

Abstract
Formation of foam cells is a hallmark at the initial stages of atherosclerosis. Monocytes attracted by pro‐inflammatory stimuli attach to the inflamed vascular endothelium and penetrate to the arterial intima where they differentiate to macrophages. Intimal macrophages phagocytize oxidized low‐density lipoproteins (oxLDL). Several scavenger receptors (SR), including CD36, SR‐A1 and lectin‐like oxLDL receptor‐1 (LOX‐1), mediate oxLDL uptake. In late endosomes/lysosomes of macrophages, oxLDL are catabolysed. Lysosomal acid lipase (LAL) hydrolyses cholesterol esters that are enriched in LDL to free cholesterol and free fatty acids. In the endoplasmic reticulum (ER), acyl coenzyme A: cholesterol acyltransferase‐1 (ACAT1) in turn catalyses esterification of cholesterol to store cholesterol esters as lipid droplets in the ER of macrophages. Neutral cholesteryl ester hydrolases nCEH and NCEH1 are involved in a secondary hydrolysis of cholesterol esters to liberate free cholesterol that could be then out‐flowed from macrophages by cholesterol ATP‐binding cassette (ABC) transporters ABCA1 and ABCG1 and SR‐BI. In atherosclerosis, disruption of lipid homoeostasis in macrophages leads to cholesterol accumulation and formation of foam cells.
Keywords: macrophages, atherosclerosis, atherogenesis, cholesterol, lipoproteins, foam cells
Introduction
Macrophages are key players in all stages of atherosclerosis. Initially, monocytes attracted by pro‐inflammatory signals coming from the inflamed endothelium attach to the problematic arterial sites and infiltrate the intima 1. To be attractive for monocytes and other immune cells, the problematic vascular sites should be injured or atheroprone because of the abnormal haemodynamic forces and/or accumulation of oxidized lipids in the arterial wall 1. In the subendothelial layer, monocytes differentiate to macrophages that in turn polarize to pro‐inflammatory/anti‐inflammatory phenotype depending on the local stimuli and transform to foam cells 2.
Formation of foam cells that occurs in the initial stages of atherogenesis is a hallmark of atherosclerotic disease 3, 4. Increased uptake of oxidized low‐density lipoprotein (oxLDL) and/or reduced cholesterol efflux leads to the deposition of esterified cholesterol in the cytoplasm of macrophages and generation of foam cells 5. In macrophages, oxLDL is taken up with help of scavenger receptors (SR) such as CD36 (also known as fatty acid translocase), SR‐A1 and lectin‐like oxLDL receptor‐1 (LOX‐1) (Fig. 1) 6. Acyl coenzyme A: cholesterol acyltransferase‐1 (ACAT1) and neutral cholesteryl ester hydrolase (nCEH) are involved in the formation of cholesterol esters 7. ATP‐binding cassette (ABC) transporters ABCA1 and ABCG1 and SR‐BI contribute to the reverse transport of cholesterol from macrophages 8.
Figure 1.
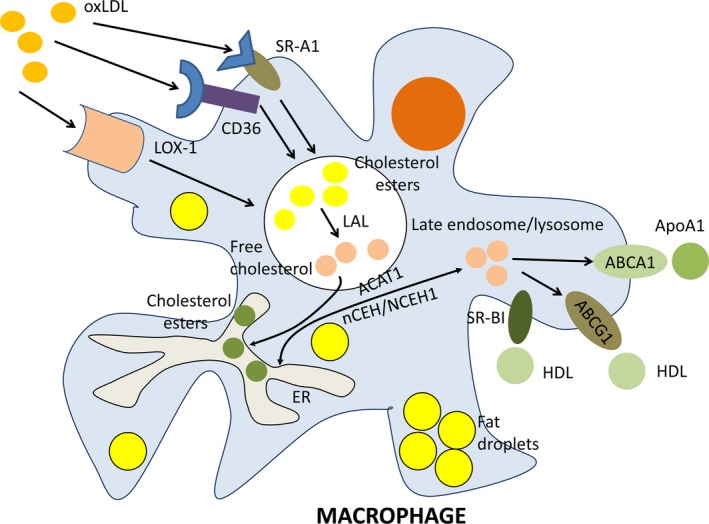
Cholesterol metabolism in macrophages. Macrophages engulf oxidized cholesterol (oxLDL) with help of several scavenger receptors (SR) including CD36, SR‐A1, and lectin‐like oxLDL receptor‐1 (LOX‐1). In late endosomes/lysosomes of macrophages, oxLDL are catabolysed. Lysosomal acid lipase (LAL) hydrolyses cholesterol esters that are enriched in LDL to free cholesterol and free fatty acids. In endoplasmic reticulum (ER), acyl coenzyme A: cholesterol acyltransferase‐1 (ACAT1) in turn catalyses esterification of cholesterol to store cholesterol esters as lipid droplets in the ER of macrophages. Neutral cholesteryl ester hydrolases nCEH and NCEH1 are involved in a secondary hydrolysis of cholesterol esters to liberate free cholesterol that could be then out‐flowed from macrophages by cholesterol ATP‐binding cassette (ABC) transporters ABCA1 and ABCG1 and scavenger receptor SR‐BI. The main acceptors of free cholesterol from ABCG1 and SR‐BI are high density lipoprotein (HDL) and from ABCA1 is apolipoprotein A1 (ApoA1). In atherosclerosis, disruption of lipid homoeostasis in macrophages leads to cholesterol accumulation and formation of foam cells.
The purpose of this review is to characterize key mechanisms of cholesterol efflux, specifically in macrophages. In this work we briefly consider all constituents of the system responsible for cholesterol metabolism in macrophages.
Cholesterol uptake
Cholesterol enters the macrophage cytoplasm through mechanisms of SR‐dependent phagocytosis and pinocytosis 9. CD36 and SR‐A are principal contributors to cholesterol uptake accounting up to 90% of oxLDL loading in macrophages 10.
Scavenger receptor CD36
CD36 is a 88 kD glycoprotein that is a member of SR class B family 11. The human CD36 gene is located on chromosome 7q11.2 11. This receptor includes two extracellular and two transmembrane domains that flank the extracellular domain 12. CD36 binds oxLDL with high affinity. A major oxLDL‐binding site is located between amino acids (a.a.) 127 and 279, with an additional site located between a.a. 28–93 13. Interaction of oxLDL with CD36 causes endocytosis of CD36‐oxLDL complex via lipid raft‐dependent mechanism 13.
The pro‐atherogenic role of oxLDL depends on the abundance of CD36. Handberg et al. 14 found a correlation between the plasma levels of soluble CD36 (sCD36) and indices of insulin resistance, carotid atherosclerosis and fatty liver. Furthermore, increased levels sCD36 were associated with insulin resistance and higher risk of type 2 diabetes 15, 16. Elevated sCD36 levels were detected in the monocytes of patients with coronary artery disease 17 and acute coronary syndrome 18. Statin therapy reduces sCD36 concentrations and suppresses oxLDL uptake by monocytes and macrophages 19. Similarly, low molecular CD36 inhibitors were shown to reduce lipid deposits in the arterial wall and improve insulin sensitivity and glucose tolerance 20. However, patients deficient in CD36, had advanced atherosclerosis 21. Indeed, the role of CD36 in atherosclerosis is complicated and should be further studied in detail.
In macrophages, expression of CD36 is regulated by many factors. An antioxidant curcumin up‐regulates the CD36 expression through stimulating nuclear erythroid‐related factor 2 (Nrf2) 22. Astaxanthin, an oxidized carotenoid, increases CD36 expression by the activation of the peroxisome proliferator‐activated receptor‐γ (PPAR‐γ) 23. Palmitic acid stimulates monocyte production of CD36 by the induction of de novo ceramide synthesis 24 as ceramides reduce CD36 expression and decrease oxLDL uptake by monocytes 25. Similarly, lipopolysaccharide from the bacterium Porphyromonas gingivalis, a causative agent of gingivitis, up‐regulates CD36 in macrophages by stimulating c‐Jun/activator protein‐1 (AP‐1) pathway 26. Interestingly, a 12‐week dietary intake of champignon, Agaricus blazei, by Apolipoprotein E (ApoE)‐deficient mice resulted in elevated levels of CD36 and higher lesion instability suggesting for pro‐atherogenic properties of this mushroom 27.
Some compounds were shown to down‐regulate CD36 expression. For example, tanshinone IIA, a diterpene from a sage herb Salvia miltiorrhiza Bunge, reduces CD36 expression by antagonizing PPAR‐γ 28. Squalene (a component of olive oil) and endomorphin‐1 (an opioid peptide) decrease CD36 and disrupt lipid overload in macrophages 29, 30. Quercitrin, a plant pigment glycoside, also suppresses CD36 expression in macrophages by altering protein kinase C (PKC)/PPAR‐γ signalling pathway 31. Exposure of the macrophages to another flavonoid and antioxidant (kaempferol) prevents nuclear translocation of transcription factor AP‐1, thereby inhibiting expression of CD36 32. Finally, walnut that is rich in γ‐3 polyunsaturated fatty acids and antioxidants was shown to display an anti‐atherosclerotic activity in ApoE‐deficient mice by decreasing the CD36 expression 33. Indeed, CD36 expression could be nutritionally modulated such that it could represent a valuable tool for the prevention of atherosclerosis.
Scavenger receptor A1
Scavenger receptor‐A1 (also known as macrophage SR MSR1 or CD204) belongs to the class A SR family 34. The human SCARA1 gene encoding this receptor is situated on chromosome 8p22 35. Three alternate transcripts are produced from this gene. Isoforms 1 and 2 are functional and able to mediate endocytosis of modified LDL. The isoform 3 cannot internalize oxLDL because of altered processing that leads to the localization of this isoform in the endoplasmic reticulum (ER) making it unable to perform endocytosis. This isoform suppresses the activity of the isoforms 1 and 2 when co‐expressed, thereby acting as a negative regulator of SR‐A1 expression in macrophages 36. SR‐A1 functions as a homotrimer consisting of three 77‐kD glycosylated subunits.
