant association(s).
Despite the feasibility of the GWAS approach in the field of antihypertensive pharmacogenomics, which has led to successful discoveries of novel genetic loci for BP response [16, 18, 19], its application in RHTN pharmacogenomics may be more complicated owing to the presence of multiple drugs, which is what defines the RHTN phenotype. A possible solution is the creation of a cohort of RHTN cases and a cohort of easily controlled HTN patients (non-RHTN), followed by GWAS analysis, evaluating the effect of the genetic variants on RHTN, conducted separately by drug class exposure. Although the analysis by individual drug class has the potential advantage of identifying genotypes that may harbor increased or reduced risks of RHTN within those exposed to a specific drug class, it can significantly decrease the number of cases and controls within each drug class, which can reduce the power to identify significant associations. However, the presence of large cohorts or clinical trials with existing data on multiple antihypertensive classes and BP response may overcome the problem of reduced power in GWAS of RHTN. Ideally, the GWAS analysis should be performed using a dataset that was designed for the purpose of evaluating RHTN, an example of such dataset is the ongoing clinical trial of Resistant Hypertension Optimal Treatment Trial (ReHOT) [89]. ReHOT is an ongoing prospective clinical trial designed to evaluate the prevalence of RHTN and the optimum fourth antihypertensive added to an optimized three-drug regimen in patients with stage II HTN from 26 sites in Brazil. Adherence to drug therapy will be confirmed by pill count and RHTN will be determined and confirmed by ambulatory blood-pressure monitoring (ABPM) after the maximum titration of the three antihypertensive drugs (diuretic “chlorthalidone”, angiotensin-converting enzyme inhibitor “enalapril” or angiotensin receptor blocker “losartan”, and calcium channel blocker “amlodipine”). Patients with confirmed RHTN will be randomized in an open label to either a MRA “spironolactone” or a sympatholytic “clonidine” and the primary outcome is effective BP reduction assessed in the office and with ABPM after 12 weeks of drug administration. Several biological samples, including DNA, will be collected to determine markers associated with RHTN and its successful treatment. While this dataset or datasets with similar design are considered by far the best to test RHTN genetic associations, identification of such datasets has been difficult, and thus it has been necessary to create RHTN case-control cohorts from within datasets from well-conducted large HTN clinical trials.
Because the GWAS analysis approach assesses the associations of millions of SNPs in the genome, the chances of false positive associations are enormous, and therefore replication of identified associations in independent cohorts is essential to have confidence in GWAS level signals with RHTN. Therefore, identification of suitable replication cohorts, with well-matched RHTN phenotypes, is one of the largest challenges for successful replications of GWAS signals in RHTN. Additionally, the lack of a consensus definition for RHTN among different studies poses another problem, which highlights the need for harmonization of the RHTN phenotype. To address the need for increased sample size and suitable replication cohorts, researchers in the same discipline are establishing collaborative efforts in the form of consortiums that contain combined genetic and clinical data to facilitate the GWAS analyses among the research groups.
Several pharmacogenomics consortia currently exist and have documented the ability to overcome the shortcomings of individual cohort efforts through this important type of collaboration. The International Consortium for Antihypertensive Pharmacogenomics Studies (ICAPS), icaps-htn.org, is one such consortium and represents an ongoing collaborative effort among research groups that own genetic and clinical data on BP response, CV outcomes, and adverse metabolic effects to antihypertensive medications from small and large clinical trials, as well as epidemiologic studies. ICAPS aims to advance the pharmacogenomics of antihypertensive medications facilitated through conducting GWAS in larger studies, performing meta-analysis, and replicating results across multiple ethnic groups. ICAPS offers the opportunity to conduct a large-scale RHTN GWAS analysis using the HTN treatment trials with genome-wide genotypic data and constructed RHTN phenotype including INVEST [82] and the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT) [90, 91].
Lastly, RHTN, which is a complex phenotype, is likely caused by a collection of common and rare variants, which together may explain the genetics of the phenotype. While the common variants can be identified by the described GWAS approach, identification of the less frequent or rare variants requires a comprehensive sequencing of the genome using next-generation sequencing methods that are becoming more widely used in the field of genomics and pharmacogenomics [92]. Therefore, the identification of the full spectrum of genetic variants will likely advance the RHTN pharmacogenomics through improving knowledge of the genetic underpinnings of the phenotype and understanding the relationship between genetic variants and drugs and how they work together to affect the phenotype i.e., the favorable or risk drugs for RHTN by genotype. The ultimate translation of pharmacogenomics findings in RHTN into clinically actionable information is important for improved BP response and CV outcomes, and requires new knowledge of the biological/pathogenic effects of the discovered variants.
