

Abstract
BACKGROUND & AIMS
Long-chain fatty acid receptors G-protein–coupled receptor 40 (GPR40) (FFAR1) and GPR120 have been implicated in the chemosensation of dietary fats. I cells in the intestine secrete cholecystokinin (CCK), a peptide hormone that stimulates digestion of fat and protein, but these cells are rare and hard to identify. We sought to determine whether dietary fat-induced secretion of CCK is directly mediated by GPR40 expressed on I cells.
METHODS
We used fluorescence-activated cell sorting to isolate a pure population of I cells from duodenal mucosa in transgenic mice that expressed green fluorescent protein under the control of the CCK promoter (CCK–enhanced green fluorescent protein [eGFP] bacterial artificial chromosome mice). CCK-eGFP cells were evaluated for GPR40 expression by quantitative reverse transcription polymerase chain reaction and immunostaining. GPR40−/− mice were bred with CCK-eGFP mice to evaluate functional relevance of GPR40 on long-chain fatty acid–stimulated increases in [Ca2+]i and CCK secretion in isolated CCK-eGFP cells. Plasma levels of CCK after olive oil gavage were compared between GPR40+/+ and GPR40−/− mice.
RESULTS
Cells that expressed eGFP also expressed GPR40; expression of GPR40 was 100-fold greater than that of cells that did not express eGFP. In vitro, linoleic, oleic, and linolenic acids increased [Ca2+]i; linolenic acid increased CCK secretion by 53% in isolated GPR40+/+ cells that expressed eGFP. In contrast, in GPR40−/− that expressed eGFP, [Ca2+]i response to linoleic acid was reduced by 50% and there was no significant CCK secretion in response to linolenic acid. In mice, olive oil gavage significantly increased plasma levels of CCK compared with pregavage levels: 5.7-fold in GPR40+/+ mice and 3.1-fold in GPR40−/− mice.
CONCLUSIONS
Long-chain fatty acid receptor GPR40 induces secretion of CCK by I cells in response to dietary fat.
Keywords: Fat Metabolism, Hormone Secretion, FACS Analysis, Digestion
Lipids and digested lipid products have long been known to stimulate satiety, which is partially mediated by secretion of the neuropeptide hormone cholecystokinin (CCK) from duodenal enteroendocrine I cells. The postprandial satiety effect of CCK is vagally mediated upon binding of CCK1 receptors on afferent nerve terminals that are closely associated with the enteroendocrine cell; gastrointestinal feedback functions such as inhibition of gastric emptying and gastric acid secretion and stimulation of exocrine pancreas secretion in response to luminal nutrients are also regulated in this manner,1 whereas CCK regulates gall bladder contraction through a hormonal route.2
Products of lipid hydrolysis, particularly long-chain fatty acids (LCFAs) of at least 12-carbon length, are required to stimulate CCK secretion in the murine enteroendocrine cell line STC-13 and to elevate plasma CCK and reduce gastric motility in humans.4,5 The sensing mechanism by which the I cell detects LCFAs and triggers CCK secretion is unknown, but given that the apical membrane of the intestinal I cell communicates with the lumen, it is possible that a direct sensing mechanism exists. Several G-protein–coupled receptors (GPRs) have been identified as fatty acid sensors with nutrient-sensing capabilities by endocrine cells6,7; in particular, the LCFA receptors GPR40 and GPR120 have been suggested as possible nutrient detectors mediating CCK secretion.8,9
GPR40 is a recently deorphanized GPR that is activated by a range of medium- to long-chain saturated and unsaturated fatty acids of chain lengths >6 carbons.10 GPR40 is highly expressed in pancreatic islets and has been extensively studied for its role in insulin secretion by mouse pancreatic β cells (MIN6) in response to unsaturated LCFAs, oleic, linoleic, and linolenic acids.11,12 Multiple other organs, including the brain and intestine in rats and humans, have also been shown to express the transcript for GPR40.10,12 Using GPR40 reporter mice and in situ hybridization, GPR40 has recently been colocalized with several enteroendocrine cell types throughout the intestine—including glucagon-like peptide 1, glucose insulinotropic peptide, and CCK-expressing cells—and although disruption of GPR40 attenuates incretin secretion in mice fed a high-fat diet,13 the role of GPR40 on CCK secretion in the native I cell has yet to be evaluated.