In macrophages, SR‐A1 is involved in the uptake of modified LDL. In ApoE mice, SR‐A1 knock‐down reduces the generation of foam cells and atherosclerosis progression 37. In LDL receptor‐deficient mice, inhibition of either CD36 or SR‐A1 alone had atheroprotective effect. However, suppression of both SRs showed no positive effect on atherosclerosis suggesting that compensatory activation of these receptors is sufficient for the intake of modified LDL 38. Similarly, in hyperlipidemic ApoE‐deficient mice, deletion of either CD36 or SR‐A1 significantly reduces lipid accumulation in macrophages, but does not diminish atherosclerosis 39. These data suggest for the existence of alternative mechanisms of lipid uptake by macrophages that are independent of CD36 or SR‐A1.
Like CD36, expression of SR‐A1 could be influenced by a variety of modulators. Pro‐inflammatory cytokines such as tumour necrosis factor α (TNF‐α) and interleukin‐6 (IL‐6) promote SR‐A1 expression by activation of transcription nuclear factor (NF)‐κB 40. Indeed, pharmacological inhibition of both cytokines decreases oxLDL accumulation and foam cell formation 41. Berberine, a plant alkaloid from Berberis sp., was shown to have pro‐atherogenic effects on culture mouse monocytes by up‐regulating SR‐A1 and increasing the cholesterol uptake by suppressing negative cell cycle regulator phosphatase and tensin homologue (PTEN) and thereby preserving protein kinase Akt from PTEN‐dependent dephosphorylation 42.
Yang et al. 43 showed that voltage‐gated potassium channel Kv1.3 is involved in the modulation of activity of SR‐A1. Kv1.3 activation led to increased activity of SR‐A1 and enhanced uptake of oxLDL. Indeed, antibody‐dependent Kv1.3 blockade resulted in reduced oxLDL entrance, decreased cholesterol esterification and enhanced apoA‐I‐mediated cholesterol efflux 44. This finding may have important pharmaceutical consequences as it allows specific targeting and modulation of lipid accumulation in macrophages.
Modulators such as curcumin, polyphenolic extracts of mulberry leaves and hydrogen sulphide (H2S) were shown to decrease SR‐A1 levels 45. In ApoE‐deficient mice, curcumin induced SR‐A1 ubiquitination and calpain‐mediated proteolysis in macrophages 45. Mulberry‐derived polyphenols down‐regulate SR‐A1 through the suppression of PPAR‐γ 46. In the vascular system, H2S is mainly produced by cystathionine γ‐lyase (CSE). However, in apoE‐deficient mice, this pathway is impaired resulting in reduced H2S production. Deregulation of CSE‐dependent generation of H2S contributes to oxLDL‐mediated inflammation in macrophages 47. H2S possesses anti‐atherosclerotic properties by decreasing plaque size and production of intercellular adhesion molecule‐1 48. H2S also inhibits foam cell formation by suppressing SR‐A1 via KATP/Erk 1/2 mechanism 49.
Lectin‐like oxidized low‐density lipoprotein receptor‐1
Lectin‐like oxidized low‐density lipoprotein receptor‐1 (LOX‐1) is a membrane glycoprotein that contains a short N‐terminal cytoplasmic domain, a transmembrane domain, a neck region that controls receptor oligomerization and an extracellular C‐type lectin‐like extracellular domain, involved in oxLDL binding 50, 51. Human LOX‐1 is encoded by the OLR1 gene located on chromosome 12p13.2‐p12.3 52. The gene encodes several LOX‐1 isoforms of which the longest isoform contains a 273‐a.a. polypeptide 53. Another variant lacks an exon that results in a frameshift and early stop codon. Indeed, the isoform 2 is shorter than isoform 1 and contains a distinct C‐terminus 54.
Compared with CD36, LOX‐1 is able to bind moderately modified, but not fully oxidized LDL suggesting for crucial role of this receptor in initial stages of atherosclerosis 55. LOX‐1 is a major oxLDL‐binding receptor in endothelial cells, but it could be up‐regulated in macrophages in atherosclerosis 56. LOX‐1 could not be found in monocytes, but may be induced in differentiated macrophages 54.
Multiple regulators are involved in the up‐regulation of this receptor especially in inflammation. Pro‐inflammatory cytokines such as IL‐1, interferon (IFN)‐γ and TNF‐α stimulate LOX‐1 expression in macrophages 57. Modified lipids such as oxLDL and products of its hydrolysis (lysophosphatidylcholine) were shown to activate LOX‐1 expression in macrophages 51. Hypertension‐related stimuli including angiotensin II and endothelin‐1 were found to induce the expression of LOX‐1 in macrophages 58. In diabetic patients, hyperglycaemia and advanced glycation end‐products (AGEs) play a key role in the up‐regulation of LOX‐1 expression in macrophages 59, 60.
Macrophage‐specific deletion of LOX‐1 did not reveal significant changes in oxLDL uptake compared with wild‐type cells 61. Therefore, in normal conditions, impact of LOX‐1 to intake and catabolism of oxLDL by macrophages is small probably because of the high contribution of other SRs. LOX‐1 knock‐down does not greatly influence oxLDL uptake by non‐stimulated macrophages as this receptor contributes for only 5–10% of a total oxLDL intake by macrophages, whereas in pro‐inflammatory‐stimulated macrophages, the impact of LOX‐1 could increase up to 40% 61.
Monocytes were shown to differentiate to dendritic cells (DCs) 62, a common event in atherosclerosis 63. High levels of LOX‐1 expression were found in mature DCs suggesting for a significant contribution to oxLDL uptake because an antibody against LOX‐1 decreases oxLDL influx by 48% 64.
A markedly increased expression of LOX‐1 was detected in atherosclerotic lesions compared to the normal vascular tissue 55. In LDL‐deficient mice with genetic deletion of LOX‐1, a less pronounced development of atherosclerotic plaques was observed in comparison with LDL‐deficient mice fed on high‐cholesterol diet. In mice, LOX‐1 knockout was accompanied by the decreased expression of the pro‐inflammatory transcription factor NF‐κB and variable pro‐inflammatory markers 65. In contrast, ApoE‐deficient mice transgenic for LOX‐1 showed advanced atherosclerosis 66. In rabbits with experimental hypercholesterolaemia, LOX‐1 was found to be predominantly expressed in unstable lesions 67. Intraplaque LOX1 levels correlated with the expression of tissue factor and apoptosis suggesting a possible involvement of this SR to plaque instability 67. All these observations suggest the pro‐atherogenic role of LOX‐1.
Formation of cholesterol esters
The rate of cholesterol esterification could determine the fate of macrophages to be transformed to foam cells or not. After engulfment, lipoproteins are transferred to late endosomes/lysosomes where LAL hydrolyses cholesterol esters with the generation of free cholesterol. Free cholesterol in turn is de novo esterified by ACAT1 and stored as lipids in the ER. In case of overload with cholesterol esters, macrophages could be transformed to foam cells (Fig. 2). Indeed, cholesterol excess should be removed from the cell. The nCEH is responsible for the secondary hydrolysis of cholesterol esters 68. Free cholesterol is then transferred out with cholesterol transporters residing on the plasma membrane. Therefore, balance between hydrolysis and esterification of cholesterol plays a key role in maintaining cholesterol homoeostasis and prevention of generation of foam cells. Two enzymes, nCEH and ACAT1, are crucially involved in this mechanism.
Figure 2.
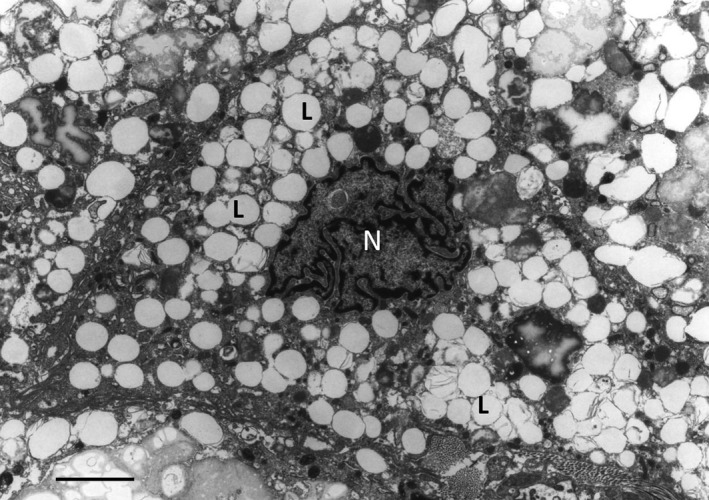
Electron micrograph showing a macrophage foam cell, the cytoplasm of which filled with a large number of ‘lipid droplets’ (L). N – nucleus. Atherosclerotic plaque tissue specimen of the human aorta. Transmission electron microscopy (TEM); scale bar = 5 μm.