Equally important is the functional validation of the discovered variants by testing the effect of the drugs on the BP response in the context of RHTN. Identifying the appropriate in vitro modeling system to evaluate the effect of identified variants, as well as the ability of drugs to modulate the RHTN phenotype is challenging. The induced pluripotent stem cells (iPSC), a novel technology currently used in the field of genomics and pharmacogenomics, holds promise in modeling the functional consequences of discovered GWAS associations [93]. Using this technology, a somatic cell from a specific patient can be transformed into a pluripotent state that has the potential to differentiate into the relevant cellular model system for the phenotype [94]. For RHTN pharmacogenomics, iPSC-derived cells from patients with RHTN and the genotype of interest can be transformed into the appropriate tissue. These iPSC-derived cells will retain the genomic information of the patient and can be used as a platform to evaluate the effect of an antihypertensive drug at the cellular level. Another way to understand the pathogenic effect of the variants is to genetically edit the variant in the iPSC derived from a patient, for instance, convert the variant into a wild type and evaluate if the phenotype can be reversed.
Conclusions
RHTN pharmacogenomics candidate gene studies have identified some SNPs that impact response to MRAs and amiloride but have yet to achieve the level of significance necessary for clinical implementation. While the advancement in genotyping technology has made possible the characterization of genetic variants across individuals in genetic studies, RHTN has not yet reaped the benefits of more comprehensive genetic approaches such as GWAS, genome sequencing, and others used in the pharmacogenomics research. It is anticipated that well-designed GWAS studies will indeed identify genetic loci for RHTN. Additional recommendations to reveal the genetic basis of RHTN include, but are not limited to, standardization of the phenotype, identifying less common variants using sequencing methods, and understanding the biological role of the discovered variants using in vitro modeling systems like iPSCs. Although the path to identify RHTN risk loci remains challenging, it is certainly worthwhile. Gaining a thorough understanding of the genetic background of RHTN is crucial in many ways, including the ability to predict an individual’s risk for RHTN, as well as improving an individual’s outcomes through optimization of drug therapy tailored to clinical and genetic risk factors. The potential for pharmacogenomics research in RHTN is likely to progress, especially if the above recommendations are considered.
Footnotes
Conflict of Interest Dr. Cooper-DeHoff declares current grant funding from NIH, NIGMS U01 GM074492 and U01 GM092586; and NIH, NHGRI U01 HG007269. Dr. Rouby declares no conflict of interest.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
- 1.Nwankwo S, Yoon S, Burt V, Gu Q. Hypertension among adults in the United States: National Health and Nutrition Examination Survey, 2011–2012. NCHS Data Brief. 2013;133:1–8. [PubMed] [Google Scholar]
- 2.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M. Executive summary: Heart Disease and Stroke Statistics—2015 update: a report from the American Heart Association. Circulation. 2015;131(4):434–41. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 3.Joffres M, Falaschetti E, Gillespie C, Robitaille C, Loustalot F, Poulter N, et al. Hypertension prevalence, awareness, treatment and control in national surveys from England, the USA and Canada, and correlation with stroke and ischaemic heart disease mortality: a cross-sectional study. BMJ Open. 2013;3(8):e003423. doi: 10.1136/bmjopen-2013-003423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, et al. Resistant hypertension: diagnosis, evaluation, and treatment: a scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Circulation. 2008;117(25):e510–26. doi: 10.1161/CIRCULATIONAHA.108.189141. [DOI] [PubMed] [Google Scholar]