The purpose of this study was to elucidate the basis for the chemosensation of dietary fat-stimulated release of CCK by intestinal I cells. Using GPR40−/− mice, we provide both in vitro and in vivo evidence that CCK secretion is stimulated by dietary luminal LCFAs directly sensed by GPR40 expressed on duodenal enteroendocrine I cells.
Materials and Methods
Experimental Animals
Transgenic mice with CCK promoter-driven enhanced green fluorescent protein (eGFP) were developed by the GenSat Bacterial Artificial Chromosomes (BAC) Transgenic project14 and obtained from the Mutant Mouse Regional Resource Center (Davis, CA).
A GPR40-targeted knockout mouse was developed by replacing the GPR40 coding region with a 21-nucleotide DNA fragment encoding genes for 9 amino acids of influenza hemagglutinin antigen, eGFP, and neomycin (Supplementary Figure 1A). eGFP was inserted with the intended use as a reporter for GPR40 expression within the intestine; however, histological examination of GPR40−/− mice did not yield any intestinal eGFP signals by epifluorescent microscopy or by flow cytometric analysis of an intestinal mucosal cell preparation (data not shown). Weakly detectable eGFP in pancreatic β cells suggested that undetectable I cell eGFP was due to lower copy number. Homozygous deletion of GPR40 was confirmed by polymerase chain reaction and Southern blot of genomic tail DNA and by Taqman reverse transcriptase polymerase chain reaction (RT-PCR) of duodenal mucosal scrapings (Supplementary Figure 1B–D), validating this mouse strain to be a GPR40 knockout model.
The GPR40−/− mice had no obvious phenotype when fed a regular chow diet. Knockout animals were fertile and had body weights and body compositions similar to their wild-type littermates. Metabolically there was no difference in fasting serum glucose, triglyceride, and insulin. In addition, consistent with findings reported by others,15–18 glucose tolerance and insulin levels were similar between GPR40+/+ and GPR40−/− mice fed a regular chow diet.
For functional studies, GPR40−/− mice were bred to CCK-eGFP mice to produce CCK-Egfp+ GPR40+/− pups. CCK-eGFP+ GPR40+/− mice were bred to produce GPR40−/− and GPR40+/+ mice expressing CCK-eGFP cells. See Supplementary Methods for genotyping details. Animals were bred and maintained on regular chow according to the National Institutes of Health Institutional Animal Care and Use Committee guidelines.
Isolation of Intestinal Endocrine Cells
Adult mice were euthanized and the proximal 5 to 6 cm of duodenum collected and confirmed for eGFP expression by epifluorescent microscopy. Intestines were washed with cold Dulbecco’s phosphate-buffered saline (PBS) and incubated in 1 mM EDTA-Dulbecco’s PBS, followed by 75 U/mL collagenase (CLPSA grade; Worthington Biochemical, Lakewood, NJ,) in a shaking water bath (20′, 37°C each). Cells were resuspended in 10% fetal bovine serum in medium, filtered through 30-μm and 20-μm nylon filters (Spectrum Laboratories, Laguna Hills, CA) to obtain a single-cell population and resuspended in a fluorescence-activated cell sorting (FACS) buffer (5% fetal bovine serum, 50 ug/mL DNase I in phenol-free Dulbecco’s modified Eagle’s medium). Cells were ~90% viable based on trypan blue exclusion.
The sample underwent FACS using the BD FACS ARIA machine (BD Biosciences, San Jose CA). Cells were sorted based on initial gating for GFP (fluorescein isothiocyanate) with sequential gating based on side and forward scatter to exclude dead cells and doublets. For RT-PCR studies, a non-eGFP cell population (fluorescein isothiocyanate <102)–comprising a mixture primarily of enterocytes, but also of goblet cells, lymphocytes, and non-GFP endocrine cells–was simultaneously sorted into a separate collection tube. Immediately after FACS, cells were found to be ~78% live using 7-aminoactinomycin D (7-AAD) dead-cell exclusion. Of the live-cell population, post-sorted cells were at least 98.5% eGFP+, which was confirmed by trypan blue exclusion and direct epifluorescent microscopy.
Immunofluorescent Staining
Single-cell preparations obtained before and after FACS were fixed in fresh 4% paraformaldehyde/PBS. Cells were dried on positively charged slides, incubated with rabbit anti-CCK antibody (1:2000; Peninsula Labs, Torrance, CA), and subsequently AlexaFluor 594 goat anti-rabbit IgG secondary antibody (Invitrogen, Carlsbad, CA).