Acyl coenzyme A: cholesterol acyltransferase‐1
In macrophages, ACAT1 [or sterol O‐acyltransferase 1 (SOAT1)] (EC 2.3.1.26) is mainly located in the tubular rough ER 69. The human SOAT1 gene resides on chromosome 1q25 and encodes a 550 a.a. polypeptide. The enzyme contains two transmembrane domains 70. In murine peritoneal macrophages, lack of ACAT1 was reported to elevate free cholesterol levels and increase cholesterol efflux 71. In opposite, ACAT1 overproduction resulted in an enhanced accumulation of cholesterol esters followed by the formation of foam cells 72. However, other investigations showed that the role of ACAT1 in atherogenesis is not ordinary. For example, in ApoE‐deficient mice, ACAT1 suppression by a small molecular inhibitor F‐1394 had an atheroprotective effect 73. In contrast, macrophage‐specific depletion of ACAT1 in mice caused advanced atherosclerosis 74, 75. The discrepancy in these results could be explained by the non‐specific action of F‐1394 that inhibits both ACAT isoforms (ACAT1 and ACAT2) and therefore has a systemic effect 74. Human ACAT2 is mainly expressed by intestinal cells and hepatocytes where it is responsible for lipoprotein assembly 76. In addition, free cholesterol produced by ACAT1‐catalysed reaction could be seriously cytotoxic for macrophages and other vascular cells. In macrophages, free cholesterol overload could lead to the formation of cholesterol crystals (Fig. 3) that are highly cytotoxic and may not only impair intracellular cholesterol metabolism but also enhance release of pro‐inflammatory cytokines IL‐1β and IL‐18 77. These processes might progress leading to cell death, accompanied by the release and accumulation of ‘free’ cholesterol crystals in the extracellular matrix of atherosclerotic lesions (Fig. 4).
Figure 3.
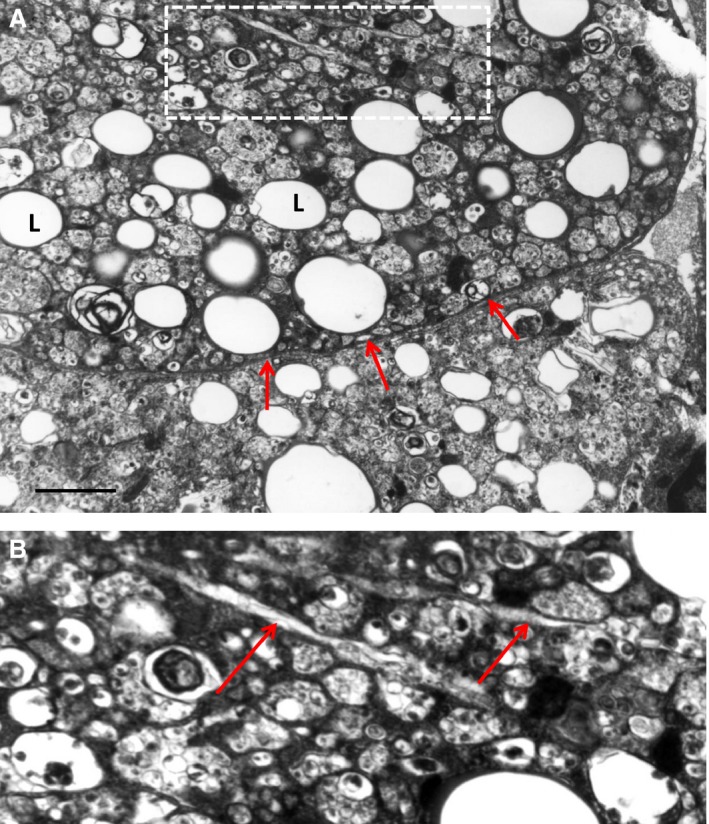
Formation of cholesterol crystals in the cytoplasm of a foam cell (A and B). (B) It is a detail of (A). L ‐ ‘lipid droplet’. In (A), arrows show the plasma membrane of the foam cells. In (B), arrows show cholesterol crystals. Atherosclerotic plaque tissue specimen of the human aorta. TEM; scale bar = 1 μm (A).
Figure 4.
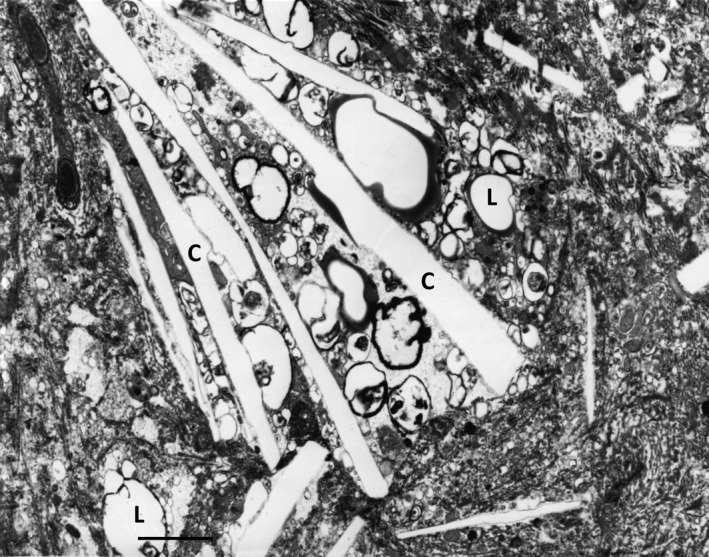
Cholesterol crystals (C) and ‘lipid droplets’ (L) located in the extracellular matrix of an atherosclerotic plaque of the human aorta. TEM; scale bar = 1 μm.
The ACAT1 expression and activity are extensively modulated by different signalling messengers. Leptin, a hormone produced by adipose tissue, stimulates ACAT1 expression through Janus‐activated kinase 2 (Jak2)/phosphatidylinositide 3‐kinase (PI3K) mechanism 78. Insulin also up‐regulates ACAT1 expression in macrophages via extracellular signal‐regulated kinase (Erk)/p38MAP kinase/Jnk‐dependent activation of CCAAT/enhancer binding protein α, a transcriptional regulator 79, 80. Except regulation of SR‐A1 activity, voltage‐gated potassium channel Kv1.3 is involved in the up‐regulation of ACAT1 that leads to enhanced uptake of oxLDL and accumulation of cholesterol ethers in macrophages 44.
In contrast, glucagon‐like peptide‐1 (GLP‐1) and glucose‐dependent insulinotropic polypeptide (GIP) reduce ACAT1 levels through PKA‐mediated pathway 81. Dipeptidylpeptidase 4 (DPP4) is known to degrade GLP‐1 82. Vildagliptin and other DPP4 inhibitors were shown to be atheroprotective in non‐diabetic and diabetic ApoE‐deficient mice through the activation of both incretin hormones (GLP‐1 and GIP) and restoring insulin production 83. Similarly with the suppression of SR‐A1, H2S utilizes the same mechanism (e.g. KATP/Erk 1/2‐dependent signalling) to inhibit ACAT1 expression in macrophages 49. Ghrelin, a ‘hunger’ hormone, produced by ghrelin cells in the gastrointestinal tract 84 also reduces the expression of ACAT1 by binding to the growth hormone secretagogue receptor and suppression of PPARγ 85.
Neutral cholesteryl ester hydrolase
nCEH also known as hormone‐sensitive lipase (EC 3.1.1.79) is responsible for the hydrolysis of cholesterol esters to generate free cholesterol for further release from the cell. The human enzyme is encoded by the LIPE gene located on chromosome 19q13.2 86. Two transcripts (long and short nCEH isoforms) are produced from this gene. The long isoform is expressed in testis and other steroidogenic tissues where it converts cholesteryl esters to free cholesterol for steroid hormone production. The short isoform is present in adipocytes, macrophages and other cells 87. nCEH is a 1076‐a.a. membrane polypeptide that contains three domains (lipid‐binding, catalytic and N‐terminal). The N‐terminal domain serves as an anchor to recruit nCEH to the ER membrane where the enzyme exposes its catalytic domain to the lumen. nCEH is a glycoprotein, with three N‐glycosylated sites at N270, N367 and N389. All these sites are glycosylated, and glycosylation at N270 is essential for catalytic activity 88.
Inhibition of nCEH greatly accelerates formation of foam cells 89. Overexpression of nCEH results in the increased hydrolysis of cholesterol esters in lipid‐laden macrophages 90. However, nCEH overexpression alone without decreasing the expression of ACAT1 and stimulation of cholesterol efflux was not enough to protect against atherosclerosis 90. Transgenic mice overexpressing nCEH and cholesterol acceptor such as ApoA4 do have reduced atherosclerosis 91. Macrophage‐specific overexpression of nCEH also diminished atherosclerosis and decreased plaque necrotic core size in LDL receptor‐deficient mice 92 suggesting a critical role of macrophages in lipid handling in atherosclerosis.
Recently, a new neutral cholesterol ester hydrolase (NCEH1; also known as arylacetamide deacetylase‐like 1, AADACL1) was found 93. Human enzyme is encoded by the NCEH1 gene located on chromosome 3 94. Like nCEH, NCEH1 resides in the ER membrane and contributes to the hydrolysis of cholesterol esters in macrophages. Knockout of NCEH1 in ApoE‐deficient mice promoted the development of atherosclerosis without altering the serum lipid profile 95 suggesting the atheroprotective role of this enzyme. Indeed, both nCEH and NCEH1 are involved in the generation of free cholesterol from cholesterol esters, thereby preventing formation of foam cells from macrophages.
Insulin was found to be involved in the regulation of nCEH expression in macrophages. In the initial stages of type 2 diabetes associated with the hyperfunction of insulin‐producing β‐cells, increased levels of insulin could down‐regulate the nCEH expression in macrophages, thereby contributing to atherogenesis 96. Interleukin 33, a member of the IL‐1 cytokine family, suppresses nCEH in macrophages through the stimulation of ST‐2/NF‐κB signalling. Furthermore in ApoE‐deficient mice, IL‐33 significantly reduced accumulation of macrophages in atherosclerotic plaques and generation of foam cells 97.
Cholesterol efflux
Free cholesterol could be removed from macrophages through active transfer mediated by cholesterol transporters or by passive transmembrane diffusion. High density lipoprotein (HDL) or ApoA1 then capture the released cholesterol. Cholesterol transporters such as ABCA1, ABCG1 and SR‐Bi play the major role in active free cholesterol efflux.