- 5.Calhoun DA, Nishizaka MK, Zaman MA, Thakkar RB, Weissmann P. Hyperaldosteronism among black and white subjects with resistant hypertension. Hypertension. 2002;40(6):892–6. doi: 10.1161/01.hyp.0000040261.30455.b6. [DOI] [PubMed] [Google Scholar]
- 6.Gaddam KK, Nishizaka MK, Pratt-Ubunama MN, Pimenta E, Aban I, Oparil S, et al. Characterization of resistant hypertension: association between resistant hypertension, aldosterone, and persistent intravascular volume expansion. Arch Intern Med. 2008;168(11):1159–64. doi: 10.1001/archinte.168.11.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oliva RV, Bakris GL. Sympathetic activation in resistant hypertension: theory and therapy. Semin Nephrol. 2014;34(5):550–9. doi: 10.1016/j.semnephrol.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 8.Tsioufis C, Kordalis A, Flessas D, Anastasopoulos I, Tsiachris D, Papademetriou V, et al. Pathophysiology of resistant hypertension: the role of sympathetic nervous system. Int J Hypertens. 2011;2011:642416. doi: 10.4061/2011/642416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Modolo R, de Faria AP, Moreno H. Resistant hypertension: a volemic or nervous matter? J Am Soc Hypertens. 2015 Feb; doi: 10.1016/j.jash.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 10.Barbaro NR, Fontana V, Modolo R, De Faria AP, Sabbatini AR, Fonseca FH, Anhê GF, Moreno H. Increased arterial stiffness in resistant hypertension is associated with inflammatory biomarkers. Blood Press; Jul, 2014. pp. 1–7. [DOI] [PubMed] [Google Scholar]
- 11.Salles GF, Fiszman R, Cardoso CRL, Muxfeldt ES. Relation of left ventricular hypertrophy with systemic inflammation and endothelial damage in resistant hypertension. Hypertension. 2007;50(4):723–8. doi: 10.1161/HYPERTENSIONAHA.107.093120. [DOI] [PubMed] [Google Scholar]
- 12.Whaley-Connell A, Johnson MS, Sowers JR. Aldosterone: role in the cardiometabolic syndrome and resistant hypertension. Prog Cardiovasc Dis. Jan;52(5):401–9. doi: 10.1016/j.pcad.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinshilboum R, Wang L. Pharmacogenomics: bench to bedside. Nat Rev Drug Discov. 2004;3(9):739–48. doi: 10.1038/nrd1497. [DOI] [PubMed] [Google Scholar]
- 14.Weinshilboum R. Inheritance and drug response. N Engl J Med. 2003;348(6):529–37. doi: 10.1056/NEJMra020021. [DOI] [PubMed] [Google Scholar]
- 15.Duarte JD, Turner ST, Tran B, Chapman AB, Bailey KR, Gong Y, et al. Association of chromosome 12 locus with antihypertensive response to hydrochlorothiazide may involve differential YEATS4 expression. Pharmacogenom J. 2013;13(3):257–63. doi: 10.1038/tpj.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turner ST, Bailey KR, Schwartz GL, Chapman AB, Chai HS, Boerwinkle E. Genomic association analysis identifies multiple loci influencing antihypertensive response to an angiotensin II receptor blocker. Hypertension. 2012;59(6):1204–11. doi: 10.1161/HYP.0b013e31825b30f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gong Y, McDonough CW, Wang Z, Hou W, Cooper-DeHoff RM, Langaee TY, et al. Hypertension susceptibility loci and blood pressure response to antihypertensives: results from the pharmacogenomic evaluation of antihypertensive responses study. Circ Cardiovasc Genet. 2012;5(6):686–91. doi: 10.1161/CIRCGENETICS.112.964080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turner ST, Boerwinkle E, O’Connell JR, Bailey KR, Gong Y, Chapman AB, et al. Genomic association analysis of common variants influencing antihypertensive response to hydrochlorothiazide. Hypertension. 2013;62(2):391–7. doi: 10.1161/HYPERTENSIONAHA.111.00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hiltunen TP, Donner KM, Sarin A-P, Saarela J, Ripatti S, Chapman AB, et al. Pharmacogenomics of hypertension: a genome-wide, placebo-controlled cross-over study, using four classes of antihypertensive drugs. J Am Heart Assoc. 