For GPR40 immunostaining, villus-enriched fractions were obtained from the proximal duodenum by incubation in Dulbecco’s PBS containing 2 mM EDTA (30 minutes, 4°C) followed by gentle pipetting and centrifugation (250g, 2 minutes). Villus samples were immediately incubated with mouse anti-GPR40 monoclonal antibody (1:10; 30 minutes, 4°C; TransGenic Inc, Kumamoto, Japan), washed, incubated with AlexaFluor 633 goat anti-mouse IgG antibody (4°C, 30 minutes; Invitrogen), washed, and fixed with 4% paraformaldehyde/PBS.
For whole tissue sections, duodenum and pancreas tissue were collected from 4% paraformaldehyde/PBS perfused mice. Ten-micrometer cryosections were permeabilized with 0.2% TritonX-100, blocked in 5% bovine serum albumin, and incubated with rabbit anti-CCK (1:1000; Peninsula Labs, Torrance, CA) followed by AlexaFluor 594 goat anti-rabbit IgG antibody (Invitrogen). Slides were viewed with a Zeiss LSM510 Meta confocal microscope.
RNA Extraction and RT-PCR
RNA was extracted from FACS CCK-eGFP+ cells (30,000–100,000 cells) and non-eGFP cells (106 cells) using Trizol Reagent (Invitrogen). See Supplementary Methods for RT-PCR details and Supplementary Table 2 for a list of targeted PCR primers.
Confocal Ca2+ Imaging of CCK-eGFP Cells
Changes in intracellular Ca2+ concentration ([Ca2+]i) in CCK-eGFP cells were measured using the Ca2+-sensitive indicator dye Quest Rhod4 (AAT Bioquest, Inc., Sunnyvale, CA). FACS eGFP+ cells were loaded with Quest-Rhod4 in Hank’s Balanced Salt Solution supplemented with 10 mM HEPES (with 1.26 mM calcium and ~1 mM magnesium [pH 7.4]), incubated in subdued light (30 minutes, room temperature), plated on a 15-well μ-slide (ibidi LLC, Verona, WI), and imaged by confocal microscopy. Relative fluorescence intensity (FI) of Quest Rhod4 from each cell was measured using a 560- to 615-nm emission band pass filter at 488-nm excitation. Time-dependent acquisition was performed at 1-second intervals for over 10 minutes at room temperature. Quest Rhod4 was chosen to minimize overlapping interference with eGFP; however, we also confirmed that the ligands used in this study had no effect on eGFP FI. Thus, changes in FI detected within a 560- to 615-nm emission range were exclusively due to the changes in Quest Rhod4 fluorescence. For data analysis, maximum percent increase over baseline was calculated as follows: [A peak FI-basal FI]/basal FI × 100.
CCK Releasing Assay
Approximately 30,000 CCK-eGFP cells were sorted from a total of ~3 × 107 cells for each releasing assay. Twice-washed cells were resuspended in HEPES-buffered Hank’s Balanced Salt Solution (final concentration: 104 cells/mL). Approximately 1000 cells/well were aliquoted into a 96-well plate coated overnight with 0.2 mg/mL rat tail collagen loaded with a freshly made 2× working dilution of either α-linolenic acid or methyl linolenate (Sigma Aldrich, St Louis, MO) suspended in 1% ethyl alcohol–HEPES-buffered Hank’s Balanced Salt Solution (vehicle). A 4-μM linolenic acid concentration was selected based on 50% effective concentration ranges in GPR40-expressing cell lines10,19 and confirmed with preliminary dose response curves. Cells were incubated (total volume: 200 μL) for 30 minutes at 37°C, 5% CO2. Each treatment was tested in triplicate. Total CCK content was determined by adding 0.2% TritonX-100 in distilled water in designated wells. Secretion was halted by placing the plate on ice for 5 minutes. Cells were pelleted by centrifugation (720g, 10 minutes, 4°C) and 20 to 30 uL supernatant was retrieved and saved at −20°C until measured.
Cells were assessed for viability after each experiment by light microscopy, trypan blue exclusion, and eGFP expression under epifluorescent microscopy. Cell numbers were subjectively assessed to ensure the absence of marked differences in aliquoting and to check for cell lysis. On rare occasions, suboptimal I-cell preparations resulted in cell lysis after treatment, and data from these experiments were not included in the study.
