

Abstract
In the years preceding a diagnosis of Alzheimer’s disease (AD), hyperexcitability of the hippocampus is a commonly observed phenomenon in those at risk for AD. Our previous work suggests a dysregulation in glutamate neurotransmission may mediate this hyperexcitability, and glutamate dysregulation correlates with cognitive deficits in the rTg(TauP301L)4510 mouse model of AD. To determine whether improving glutamate regulation would attenuate cognitive deficits and AD-related pathology, TauP301L mice were treated with riluzole (~ 12.5 mg/kg/day p.o.), an FDA-approved drug for ALS that lowers extracellular glutamate levels. Riluzole-treated TauP301L mice exhibited improved memory performance that was associated with a decrease in glutamate release and an increase in glutamate uptake in the dentate gyrus (DG), cornu ammonis 3(CA3), and cornu ammonis 1(CA1) regions of the hippocampus. Riluzole treatment also attenuated the TauP301L-mediated increase in hippocampal vesicular glutamate transporter (vGLUT1), and the TauP301L-mediated decrease in hippocampal glutamate transporter 1 (GLT-1) and PSD-95 expression. Riluzole treatment also reduced tau pathology. These findings further elucidate the changes in glutamate regulation associated with tau pathology and open new opportunities for the development of clinically applicable therapeutic approaches to regulate glutamate in vulnerable circuits for those at risk for the development of AD.
Keywords: Alzheimer, tau, glutamate clearance, in vivo electrochemistry, synaptic release, hippocampus, riluzole, memory, cognition
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder that targets vulnerable neural networks, particularly those involved in learning and memory (Brier et al. 2012, Palop et al. 2006). In the years preceding AD diagnosis, a hyperactivity of the distributed memory network is often observed in those at risk for AD (Sperling et al. 2010, Bookheimer et al. 2000, Quiroz et al. 2010, Bondi et al. 2005, Bassett et al. 2006, Filippini et al. 2009). Though originally this hyperactivity was believed to serve a compensatory function for deteriorating circuitry (Bondi et al. 2005), more recent evidence suggests this hyperactivity may be indicative of excitotoxicity, could directly contribute to cognitive impairment, and may even be permissive for the development of AD (Vossel et al. 2013, Koh et al. 2010, Bakker et al. 2012b, Kamenetz et al. 2003, Busche et al. 2008, Mackenzie & Miller 1994, Yamada et al. 2014).
Using a tau mouse model of AD (rTg(TauP301L)4510), we recently showed (Hunsberger et al. 2014a) that P301L tau expression is associated with increased hippocampal glutamate release and decreased glutamate uptake, and these alterations in glutamate signaling correlated with cognitive deficits in the hippocampal-dependent Barnes maze task. The dysregulation of glutamate in mice expressing P301L tau was observed at a time when tau pathology was subtle and before readily detectable neuron loss. Here, we sought to determine whether reducing extracellular glutamate levels would alleviate cognitive deficits associated with P301L tau expression. To test this hypothesis, TauP301L mice were given riluzole, an FDA-approved disease-modifying drug for amyotrophic lateral sclerosis (ALS), that modulates glutamatergic signaling. At physiologically relevant drug concentrations, riluzole’s in vivo mechanisms of action include a stabilization of the inactivate state of the voltage-gated sodium channel, leading to a decrease in glutamate release, and a potentiation of glutamate uptake via an increase in glutamate transporter expression (Gourley et al. 2012, Azbill et al. 2000, Frizzo et al. 2004, Fumagalli et al. 2008). Though other effects have been noted in in vitro studies, these effects only occur at unrealistically high concentrations, which are unlikely to be achieved in animals or patients (see (Pittenger et al. 2008) for review).
We assessed the effects of riluzole administration on hippocampal-dependent learning and memory, glutamate regulation in the hippocampus (DG, CA3, and CA1), and tau pathology in the hippocampus of TauP301L mice. Focus was given to the hippocampus due to its role in cognitive functions such as learning and memory, and because it is one of the first structures affected in AD (Braak & Braak 1998, Du et al. 2004, van de Pol et al. 2007). This increased vulnerability may be due to the high concentration of glutamate receptors that mediate communication of the trisynaptic circuit (DG, CA1, CA3) of the hippocampus (Greenamyre & Young 1989). Though the sub-regions of this circuit are connected, they differ in terms of synaptic connectivity, surface expression of glutamate receptors, gene expression profiles, and levels of glutamate release and clearance following evoked release (Gegelashvili & Schousboe 1998, Wilson et al. 2005b, Greene et al. 2009, Talauliker 2010). For these reasons, we examined the sub-regions of the trisynaptic circuit separately. Riluzole’s effects on glutamate regulation in these sub-regions were compared in vivo using MEAs coupled with amperometry. This is the first time riluzole’s effects on glutamate have been examined using this approach, which allows for a high-resolution spatio-temporal study of the complex connections of the trisynaptic loop of the hippocampus in vivo without disrupting extrinsic and intrinsic connections. Results from our work suggest targeting excess hippocampal activity using riluzole may have therapeutic potential for the prevention of AD.
Materials & Methods
Mice
Mice expressing P301L mutant human tau linked to a hereditary tauopathy were created by crossing mice harboring a responder transgene with mice harboring an activator transgene, as previously described (Paulson et al. 2008, SantaCruz et al. 2005). Briefly, activator mice (129s6 background strain) were crossed with responder mice (FVB/N background strain) to create regulatable transgenic mice expressing human four-repeat tau lacking the N-terminal sequences (4R0N) with the P301L mutation. The necessary mice to maintain activator and responder lines were generously donated by Dr. Karen Ashe at the University of Minnesota. Because previously published work suggests developmental P301L tau expression produces alterations not observed following adult-onset tau expression (Caouette et al. 2013), possibly due to the important role of tau in brain development (Wang & Liu 2008), P301L tau expression was suppressed during brain development (Hunsberger et al. 2014b, Hunsberger et al. 2014a). To avoid mutant tau expression during the perinatal and early postnatal stages, 40 ppm doxycycline hyclate (DOX) was administered via water bottles to breeder dams for three weeks prior to mating and to all experimental mice from birth until 2.5 months of age (Hölscher 1999). All mice were housed, between two and five per cage, in a temperature and humidity-controlled colony room with a 12:12 light/dark cycle. All experimental procedures were conducted in accordance with the standards of International Animal Care and Use Committee, and the West Virginia University Animal Care and Use Committee approved all experimental procedures used in the study.
Experimental design
At 2.5 months of age, while still on DOX to suppress tau expression, mice underwent behavioral testing in the water radial arm maze (WRAM) to establish that cognitive deficits were dependent upon tau expression as previously described (Hunsberger et al. 2014b) and to pseudo-randomly assign TauP301L mice to treatment groups (vehicle or riluzole) based on pre-tau behavioral performance (Figure 1). After pre-tau behavioral testing, DOX was removed from the drinking water, and three groups of mice were established: Vehicle-Controls (N = 21; n = 10 female, n= 11 males), Vehicle-TauP301L (N = 24; n = 9 female, n= 15 males), and Riluzole-TauP301L (N = 19; n = 11 female, n= 8 males).
Figure 1. Experimental design.
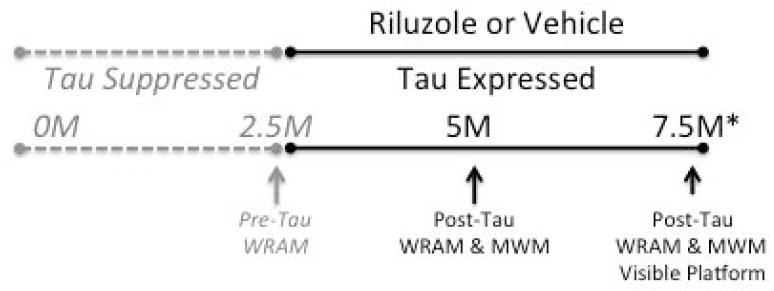
Tau expression was suppressed from conception until 2.5 months (M) of age. Mice underwent testing in the water radial arm maze (WRAM) at 2.5M of age, prior to the onset of tau expression. After WRAM testing at 2.5M, tau expression began, and riluzole or vehicle were administered via the drinking water. Mice underwent behavioral testing at 5M and 7.5M of age, after 2.5M and 5M of tau expression, in the WRAM and Morris water maze (MWM). At the end of MWM testing at 7.5 months of age, the visible platform test was conducted to ensure no differences in visual or motoric function. At the end of visible platform testing, mice underwent anesthetized glutamate recordings (*).
At 5 and 7.5 months of age, after 2.5 and 5 months of tau expression, respectively, mice underwent post-tau cognitive testing in the WRAM and Morris water maze (MWM). One male Vehicle-TauP301L male died during the course of the experiment, thus reducing the total sample size for the final round of behavioral testing at 7.5 months of age. WRAM and MWM testing were separated by one day. Mice underwent visible platform training approximately 30 minutes after the last MWM probe trial at 7.5 months of age. Following behavioral testing, mice underwent in vivo anesthetized glutamate recordings and were euthanized immediately afterwards.