2015;4(1):e001521. doi: 10.1161/JAHA.114.001521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bubien JK. Epithelial Na+ channel (ENaC), hormones, and hypertension. J Biol Chem. 2010;285(31):23527–31. doi: 10.1074/jbc.R109.025049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Studer RA, Person E, Robinson-Rechavi M, Rossier BC. Evolution of the epithelial sodium channel and the sodium pump as limiting factors of aldosterone action on sodium transport. Physiol Genomics. 2011;43(13):844–54. doi: 10.1152/physiolgenomics.00002.2011. [DOI] [PubMed] [Google Scholar]
- 22.Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med. 2009;150(11):776–83. doi: 10.7326/0003-4819-150-11-200906020-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hummler E. Epithelial sodium channel, salt intake, and hypertension. Curr Hypertens Rep. 2003;5(1):11–8. doi: 10.1007/s11906-003-0005-1. [DOI] [PubMed] [Google Scholar]
- 24.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997;77(2):359–96. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 25.Rayner BL, Owen EP, King JA, Soule SG, Vreede H, Opie LH, et al. A new mutation, R563Q, of the beta subunit of the epithelial sodium channel associated with low-renin, low-aldosterone hypertension. J Hypertens. 2003;21(5):921–6. doi: 10.1097/00004872-200305000-00016. [DOI] [PubMed] [Google Scholar]
- 26.Eide IK, Torjesen PA, Drolsum A, Babovic A, Lilledahl NP. Low-renin status in therapy-resistant hypertension: a clue to efficient treatment. J Hypertens. 2004;22(11):2217–26. doi: 10.1097/00004872-200411000-00026. [DOI] [PubMed] [Google Scholar]
- 27.Spence JD. Physiologic tailoring of treatment in resistant hypertension. Curr Cardiol Rev. 2010;6(2):119–23. doi: 10.2174/157340310791162695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16(11 Pt 1):925–30. doi: 10.1016/s0895-7061(03)01032-x. [DOI] [PubMed] [Google Scholar]
- 29.Ori Y, Chagnac A, Korzets A, Zingerman B, Herman-Edelstein M, Bergman M, et al. Regression of left ventricular hypertrophy in patients with primary aldosteronism/low-renin hypertension on low-dose spironolactone. Nephrol Dial Transplant. 2013;28(7):1787–93. doi: 10.1093/ndt/gfs587. [DOI] [PubMed] [Google Scholar]
- 30.Oxlund CS, Henriksen JE, Tarnow L, Schousboe K, Gram J, Jacobsen IA. Low dose spironolactone reduces blood pressure in patients with resistant hypertension and type 2 diabetes mellitus: a double blind randomized clinical trial. J Hypertens. 2013;31(10):2094–102. doi: 10.1097/HJH.0b013e3283638b1a. [DOI] [PubMed] [Google Scholar]
- 31.Spence JD. Lessons from Africa: the importance of measuring plasma renin and aldosterone in resistant hypertension. Can J Cardiol. 2012;28(3):254–7. doi: 10.1016/j.cjca.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 32.Jones ESW, Owen EP, Rayner BL. The association of the R563Q genotype of the ENaC with phenotypic variation in Southern Africa. Am J Hypertens. 2012;25(12):1286–91. doi: 10.1038/ajh.2012.125. [DOI] [PubMed] [Google Scholar]
- 33.Strushkevich N, Gilep AA, Shen L, Arrowsmith CH, Edwards AM, Usanov SA, et al. Structural insights into aldosterone synthase substrate specificity and targeted inhibition. Mol Endocrinol. 2013;27(2):315–24. doi: 10.1210/me.2012-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alvarez-Madrazo S, Mackenzie SM, Davies E, Fraser R, Lee W-K, Brown M, et al. Common polymorphisms in the CYP11B1 and CYP11B2 genes: evidence for a digenic influence on hypertension. Hypertension. 2013;61(1):232–9. doi: 10.1161/HYPERTENSIONAHA.112.200741. [DOI] [PubMed] [Google Scholar]