ATP‐binding cassette transporter, ABCA1
The ABCA1, also known as cholesterol efflux regulatory protein, regulates cholesterol efflux and phospholipid homoeostasis 98. The human ABCA1 gene is located on chromosome 9q31.1 99. The 200‐kDa transporter is ubiquitously expressed throughout the body. Interestingly, mice deficient for ABCA1 and SR‐BI had severe hypocholesterolaemia mainly as a result of HDL loss, but showed no atherosclerosis because of the absence of pro‐atherogenic lipids 100. ApoA1, the major protein component of HDL, serves as an acceptor of cholesterol released by ABCA1 101. Unexpectedly, in LDL receptor‐deficient mice, hepatic ABCA1 overexpression led to the deposition of pro‐atherogenic lipids and advanced atherosclerosis because of the enhanced transfer of HDL cholesterol to LDL and delayed catabolism of cholesterol‐enriched LDL 102.
As a result of the key role in cholesterol reverse transport, ABCA1 expression is controlled by various regulators and bioactive molecules. In macrophages, the expression of ABCA1 is regulated by liver X receptor α (LXRα), a nuclear transcription factor 103. Quercetin was observed to stimulate LXRα through the activation of PPARγ that finally results in the up‐regulation of ABCA1 transcription and increased cholesterol efflux from macrophages 104. Apelin‐13, a vasoactive peptide, enhances cholesterol out‐flow from foam cells through the stimulation of PKCα and inhibition of calpain, a protease that is involved in ubiquitination‐mediated ABCA1 degradation 105, 106. Similarly, various proteasome inhibitors and ApoA1, an acceptor of cholesterol transferred by ABCA1, restore cholesterol efflux in macrophages by suppression of ABCA1 degradation 107, 108. S‐allylcysteine, a major garlic extract constituent, possesses the atheroprotective activity by increasing ABCA1 expression through unknown mechanism 109. However, lipid‐lowering properties of S‐allylcysteine appear to play a secondary role in a summary of its anti‐atherogenic effects as this cysteine derivative only moderately inhibited lipid accumulation in macrophages 110. The main beneficial effects of S‐allylcysteine on macrophage function in atherosclerosis are inhibiting inducible nitric oxide synthase and suppressing the production of hydroxyl radical, thereby suggesting for the primary role of the antioxidant activity 111.
Unsaturated free fatty acids such as palmitic acid or linoleic acid down‐regulate ABCA1 expression in macrophages by epigenetic repression of LXR genes and in LXR‐independent post‐translational level involving ABCA1 phosphorylation and destabilization by protein kinase Cδ that induces degradation of the transporter 112, 113. Pro‐inflammatory cytokines IL‐12 and IL‐18 suppress ABCA1 expression through IL‐18 receptor/NF‐κB‐mediated induction of zinc finger protein 202 (ZNF202), a transcriptional repressor 114. MicroRNA (miR)‐26 that is critically involved in the regulation of vascular smooth muscle cell differentiation 115 was shown to target LXRα and therefore down‐regulate ABCA1 expression 116. MiR‐144 directly decreases ABCA1 mRNA levels in macrophages and liver as two miR‐144‐binding sites were found in the 3′ untranslated region of the ABCA1 mRNA 117. Indeed, development of miR‐26 and miR‐144 inhibitors has a therapeutic potential to suppress the formation of foam cells in atherosclerosis.
ATP‐binding cassette transporter ABCG1
This transporter transfers cholesterol to HDL particles, but not to ApoA1. The human ABCG1 gene is mapped to chromosome 21q22.3 118. The gene contains alternative start codons that result in the production of multiple transcripts in a tissue‐specific manner 119. In LDL receptor‐deficient mice, ABCG1 knock‐down inhibition, results in a moderate rise of atherosclerotic plaques 120. In contrast, a study shows the atheroprotective effect of genetic deletion of ABCG1 in the LDL receptor‐deficient mouse 121. Indeed, as ABCA1 is a primary cholesterol transporter in macrophages and it could compensate lipid efflux.
In fact, the regulation of expression of ABCA1 and ABCG1 is shared between LXRα and LXRβ that control tissue lipid intake 8. Notably, ABCG1 expression could be modulated by dietary components. Cultured macrophages exposed to extra‐virgin olive oil developed increased ABCG1 expression and enhanced lipid overflow 122. Cineole, a monoterpenoid of Eucalyptus sp. was shown to stimulate ABCG1 expression via activation of LXR receptors 123. Cyanidin‐3‐O‐β‐glucoside (Cy‐3‐G), an anthocyanin derived from blackberry, blueberry, bilberry, cranberry and other herbs is known to have remarkable atheroprotective properties 124. In the intestine, Cy‐3‐G could be converted by gut microflora to protocatechuic acid (PCA), a bioactive metabolite that is able to reduce expression of miR‐10b, which represses ABCA1 and ABCG1 125. Except for inhibiting lipid accumulation in macrophages, PCA exhibits anti‐inflammatory properties by decreasing subendothelial monocyte infiltration and recruitment in ApoE‐deficient mice 126. Therefore, regular intake of a proper food could prevent lipid misbalance and cholesterol deposition in macrophages and other cells.
Scavenger receptor BI
Scavenger receptor‐BI is responsible for cholesterol transfer to HDL. The human receptor is encoded by the SCARB1 gene located on chromosome 12g24.31 44. Two SR‐BI isoforms could be translated from the human SCARB1 gene. The longest isoform contains 509 a.a., with a C‐terminal distinct from the shortest isoform (506 a.a.) 127. Both isoforms have two transmembrane domains, two short cytoplasmic tails and a large extracellular loop 128.
In ApoE‐deficient mice, macrophage‐specific deletion of SR‐BI resulted in advanced atherosclerosis, with an 86% increase in average lesion area. Overexpression of SR‐BI delayed formation of atherosclerotic plaques in ApoE‐deficient mice 129. However, in LDL receptor‐deficient mice, macrophage‐specific overproduction of SR‐BI protected against advanced atherosclerosis, but contributed to early plaque formation 130. The extraordinary role of SR‐BI in atherosclerosis could be explained by ability of this transporter to move cholesterol bidirectionally 131. At early atherosclerosis stages, SR‐BI in macrophages acts like SR‐A1 to conduct cholesterol and phospholipid influx to decrease excessive serum cholesterol levels. Furthermore, SR‐B1 was shown to block ABCA1‐dependent cholesterol efflux in macrophages suggesting for distinct and competing roles of these transporters in mediating cholesterol flux 132. Local HDL phospholipid composition could greatly influence the reverse cholesterol transport, with increased phosphatidylcholine enrichment of HDL that stimulates cholesterol efflux 131.
Numerous bioregulators modulate SR‐BI expression. Dietary components such as polyphenol resveratrol and 13‐hydroxy linoleic acid activate PPARγ that in turn up‐regulates LXRα and SR‐BI 133, 134. Similarly, caffeic acid and ferulic acid, two major phenolic acids of coffee, increase cholesterol efflux from macrophages by stimulation of SR‐BI and ABCG1, e.g. carriers that deliver cholesterol to HDL, but not to ApoA1 135. In contrast, pappalysin‐1 (or pregnancy‐associated plasma protein A; PAPPA), a metalloproteinase that cleaves insulin‐like growth factor binding proteins could decrease expression of ABCA1, ABCG1, and SR‐BI by inhibiting IGF‐I‐mediated (IGF/PI3‐K/Akt) stimulation of LXRα 136.
Concluding remarks
Macrophages are master regulators of plasma lipid balance being involved in ingestion and storage of excessive lipids that could be then released in a case hypolipidaemia. However, in atherosclerosis, there is a deregulation of cholesterol uptake and reverse transport by macrophages. OxLDL, which is a major source of cholesterol, contributes to the suppression of cholesterol efflux, whereas expression of SRs especially LOX‐1 becomes significantly up‐regulated. Indeed, it is not surprising that the genetic deletion of either SR‐A1 or CD36 has no beneficial effects in ApoE‐deficient mice 38, 39 because oxLDL uptake by macrophages could be compensated by SR‐BI (in early atherosclerosis) and LOX‐1.
It should be stressed that lipid metabolism in macrophages could be modulated by multiple dietary factors that provide a good option to prevent lipid accumulation or improve macrophage function in atherosclerotic patients. For example, several polyphenolic acids derived from blueberries were shown to efficiently suppress generation of foam cells through the down‐regulation of CD36 and stimulation of ABCA1 137.
Targeting components of cholesterol uptake/esterification/efflux should be of great therapeutic value. Statins (lipid‐lowering agents) were shown to possess a wide range of anti‐inflammatory activities including improvement of lipid handling by macrophages in patients with cardiovascular disease. Simvastatin showed beneficial effects in the treatment of stroke‐prone hypertensive rats via decreasing macrophage infiltration and lipid deposition and inhibition of LOX‐1 expression 138. Similarly, pravastatin also reduced LOX‐1 expression in intimal macrophages and decreased lipid core size in the atherosclerotic plaques of Watanabe heritable hyperlipidaemic rabbits 139. In ApoE‐deficient mice, K‐604 and rimonabant, recently developed ACAT1 inhibitors, suppressed foam cell formation and reduced atherosclerosis 140, 141.
Pro‐protein convertase subtilisin/kexin type 9 (PCSK9) belongs to the family of PCSKs involved in the activation of peptide hormones and receptors from precursors. Specifically, PCSK9 decreases LDL cholesterol uptake by liver by stimulating degradation of LDL receptors 142. As a consequence, plasma LDL cholesterol (LDL‐C) concentrations become elevated which could increase cardiovascular risk. Increased expression of PCSKs was found in human and mouse atherosclerotic plaques suggesting the involvement of PCSKs in atherogenesis 143. To date, multiple PCSK9 inhibitors including low‐molecular inhibitors and monoclonal antibodies such as alirocumab (Aventis/Regeneron), bococizumab (Pfizer) and evolocumab (Amgen) have been developed 144. In animal models of atherosclerosis, PCSK9 inhibition resulted in significant lowering of plasma LDL‐C levels and diminished atherosclerosis progression 142. Clinical trials involving anti‐PCSK9 monoclonal antibodies showed a notable atheroprotective effect associated with cholesterol decrease, cardiac events and mortality 145.