Riluzole administration
Riluzole-TauP301L mice received riluzole (Sigma Aldrich, St. Louis, MO) + 1% w/v saccharin (vehicle) in the drinking water. Riluzole was dissolved by stirring the compound in room temperature tap water, and solutions were changed every 72 h. Light-protectant water bottles were weighed daily, and the concentration of riluzole adjusted every 72 h such that intake remained at ~ 12.5 mg/kg/day (per os) for each cage, a dose previously tested in mice (e.g., Ishiyama et al. 2004, Gourley et al. 2012). The benefit of dissolving riluzole in the drinking water, rather than administering daily injections, is that this is a more robust and easily-replicable administration protocol, administration via drinking water minimizes the stress associated with daily drug injections, and oral administration is the route riluzole is administered in humans. Vehicle-Controls and Vehicle-TauP301L mice consumed saccharin (vehicle) alone. All animals received food ad libitum.
Riluzole has an ideal pharmacokinetic profile (Wagner & Landis 1997), including an absorption of 90% following oral administration, a bioavailability of 60%, peak concentrations within 1-1.5 hours, a 12 hour half-life, and minor side effects. The most commonly reported adverse effects in humans include nausea and asthenia (Miller et al. 2003). The dose used here (12.5 mg/kg/day) was chosen because previous research using mice administered riluzole via their drinking water suggests this dose increases glutamate glial transporter 1 (GLT-1) expression without significantly affecting baseline locomotor activity, thymus and adrenal gland weights, or blood serum corticosterone (Gourley et al. 2012). Similarly, we observed no differences in water consumption or bodyweight among the treatment Groups at 5 or 7.5 months of age (ps > 0.1), and swim speeds in the MWM did not differ at 5 or 7.5 months of age (ps > 0.1), suggesting this dose of riluzole was not toxic and did not produce nausea or asthenia.
Water radial arm maze (WRAM)
The WRAM was performed as previously described (Bimonte-Nelson et al. 2003). The maze was filled with room temperature water (22 °C) and made opaque with nontoxic white paint. The distance from the water level to the top of the maze was approximately 5 cm. Four of the eight arms contained hidden platforms (8 cm × 8 cm) with wire mesh tops located 1 cm below the surface of the water. The location of the platforms were counterbalanced across the groups, but remained fixed for a particular mouse for the duration of testing at a particular age. A platform was never in more than two adjacent arms or in the “start arm” from which a mouse was released. Salient extra-maze cues remained constant for the duration of testing at a particular age. However, both the location of the platform arms and the cues were changed at subsequent ages.
Once released from the start arm, the mouse had 2 minutes to locate a hidden platform. If the allotted time expired, the mouse was guided to the nearest platform. Once a platform was found, the mouse remained on the platform for 15 seconds. At that point, the mouse was removed and placed in the holding cage lined with paper towels and warmed to ~31 °C by a heating pad and heat lamp for 30 seconds to prevent hypothermia. During the interval, the just-chosen platform was removed. The mouse was then placed in the start arm again and allowed to locate another platform. Each mouse was given 4 trials per session and one session per day. One platform was removed after each trial until only one platform remained in trial 4. Thus, each subsequent trial resulted in an increase in memory load, as the mice had to remember not only the locations of the remaining platforms, but also the platforms that had already been found (Bimonte-Nelson et al. 2003).
Each mouse was given 1 session a day for 11 consecutive days. Day 1 was considered a training session because the mice did not have previous experience in the maze, whereas days 2-11 were considered testing sessions for acquisition (Bimonte-Nelson et al. 2003). On day 12, a four-hour delay was inserted between trials 2 and 3. On day 13, a six-hour delay was inserted between trials 2 and 3. Delays were inserted to increase memory demand for the trials following the delay (Engler-Chiurazzi et al. 2011).
An arm entry was defined as all four paws entering into an arm of the maze. Reference memory (REF) errors were defined as the number of first entries into any arm that never contained a platform. Working memory incorrect (WMI) errors were defined as the number of repeat entries into an arm that never contained a platform (i.e., repeat entries into a reference memory arm). Working memory correct (WMC) errors were defined as the number of first and repeat entries into any arm where a platform had been during a previous trial. Errors were analyzed for each daily session for days 2-11. For days 12 and 13, the average number of errors on trials after the delay (trials 3 and 4) was analyzed.
Morris water maze (MWM)
The MWM was performed as previously described (Zhang et al. 2008). Briefly, at 5 months of age, each mouse received 4 days of total testing (6 trials × 2 days + 4 trials × 1 day + 1 probe × 1 day). During hidden platform training, the pool was filled with water room temperature water (22 °C). A platform was hidden 1 cm under the opaque water in one of four quadrants. During hidden platform training (Days 1-3), the mouse was released from pre-determined, semi-random starting locations, and swam for either 60 seconds, or until it reached a hidden platform. Once on the platform for 15 seconds, the mouse was removed and placed in the holding cage lined with paper towels and warmed to ~31 °C by a heating pad and heat lamp. The intertrial interval was 20 minutes. For hidden platform training, pathlength (i.e., distance to the platform) was compared. On Day 4, a probe trial, where the platform was removed, was conducted, and the platform crossing index (PCI) and percent time in the target quadrant were recorded during the 60-second test. A second probe trial was conducted 24 hours later. Salient extra-maze cues remained constant for the duration of testing at a particular age. However, both the location of the platform and the cues were changed at 7.5 months of age.
The initial training at 7.5 months of age was the same as that at 5 months of age (6 trials × 2 days + 4 trials × 1 day), but additional training and probe trials were conducted to determine if further training might reveal greater differences among the Groups. After the probe trial on Day 4, four additional training trials occurred. On Days 5 and 6, a probe trail was conducted followed by four training trials. On Day 7, each mouse underwent four training trials without a probe trial. On Day eight, the last day of MWM testing, each mouse underwent a probe trial only (Hunsberger et al. 2014b). Two mice (one Ril-TauP301L and one Veh-Control) were identified as outliers due to an average hidden pathlength and swim speeds greater than two standard deviations above their group means; these mice were removed from MWM analyses.
Visible platform test
To ensure any deficits observed in the WRAM or MWM were not due to visual or motor deficits, the visible platform test was performed as previously described (Bimonte-Nelson et al. 2003) at the end of MWM testing at 7.5 months of age. Testing was conducted in a tub partially filled with room temperature water (22 °C) and made opaque with non-toxic white paint. A black platform with a flag raised was placed approximately 2.5 cm above the water level. Each mouse was placed into the tub facing the wall opposite the platform and given 2 minutes to swim to the platform. If the mouse found the platform within the allotted time limit, the mouse remained on the platform for 20 seconds; if the mouse did not find the platform, the mouse was gently guided to the platform. After each mouse completed the first trial, the platform was moved to a new location along the back wall of the tub. All mice were given 5 independent trials with approximately 10 minutes between each trial.
Enzyme-based microelectrode arrays (MEAs)
Ceramic-based MEAs, consisting of a ceramic-based multisite microelectrode with 4 platinum recording sites (Burmeister & Gerhardt 2001), were used to examine glutamate regulation and were purchased from Quanteon, L.L.C. (Nicholasville, KY). These sites were arranged in dual pairs to allow interfering agents to be detected and removed from the analyte signal (Burmeister & Gerhardt 2001). Coating of the microelectrodes has been described previously (Hinzman et al. 2010). Briefly, the recording sites were covered with glutamate-oxidase (GluOx) to oxidize glutamate to alpha-ketoglutarate and hydrogen peroxide (H202), the reporter molecule (Burmeister & Gerhardt 2001). For the other sites (sentinel sites), an inactive protein matrix was used to block larger molecules such as ascorbic acid or monoamines. Small molecules, like H202, can diffuse through the mPD exclusion layer. The background current from the sentinel sites was then subtracted from the recording sites to produce a selective measure of extracellular glutamate. A reference electrode Ag/AgCl was implanted into a remote site from the recording area (Burmeister & Gerhardt 2001).
MEA Calibration
To ensure sensitivity and selectivity and to create a standard curve for the conversion of current to glutamate concentration, calibrations were conducted on the MEAs prior to their use. Using the FAST-16 mkII system (Quanteon), a constant potential of + 0.7 V versus an Ag/AgCl reference was applied to the MEA to oxidize the reporter molecule. The resulting current was amplified, digitized, and filtered by the FAST 16 mkII system. The MEA tip was submerged in 40 mL of a 0.05 M phosphate-buffered saline maintained at 37°C. A standard curve was determined by adding successive aliquots of 20 μL glutamate to achieve concentrations of 20, 40, and 60 μM. The increase in current (nA) produced by oxidation was used to calculate the calibration slope to a known concentration of glutamate (Burmeister, et al., 2002). Ascorbic acid (250 μM) and dopamine (2 μM) were added to the solution to determine selectivity for glutamate (Hinzman et al. 2012). To determine the limit of detection (LOD), the smallest concentration of glutamate that can be measured by the device, the slope of the standard curve was used, as well as the noise or relative standard deviation of the baseline signal (Hinzman et al. 2012).