- 35.Fontana V, de Faria APC, Barbaro NR, Sabbatini AR, Modolo R, Lacchini R, et al. Modulation of aldosterone levels by −344 C/T CYP11B2 polymorphism and spironolactone use in resistant hypertension. J Am Soc Hypertens. 2014;8(3):146–51. doi: 10.1016/j.jash.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 36.Ubaid-Girioli S, de Souza LA, Yugar-Toledo JC, Cláudio Martins L, Ferreira-Melo S, Rizzi Coelho O, et al. Aldosterone excess or escape: treating resistant hypertension. J Clin Hypertens. 2009;11(5):245–52. doi: 10.1111/j.1751-7176.2009.00110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zordoky BNM, El-Kadi AOS. Effect of cytochrome P450 polymorphism on arachidonic acid metabolism and their impact on cardiovascular diseases. Pharmacol Ther. 2010;125(3):446–63. doi: 10.1016/j.pharmthera.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 38•.Laffer CL, Elijovich F, Eckert GJ, Tu W, Pratt JH, Brown NJ. Genetic variation in CYP4A11 and blood pressure response to mineralocorticoid receptor antagonism or ENaC inhibition: an exploratory pilot study in African Americans. J Am Soc Hypertens. 2014;8(7):475–80. doi: 10.1016/j.jash.2014.04.011. A recent research evaluating the effect of variants in CYP4A11 on the effectiveness of spironolactone and amiloride. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar N, Calhoun DA, Dudenbostel T. Management of patients with resistant hypertension: current treatment options. Integr Blood Press Control. 2013;6:139–51. doi: 10.2147/IBPC.S33984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vongpatanasin W. Resistant hypertension. JAMA. 2014;311(21):2216. doi: 10.1001/jama.2014.5180. [DOI] [PubMed] [Google Scholar]
- 41.Pimenta E, Gaddam KK, Oparil S, Aban I, Husain S, Dell’Italia LJ, et al. Effects of dietary sodium reduction on blood pressure in subjects with resistant hypertension: results from a randomized trial. Hypertension. 2009;54(3):475–81. doi: 10.1161/HYPERTENSIONAHA.109.131235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hall JE, Granger JP, do Carmo JM, da Silva AA, Dubinion J, George E, et al. Comprehensive physiology. 4. Vol. 2. Hoboken: Wiley; 2012. [DOI] [PubMed] [Google Scholar]
- 43.Armando I, Villar VAM, Jose PA. Genomics and pharmacogenomics of salt-sensitive hypertension. Curr Hypertens Rev. 2015;11(1):49–56. [PubMed] [Google Scholar]
- 44.Sanada H, Jones JE, Jose PA. Genetics of salt-sensitive hypertension. Curr Hypertens Rep. 2010;13(1):55–66. doi: 10.1007/s11906-010-0167-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cusi D, Barlassina C, Azzani T, Casari G, Citterio L, Devoto M, et al. Polymorphisms of alpha-adducin and salt sensitivity in patients with essential hypertension. Lancet (London, England) 1997;349(9062):1353–7. doi: 10.1016/S0140-6736(97)01029-5. [DOI] [PubMed] [Google Scholar]
- 46.Kelly TN, Rice TK, Gu D, Hixson JE, Chen J, Liu D, et al. Novel genetic variants in the alpha-adducin and guanine nucleotide binding protein beta-polypeptide 3 genes and salt sensitivity of blood pressure. Am J Hypertens. 2009;22(9):985–92. doi: 10.1038/ajh.2009.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schunkert H, Hense HW, Döring A, Riegger GA, Siffert W. Association between a polymorphism in the G protein beta3 subunit gene and lower renin and elevated diastolic blood pressure levels. Hypertension. 1998;32(3):510–3. doi: 10.1161/01.hyp.32.3.510. [DOI] [PubMed] [Google Scholar]
- 48.Turner ST, Schwartz GL, Chapman AB, Boerwinkle E. C825T polymorphism of the G protein 3-subunit and antihypertensive response to a thiazide diuretic. Hypertension. 2001;37(2):739–43. doi: 10.1161/01.hyp.37.2.739. [DOI] [PubMed] [Google Scholar]
- 49.Schelleman H, Stricker BHC, Verschuren WMM, de Boer A, Kroon AA, de Leeuw PW, et al. Interactions between five candidate genes and antihypertensive drug therapy on blood pressure. Pharmacogenom J. 2005;6(1):22–6. doi: 10.1038/sj.tpj.6500339. [DOI] [PubMed] [Google Scholar]
- 50.Gong Y, Mcdonough CW, Padmanabhan S, Johnson JA. Handbook of pharmacogenomics and stratified medicine. Elsevier; 2014. [Google Scholar]