In cultured human THP‐1 macrophages, PCSK9 expression was shown to be up‐regulated by oxLDL. Knock‐down of PCSK9 expression with PCSK9‐specific small interfering RNA resulted in the inhibition of oxLDL‐induced degradation of IκB‐α, an inhibitor of nuclear transcription factor NF‐κB that primes expression of a set of pro‐inflammatory genes 146. Indeed, PCSK9 inhibition prevents pro‐inflammatory activation of macrophages.
In addition to SRs, enzymes and transporters characterized in this review, it is necessary to investigate other biomolecules that could contribute to cholesterol handling in macrophages. Further studies are required to evaluate mechanisms that control expression and function of these biomolecules and investigate their involvement in cardiovascular disease. It is important to note here that in atherosclerosis, the disruption of lipid homoeostasis in macrophages, which leads to cholesterol accumulation and formation of foam cells, is only one of the well‐known mechanisms responsible for atherosclerotic plaque formation 1, 2, 3, 4, 5. Accumulated evidence indicate that the multifactorial inflammatory processes are responsible for the progression of atherosclerosis and that different cell types are simultaneously involved in all stages of plaque development 1, 2, 3, 4, 5.
Disclosure
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the Russian Scientific Foundation (grant 14‐15‐00112) for support of our work.
References
- 1. Gimbrone MA Jr, García‐Cardeña G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc Pathol. 2013; 22: 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Colin S, Chinetti‐Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. 2014; 262: 153–66. [DOI] [PubMed] [Google Scholar]
- 3. Stary HC, Chandler AB, Glagov S, et al A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1994; 89: 2462–78. [DOI] [PubMed] [Google Scholar]
- 4. Bobryshev YV. Monocyte recruitment and foam cell formation in atherosclerosis. Micron. 2006; 37: 208–22. [DOI] [PubMed] [Google Scholar]
- 5. Yu XH, Fu YC, Zhang DW, et al Foam cells in atherosclerosis. Clin Chim Acta. 2013; 424: 245–52. [DOI] [PubMed] [Google Scholar]
- 6. Collot‐Teixeira S, Martin J, McDermott‐Roe C, et al CD36 and macrophages in atherosclerosis. Cardiovasc Res. 2007; 75: 468–77. [DOI] [PubMed] [Google Scholar]
- 7. Ghosh S, Zhao B, Bie J, et al Macrophage cholesteryl ester mobilization and atherosclerosis. Vascul Pharmacol. 2010; 52: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Westerterp M, Bochem AE, Yvan‐Charvet L, et al ATP‐binding cassette transporters, atherosclerosis, and inflammation. Circ Res. 2014; 114: 157–70. [DOI] [PubMed] [Google Scholar]
- 9. Kruth HS. Fluid‐phase pinocytosis of LDL by macrophages: a novel target to reduce macrophage cholesterol accumulation in atherosclerotic lesions. Curr Pharm Des. 2013; 19: 5865–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. de Villiers WJ, Smart EJ. Macrophage scavenger receptors and foam cell formation. J Leukoc Biol. 1999; 66: 740–6. [DOI] [PubMed] [Google Scholar]
- 11. Armesilla AL, Vega MA. Structural organization of the gene for human CD36 glycoprotein. J Biol Chem. 1994; 269: 18985–91. [PubMed] [Google Scholar]
- 12. Van Berkel TJ, Van Eck M, Herijgers N, et al Scavenger receptor classes A and B. Their roles in atherogenesis and the metabolism of modified LDL and HDL. Ann N Y Acad Sci. 2000; 902: 113–27. [DOI] [PubMed] [Google Scholar]
- 13. Tarhda Z, Semlali O, Kettani A, et al Three dimensional structure prediction of fatty acid binding site on human transmembrane receptor CD36. Bioinform Biol Insights. 2013; 7: 369–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Handberg A, Højlund K, Gastaldelli A, et al Plasma sCD36 is associated with markers of atherosclerosis, insulin resistance and fatty liver in a nondiabetic healthy population. J Intern Med. 2012; 271: 294–304. [DOI] [PubMed] [Google Scholar]
- 15. Handberg A, Levin K, Højlund K, et al Identification of the oxidized low‐density lipoprotein scavenger receptor CD36 in plasma: a novel marker of insulin resistance. Circulation. 2006; 114: 1169–76. [DOI] [PubMed] [Google Scholar]
- 16. Handberg A, Norberg M, Stenlund H, et al Soluble CD36 (sCD36) clusters with markers of insulin resistance, and high sCD36 is associated with increased type 2 diabetes risk. J Clin Endocrinol Metab. 2010; 95: 1939–46. [DOI] [PubMed] [Google Scholar]
- 17. Teupser D, Mueller MA, Koglin J, et al CD36 mRNA expression is increased in CD14+ monocytes of patients with coronary heart disease. Clin Exp Pharmacol Physiol. 2008; 35: 552–6. [DOI] [PubMed] [Google Scholar]
- 18. Piechota M, Banaszewska A, Dudziak J, et al Highly upregulated expression of CD36 and MSR1 in circulating monocytes of patients with acute coronary syndromes. Protein J. 2012; 31: 511–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fuhrman B, Koren L, Volkova N, et al Atorvastatin therapy in hypercholesterolemic patients suppresses cellular uptake of oxidized‐LDL by differentiating monocytes. Atherosclerosis. 2002; 164: 179–85. [DOI] [PubMed] [Google Scholar]
- 20. Geloen A, Helin L, Geeraert B, et al CD36 inhibitors reduce postprandial hypertriglyceridemia and protect against diabetic dyslipidemia and atherosclerosis. PLoS ONE. 2012; 7: e37633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yuasa‐Kawase M, Masuda D, Yamashita T, et al Patients with CD36 deficiency are associated with enhanced atherosclerotic cardiovascular diseases. J Atheroscler Thromb. 2012; 19: 263–75. [DOI] [PubMed] [Google Scholar]
- 22. Mimche PN, Thompson E, Taramelli D, et al Curcumin enhances non‐opsonic phagocytosis of Plasmodium falciparum through up‐regulation of CD36 surface expression on monocytes/macrophages. J Antimicrob Chemother. 2012; 67: 1895–904. [DOI] [PubMed] [Google Scholar]
- 23. Inoue M, Tanabe H, Matsumoto A, et al Astaxanthin functions differently as a selective peroxisome proliferator‐activated receptor γ modulator in adipocytes and macrophages. Biochem Pharmacol. 2012; 84: 692–700. [DOI] [PubMed] [Google Scholar]
- 24. Gao D, Pararasa C, Dunston CR, et al Palmitate promotes monocyte atherogenicity via de novo ceramide synthesis. Free Radic Biol Med. 2012; 53: 796–806. [DOI] [PubMed] [Google Scholar]
- 25. Luan Y, Griffiths HR. Ceramides reduce CD36 cell surface expression and oxidised LDL uptake by monocytes and macrophages. Arch Biochem Biophys. 2006; 450: 89–99. [DOI] [PubMed] [Google Scholar]
- 26. Li XY, Wang C, Xiang XR, et al Porphyromonas gingivalis lipopolysaccharide increases lipid accumulation by affecting CD36 and ATP‐binding cassette transporter A1 in macrophages. Oncol Rep. 2013; 30: 1329–36. [DOI] [PubMed] [Google Scholar]
- 27. Gonçalves JL, Roma EH, Gomes‐Santos AC, et al Pro‐inflammatory effects of the mushroom Agaricus blazei and its consequences on atherosclerosis development. Eur J Nutr. 2012; 51: 927–37. [DOI] [PubMed] [Google Scholar]
- 28. Tang FT, Cao Y, Wang TQ, et al Tanshinone IIA attenuates atherosclerosis in ApoE(‐/‐) mice through down‐regulation of scavenger receptor expression. Eur J Pharmacol. 2011; 650: 275–84. [DOI] [PubMed] [Google Scholar]
- 29. Chiurchiù V, Izzi V, D'Aquilio F, et al Endomorphin‐1 prevents lipid accumulation via CD36 down‐regulation and modulates cytokines release from human lipid‐laden macrophages. Peptides. 2011; 32: 80–5. [DOI] [PubMed] [Google Scholar]
- 30. Granados‐Principal S, Quiles JL, Ramirez‐Tortosa CL, et al Squalene ameliorates atherosclerotic lesions through the reduction of CD36 scavenger receptor expression in macrophages. Mol Nutr Food Res. 2012; 56: 733–40. [DOI] [PubMed] [Google Scholar]
- 31. Choi JS, Bae JY, Kim DS, et al Dietary compound quercitrin dampens VEGF induction and PPARgamma activation in oxidized LDL‐exposed murine macrophages: association with scavenger receptor CD36. J Agric Food Chem. 2010; 58: 1333–41. [DOI] [PubMed] [Google Scholar]
- 32. Li XY, Kong LX, Li J, et al Kaempferol suppresses lipid accumulation in macrophages through the downregulation of cluster of differentiation 36 and the upregulation of scavenger receptor class B type I and ATP‐binding cassette transporters A1 and G1. Int J Mol Med. 2013; 31: 331–8. [DOI] [PubMed] [Google Scholar]
- 33. Nergiz‐Ünal R, Kuijpers MJ, de Witt SM, et al Atheroprotective effect of dietary walnut intake in ApoE‐deficient mice: involvement of lipids and coagulation factors. Thromb Res. 2013; 131: 411–7. [DOI] [PubMed] [Google Scholar]