Samples were diluted 1:5, and total CCK content samples were diluted 1:50. Dilution factors were based on previous dilution experiments that enabled detection on the standard curve. Each sample was measured in duplicate using the 4-day incubation protocol of a commercially available CCK Radioimmunoassay Kit (Alpco Diagnostics, Salem, NH), which has a sensitivity of 0.3 pmol/L and has <0.5% cross-reactivity with sulfated gastrin.
In Vivo Free Fatty Acid (FFA) Gavage and Plasma Collection
GPR40+/+ and GPR40−/− mice age-matched littermates (4–10 weeks old) weighing 20–35 g were fasted overnight on wire-bottom cages. Mice were anesthetized by isofluorane and a fasting whole blood sample (T = 0 minutes) was collected retro-orbitally using heparinized hematocrit tubes, and stored in fresh tubes containing 2 uL aprotinin (MP Biomedicals, Solon, OH; 10 mg/mL, 6000 KIU/mg). Mice were gavaged ~0.5 mL olive oil, and blood was resampled under anesthesia at 30 and 60 minutes. Plasma was collected and ethanol precipitated to remove plasma proteins. The supernatant was concentrated in a Speed Vac Concentrator at 37°C, restored to original volume with diluents, and stored at −20°C until radioimmunoassayed (Alpco Diagnostics, Salem, NH). Standard curves were produced by diluting the 50-pM standard with plasma-protein extracted serum from CCK knockout mice (Mutant Mouse Regional Resource Center) rather than the provided diluent, which improved sensitivity so samples could be measured within the standard curve. The few fasting plasma samples below the detection of the assay were assigned a value of 0 pM.
Data Analysis and Statistics
Results are expressed as mean ± standard error of mean. Differences in CCK secretion in acutely isolated cells were determined by 1-sample t test. Plasma CCK secretion differences between GPR40+/+ and GPR40−/− mice were determined by 2-way analysis of variance and Bonferroni post-hoc test. Statistical analysis was performed using GraphPad Prism version 3, and significance was accepted at P < .05.
Results
Validation of Purity of CCK-eGFP Cells From the CCK-eGFP BAC Transgenic Mouse
Rare eGFP-expressing cells from CCK-eGFP BAC transgenic mice were sparsely distributed along the epithelium of the duodenal mucosa, displaying typical teardrop enteroendocrine cell morphology and co-immunoreactivity with CCK (Figure 1A). Roughly 0.1%–0.2% of the preparation of singly dispersed duodenal mucosal cells were eGFP+ cells, as determined by flow cytometric analysis (data not shown). Enzymatically dispersed eGFP+ cells continued to be immunoreactive with CCK antiserum (Figure 1B and E). After FACS, eGFP+ cells excluding the dead-cell marker 7-AAD were 98.5% pure, which was also confirmed by direct microscopy (Figure 1C and F). As seen in Figure 2, CCK transcript is very highly expressed in eGFP+ cells and is not detectable in the eGFP− cells. Furthermore, the enterocyte-specific brush border enzyme alkaline phosphatase and the goblet cell-exclusive secretory granule marker calcium-activated chloride channel 3 (ClCa3; also known as gob520) were barely detectable in eGFP cells, confirming that sorted eGFP cells were CCK-expressing cells and not contaminating goblet cells or enterocytes. Therefore, in the duodenum of CCK-eGFP BAC transgenic mice, eGFP-expressing cells were considered CCK-expressing I cells and will be referred to as CCK-eGFP cells.
Figure 1.