MEA/Micropipette assembly
A glass micropipette with an inner diameter tip of 10-15 μm (Quanteon) was attached to the MEA for intracranial drug deliveries. The micropipette was centered between the dorsal and ventral platinum recording pairs and positioned 80-100 μm away from the MEA surface. Location of the micropipette to the MEA was verified post-surgery to ensure that the pipette did not move. The micropipette was back-filled with sterile-filtered isotonic 70 mM KCl solution or 200 μM glutamate solution. The micropipette was attached to a Picospritzer III (Parker-Hannifin, Cleveland, OH) and set to consistently deliver volumes of 50-100 nL. Pressure was applied from 0.138 – 1.38 bar for .30 - 2.5 sec. A stereomicroscope fitted with a reticule was used to monitor volume displacement (Friedemann & Gerhardt 1992).
In vivo anesthetized recordings
Mice were anesthetized with isoflurane (1-4% inhalation; continuous) and placed into a stereotaxic apparatus. Isoflurane was chosen because other anesthetics have been shown to alter resting glutamate levels, whereas isoflurane does not (Mattinson et al. 2011). Though initial reports suggested isoflurane increases tau phosphorylation (Planel et al. 2004), more recent reports suggest that when anesthesia-induced hypothermia is controlled for, isoflurane does not increase tau phosphorylation (Tan et al. 2010). To ensure our mice did not become hypothermic while under anesthesia, body temperature was continuously measured using a rectal probe and maintained at 37°C with a water pad connected to a recirculating water bath (Gaymar Industries Inc., Orchard Park, NY).
The MEA/micropipette array was placed into the DG, CA3, and CA1 of the hippocampus. Stereotaxic coordinates for the different sub-regions of the hippocampus were calculated using the mouse brain atlas (Paxinos & Watson 2004) [DG (AP: -2.3mm, ML: +/-1.5mm, DV: 2.1mm), CA3 (AP: -2.3mm, ML: +/-2.7mm, DV: 2.25mm), CA1 (AP: -2.3mm, ML: +/-1.7mm, DV: 1.4mm)].
Prior studies have shown that the MEAs produce minimal effects both acutely and chronically (Hascup et al. 2009). To confirm the stereotaxic coordinates targeted the regions of interest, an MEA with an attached micropipette was used to locally apply Fluoro-Ruby (Millipore), and MEA placement following brain sectioning was confirmed, as previously shown (Hunsberger et al. 2014a). However, because brain tissue was used for immunoblotting, the MEA placement was not confirmed for each mouse. All MEA recordings were performed at 10 Hz using constant potential amperometry recordings with the FAST-16. After the MEA reached a stable baseline (10-20min), tonic glutamate levels (μM) were calculated by averaging extracellular glutamate levels over 10 seconds prior to any application of solutions. In all three sub-regions of one hemisphere, evoked release (i.e., amplitude) was measured by local application of KCl delivered every 2-3 minutes. KCl-evoked release of glutamate is highly reproducible and indicative of the intact glutamate neuronal system that is detected by the MEAs (Day et al. 2006). After 10 reproducible signals, the results were averaged for each group and the average amplitude compared (Hinzman et al. 2010, Hinzman et al. 2012, Nickell et al. 2007). KCl-evoked release of glutamate was measured to determine the “capacity” of the nerve terminals to release glutamate (Hinzman et al. 2010).
To examine glutamate uptake, exogenous glutamate was applied in the opposite hemisphere. After the MEA reached a stable baseline (10-20 min), varying volumes of 200 μM sterile-filtered glutamate solution were applied into the extracellular space every 2-3 minutes. The net area under the curve (AUC) was used as a measure of glutamate uptake. The hemispheres used for KCl and glutamate application were counterbalanced, as was the order of sub-regions within a hemisphere. Data from some hippocampal regions were excluded for reasons including death during surgery, failure of the MEA, or clogging of the micropipette. For each glutamate-related measure, the number of mice per treatment group is indicated in the corresponding figure caption.
Immunoblotting
Immunoblotting steps have been described in detail previously (Hunsberger et al. 2014a). Briefly, hippocampal tissue was prepared for immunoblotting using 500 ul extraction buffer with protease inhibitors. Protein concentrations were determined with a BCA protein assay using BSA as a standard. Hippocampal tissue samples were thawed and 10 μg aliquots were mixed with loading buffer. Before loading, samples were either heated to 70°C (vGLUT1 and synaptophysin) or 95°C (for all other proteins) for 5 min and then separated on 10% criterion gels (Biorad), and transferred onto .45 μm PVDF membranes (Millipore, Bedford, MA). Membrane blots were blocked for 1 h at RT in 5% BSA in 0.1% TTBS or 5% milk in TTBS. After blocking, membranes were incubated with an antibody directed against the protein of interest overnight at 4°C. The next day, membranes were incubated with Streptactin-HRP (Biorad) and the appropriate biotinylated or HRP-conjugated secondary antibody for 1.5 h at RT. Blots were then incubated with SuperSignal West Pico chemiluminescent substrate (Thermo Scientific) or Novex AP chemiluminescent substrate (Invitrogen) for 5 minutes, and visualized using Fluorchem E imager (Cell Biosciences). Membranes were stripped for 15 minutes with Restore PLUS western blot stripping buffer (Peirce Chemical) and re-probed and normalized to synaptophysin or actin. Band density was measured using AlphaView software (Proteinsimple, Santa Clara, California, USA).
Data analysis
Amperometric data were analyzed using a custom Microsoft excel software program (MatLab). To determine concentrations of glutamate in the hippocampus, the background current from the sentinel sites was subtracted from the signal obtained from the GluOx recording sites. The resting current (pA) was divided by the slope (μM/pA) obtained during calibration and reported as a concentration of glutamate.
All statistical analyses were performed using JMP (SAS, Cary, NC 27513). Statistical analysis consisted of ANOVA and repeated-measures ANOVA (RMANOVA). For all measures, the main effects of Group (Veh-Controls, Veh-P301L, Ril-P301L) and Sex (male, female), as well as the interaction between the two (Group*Sex) were assessed. For the RMANOVA of behavioral data, Trial or Probe served as the within-subject variables. All significant omnibus tests were followed by Tukey post hoc comparisons. Using Pearson r correlations, KCl-evoked glutamate release (amplitude) and glutamate uptake (net AUC) in the DG, CA3, and CA1 were correlated separately with performance in the MWM. Correlations were run only for those mice in which data for both behavior and glutamate were analyzed.
The critical alpha level was set to 0.05, and all values in the text and figures represent means ± SEM. Unless otherwise noted, there were no differences between the sexes and no Group*Sex interactions, and thus, focus is given to the effect of Group. Due to limitation of space, only significant results are graphed.
Results
Riluzole rescues cognitive deficits in TauP301L Mice
Pre-Tau WRAM performance
At 2.5 months of age, prior to the onset of tau expression, mice underwent WRAM testing to ensure no pre-tau differences in behavior and to assign TauP301L mice to treatment (riluzole or vehicle) groups. For acquisition, there was a within-subject effect of Trial for all 3 dependent measures, i.e., reference memory errors (REF), working memory incorrect (WMI), or working memory correct (WMC), but no main effects of Group, Sex, or Group*Sex interactions, nor any interactions with Trial (ps > 0.1), suggesting similar acquisition across the groups. Similarly, during delay trials, there were no main effects of Group, Sex, or Group*Sex interactions for REF, WMI, or WMC (ps > 0.1), indicating similar performance prior to tau expression.
Post-tau WRAM acquisition
At 5 and 7.5 months of age, after assignment to either vehicle or riluzole treatment and 2.5 and 5 months of tau expression, respectively, mice were again tested in the WRAM with new extra-maze cues and a reassignment of arms containing platforms at each age. During acquisition at 5 months of age, there was a main effect of Sex for REF [F(1,58) = 5.86, p = .02], WMI [F(1,58) = 5.10, p = .03], and WMC [F(1,58) = 4.21, p = .04], such that females performed significantly better than males. However, there was not a main effect of Group or a Group*Sex interaction, or any interactions with Trial, for any measure (ps > 0.1), suggesting similar acquisition. When mice underwent testing again at 7.5 months of age, acquisition was similar across the groups, including the sexes, for all measures (ps > 0.1).
Post-tau WRAM delay trials
To increase memory demand, a 4-hour or 6-hour delay was inserted between Trials 2 and 3 on Day 12 and Day 13, respectively. Memory retention for the post-delay trials (Trials 3 and 4) within that day was assessed, as previously described (Engler-Chiurazzi et al. 2011, Braden et al. 2010). We observed no impairing effects of the 4-hour delay among the groups for any error measure at either age (ps > 0.1). However, insertion of a longer delay (6-hours) between Trials 2 and 3 on Day 13 revealed impaired performance for WMI in Veh-TauP301L mice at 5 [F(2,58) = 4.18, p = .02] and 7.5 [F(2,57) = 3.19, p = .05] months of age, an effect rescued by riluzole treatment (Figure 2).
Figure 2. Riluzole rescues cognitive deficits associated with P301L tau expression in the water radial arm maze.