- 51.Dahlberg J, Nilsson LO, von Wowern F, Melander O. Polymorphism in NEDD4L is associated with increased salt sensitivity, reduced levels of P-renin and increased levels of Nt-proANP. PLoS One. 2007;2(5):e432. doi: 10.1371/journal.pone.0000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Svensson-Färbom P, Wahlstrand B, Almgren P, Dahlberg J, Fava C, Kjeldsen S, et al. A functional variant of the NEDD4L gene is associated with beneficial treatment response with β-blockers and diuretics in hypertensive patients. J Hypertens. 2011;29(2):388–95. doi: 10.1097/HJH.0b013e3283410390. [DOI] [PubMed] [Google Scholar]
- 53.McDonough CW, Burbage SE, Duarte JD, Gong Y, Langaee TY, Turner ST, et al. Association of variants in NEDD4L with blood pressure response and adverse cardiovascular outcomes in hypertensive patients treated with thiazide diuretics. J Hypertens. 2013;31(4):698–704. doi: 10.1097/HJH.0b013e32835e2a71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yugar-Toledo JC, Martin JFV, Krieger JE, Pereira AC, Demacq C, Coelho OR, Pimenta E, Calhoun DA, Júnior HM. Gene variation in resistant hypertension: multilocus analysis of the angiotensin 1-converting enzyme, angiotensinogen, and endothelial nitric oxide synthase genes. 2011 Jul; doi: 10.1089/dna.2010.1156. [DOI] [PubMed] [Google Scholar]
- 55.Sandrim VC, de Syllos RWC, Lisboa HRK, Tres GS, Tanus-Santos JE. Influence of eNOS haplotypes on the plasma nitric oxide products concentrations in hypertensive and type 2 diabetes mellitus patients. Nitric Oxide. 2007;16(3):348–55. doi: 10.1016/j.niox.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 56.Giles TD, Sander GE, Nossaman BD, Kadowitz PJ. Impaired vasodilation in the pathogenesis of hypertension: focus on nitric oxide, endothelial-derived hyperpolarizing factors, and prostaglandins. J Clin Hypertens (Greenwich) 2012;14(4):198–205. doi: 10.1111/j.1751-7176.2012.00606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilcox JN, Subramanian RR, Sundell CL, Tracey WR, Pollock JS, Harrison DG, et al. Expression of multiple isoforms of nitric oxide synthase in normal and atherosclerotic vessels. Arterioscler Thromb Vasc Biol. 1997;17(11):2479–88. doi: 10.1161/01.atv.17.11.2479. [DOI] [PubMed] [Google Scholar]
- 58.Ballinger SW, Patterson C, Yan C-N, Doan R, Burow DL, Young CG, et al. Hydrogen peroxide- and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ Res. 2000;86(9):960–6. doi: 10.1161/01.res.86.9.960. [DOI] [PubMed] [Google Scholar]
- 59.Oliveira-Paula GH, Lacchini R, Coeli-Lacchini FB, Junior HM, Tanus-Santos JE. Inducible nitric oxide synthase haplotype associated with hypertension and responsiveness to antihypertensive drug therapy. Gene. 2013;515(2):391–5. doi: 10.1016/j.gene.2012.12.059. [DOI] [PubMed] [Google Scholar]
- 60.Wang SS, Davis S, Cerhan JR, Hartge P, Severson RK, Cozen W, et al. Polymorphisms in oxidative stress genes and risk for non-Hodgkin lymphoma. Carcinogenesis. 2006;27(9):1828–34. doi: 10.1093/carcin/bgl013. [DOI] [PubMed] [Google Scholar]
- 61.Kaise M, Miwa J, Suzuki N, Mishiro S, Ohta Y, Yamasaki T, et al. Inducible nitric oxide synthase gene promoter polymorphism is associated with increased gastric mRNA expression of inducible nitric oxide synthase and increased risk of gastric carcinoma. Eur J Gastroenterol Hepatol. 2007;19(2):139–45. doi: 10.1097/01.meg.0000252637.11291.1d. [DOI] [PubMed] [Google Scholar]
- 62.Fu L, Zhao Y, Lu J, Shi J, Li C, Liu H, et al. Functional single nucleotide polymorphism-1026C/A of inducible nitric oxide synthase gene with increased YY1-binding affinity is associated with hypertension in a Chinese Han population. J Hypertens. 2009;27(5):991–1000. doi: 10.1097/hjh.0b013e3283294bec. [DOI] [PubMed] [Google Scholar]
- 63.Li W, Liu H, Fu L, Li D, Zhao Y. Identification of Yin Yang 1-interacting partners at -1026C/A in the human iNOS promoter. Arch Biochem Biophys. 2010;498(2):119–26. doi: 10.1016/j.abb.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 64.Brown KE, Dhaun N, Goddard J, Webb DJ. Potential therapeutic role of phosphodiesterase type 5 inhibition in hypertension and chronic kidney disease. Hypertension. 2014;63(1):5–11. doi: 10.1161/HYPERTENSIONAHA.113.01774. [DOI] [PubMed] [Google Scholar]