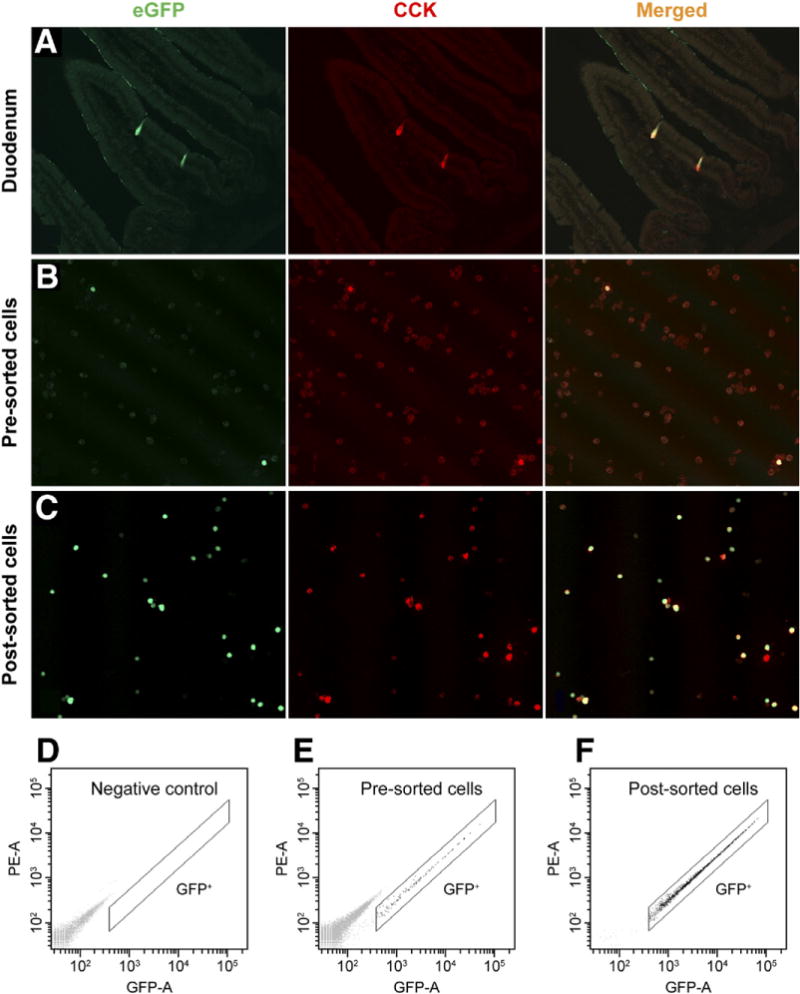
Validation of cholecystokinin (CCK)–enhanced green fluorescent protein (eGFP) transgene specificity and fluorescence-activated cell sorting (FACS) purification. All eGFP cells were CCK immunoreactive in (A) duodenal tissue, (B) dispersed single cells (20×), and (C) post-sorted cells (20×). FACS gating for GFP expression in singly dispersed cells from (D) CCK-eGFP− mice (negative control), (E) presorted, and (F) post-sorted singly dispersed cells from CCK-eGFP+ mice.
Figure 2.
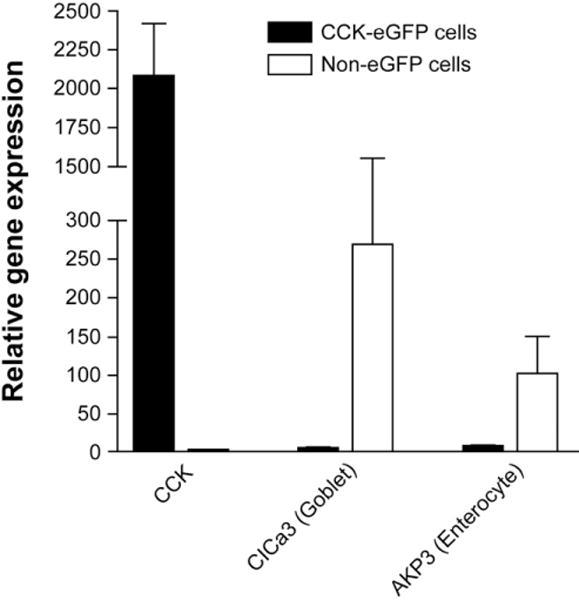
Verification of fluorescence-activated cell sorting (FACS) purification of cholecystokinin (CCK)–enhanced green fluorescent protein (eGFP) cells by gene expression using Taqman reverse transcriptase polymerase chain reaction. Expression of CCK messenger RNA was high relative to β-actin (×103) in CCK-eGFP cells with nearly undetectable contaminating transcripts for enterocyte (alkaline phosphatase [AKP3]) and goblet cell (calcium/chloride channel [CLCA3]) markers compared to a simultaneously sorted non-eGFP population (n = 5–6 separate cell isolations).
Acutely Isolated CCK-eGFP Cells Highly Express LCFA Receptor GPR40
Gene expression for GPR40 and GPR120 was approximately 100-fold (P = .003, n = 4) and 7-fold (P = .02, n = 5) greater, respectively, in the CCK-eGFP cell population relative to the non-eGFP cell population (Figure 3).
Figure 3.