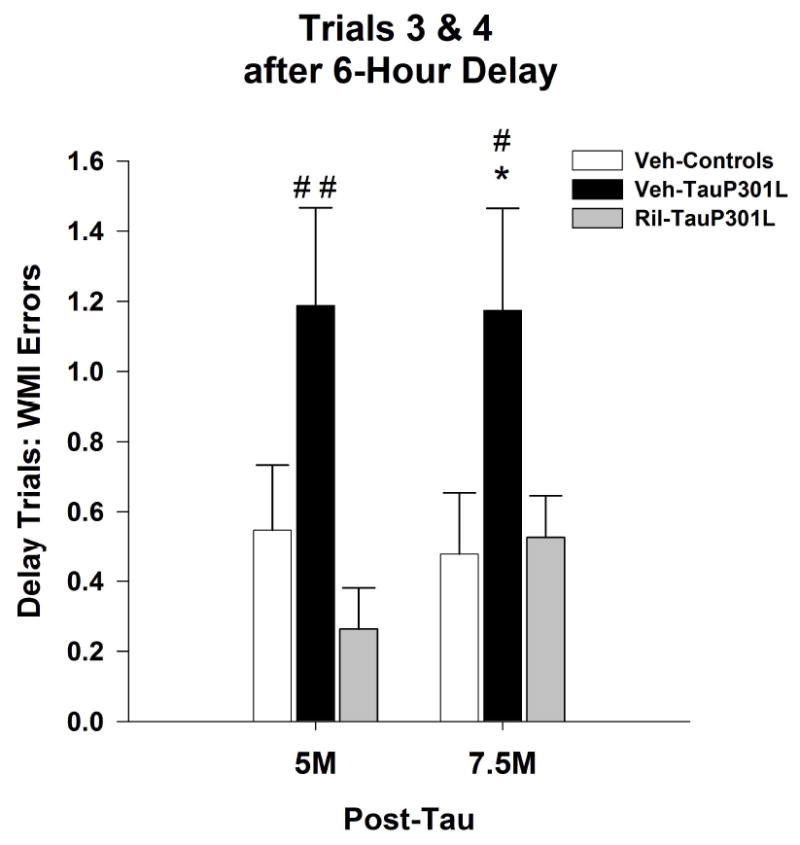
Following the insertion of a 6-hour delay between trials 2 and 3, TauP301L mice exhibited significantly more working memory incorrect (WMI) errors at 5 and 7.5M of age, an effect attenuated by riluzole treatment. (Mean ± SEM; * p<.05 Veh-Control vs. Veh-TauP301L, # p<.05 Ril-Tau-301L vs.Veh-TauP301L, ## p<.01 Ril-Tau-301L vs. Veh-TauP301L).
Morris water maze
Swim speed did not differ among the groups at any age tested (ps > 0.1), suggesting similar motoric functioning. At 5 months of age, Veh-TauP301L mice exhibited significantly longer pathlengths to the hidden platform [Group: F(2,56) = 3.72, p = .03; Figure 3A]. For the two probe trials employed at 5 months, there were no differences among the groups for the percent time in the target quadrant [F(2,56) = 0.84, p = .44] or the platform crossing index [F(2,56) = .66, p = .52].
Figure 3. Riluzole improves performance of TauP301L mice in the Morris water maze (MWM).
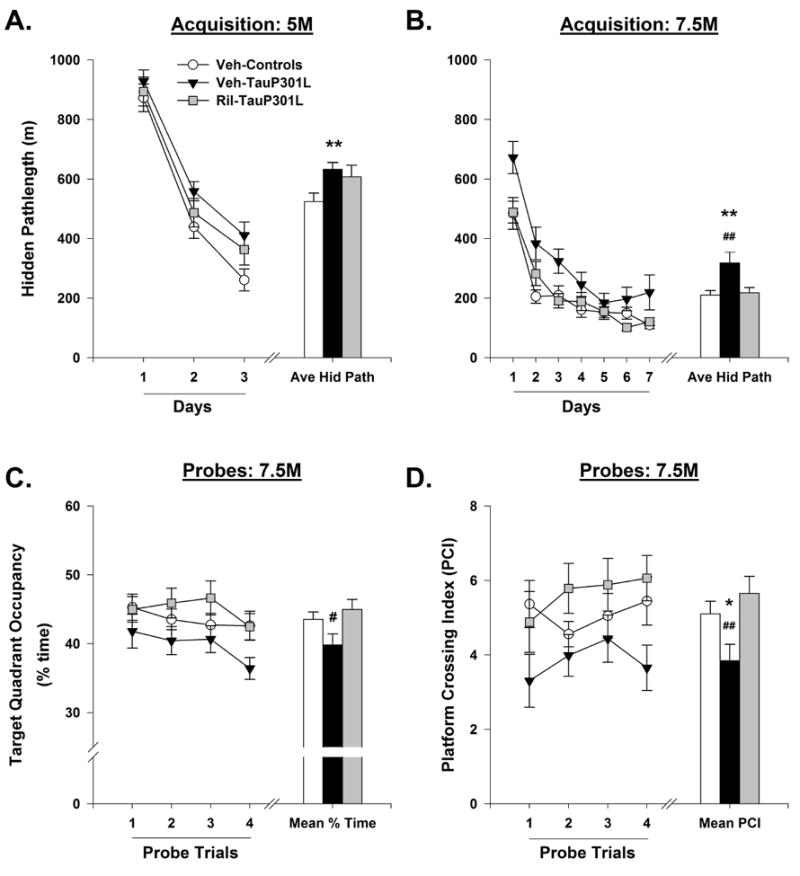
Veh-TauP301L mice exhibited longer pathlengths during hidden platform training at 5 (A) and 7.5 (B) months of age. At 7.5 months of age, riluzole treatment rescued the deficits in spatial reference memory, as indicated by an increase in time spent in the target quadrant (C) and the platform crossing index (D). Average hidden pathlength; Ave Hid Path; Months, M; Platform crossing index, PIC. (Mean ± SEM; * p<.05 Veh-Control vs. Veh-TauP301L, ** p<.01 Veh-Control vs. Veh-TauP301L, # p<.05 Ril-Tau-301L vs.Veh-TauP301L, ## p<.01 Ril-Tau-301L vs.Veh-TauP301L).
At 7.5 months of age, comparison of the pathlength to the hidden platform revealed a between-subject effect of Group; the average hidden pathlength for Veh-TauP301L mice was significantly longer than that of Veh-Controls and Ril-TauP301L mice [F(2,55) = 5.48, p = .007; Figure 3B]. Analysis of spatial reference memory during the 4 probe trials employed at 7.5 months of age revealed a significant main effect of Group for the percent time in the target quadrant [F(2,55) = 3.33, p = .04; Figure 3C], as well as the platform crossing index [F(2,55) = 3.54, p = .04; Figure 3D]; riluzole improved probe trial performance in TauP301L mice.
Visible platform
To determine whether visual or motor deficits were present in any mice, thus affecting the results and interpretations of behavioral testing, visible platform training was performed at the conclusion of WRAM and MWM behavioral testing. The average time to locate the visible platform did not differ among groups [Group: F(2,55) = 1.92, p = .16; Sex: F(1,55) = 1.33, p = .25; Group*Sex: F(2,55) = 2.45, p = .10], suggesting similar orientation, visual, and motoric functioning.
Riluzole rescues glutamate alterations in TauP301L mice
Tonic glutamate levels were not significantly different in the DG [F(2,44) = 1.53; p = .23] among the groups. However, in the CA3 [F(2,47) = 3.42; p = .04] and CA1 [F(2,47) = .8.29; p < .001] regions, Veh-TauP301L mice exhibited increased tonic glutamate levels, an effect rescued by riluzole treatment (Figure 4A). To examine the capacity for glutamate release, KCl was delivered via a micropipette. Local application of 50-100 nL of 70 mM KCl produced reproducible glutamate release in all regions of the hippocampus (Figure 4B). The amplitudes of KCL-evoked-glutamate release were significantly increased in Veh-TauP301L mice in the DG [F(2,44) = 5.60; p = .007], CA3 [F(2,46) = 13.49; p < .0001] and CA1 [F(2,44) = 5.64, p = .007]. Riluzole treatment rescued the P301L-mediated increase in KCl-evoked glutamate release in all 3 sub-regions (Figure 4C).
Figure 4. Extracellular tonic and potassium chloride (KCl)-evoked release of glutamate in the DG, CA3, and CA1 regions of the hippocampus.