- 34. Prabhudas M, Bowdish D, Drickamer K, et al Standardizing scavenger receptor nomenclature. J Immunol. 2014; 192: 1997–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Emi M, Asaoka H, Matsumoto A, et al Structure, organization, and chromosomal mapping of the human macrophage scavenger receptor gene. J Biol Chem. 1993; 268: 2120–5. [PubMed] [Google Scholar]
- 36. Matsumoto A, Naito M, Itakura H, et al Human macrophage scavenger receptors: primary structure, expression, and localization in atherosclerotic lesions. Proc Natl Acad Sci USA. 1990; 87: 9133–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dai XY, Cai Y, Mao DD, et al Increased stability of phosphatase and tensin homolog by intermedin leading to scavenger receptor A inhibition of macrophages reduces atherosclerosis in apolipoprotein E‐deficient mice. J Mol Cell Cardiol. 2012; 53: 509–20. [DOI] [PubMed] [Google Scholar]
- 38. Mäkinen PI, Lappalainen JP, Heinonen SE, et al Silencing of either SR‐A or CD36 reduces atherosclerosis in hyperlipidaemic mice and reveals reciprocal upregulation of these receptors. Cardiovasc Res. 2010; 88: 530–8. [DOI] [PubMed] [Google Scholar]
- 39. Moore KJ, Kunjathoor VV, Koehn SL, et al Loss of receptor‐mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest. 2005; 115: 2192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hashizume M, Mihara M. Blockade of IL‐6 and TNF‐α inhibited oxLDL‐induced production of MCP‐1 via scavenger receptor induction. Eur J Pharmacol. 2012; 689: 249–54. [DOI] [PubMed] [Google Scholar]
- 41. Hashizume M, Mihara M. Atherogenic effects of TNF‐α and IL‐6 via up‐regulation of scavenger receptors. Cytokine. 2012; 58: 424–30. [DOI] [PubMed] [Google Scholar]
- 42. Li K, Yao W, Zheng X, et al Berberine promotes the development of atherosclerosis and foam cell formation by inducing scavenger receptor A expression in macrophage. Cell Res. 2009; 19: 1006–17. [DOI] [PubMed] [Google Scholar]
- 43. Yang XF, Yang Y, Lian YT, et al The antibody targeting the E314 peptide of human Kv1.3 pore region serves as a novel, potent and specific channel blocker. PLoS ONE. 2012; 7: e36379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang Y, Wang YF, Yang XF, et al Specific Kv1.3 blockade modulates key cholesterol‐metabolism‐associated molecules in human macrophages exposed to ox‐LDL. J Lipid Res. 2013; 54: 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhao JF, Ching LC, Huang YC, et al Molecular mechanism of curcumin on the suppression of cholesterol accumulation in macrophage foam cells and atherosclerosis. Mol Nutr Food Res. 2012; 56: 691–701. [DOI] [PubMed] [Google Scholar]
- 46. Yang MY, Huang CN, Chan KC, et al Mulberry leaf polyphenols possess antiatherogenesis effect via inhibiting LDL oxidation and foam cell formation. J Agric Food Chem. 2011; 59: 1985–95. [DOI] [PubMed] [Google Scholar]
- 47. Wang XH, Wang F, You SJ, et al Dysregulation of cystathionine γ‐lyase (CSE)/hydrogen sulfide pathway contributes to ox‐LDL‐induced inflammation in macrophage. Cell Signal. 2013; 25: 2255–62. [DOI] [PubMed] [Google Scholar]
- 48. Wang Y, Zhao X, Jin H, et al Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2009; 29: 173–9. Doi:10.1161/ATVBAHA.108.179333. [DOI] [PubMed] [Google Scholar]
- 49. Zhao ZZ, Wang Z, Li GH, et al Hydrogen sulfide inhibits macrophage‐derived foam cell formation. Exp Biol Med. 2011; 236: 169–76. [DOI] [PubMed] [Google Scholar]
- 50. Dunn S, Vohra RS, Murphy JE, et al The lectin‐like oxidized low‐density‐lipoprotein receptor: a pro‐inflammatory factor in vascular disease. Biochem J. 2008; 409: 349–55. [DOI] [PubMed] [Google Scholar]
- 51. Pirillo A, Norata GD, Catapano AL. LOX‐1, OxLDL, and atherosclerosis. Mediators Inflamm. 2013; 2013: 152786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yamanaka S, Zhang XY, Miura K, et al The human gene encoding the lectin‐type oxidized LDL receptor (OLR1) is a novel member of the natural killer gene complex with a unique expression profile. Genomics. 1998; 54: 191–9. [DOI] [PubMed] [Google Scholar]
- 53. Nagase M, Abe J, Takahashi K, et al Genomic organization and regulation of expression of the lectin‐like oxidized low‐density lipoprotein receptor (LOX‐1) gene. J Biol Chem. 1998; 273: 33702–7. [DOI] [PubMed] [Google Scholar]
- 54. Yoshida H, Kondratenko N, Green S, et al Identification of the lectin‐like receptor for oxidized low‐density lipoprotein in human macrophages and its potential role as a scavenger receptor. Biochem J. 1998; 334: 9–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kataoka H, Kume N, Miyamoto S, et al Expression of lectinlike oxidized low‐density lipoprotein receptor‐1 in human atherosclerotic lesions. Circulation. 1999; 99: 3110–7. [DOI] [PubMed] [Google Scholar]
- 56. Kume N, Kita T. Lectin‐like oxidized low‐density lipoprotein receptor‐1 (LOX‐1) in atherogenesis. Trends Cardiovasc Med. 2001; 11: 22–5. [DOI] [PubMed] [Google Scholar]
- 57. Kume N, Moriwaki H, Kataoka H, et al Inducible expression of LOX‐1, a novel receptor for oxidized LDL in macrophages and vascular smooth muscle cells. Ann N Y Acad Sci. 2000; 902: 323–7. [DOI] [PubMed] [Google Scholar]
- 58. Mitra S, Deshmukh A, Sachdeva R, et al Oxidized low‐density lipoprotein and atherosclerosis implications in antioxidant therapy. Am J Med Sci. 2011; 342: 135–42. [DOI] [PubMed] [Google Scholar]
- 59. Li L, Sawamura T, Renier G. Glucose enhances human macrophage LOX‐1 expression: role for LOX‐1 in glucose‐induced macrophage foam cell formation. Circ Res. 2004; 94: 892–901. [DOI] [PubMed] [Google Scholar]
- 60. Rudijanto A. The expression and down stream effect of lectin like‐oxidized low density lipoprotein 1 (LOX‐1) in hyperglycemic state. Acta Med Indones. 2007; 39: 36–43. [PubMed] [Google Scholar]
- 61. Schaeffer DF, Riazy M, Parhar KS, et al LOX‐1 augments oxLDL uptake by lysoPC‐stimulated murine macrophages but is not required for oxLDL clearance from plasma. J Lipid Res. 2009; 50: 1676–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Randolph GJ, Beaulieu S, Lebecque S, et al Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science. 1998; 282: 480–3. [DOI] [PubMed] [Google Scholar]
- 63. Bobryshev YV. Dendritic cells in atherosclerosis: current status of the problem and clinical relevance. Eur Heart J. 2005; 26: 1700–4. [DOI] [PubMed] [Google Scholar]
- 64. Nickel T, Schmauss D, Hanssen H, et al oxLDL uptake by dendritic cells induces upregulation of scavenger‐receptors, maturation and differentiation. Atherosclerosis. 2009; 205: 442–50. [DOI] [PubMed] [Google Scholar]
- 65. Mehta JL, Sanada N, Hu CP, et al Deletion of LOX‐1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ Res. 2007; 100: 1634–42. [DOI] [PubMed] [Google Scholar]
- 66. Inoue K, Arai Y, Kurihara H, et al Overexpression of lectin‐like oxidized low‐density lipoprotein receptor‐1 induces intramyocardial vasculopathy in apolipoprotein E‐null mice. Circ Res. 2005; 97: 176–84. [DOI] [PubMed] [Google Scholar]
- 67. Kuge Y, Kume N, Ishino S, et al Prominent lectin‐like oxidized low density lipoprotein (LDL) receptor‐1 (LOX‐1) expression in atherosclerotic lesions is associated with tissue factor expression and apoptosis in hypercholesterolemic rabbits. Biol Pharm Bull. 2008; 31: 1475–82. [DOI] [PubMed] [Google Scholar]
- 68. Ghosh S. Early steps in reverse cholesterol transport: cholesteryl ester hydrolase and other hydrolases. Curr Opin Endocrinol Diabetes Obes. 2012; 19: 136–41. [DOI] [PubMed] [Google Scholar]
- 69. Sakashita N, Miyazaki A, Takeya M, et al Localization of human acyl‐coenzyme A: cholesterol acyltransferase‐1 (ACAT‐1) in macrophages and in various tissues. Am J Pathol. 2000; 156: 227–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chang CC, Huh HY, Cadigan KM, et al Molecular cloning and functional expression of human acyl‐coenzyme A: cholesterol acyltransferase cDNA in mutant Chinese hamster ovary cells. J Biol Chem. 1993; 268: 20747–55. [PubMed] [Google Scholar]
- 71. Dove DE, Su YR, Swift LL, et al ACAT1 deficiency increases cholesterol synthesis in mouse peritoneal macrophages. Atherosclerosis. 2006; 186: 267–74. [DOI] [PubMed] [Google Scholar]
- 72. Yang L, Yang JB, Chen J, et al Enhancement of human ACAT1 gene expression to promote the macrophage‐derived foam cell formation by dexamethasone. Cell Res. 2004; 14: 315–23. [DOI] [PubMed] [Google Scholar]