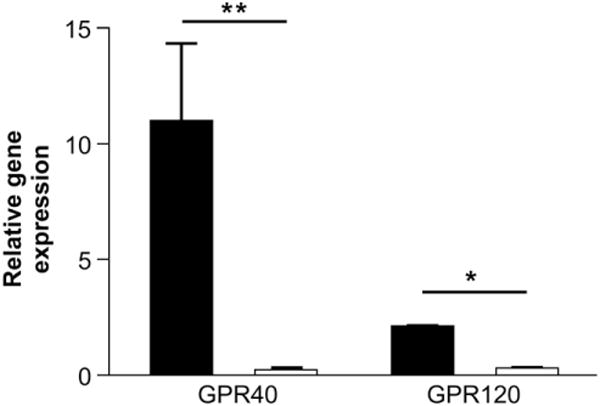
Gene expression of putative fatty-acid sensors in acutely isolated cholecystokinin (CCK)–enhanced green fluorescent protein (eGFP) cells. Taqman reverse transcriptase polymerase chain reaction results for G-protein–coupled receptor (GPR) 40 and GPR120 gene transcripts in the CCK-eGFP and non-eGFP cell population relative to housekeeping gene β-actin (×103). Statistical significance was determined by paired t test between the ΔCt values of the GFP and non-GFP cell populations. n = 4–5 separate cell isolations. *P < .05, **P < 001.
GPR40 protein expression was confirmed by positive immunostaining of CCK-eGFP cells within the duodenal villi of GPR40+/+ CCK-eGFP transgenic mice (Figure 4A, Supplementary Figure 2), which was absent in GPR40−/− mice (Figure 4B) and also in control staining with secondary antibody alone (Figure 4C). GPR40 immunoreactivity was also observed in rare CCK-eGFP− cells (Figure 4D). Together, this suggests that GPR40 is expressed on CCK-eGFP cells and other non–CCK-producing cells known to express GPR40.13
Figure 4.
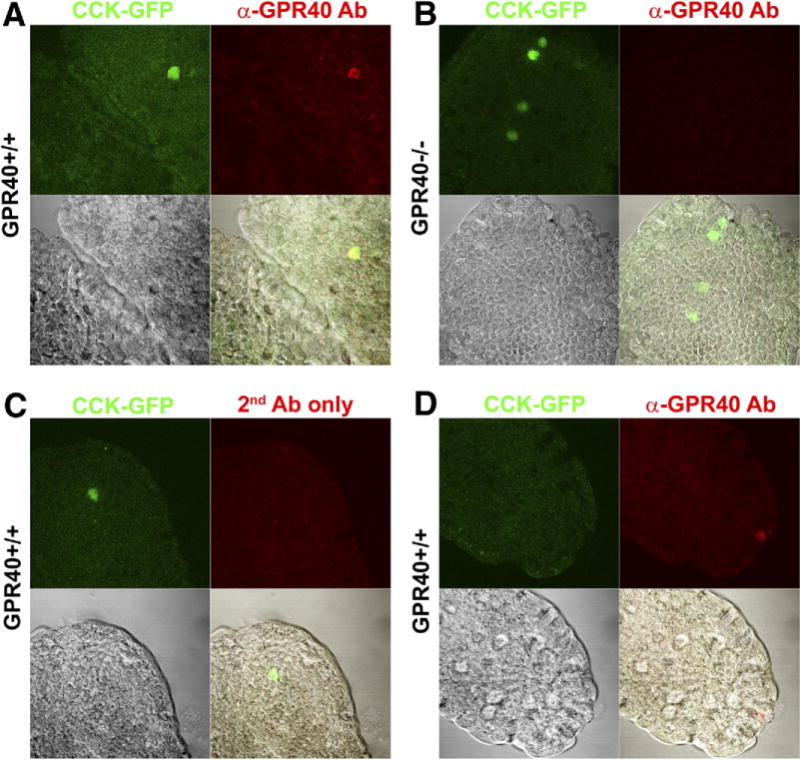
G-protein–coupled receptor 40 (GPR40) expression in cholecystokinin (CCK)–enhanced green fluorescent protein (eGFP) cells of primary duodenal villi. GPR40 immunoreactivity (red) was present in CCK-eGFP cells (green) from (A) GPR40+/+ mice, but not (B) GPR40−/− mice or (C) in the absence of primary antibody (negative control). (D) GPR40 immunoreactivity was present in rare eGFP-negative cells in GPR40+/+ mice. Images in each panel (63×) include CCK-eGFP only (top left; green), α-GPR40 or AlexaFluor 633 secondary antibody only (top right; red), transmitted light (bottom left), and merged panels (bottom right).