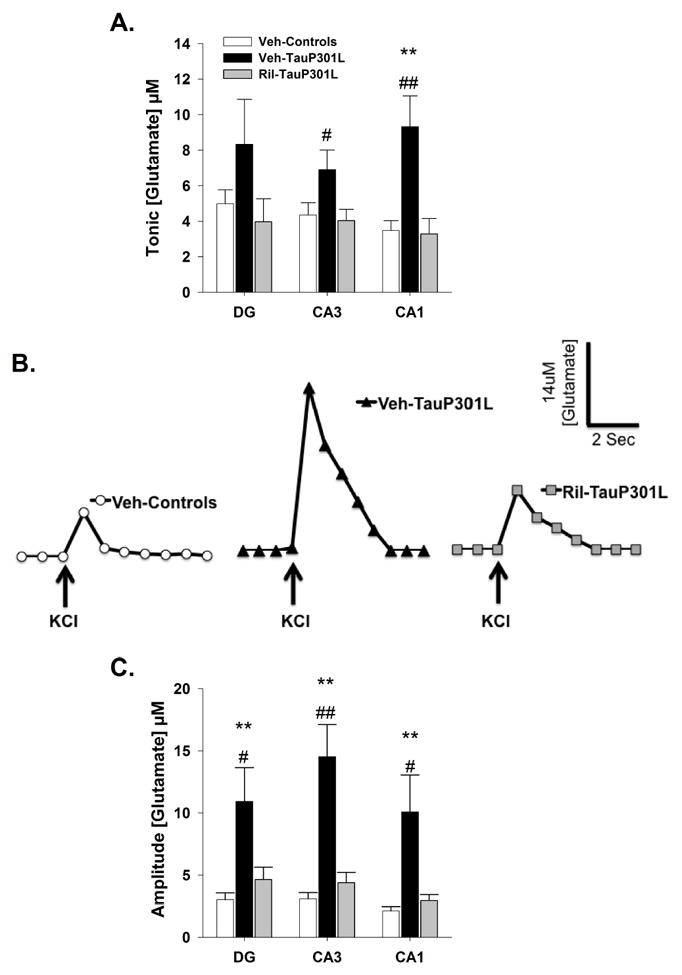
(A) In the CA3 and CA1 regions of the hippocampus, tonic glutamate levels were significantly increased in Veh-TauP301L mice, an effect attenuated by riluzole treatment. (B) Baseline-matched representative recordings of KCl-evoked glutamate release in the CA3 showed riluzole-treatment attenuated the significant increase in the amplitude of glutamate release observed in Veh-TauP301L mice. Local application of KCl (↑) produced a robust increase in extracellular glutamate that rapidly returned to tonic levels. (C) The significantly increased KCl-evoked glutamate release observed in Veh-TauP301L mice in the DG, CA3, and CA1 after local application of KCl was attenuated with riluzole treatment. (Mean ± SEM; ** p<.01 Veh-Control vs. Veh-TauP301L, # p<.05 Ril-Tau-301L vs.Veh-TauP301L, ## p<.01 Ril-Tau-301L vs.Veh-TauP301L; n = 14-19/group).
Rapid application of glutamate into the extracellular space allowed us to mimic endogenous glutamate release and to examine glutamate uptake in vivo. To ensure differences in net area under the curve (AUC) among the groups following application of endogenous glutamate were due to alterations in uptake and not differences in the amount of endogenous glutamate applied, we first compared the amplitude of glutamate signals following administration of exogenous glutamate; no differences in amplitude were observed among the mice in the DG [F(2,37) = 2.57; p = .09], CA3 [F(2,40) = 0.55; p = .58], and CA1 [F(2,41) = 2.77; p = .08] (Figure 5A), suggesting similar applications of exogenous glutamate. We next examined Trise, the time for the signal to reach maximum amplitude, to ensure no differences in the diffusion of glutamate in the extracellular space; Trise was not significantly different among the groups in the DG [F(2,37) = 1.21; p = .31], CA3 [F(2,40) = 1.55; p = .23], and CA1 [F(2,41) =1.54; p = .23] (Figure 5B), suggesting any reductions in glutamate uptake were not due to diffusion from the point source (micropipette) to the MEA (Sykova et al. 1998). Because neither amplitude nor Trise differed among the groups, any differences in net AUC likely results from decreases in glutamate uptake. Following exogenous application of glutamate, Veh-TauP301L mice exhibited an increased net AUC in the DG [F(2,37) = 6.49, p = .004], CA3 [F(2,40) = 7.63; p = .0016], and CA1 [F(2,41) = 4.23; p = .02], suggesting reduced glutamate uptake in all 3 sub-regions of the hippocampus. Riluzole treatment improved glutamate uptake in all 3 regions (Figure 5C,D).
Figure 5. Glutamate uptake following exogenous glutamate application in the DG, CA3, and CA1 regions of the hippocampus.

(A) The amplitude of glutamate signal was similar among groups in each region. (B) Trise, an indicator of glutamate diffusion, was similar among the groups in each region. (C) Representative glutamate signals in the CA3 from local application of glutamate in Veh-Controls, Veh-TauP301L, and Ril-TauP301L mice. (D) Riluzole treatment reduced the significant increases in the net area under the curve (AUC) observed in Veh-TauP301L mice in all 3 regions of the hippocampus, indicating improved glutamate uptake in riluzole-treated TauP301L mice. (Mean ± SEM; * p<.05 Veh-Control vs. Veh-TauP301L, ** p<.01 Veh-Control vs. Veh-TauP301L, # p<.05 Ril-Tau-301L vs.Veh-TauP301L, ## p<.01 Ril-Tau-301L vs.Veh-TauP301L; n = 13-15/group).
Glutamate alterations correlate with cognitive deficits in TauP301L mice
To determine whether glutamate (baseline, evoked release, and clearance) correlated with cognitive performance at 7.5 months of age, we first identified the behavioral outcome most sensitive to P301L tau expression. Calculating the effect size, we determined that the average hidden pathlength in the MWM was the most sensitive measure (η2 = 0.16) compared to other MWM measures, including percent time in the target quadrant (η2 = 0.11) and PCI (η2 = 0.15), as well as working memory incorrect errors in the WRAM (η2 = 0.09).
The average hidden pathlength was significantly correlated with tonic glutamate levels in the CA3 (p = .0025) and CA1 (p = .023) but not the DG (p = .08) of TauP301L mice. As previously reported (Hunsberger et al. 2014a), performance was significantly correlated with glutamate uptake (net AUC) in the DG (p = .028) and CA1 (p = .0056) but not the CA3 (p = .22), while the opposite pattern was observed for amplitude of evoked glutamate release. KCl-evoked release in the CA3 was significantly correlated with MWM performance (p = .0116), whereas for the DG and CA1 regions, there was no relationship between release and performance (p = .68 and p = .22, respectively).
Riluzole rectifies alterations of the tripartite synapse
As previously reported (Hunsberger et al. 2014a), hippocampal vGLUT1 expression was significantly increased in Veh-TauP301L mice [F(2,35) = 9.09; p = .0007], an effect rescued by riluzole treatment (Figure 6A). This difference in vGLUT1 expression was not due to a widespread increase in pre-synaptic terminals, as indicated by similar synaptophysin expression among the groups [F(2,35) = 0.19; p = .82]. Riluzole treatment also increased the P301L-mediated decrease in GLT-1 expression previously observed in TauP301L mice [F(2,35) = 8.37, p = .0011; Figure 6B]. There were no differences among the groups for the loading control, beta-actin [F(2,35) = 2.29, p = .12]. PSD-95, a major postsynaptic scaffold protein at excitatory synapses, was used as a marker of excitatory synapses in the hippocampus. Riluzole treatment rescued the reduction in PSD-95 expression observed in Veh-TauP301L mice ([F(2,35) = 5.32, p = .01]; Figure 6C).
Figure 6. Riluzole rectifies alterations in tripartite synapse associated with P301L tau expression.
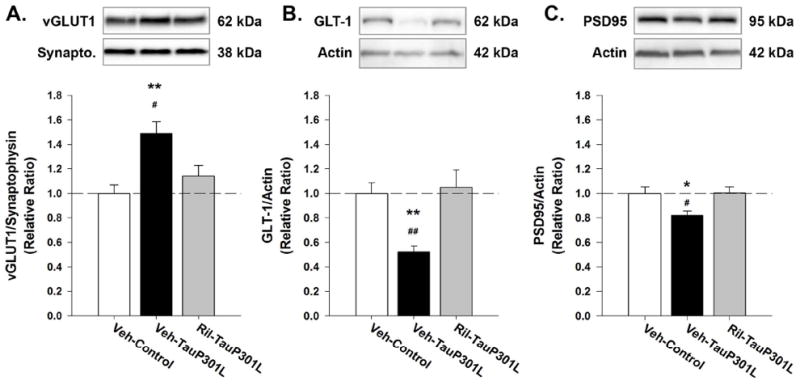
(A) Riluzole treatment rescued the P301L-mediated increase in vGLUT1 expression observed in TauP301L mice. (B) Riluzole increased GLT-1 expression in TauP301L mice. (C) PSD-95 expression was increased in riluzole-treated TauP301L mice. (Mean ± SEM; * p<.05 Veh-Control vs. Veh-TauP301L, ** p<.01 Veh-Control vs. Veh-TauP301L, # p<.05 Ril-TauP301L vs.Veh-TauP301L, ## p<.01 Ril-Tau-301L vs.Veh-TauP301L; n = 12-14/group).
Riluzole attenuates tau pathology
We next determined whether riluzole treatment could attenuate tau pathology. Early changes in tau were examined using CP-13 and MC-1, which detect phosphorylation (pSer202) and conformation-specific changes, respectively. Both hippocampal CP-13 [F(2,34) = 25.72, p < .0001] and MC-1 [F(2,35) = 113.95, p < .0001] were increased in Veh-TauP301L mice. Riluzole treatment significantly decreased the immunoreactive signal for tau hyperphosphorylated at residue S202 (CP-13) and the conformational epitope (7–9 and 326–330 aa) recognized by MC-1 (Figure 7A, B). In addition, detection of total tau (human and mouse) with the Tau-5 antibody revealed a significant reduction in total tau levels in the hippocampus following riluzole treatment [F(2,35) = 42.37, p < .0001; Figure 7C].