- 73. Kusunoki J, Hansoty DK, Aragane K, et al Acyl‐CoA:cholesterol acyltransferase inhibition reduces atherosclerosis in apolipoprotein E‐deficient mice. Circulation. 2001; 103: 2604–9. [DOI] [PubMed] [Google Scholar]
- 74. Fazio S, Major AS, Swift LL, et al Increased atherosclerosis in LDL receptor‐null mice lacking ACAT1 in macrophages. J Clin Invest. 2001; 107: 163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Su YR, Dove DE, Major AS, et al Reduced ABCA1‐mediated cholesterol efflux and accelerated atherosclerosis in apolipoprotein E‐deficient mice lacking macrophage‐derived ACAT1. Circulation. 2005; 111: 2373–81. [DOI] [PubMed] [Google Scholar]
- 76. Chang CC, Sakashita N, Ornvold K, et al Immunological quantitation and localization of ACAT‐1 and ACAT‐2 in human liver and small intestine. J Biol Chem. 2000; 275: 28083–92. [DOI] [PubMed] [Google Scholar]
- 77. Duewell P, Kono H, Rayner KJ, et al NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010; 464: 1357–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hongo S, Watanabe T, Arita S, et al Leptin modulates ACAT1 expression and cholesterol efflux from human macrophages. Am J Physiol Endocrinol Metab. 2009; 297: E474–82. [DOI] [PubMed] [Google Scholar]
- 79. Xin C, Yan‐Fu W, Ping H, et al Study of the insulin signaling pathways in the regulation of ACAT1 expression in cultured macrophages. Cell Biol Int. 2009; 33: 602–6. [DOI] [PubMed] [Google Scholar]
- 80. Ge J, Zhai W, Cheng B, et al Insulin induces human acyl‐coenzyme A: cholesterol acyltransferase1 gene expression via MAP kinases and CCAAT/enhancer‐binding protein α. J Cell Biochem. 2013; 114: 2188–98. [DOI] [PubMed] [Google Scholar]
- 81. Nagashima M, Watanabe T, Terasaki M, et al Native incretins prevent the development of atherosclerotic lesions in apolipoprotein E knockout mice. Diabetologia. 2011; 54: 2649–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Darsalia V, Larsson M, Nathanson D, et al Glucagon‐like receptor 1 agonists and DPP‐4 inhibitors: potential therapies for the treatment of stroke. J Cereb Blood Flow Metab. 2015; 35: 718–23. doi: 10.1038/jcbfm.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Terasaki M, Nagashima M, Nohtomi K, et al Preventive effect of dipeptidyl peptidase‐4 inhibitor on atherosclerosis is mainly attributable to incretin's actions in nondiabetic and diabetic apolipoprotein E‐null mice. PLoS ONE. 2013; 8: e70933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Inui A, Asakawa A, Bowers CY, et al Ghrelin, appetite, and gastric motility: the emerging role of the stomach as an endocrine organ. FASEB J. 2004; 18: 439–56. [DOI] [PubMed] [Google Scholar]
- 85. Cheng B, Wan J, Wang Y, et al Ghrelin inhibits foam cell formation via simultaneously down‐regulating the expression of acyl‐coenzyme A: cholesterol acyltransferase 1 and up‐regulating adenosine triphosphate‐binding cassette transporter A1. Cardiovasc Pathol. 2010; 19: e159–66. [DOI] [PubMed] [Google Scholar]
- 86. Levitt RC, Liu Z, Nouri N, et al Mapping of the gene for hormone sensitive lipase (LIPE) to chromosome 19q13.1‐>q13.2. Cytogenet Cell Genet. 1995; 69: 211–4. [DOI] [PubMed] [Google Scholar]
- 87. Kraemer FB, Shen WJ. Hormone‐sensitive lipase: control of intracellular tri‐(di‐) acylglycerol and cholesteryl ester hydrolysis. J Lipid Res. 2002; 43: 1585–94. [DOI] [PubMed] [Google Scholar]
- 88. Igarashi M, Osuga J, Isshiki M, et al Targeting of neutral cholesterol ester hydrolase to the endoplasmic reticulum via its N‐terminal sequence. J Lipid Res. 2010; 51: 274–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Igarashi M, Osuga J, Uozaki H, et al The critical role of neutral cholesterol ester hydrolase 1 in cholesterol removal from human macrophages. Circ Res. 2010; 107: 1387–95. [DOI] [PubMed] [Google Scholar]
- 90. Escary JL, Choy HA, Reue K, et al Paradoxical effect on atherosclerosis of hormone‐sensitive lipase overexpression in macrophages. J Lipid Res. 1999; 40: 397–404. [PubMed] [Google Scholar]
- 91. Choy HA, Wang XP, Schotz MC. Reduced atherosclerosis in hormone‐sensitive lipase transgenic mice overexpressing cholesterol acceptors. Biochim Biophys Acta. 2003; 1634: 76–85. [DOI] [PubMed] [Google Scholar]
- 92. Zhao B, Song J, Chow WN, et al Macrophage‐specific transgenic expression of cholesteryl ester hydrolase significantly reduces atherosclerosis and lesion necrosis in Ldlr mice. J Clin Invest. 2007; 117: 2983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Okazaki H, Igarashi M, Nishi M, et al Identification of neutral cholesterol ester hydrolase, a key enzyme removing cholesterol from macrophages. J Biol Chem. 2008; 283: 33357–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sekiya M, Osuga J, Igarashi M, et al The role of neutral cholesterol ester hydrolysis in macrophage foam cells. J Atheroscler Thromb. 2011; 18: 359–64. [DOI] [PubMed] [Google Scholar]
- 95. Sekiya M, Osuga J, Nagashima S, et al Ablation of neutral cholesterol ester hydrolase 1 accelerates atherosclerosis. Cell Metab. 2009; 10: 219–28. [DOI] [PubMed] [Google Scholar]
- 96. Yamashita M, Tamasawa N, Matsuki K, et al Insulin suppresses HDL‐mediated cholesterol efflux from macrophages through inhibition of neutral cholesteryl ester hydrolase and ATP‐binding cassette transporter G1 expressions. J Atheroscler Thromb. 2010; 17: 1183–9. [DOI] [PubMed] [Google Scholar]
- 97. McLaren JE, Michael DR, Salter RC, et al IL‐33 reduces macrophage foam cell formation. J Immunol. 2010; 185: 1222–9. [DOI] [PubMed] [Google Scholar]
- 98. Van Eck M. ATP‐binding cassette transporter A1: key player in cardiovascular and metabolic disease at local and systemic level. Curr Opin Lipidol. 2014; 25: 297–303. [DOI] [PubMed] [Google Scholar]
- 99. Luciani MF, Denizot F, Savary S, et al Cloning of two novel ABC transporters mapping on human chromosome 9. Genomics. 1994; 21: 150–9. [DOI] [PubMed] [Google Scholar]
- 100. Zhao Y, Pennings M, Vrins CL, et al Hypocholesterolemia, foam cell accumulation, but no atherosclerosis in mice lacking ABC‐transporter A1 and scavenger receptor BI. Atherosclerosis. 2011; 218: 314–22. [DOI] [PubMed] [Google Scholar]
- 101. Lorenzi I, von Eckardstein A, Cavelier C, et al Apolipoprotein A‐I but not high‐density lipoproteins are internalised by RAW macrophages: roles of ATP‐binding cassette transporter A1 and scavenger receptor BI. J Mol Med. 2008; 86: 171–83. [DOI] [PubMed] [Google Scholar]
- 102. Joyce CW, Wagner EM, Basso F, et al ABCA1 overexpression in the liver of LDLr‐KO mice leads to accumulation of pro‐atherogenic lipoproteins and enhanced atherosclerosis. J Biol Chem. 2006; 281: 33053–65. [DOI] [PubMed] [Google Scholar]
- 103. Bennett DJ, Cooke AJ, Edwards AS. Non‐steroidal LXR agonists; an emerging therapeutic strategy for the treatment of atherosclerosis. Recent Pat Cardiovasc Drug Discov. 2006; 1: 21–46. [DOI] [PubMed] [Google Scholar]
- 104. Lee SM, Moon J, Cho Y, et al Quercetin up‐regulates expressions of peroxisome proliferator‐activated receptor γ, liver X receptor α, and ATP binding cassette transporter A1 genes and increases cholesterol efflux in human macrophage cell line. Nutr Res. 2013; 33: 136–43. [DOI] [PubMed] [Google Scholar]
- 105. Mizuno T, Hayashi H, Naoi S, et al Ubiquitination is associated with lysosomal degradation of cell surface‐resident ATP‐binding cassette transporter A1 (ABCA1) through the endosomal sorting complex required for transport (ESCRT) pathway. Hepatology. 2011; 54: 631–43. [DOI] [PubMed] [Google Scholar]
- 106. Liu XY, Lu Q, Ouyang XP, et al Apelin‐13 increases expression of ATP‐binding cassette transporter A1 via activating protein kinase C α signaling in THP‐1 macrophage‐derived foam cells. Atherosclerosis. 2013; 226: 398–407. [DOI] [PubMed] [Google Scholar]
- 107. Tang CK, Tang GH, Yi GH, et al Effect of apolipoprotein A‐I on ATP binding cassette transporter A1 degradation and cholesterol efflux in THP‐1 macrophage‐derived foam cells. Acta Biochim Biophys Sin. 2004; 36: 218–26. [DOI] [PubMed] [Google Scholar]
- 108. Ogura M, Ayaori M, Terao Y, et al Proteasomal inhibition promotes ATP‐binding cassette transporter A1 (ABCA1) and ABCG1 expression and cholesterol efflux from macrophages in vitro and in vivo . Arterioscler Thromb Vasc Biol. 2011; 31: 1980–7. [DOI] [PubMed] [Google Scholar]
- 109. Malekpour‐Dehkordi Z, Javadi E, Doosti M, et al S‐Allylcysteine, a garlic compound, increases ABCA1 expression in human THP‐1 macrophages. Phytother Res. 2013; 27: 357–61. [DOI] [PubMed] [Google Scholar]