- 65.Oliver JJ, Melville VP, Webb DJ. Effect of regular phosphodiesterase type 5 inhibition in hypertension. Hypertension. 2006;48(4):622–7. doi: 10.1161/01.HYP.0000239816.13007.c9. [DOI] [PubMed] [Google Scholar]
- 66.Oliver JJ, Hughes VEC, Dear JW, Webb DJ. Clinical potential of combined organic nitrate and phosphodiesterase type 5 inhibitor in treatment-resistant hypertension. Hypertension. 2010;56(1):62–7. doi: 10.1161/HYPERTENSIONAHA.109.147686. [DOI] [PubMed] [Google Scholar]
- 67.Safarinejad MR, Khoshdel A, Shekarchi B, Taghva A, Safarinejad S. Association of the T-786C, G894T and 4a/4b polymorphisms of the endothelial nitric oxide synthase gene with vasculogenic erectile dysfunction in Iranian subjects. BJU Int. 2011;107(12):1994–2001. doi: 10.1111/j.1464-410X.2010.09755.x. [DOI] [PubMed] [Google Scholar]
- 68.Sinici I, Güven EO, Serefoğlu E, Hayran M. T-786C polymorphism in promoter of eNOS gene as genetic risk factor in patients with erectile dysfunction in Turkish population. Urology. 2010;75(4):955–60. doi: 10.1016/j.urology.2009.06.063. [DOI] [PubMed] [Google Scholar]
- 69.Rosas-Vargas H, Coral-Vazquez RM, Tapia R, Borja JL, Salas RA, Salamanca F. Glu298Asp endothelial nitric oxide synthase polymorphism is a risk factor for erectile dysfunction in the Mexican Mestizo population. J Androl. Jan;25(5):728–32. doi: 10.1002/j.1939-4640.2004.tb02847.x. [DOI] [PubMed] [Google Scholar]
- 70.Lee Y-C, Wu W-J, Liu C-C, Wang C-J, Li W-M, Huang C-H, et al. The associations among eNOS G894T gene polymorphism, erectile dysfunction, and benign prostate hyperplasia-related lower urinary tract symptoms. J Sex Med. 2009;6(11):3158–65. doi: 10.1111/j.1743-6109.2009.01353.x. [DOI] [PubMed] [Google Scholar]
- 71.Metzger IF, Sertório JTC, Tanus-Santos JE. Modulation of nitric oxide formation by endothelial nitric oxide synthase gene haplotypes. Free Radic Biol Med. 2007;43(6):987–92. doi: 10.1016/j.freeradbiomed.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 72.Metzger IF, Ishizawa MH, Rios-Santos F, Carvalho WA, Tanus-Santos JE. Endothelial nitric oxide synthase gene haplotypes affect nitrite levels in black subjects. Pharmacogenom J. 2011;11(6):393–9. doi: 10.1038/tpj.2010.52. [DOI] [PubMed] [Google Scholar]
- 73.Metzger IF, Souza-Costa DC, Marroni AS, Nagassaki S, Desta Z, Flockhart DA, et al. Endothelial nitric oxide synthase gene haplotypes associated with circulating concentrations of nitric oxide products in healthy men. Pharmacogenet Genomics. 2005;15(8):565–70. doi: 10.1097/01.fpc.0000167328.85163.44. [DOI] [PubMed] [Google Scholar]
- 74.Souza-Costa DC, Belo VA, Silva PS, Metzger IF, Lanna CM, Machado MA, et al. eNOS haplotype associated with hypertension in obese children and adolescents. Int J Obes. 2010;35(3):387–92. doi: 10.1038/ijo.2010.146. [DOI] [PubMed] [Google Scholar]
- 75.Miyamoto Y, Saito Y, Nakayama M, Shimasaki Y, Yoshimura T, Yoshimura M, et al. Replication protein A1 reduces transcription of the endothelial nitric oxide synthase gene containing a -786TC mutation associated with coronary spastic angina. Hum Mol Genet. 2000;9(18):2629–37. doi: 10.1093/hmg/9.18.2629. [DOI] [PubMed] [Google Scholar]
- 76.Quinaglia T, de Faria APC, Fontana V, Barbaro NR, Sabbatini AR, Sertório JT, et al. Acute cardiac and hemodynamic effects of sildenafil on resistant hypertension. Eur J Clin Pharmacol. 2013;69(12):2027–36. doi: 10.1007/s00228-013-1571-z. [DOI] [PubMed] [Google Scholar]
- 77•.Lynch AI, Irvin MR, Davis BR, Ford CE, Eckfeldt JH, Arnett DK. Genetic and adverse health outcome associations with treatment resistant hypertension in GenHAT. Int J Hypertens. 2013;2013:578578. doi: 10.1155/2013/578578. A recent analysis from the Genetics of Hypertension Associated Treatment Study assessing the association of 78 candidate gene polymorphism with RHTN. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arnett D, Boerwinkle E, Davis B. Pharmacogenetic approaches to hypertension therapy: design and rationale for the Genetics of Hypertension Associated Treatment (GenHAT) study. 2002 doi: 10.1038/sj.tpj.6500113. [DOI] [PubMed] [Google Scholar]