Figure 7. Riluzole rescues tau pathology.
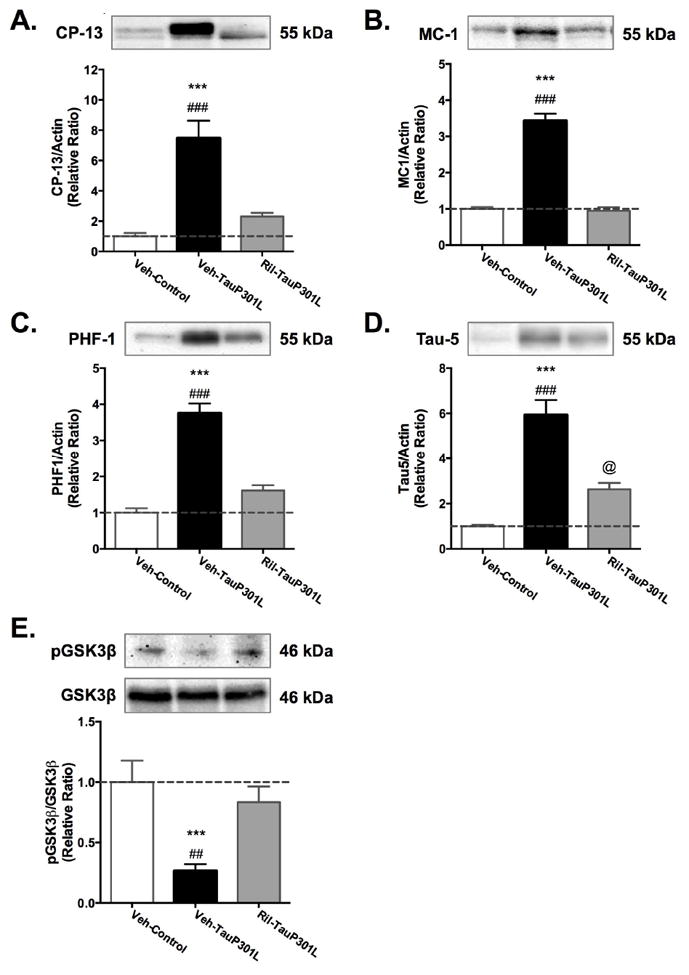
Treating mice with riluzole reduced tau phosphorylation (A) and conformational changes (B) to control levels. Total tau levels were also significantly reduced by riluzole (C). (Mean ± SEM; * p<.05 Veh-Control vs. Veh-TauP301L, ** p<.01 Veh-Control vs. Veh-TauP301L, *** p<.001 Veh-Control vs. Veh-TauP301L, # p<.05 Ril-Tau-301L vs.Veh-TauP301L, ## p<.01 Ril-Tau-301L vs.Veh-TauP301L, ### p<.01 Ril-Tau-301L vs.Veh-TauP301L; @ p<0.5 Veh-Control vs. Ril-TauP301L;n = 12-14/group).
Discussion
Recent work suggests tau may mediate hyperexcitability. For example, deletion of tau in models of epilepsy reduces hyperexcitability, seizure frequency, and duration (Holth et al. 2013, DeVos et al. 2013). Seizure severity is also reduced in tau knockout mice following convulsant administration (Ittner et al. 2010, Roberson et al. 2007). Though the exact mechanism remains to be determined, our current findings add to a body of literature suggesting that tau influences hyperexcitability through its effects on glutamate neurotransmission (Roberson et al., 2011; Roberson et al., 2007(Hunsberger et al. 2014a). Here, we also present evidence that rectifying alterations in glutamatergic circuits can rescue cognitive deficits and tau pathology associated with P301L tau expression.
Though the effects of riluzole on extracellular glutamate have been examined in vivo using microdialysis (e.g., Kwon et al. 1998), this is the first report of riluzole’s effects on the rapid time dynamics of extracellular glutamate as measured by in vivo amperometry, which has many benefits over other in vivo or ex vivo methods. For example, with studies employing microdialysis to measure glutamate, there are often spatial and temporal limitations that restrict the ability to sample dynamic changes in glutamate near the synapse (Hillered et al. 2005, Obrenovitch et al. 2000). Damage caused by the large sampling area (1–4 mm in length) limits the detection of calcium and sodium dependent neuronal release (Borland et al. 2005, Jaquins-Gerstl & Michael 2009), and the low temporal resolution (1–20 min) is inadequate to measure the fast dynamics of transient release and uptake of glutamate (Diamond 2005). The MEAs allow for such measures due to their high temporal resolution (2 Hz), low limit of detection (<0.5 μM), and high spatial resolution, allowing for selective measurement of extracellular glutamate closer to synapses (Burmeister & Gerhardt 2001, Burmeister et al. 2002, Hascup et al. 2010, Rutherford et al. 2007). Another benefit of MEAs over other ex vivo methods is the ability to study brain regions in vivo without disrupting their extrinsic and intrinsic connections, a particularly important consideration when examining the complex neural networks of the hippocampus.
Using this technique, we observed increases in both tonic and evoked glutamate release and decreases in glutamate uptake in TauP301L mice. Riluzole treatment appeared to return these shifts in glutamate regulation to control levels. In vitro studies support that a major portion of tonic glutamate is mediated by glia-dependent release of glutamate, and not vesicular glutamate release (Jabaudon et al. 1999; Cavelier and Attwell 2005; Le Meur et al. 2007). However, our MEAs appear to measure resting glutamate levels that are diminished by ~50% by inhibitors of calcium and sodium channels (Hascup et al. 2010), supporting that resting glutamate has a major neuronal component. Similarly, local application of TBOA to inhibit glutamate transporters leads to an increase in tonic glutamate levels, suggesting transporters also help maintain normal tonic glutamate levels (Day et al. 2006, Hascup et al. 2010). Delineation of the possible mechanisms mediating the increase tonic levels in TauP301L mice will be carried out in future studies. Interestingly, in our previous work (Hunsberger et al. 2014a), we did not observe differences in tonic (resting) glutamate levels between Controls and TauP301L mice. However, in the present study, TauP301L mice exhibited increased levels of tonic glutamate, particularly in the CA3 and CA1 regions. This difference in findings between studies may be due to the duration of tau expression at the time of testing. In the original study (Hunsberger et al. 2014a), mice expressed tau for approximately 3 months, whereas in the current study, mice expressed tau for approximately 5 months. Thus, with longer durations of tau expression, tonic glutamate may also become deregulated by P301L tau expression, a hypothesis that warrants further testing.
An interesting phenomenon, observed in both our previous study (Hunsberger et al. 2014a) and the current study, is the sub-regional relationships between extracellular glutamate alterations and behavioral deficits. In both studies, glutamate release in the CA3, but not the DG or CA1, was correlated with cognitive performance in TauP301L mice, whereas glutamate uptake in the DG and CA1, but not the CA3, was associated with cognitive deficits. At this time, we can only speculate as to the reason for these sub-regional relationships with cognitive performance, though the circuitry of the hippocampus may offer some clues, particularly for the negative correlation between glutamate release in the CA3 and cognitive performance. In the trisynaptic loop of the hippocampus, flow is mainly unidirectional with information entering the loop via the entorhinal cortex with projections running from the DG to the CA3 to the CA1 and back again to the EC. CA3 neurons also receive more than 95% of their input from recurrent CA3 collaterals, referred to as “auto-associative” tracts. It is these recurrent CA3 collaterals that may make the CA3 particularly susceptible to increases in glutamate release. Support for this comes from studies examining hippocampal activity in cognitively impaired, aged rodents and humans; the CA3 is notably the most hyperexcitable region and this hyperexcitability correlates with cognitive performance (Wilson et al. 2005a, Bakker et al. 2012a, Yassa et al. 2010). Reducing CA3 hyperactivity improves memory in aged rats (Koh et al. 2010). Examination of the effects of sub-regional manipulations of glutamatergic activity on cognitive performance will help address these issues, as will studies examining the temporal relations of these circuits with aging and longer durations of P301L tau expression.
Increased hippocampal activation in MCI is predictive of the degree and rate of cognitive decline, as well as the conversion to AD (Mackenzie & Miller 1994). Recent work sheds light on one way in which hyperactivity might be permissive for the development of AD. In AD, tau - typically an intracellular protein - is released into the extracellular space and endocytosed by neighboring neurons (Liu et al. 2012). This spread occurs along synaptically connected circuits, resulting in a prion-like cell-to-cell transmission of tau pathology. Relevant to the current paper is the finding that glutamate release and stimulation of glutamate receptors induces tau release from neurons into the extracellular space (Yamada et al. 2014, Pooler et al. 2013). Thus, glutamate-mediated exocytosis of tau may indicate one mechanism for the trans-synaptic spread of tau pathology associated with synaptic activity. This could also result in a vicious feed-forward cycle whereby tau pathology increases glutamate signaling, which then propagates the spread of tau pathology. Prevention of this spread may be one means by which riluzole-treatment reduced total tau levels. Further studies are needed to establish the relevance of increased glutamate signaling to the spread of tau pathology.