- 110. Campbell JH, Efendy JL, Smith NJ, et al Molecular basis by which garlic suppresses atherosclerosis. J Nutr. 2001; 131: 1006S–9S. [DOI] [PubMed] [Google Scholar]
- 111. Kim KM, Chun SB, Koo MS, et al Differential regulation of NO availability from macrophages and endothelial cells by the garlic component S‐allyl cysteine. Free Radic Biol Med. 2001; 30: 747–56. [DOI] [PubMed] [Google Scholar]
- 112. Wang Y, Oram JF. Unsaturated fatty acids phosphorylate and destabilize ABCA1 through a protein kinase C delta pathway. J Lipid Res. 2007; 48: 1062–8. [DOI] [PubMed] [Google Scholar]
- 113. Ku CS, Park Y, Coleman SL, et al Unsaturated fatty acids repress expression of ATP binding cassette transporter A1 and G1 in RAW 264.7 macrophages. J Nutr Biochem. 2012; 23: 1271–6. [DOI] [PubMed] [Google Scholar]
- 114. Yu XH, Jiang HL, Chen WJ, et al Interleukin‐18 and interleukin‐12 together downregulate ATP‐binding cassette transporter A1 expression through the interleukin‐18R/nuclear factor‐κB signaling pathway in THP‐1 macrophage‐derived foam cells. Circ J. 2012; 76: 1780–91. [DOI] [PubMed] [Google Scholar]
- 115. Leeper NJ, Raiesdana A, Kojima Y, et al MicroRNA‐26a is a novel regulator of vascular smooth muscle cell function. J Cell Physiol. 2011; 226: 1035–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Sun D, Zhang J, Xie J, et al MiR‐26 controls LXR‐dependent cholesterol efflux by targeting ABCA1 and ARL7. FEBS Lett. 2012; 586: 1472–9. [DOI] [PubMed] [Google Scholar]
- 117. de Aguiar Vallim TQ, Tarling EJ, Kim T, et al MicroRNA‐144 regulates hepatic ATP binding cassette transporter A1 and plasma high‐density lipoprotein after activation of the nuclear receptor farnesoid X receptor. Circ Res. 2013; 112: 1602–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Chen H, Rossier C, Lalioti MD, et al Cloning of the cDNA for a human homologue of the Drosophila white gene and mapping to chromosome 21q22.3. Am J Hum Genet. 1996; 59: 66–75. [PMC free article] [PubMed] [Google Scholar]
- 119. Croop JM, Tiller GE, Fletcher JA, et al Isolation and characterization of a mammalian homolog of the Drosophila white gene. Gene. 1997; 185: 77–85. [DOI] [PubMed] [Google Scholar]
- 120. Ranalletta M, Wang N, Han S, et al Decreased atherosclero sis in low‐density lipoprotein receptor knockout mice transplanted with Abcg1‐/‐ bone marrow. Arterioscler Thromb Vasc Biol. 2006; 26: 2308–15. [DOI] [PubMed] [Google Scholar]
- 121. Baldán A, Pei L, Lee R, et al Impaired development of atherosclerosis in hyperlipidemic Ldlr‐/‐ and ApoE‐/‐ mice transplanted with Abcg1‐/‐ bone marrow. Arterioscler Thromb Vasc Biol. 2006; 26: 2301–7. [DOI] [PubMed] [Google Scholar]
- 122. Helal O, Berrougui H, Loued S, et al Extra‐virgin olive oil consumption improves the capacity of HDL to mediate cholesterol efflux and increases ABCA1 and ABCG1 expression in human macrophages. Br J Nutr. 2013; 109: 1844–55. [DOI] [PubMed] [Google Scholar]
- 123. Jun HJ, Hoang MH, Yeo SK, et al Induction of ABCA1 and ABCG1 expression by the liver X receptor modulator cineole in macrophages. Bioorg Med Chem Lett. 2013; 23: 579–83. [DOI] [PubMed] [Google Scholar]
- 124. Hazen SL, Smith JD. An antiatherosclerotic signaling cascade involving intestinal microbiota, microRNA‐10b, and ABCA1/ABCG1‐mediated reverse cholesterol transport. Circ Res. 2012; 111: 948–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wang D, Xia M, Yan X, et al Gut microbiota metabolism of anthocyanin promotes reverse cholesterol transport in mice via repressing miRNA‐10b. Circ Res. 2012; 111: 967–81. [DOI] [PubMed] [Google Scholar]
- 126. Wang D, Wei X, Yan X, et al Protocatechuic acid, a metabolite of anthocyanins, inhibits monocyte adhesion and reduces atherosclerosis in apolipoprotein E‐deficient mice. J Agric Food Chem. 2010; 58: 12722–8. [DOI] [PubMed] [Google Scholar]
- 127. Calvo D, Dopazo J, Vega MA. The CD36, CLA‐1 (CD36L1), and LIMPII (CD36L2) gene family: cellular distribution, chromosomal location, and genetic evolution. Genomics. 1995; 25: 100–6. [DOI] [PubMed] [Google Scholar]
- 128. Calvo D, Vega MA. Identification, primary structure, and distribution of CLA‐1, a novel member of the CD36/LIMPII gene family. J Biol Chem. 1993; 268: 18929–35. [PubMed] [Google Scholar]
- 129. Zhang W, Yancey PG, Su YR, et al Inactivation of macrophage scavenger receptor class B type I promotes atherosclerotic lesion development in apolipoprotein E‐deficient mice. Circulation. 2003; 108: 2258–63. [DOI] [PubMed] [Google Scholar]
- 130. Van Eck M, Bos IS, Hildebrand RB, et al Dual role for scavenger receptor class B, type I on bone marrow‐derived cells in atherosclerotic lesion development. Am J Pathol. 2004; 165: 785–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Yancey PG, De la Llera‐Moya M, Swarnaker S, et al High density lipoprotein phospholipid composition is a major determinant of the bi‐directional flux and net movement of cellular free cholesterol mediated by scavenger receptor BI. J Biol Chem. 2000; 275: 36596–604. [DOI] [PubMed] [Google Scholar]
- 132. Chen W, Silver DL, Smith JD, et al Scavenger receptor‐BI inhibits ATP‐binding cassette transporter 1‐ mediated cholesterol efflux in macrophages. J Biol Chem. 2000; 275: 30794–800. [DOI] [PubMed] [Google Scholar]
- 133. Kämmerer I, Ringseis R, Biemann R, et al 13‐hydroxy linoleic acid increases expression of the cholesterol transporters ABCA1, ABCG1 and SR‐BI and stimulates apoA‐I‐dependent cholesterol efflux in RAW264.7 macrophages. Lipids Health Dis. 2011; 10: 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Voloshyna I, Hai O, Littlefield MJ, et al Resveratrol mediates anti‐atherogenic effects on cholesterol flux in human macrophages and endothelium via PPARγ and adenosine. Eur J Pharmacol. 2013; 698: 299–309. [DOI] [PubMed] [Google Scholar]
- 135. Uto‐Kondo H, Ayaori M, Ogura M, et al Coffee consumption enhances high‐density lipoprotein‐mediated cholesterol efflux in macrophages. Circ Res. 2010; 106: 779–87. [DOI] [PubMed] [Google Scholar]
- 136. Tang SL, Chen WJ, Yin K, et al PAPP‐A negatively regulates ABCA1, ABCG1 and SR‐B1 expression by inhibiting LXRα through the IGF‐I‐mediated signaling pathway. Atherosclerosis. 2012; 222: 344–54. [DOI] [PubMed] [Google Scholar]
- 137. Xie C, Kang J, Chen JR, et al Phenolic acids are in vivo atheroprotective compounds appearing in the serum of rats after blueberry consumption. J Agric Food Chem. 2011; 59: 10381–7. [DOI] [PubMed] [Google Scholar]
- 138. Tsuchiya A, Nagotani S, Hayashi T, et al Macrophage infiltration, lectin‐like oxidized‐LDL receptor‐1, and monocyte chemoattractant protein‐1 are reduced by chronic HMG‐CoA reductase inhibition. Curr Neurovasc Res. 2007; 4: 268–73. [DOI] [PubMed] [Google Scholar]
- 139. Hofnagel O, Luechtenborg B, Eschert H, et al Pravastatin inhibits expression of lectin‐like oxidized low‐density lipoprotein receptor‐1 (LOX‐1) in Watanabe heritable hyperlipidemic rabbits: a new pleiotropic effect of statins. Arterioscler Thromb Vasc Biol. 2006; 26: 604–10. [DOI] [PubMed] [Google Scholar]
- 140. Netherland C, Thewke DP. Rimonabant is a dual inhibitor of acyl CoA: cholesterol acyltransferases 1 and 2. Biochem Biophys Res Commun. 2010; 398: 671–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Yoshinaka Y, Shibata H, Kobayashi H, et al A selective ACAT‐1 inhibitor, K‐604, stimulates collagen production in cultured smooth muscle cells and alters plaque phenotype in apolipoprotein E‐knockout mice. Atherosclerosis. 2010; 213: 85–91. [DOI] [PubMed] [Google Scholar]
- 142. Urban D, Pöss J, Böhm M, et al Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J Am Coll Cardiol. 2013; 62: 1401–8. [DOI] [PubMed] [Google Scholar]
- 143. Stawowy P. Proprotein convertases in atherogenesis. Curr Opin Lipidol. 2015; 26: 338–44. [DOI] [PubMed] [Google Scholar]
- 144. Lambert G, Sjouke B, Choque B, et al The PCSK9 decade. J Lipid Res. 2012; 53: 2515–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Navarese EP, Kolodziejczak M, Schulze V, et al Effects of proprotein convertase subtilisin/kexin type 9 antibodies in adult with hypercholesterolemia: a systematic review and meta‐analysis. Ann Intern Med. 2015; 163: 40–51. [DOI] [PubMed] [Google Scholar]
- 146. Tang Z, Jiang L, Peng J, et al PCSK9 siRNA suppresses the inflammatory response induced by oxLDL through inhibition of NF‐κB activation in THP‐1‐derived macrophages. Int J Mol Med. 2012; 30: 931–8. [DOI] [PubMed] [Google Scholar]