- 79.Davis B, Cutler J, Gordon D. Rationale and design for the antihypertensive and lipid lowering treatment to prevent heart attack trial (ALLHAT) Am J. 1996 doi: 10.1016/0895-7061(96)00037-4. [DOI] [PubMed] [Google Scholar]
- 80.Barbaro NR, Fontana V, Moreno H. Angiotensinogen variants among resistant hypertensive patients. Int J Hypertens. 2014;2014:424793. doi: 10.1155/2014/424793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81•.Fontana V, McDonough CW, Gong Y, El Rouby NM, Sá ACC, Taylor KD, et al. Large-scale gene-centric analysis identifies polymorphisms for resistant hypertension. J Am Heart Assoc. 2014;3:6. doi: 10.1161/JAHA.114.001398. An analysis from the INVEST-Genetic substudy assessing the association of approximately 50,000 SNPs in genes implicated in CV, metabolic and inflammatory processes with RHTN in 1,714 participants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pepine CJ, Handberg EM, Cooper-DeHoff RM, Marks RG, Kowey P, Messerli FH, et al. A calcium antagonist vs. a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. JAMA. 2003;290(21):2805–16. doi: 10.1001/jama.290.21.2805. [DOI] [PubMed] [Google Scholar]
- 83.Smith SM, Gong Y, Handberg E, Messerli FH, Bakris GL, Ahmed A, et al. Predictors and outcomes of resistant hypertension among patients with coronary artery disease and hypertension. J Hypertens. 2014;32(3):635–43. doi: 10.1097/HJH.0000000000000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Merz C, Kelsey S. The Women’s Ischemia Syndrome Evaluation (WISE) study: protocol design, methodology and feasibility report. J. 1999 doi: 10.1016/s0735-1097(99)00082-0. [DOI] [PubMed] [Google Scholar]
- 85.Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A. Nat Genet. 2009;41(6):677–87. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ganesh SK, Tragante V, Guo W, Guo Y, Lanktree MB, Smith EN. Loci influencing blood pressure identified using a cardiovascular gene-centric array. Hum Mol Genet. 2013;22(8):1663–78. doi: 10.1093/hmg/dds555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449(7164):851–61. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Krieger EM, Drager LF, Giorgi DMA, Krieger JE, Pereira AC, Barreto-Filho JAS, et al. Resistant hypertension optimal treatment trial: a randomized controlled trial. Clin Cardiol. 2014;37(1):1–6. doi: 10.1002/clc.22228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gupta AK, Nasothimiou EG, Chang CL, Sever PS, Dahlöf B, Poulter NR. Baseline predictors of resistant hypertension in the Anglo-Scandinavian Cardiac Outcome Trial (ASCOT): a risk score to identify those at high-risk. J Hypertens. 2011;29(10):2004–13. doi: 10.1097/HJH.0b013e32834a8a42. [DOI] [PubMed] [Google Scholar]
- 91.Sever PS, Dahlöf B, Poulter NR, Wedel H, Beevers G, Caulfield M, et al. Rationale, design, methods and baseline demography of participants of the Anglo-Scandinavian Cardiac Outcomes Trial. ASCOT investigators. J Hypertens. 2001;19(6):1139–47. doi: 10.1097/00004872-200106000-00020. [DOI] [PubMed] [Google Scholar]
- 92.McCarthy JJ, McLeod HL, Ginsburg GS. Genomic medicine: a decade of successes, challenges, and opportunities. Sci Transl Med. 2013;5(189):189sr4. doi: 10.1126/scitranslmed.3005785. [DOI] [PubMed] [Google Scholar]
- 93.Hankowski KE, Hamazaki T, Umezawa A, Terada N. Induced pluripotent stem cells as a next-generation biomedical interface. Lab Invest. 2011;91(7):972–7. doi: 10.1038/labinvest.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhu H, Lensch MW, Cahan P, Daley GQ. Investigating monogenic and complex diseases with pluripotent stem cells. Nat Rev Genet. 2011;12(4):266–75. doi: 10.1038/nrg2951. [DOI] [PubMed] [Google Scholar]