Table 1.
Correlations between glutamate dysregulation & average hidden pathlength in the Morris water maze for Veh-TauP301L mice (significant p-values in bold).
| DG | CA3 | CA1 | |
|---|---|---|---|
|
|
|||
| Tonic vs. Pathlength | Path=278+3.4*Base | Path=237+4.7*Base | Path=237+6.7*Base |
| r2 (19)= .17, p = .08 | r2 (19)= .43, p = .0025 | r2 (19)= .27, p = .0231 | |
|
|
|||
| KCl-evoked Release vs. Pathlength | Path=315+1.3*Amp | Path=253+3.1*Amp | Path=263+4.1*Amp |
| r2 (18)= .01, p = .68 | r2 (18)= .64, p = .0116 | r2 (17)= .10, p = .22 | |
|
|
|||
| Clearance vs. Pathlength | Path=248+.37*AUC | Path=259+.27*AUC | Path=231+.43*AUC |
| r2(14)= .34, p = .028 | r2(14)= .12, p = .22 | r2(14)= .49, p = .0056 | |
|
|
|||
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (Reed - U54GM104942), the Alzheimer’s Association (Reed - NIRG-12-242187), a WVU Faculty Research Senate Grant, and a WVU PSCOR grant. GG is the sole proprietor of Quanteon, LLC that makes the Fast-16 recording system used for glutamate measurements in this study.
Abbreviations
- AD
Alzheimer’s Disease
- DG
Dentate gyrus
- CA3
Cornu Ammonis 3
- CA1
Cornu Ammonis 1
- vGLUT-1
vesicular glutamate transporter 1
- GLT-1
glutamate transporter 1
- MEA
microelectrode array
- ALS
amyotrophic lateral sclerosis
- tTA
Tetracycline-Controlled Transcriptional Activation
- CaMKII
calcium/calmodulin kinase II
- DOX
doxycycline
- WRAM
water radial arm maze
- MWM
Morris water maze
- WMC
working memory correct
- REF
reference
- WMI
working memory incorrect
- PCI
platform crossing index
- GluOx
glutamate-oxidase
- KCl
potassium chloride
- AUC
area under the curve
- Aβ
beta-amyloid
References
- Azbill RD, Mu X, Springer JE. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Res. 2000;871:175–180. doi: 10.1016/s0006-8993(00)02430-6. [DOI] [PubMed] [Google Scholar]
- Bakker A, Krauss Gregory L, Albert Marilyn S, et al. Reduction of Hippocampal Hyperactivity Improves Cognition in Amnestic Mild Cognitive Impairment. Neuron. 2012a;74:467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker A, Krauss GL, Albert MS, et al. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012b;74:467–474. doi: 10.1016/j.neuron.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett SS, Yousem DM, Cristinzio C, Kusevic I, Yassa MA, Caffo BS, Zeger SL. Familial risk for Alzheimer’s disease alters fMRI activation patterns. Brain. 2006;129:1229–1239. doi: 10.1093/brain/awl089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bimonte-Nelson HA, Hunter CL, Nelson ME, Granholm AC. Frontal cortex BDNF levels correlate with working memory in an animal model of Down syndrome. Behav Brain Res. 2003;139:47–57. doi: 10.1016/s0166-4328(02)00082-7. [DOI] [PubMed] [Google Scholar]
- Bondi MW, Houston WS, Eyler LT, Brown GG. fMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer disease. Neurology. 2005;64:501–508. doi: 10.1212/01.WNL.0000150885.00929.7E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, Small GW. Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med. 2000;343:450–456. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borland LM, Shi G, Yang H, Michael AC. Voltammetric study of extracellular dopamine near microdialysis probes acutely implanted in the striatum of the anesthetized rat. Journal of neuroscience methods. 2005;146:149–158. doi: 10.1016/j.jneumeth.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Argyrophilic grain disease: frequency of occurrence in different age categories and neuropathological diagnostic criteria. J Neural Transm. 1998;105:801–819. doi: 10.1007/s007020050096. [DOI] [PubMed] [Google Scholar]
- Braden BB, Talboom JS, Crain ID, Simard AR, Lukas RJ, Prokai L, Scheldrup MR, Bowman BL, Bimonte-Nelson HA. Medroxyprogesterone acetate impairs memory and alters the GABAergic system in aged surgically menopausal rats. Neurobiol Learn Mem. 2010;93:444–453. doi: 10.1016/j.nlm.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brier MR, Thomas JB, Snyder AZ, Benzinger TL, Zhang D, Raichle ME, Holtzman DM, Morris JC, Ances BM. Loss of intranetwork and internetwork resting state functional connections with Alzheimer’s disease progression. J Neurosci. 2012;32:8890–8899. doi: 10.1523/JNEUROSCI.5698-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister JJ, Gerhardt GA. Self-referencing ceramic-based multisite microelectrodes for the detection and elimination of interferences from the measurement of L-glutamate and other analytes. Analytical chemistry. 2001;73:1037–1042. doi: 10.1021/ac0010429. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Pomerleau F, Palmer M, Day BK, Huettl P, Gerhardt GA. Improved ceramic-based multisite microelectrode for rapid measurements of L-glutamate in the CNS. Journal of neuroscience methods. 2002;119:163–171. doi: 10.1016/s0165-0270(02)00172-3. [DOI] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- Caouette D, Xie Z, Milici A, Kuhn M, Bocan T, Yang D. Perinatal Suppression of Tau P301L Has a Long Lasting Preventive Effect against Neurodegeneration. International Journal of Neuropathology. 2013;1:53–69. [Google Scholar]
- Day BK, Pomerleau F, Burmeister JJ, Huettl P, Gerhardt GA. Microelectrode array studies of basal and potassium-evoked release of L-glutamate in the anesthetized rat brain. J Neurochem. 2006;96:1626–1635. doi: 10.1111/j.1471-4159.2006.03673.x. Epub 2006 Jan 1625. [DOI] [PubMed] [Google Scholar]
- DeVos SL, Goncharoff DK, Chen G, et al. Antisense reduction of tau in adult mice protects against seizures. J Neurosci. 2013;33:12887–12897. doi: 10.1523/JNEUROSCI.2107-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS. Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2005;25:2906–2916. doi: 10.1523/JNEUROSCI.5125-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du AT, Schuff N, Kramer JH, et al. Higher atrophy rate of entorhinal cortex than hippocampus in AD. Neurology. 2004;62:422–427. doi: 10.1212/01.wnl.0000106462.72282.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler-Chiurazzi E, Tsang C, Nonnenmacher S, Liang WS, Corneveaux JJ, Prokai L, Huentelman MJ, Bimonte-Nelson HA. Tonic Premarin dose-dependently enhances memory, affects neurotrophin protein levels and alters gene expression in middle-aged rats. Neurobiol Aging. 2011;32:680–697. doi: 10.1016/j.neurobiolaging.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippini N, MacIntosh BJ, Hough MG, Goodwin GM, Frisoni GB, Smith SM, Matthews PM, Beckmann CF, Mackay CE. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci U S A. 2009;106:7209–7214. doi: 10.1073/pnas.0811879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedemann MN, Gerhardt GA. Regional effects of aging on dopaminergic function in the Fischer-344 rat. Neurobiology of aging. 1992;13:325–332. doi: 10.1016/0197-4580(92)90046-z. [DOI] [PubMed] [Google Scholar]
- Frizzo ME, Dall’Onder LP, Dalcin KB, Souza DO. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell Mol Neurobiol. 2004;24:123–128. doi: 10.1023/b:cemn.0000012717.37839.07. [DOI] [PubMed] [Google Scholar]
- Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol. 2008;578:171–176. doi: 10.1016/j.ejphar.2007.10.023. Epub 2007 Oct 2025. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Schousboe A. Cellular distribution and kinetic properties of high-affinity glutamate transporters. Brain Res Bull. 1998;45:233–238. doi: 10.1016/s0361-9230(97)00417-6. [DOI] [PubMed] [Google Scholar]
- Gourley SL, Espitia JW, Sanacora G, Taylor JR. Antidepressant-like properties of oral riluzole and utility of incentive disengagement models of depression in mice. Psychopharmacology (Berl) 2012;219:805–814. doi: 10.1007/s00213-011-2403-4. Epub 2011 Jul 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenamyre JT, Young AB. Excitatory amino acids and Alzheimer’s disease. Neurobiol Aging. 1989;10:593–602. doi: 10.1016/0197-4580(89)90143-7. [DOI] [PubMed] [Google Scholar]
- Greene JG, Borges K, Dingledine R. Quantitative transcriptional neuroanatomy of the rat hippocampus: evidence for wide-ranging, pathway-specific heterogeneity among three principal cell layers. Hippocampus. 2009;19:253–264. doi: 10.1002/hipo.20502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hascup ER, af Bjerken S, Hascup KN, Pomerleau F, Huettl P, Stromberg I, Gerhardt GA. Histological studies of the effects of chronic implantation of ceramic-based microelectrode arrays and microdialysis probes in rat prefrontal cortex. Brain Res. 2009;1291:12–20. doi: 10.1016/j.brainres.2009.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hascup ER, Hascup KN, Stephens M, Pomerleau F, Huettl P, Gratton A, Gerhardt GA. Rapid microelectrode measurements and the origin and regulation of extracellular glutamate in rat prefrontal cortex. Journal of Neurochemistry. 2010;115:1608–1620. doi: 10.1111/j.1471-4159.2010.07066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillered L, Vespa PM, Hovda DA. Translational neurochemical research in acute human brain injury: the current status and potential future for cerebral microdialysis. Journal of neurotrauma. 2005;22:3–41. doi: 10.1089/neu.2005.22.3. [DOI] [PubMed] [Google Scholar]