Dose-Dependent and GPR40-Specific Stimulation of Intracellular Ca2+ in CCK-eGFP Cells to FFA
FFA-stimulated activation of GPR40 triggers [Ca2+]i signaling, which is mediated by the phospholipase C/inositol 1,4,5-triphosphate pathway and is likely a prerequisite for LCFA-stimulated CCK release from I cells.21,22 To examine whether CCK-eGFP cells can be activated by LCFA directly through GPR40, changes in [Ca2+]i in response to LCFAs was assessed using the Ca2+ indicator dye Quest Rhod4. Linoleic acid (1–100 μM), a well-characterized stimulant of GPR40,22–24 significantly induced a time- and dose-dependent increase in relative FI of Quest Rhod4 in CCK-eGFP cells (P < .003 by regression analysis; Figure 5A, C, and D). Consistent with the expected LCFA pharmacology of GPR40, both linolenic (10 μM) and oleic (10 μM) acids significantly increased the relative FI of Quest Rhod4 (P = .001 and P < .05, respectively) similar to levels induced by linoleic acid (Figure 5B–D). Furthermore, the inactive methyl ester analog of linolenic acid (methyl linolenate) had no significant effect on Quest Rhod4 FI (P > .50; Figure 5B and D).
Figure 5.
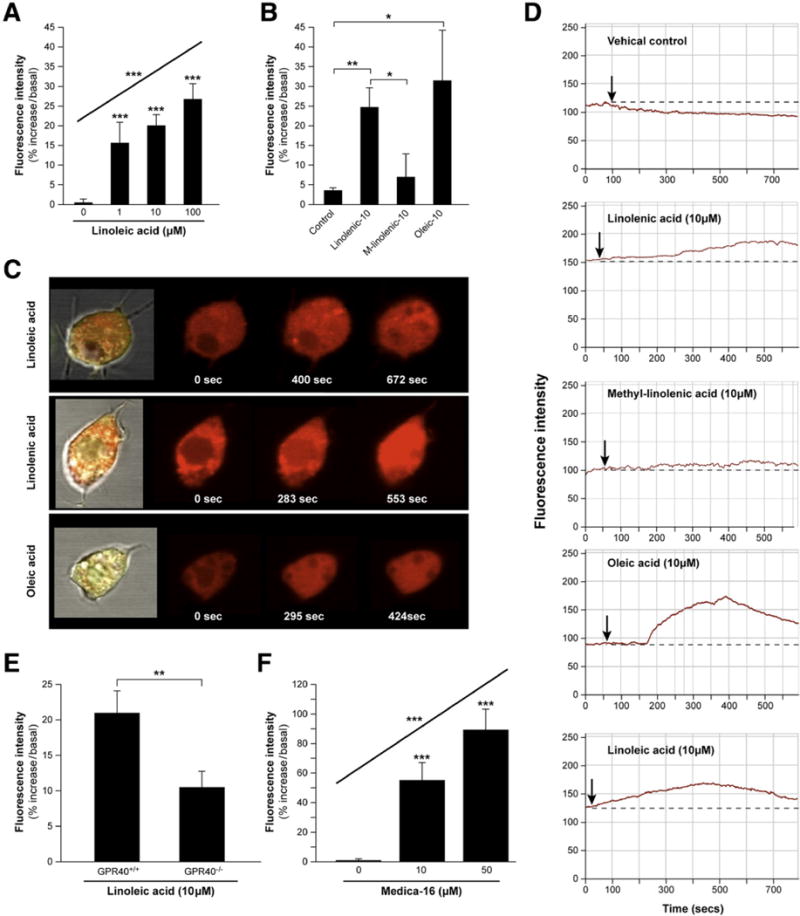
Long-chain fatty acid (LCFA) stimulated release of [Ca2+]i in fluorescence-activated cell sorting (FACS) isolated cholecystokinin (CCK-)–enhanced green fluorescent protein (eGFP) cells. (A) G-protein–coupled receptor 40 (GPR40)+/+ CCK-eGFP cells exhibit a dose-dependent response to linoleic acid (1–100 μM; n = 3–12 cells). (B) GPR40+/+ CCK-eGFP cell response to various LCFAs (all 10 μM; n = 6–7 cells). Representative (C) confocal images and (D) time-courses of isolated Quest Rhod4-loaded CCK-eGFP cell in response to various LCFAs. [Ca2+]i response to (E) linoleic acid (10 μM) in GPR40+/+ vs GPR40−/− CCK-eGFP cells (n = 10 cells, respectively), and (F) an increasing dose of the GPR40-selective agonist MEDICA 16 (0–50 μM) in GPR40+/+ CCK-eGFP cells (n = 3–12 cells). Changes in [Ca2+]i are expressed as mean ± standard error of mean of maximum percent increase in the fluorescence intensity (FI) of Quest Rhod4, relative to baseline. Cont, vehicle control. *P < .05; **P ≤ .001; ***P < .0001.