- Hinzman JM, Thomas TC, Burmeister JJ, Quintero JE, Huettl P, Pomerleau F, Gerhardt GA, Lifshitz J. Diffuse brain injury elevates tonic glutamate levels and potassium-evoked glutamate release in discrete brain regions at two days post-injury: an enzyme-based microelectrode array study. Journal of neurotrauma. 2010;27:889–899. doi: 10.1089/neu.2009.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinzman JM, Thomas TC, Quintero JE, Gerhardt GA, Lifshitz J. Disruptions in the regulation of extracellular glutamate by neurons and glia in the rat striatum two days after diffuse brain injury. Journal of neurotrauma. 2012;29:1197–1208. doi: 10.1089/neu.2011.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hölscher C. Stress impairs performance in spatial water maze learning tasks. Behavioural Brain Research. 1999;100:225–235. doi: 10.1016/s0166-4328(98)00134-x. [DOI] [PubMed] [Google Scholar]
- Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, Pautler RG, Botas J, Noebels JL. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J Neurosci. 2013;33:1651–1659. doi: 10.1523/JNEUROSCI.3191-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsberger H, Rudy C, Batten S, Gerhardt G, Reed M. P301L tau expression affects glutamate release and clearance in the hippocampal trisynaptic pathway. J Neurochem. 2014a doi: 10.1111/jnc.12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunsberger H, Rudy C, Weitzner D, Zhang C, Tosto D, Knowlan K, Xu Y, Reed M. Effect size of memory deficits in mice with adult-onset P301L tau expression. Behavioural Brain Research. 2014b;272:181–195. doi: 10.1016/j.bbr.2014.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama T, Okada R, Nishibe H, Mitsumoto H, Nakayama C. Riluzole slows the progression of neuromuscular dysfunction in the wobbler mouse motor neuron disease. Brain Res. 2004;1019:226–236. doi: 10.1016/j.brainres.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, et al. Dendritic function of tau mediates amyloid-[beta] toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Jaquins-Gerstl A, Michael AC. Comparison of the brain penetration injury associated with microdialysis and voltammetry. Journal of neuroscience methods. 2009;183:127–135. doi: 10.1016/j.jneumeth.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Koh MT, Haberman RP, Foti S, McCown TJ, Gallagher M. Treatment strategies targeting excess hippocampal activity benefit aged rats with cognitive impairment. Neuropsychopharmacology. 2010;35:1016–1025. doi: 10.1038/npp.2009.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JY, Bacher A, Zornow MH. Riluzole does not attenuate increases in hippocampal glutamate concentrations in a rabbit model of repeated transient global cerebral ischemia. Anesth Analg. 1998;86:128–133. doi: 10.1097/00000539-199801000-00026. [DOI] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Miller LA. Senile plaques in temporal lobe epilepsy. Acta Neuropathol. 1994;87:504–510. doi: 10.1007/BF00294177. [DOI] [PubMed] [Google Scholar]
- Mattinson CE, Burmeister JJ, Quintero JE, Pomerleau F, Huettl P, Gerhardt GA. Tonic and phasic release of glutamate and acetylcholine neurotransmission in sub-regions of the rat prefrontal cortex using enzyme-based microelectrode arrays. Journal of neuroscience methods. 2011;202:199–208. doi: 10.1016/j.jneumeth.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RG, Mitchell JD, Lyon M, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND) Amyotrophic lateral sclerosis and other motor neuron disorders : official publication of the World Federation of Neurology, Research Group on Motor Neuron Diseases. 2003;4:191–206. [PubMed] [Google Scholar]
- Nickell J, Salvatore MF, Pomerleau F, Apparsundaram S, Gerhardt GA. Reduced plasma membrane surface expression of GLAST mediates decreased glutamate regulation in the aged striatum. Neurobiology of aging. 2007;28:1737–1748. doi: 10.1016/j.neurobiolaging.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Obrenovitch TP, Urenjak J, Zilkha E, Jay TM. Excitotoxicity in neurological disorders--the glutamate paradox. International journal of developmental neuroscience : the official journal of the International Society for Developmental Neuroscience. 2000;18:281–287. doi: 10.1016/s0736-5748(99)00096-9. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443:768–773. doi: 10.1038/nature05289. [DOI] [PubMed] [Google Scholar]
- Paulson JB, Ramsden M, Forster C, Sherman MA, McGowan E, Ashe KH. Amyloid plaque and neurofibrillary tangle pathology in a regulatable mouse model of Alzheimer’s disease. Am J Pathol. 2008;173:762–772. doi: 10.2353/ajpath.2008.080175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger C, Coric V, Banasr M, Bloch M, Krystal JH, Sanacora G. Riluzole in the treatment of mood and anxiety disorders. CNS Drugs. 2008;22:761–786. doi: 10.2165/00023210-200822090-00004. [DOI] [PubMed] [Google Scholar]
- Planel E, Miyasaka T, Launey T, et al. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: implications for Alzheimer’s disease. J Neurosci. 2004;24:2401–2411. doi: 10.1523/JNEUROSCI.5561-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013;14:389–394. doi: 10.1038/embor.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz YT, Budson AE, Celone K, Ruiz A, Newmark R, Castrillon G, Lopera F, Stern CE. Hippocampal hyperactivation in presymptomatic familial Alzheimer’s disease. Ann Neurol. 2010;68:865–875. doi: 10.1002/ana.22105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rutherford EC, Pomerleau F, Huettl P, Stromberg I, Gerhardt GA. Chronic second-by-second measures of L-glutamate in the central nervous system of freely moving rats. J Neurochem. 2007;102:712–722. doi: 10.1111/j.1471-4159.2007.04596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SantaCruz K, Lewis J, Spires T, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Dickerson BC, Pihlajamaki M, et al. Functional alterations in memory networks in early Alzheimer’s disease. Neuromolecular Med. 2010;12:27–43. doi: 10.1007/s12017-009-8109-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykova E, Mazel T, Simonova Z. Diffusion constraints and neuron-glia interaction during aging. Exp Gerontol. 1998;33:837–851. doi: 10.1016/s0531-5565(98)00038-2. [DOI] [PubMed] [Google Scholar]
- Talauliker PM. College of Medicine PhD. University of Kentucky; 2010. Characterization and optimization of microelectrode arrays for glutamate measurements in the rat hippocampus. [Google Scholar]
- Tan W, Cao X, Wang J, Lv H, Wu B, Ma H. Tau hyperphosphorylation is associated with memory impairment after exposure to 1.5% isoflurane without temperature maintenance in rats. European journal of anaesthesiology. 2010;27:835–841. doi: 10.1097/EJA.0b013e32833a6561. [DOI] [PubMed] [Google Scholar]
- van de Pol LA, van der Flier WM, Korf ES, Fox NC, Barkhof F, Scheltens P. Baseline predictors of rates of hippocampal atrophy in mild cognitive impairment. Neurology. 2007;69:1491–1497. doi: 10.1212/01.wnl.0000277458.26846.96. [DOI] [PubMed] [Google Scholar]
- Vossel KA, Beagle AJ, Rabinovici GD, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA neurology. 2013;70:1158–1166. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner ML, Landis BE. Riluzole: a new agent for amyotrophic lateral sclerosis. Ann Pharmacother. 1997;31:738–744. doi: 10.1177/106002809703100614. [DOI] [PubMed] [Google Scholar]
- Wang JZ, Liu F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog Neurobiol. 2008;85:148–175. doi: 10.1016/j.pneurobio.2008.03.002. Epub 2008 Mar 2022. [DOI] [PubMed] [Google Scholar]
- Wilson IA, Ikonen S, Gallagher M, Eichenbaum H, Tanila H. Age-associated alterations of hippocampal place cells are subregion specific. J Neurosci. 2005a;25:6877–6886. doi: 10.1523/JNEUROSCI.1744-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G. Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci. 2005b;25:6221–6234. doi: 10.1523/JNEUROSCI.3003-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Holth JK, Liao F, et al. Neuronal activity regulates extracellular tau in vivo. J Exp Med. 2014;211:387–393. doi: 10.1084/jem.20131685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassa MA, Stark SM, Bakker A, Albert MS, Gallagher M, Stark CEL. High-resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic Mild Cognitive Impairment. NeuroImage. 2010;51:1242–1252. doi: 10.1016/j.neuroimage.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HT, Huang Y, Masood A, et al. Anxiogenic-like behavioral phenotype of mice deficient in phosphodiesterase 4B (PDE4B) Neuropsychopharmacology. 2008;33:1611–1623. doi: 10.1038/sj.npp.1301537. [DOI] [PMC free article] [PubMed] [Google Scholar]