To determine whether the linoleic acid–induced response in Ca2+ was mediated specifically through GPR40, GPR40−/− CCK-eGFP cells were compared with wild-type CCK-eGFP cells. The [Ca2+]i response to linoleic acid (10 μM) in GPR40−/− CCK-eGFP cells (n = 10) was significantly lower than that in GPR40+/+ CCK-eGFP cells (n = 10; P < .001; Figure 5E). Additionally, the selective GPR40 agonist MEDICA-1623 induced a dose-dependent increase in [Ca2+]i in CCK-eGFP cells (P < .0001; Figure 5F).
LCFA-Stimulated CCK Secretion Is Attenuated With Deletion of GPR40 in Acutely Isolated Cells
To determine whether GPR40 directly mediated LCFA-induced CCK secretion, FACS GPR40+/+ and GPR40−/− CCK-eGFP cells were exposed to either linolenic acid or its inactive methyl ester analog, methyl linolenate, and CCK was measured in the supernatant. Basal CCK secretion from acutely isolated cells was approximately 5.4% ± 0.01% of total CCK content. Compared to normalized basal secretion, linolenic acid stimulated a significant 1.5- ± 0.2-fold increase in CCK secretion in GPR40+/+ CCK-eGFP cells (P = .02, n = 12). However, there was no significant change in baseline CCK secretion in GPR40−/− CCK-eGFP cells (1.1- ± 0.1-fold increase, n = 6; P = .6). As expected, neither GPR40+/+ (n = 8) nor GPR40−/− (n = 6) CCK-eGFP cells were responsive to methyl linolenate (1.0- ± 0.1-fold and 1.0- ± 0.1-fold, respectively; P > .9), and both were equally responsive to the depolarizing effects of KCl (1.6- ± 0.2-fold, P = .04, n = 6 and 1.5- ± 0.2-fold; P = .03, n = 6, respectively), which was used as a positive control for CCK secretion (Figure 6).
Figure 6.
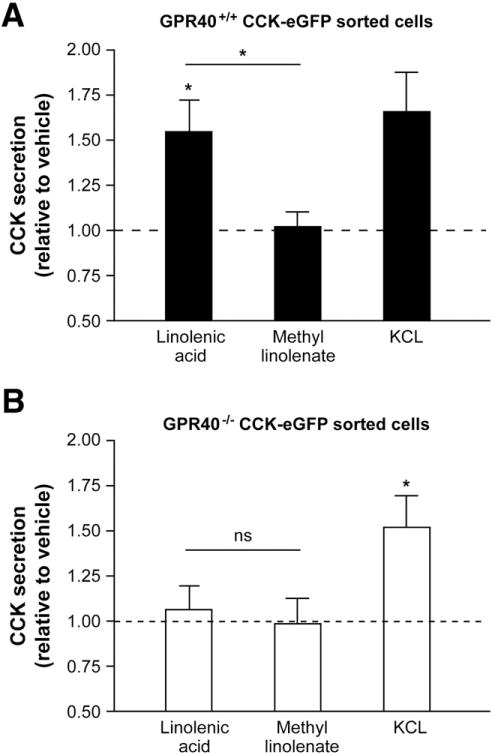
Fatty-acid stimulated cholecystokinin (CCK) secretion from acutely isolated CCK- enhanced green fluorescent protein (eGFP) cells with or without targeted disruption of G-protein–coupled receptor 40 (GPR40). CCK secretion in response to linolenic acid (4 uM) and methyl linolenate (4 uM) in (A) GPR40+/+ (black bars) and (B) GPR40−/− (clear bars) CCK-eGFP cells. KCl (50 mM) was used as a positive control. n = 6–12 separate cell preparations with each experiment performed in triplicate and assayed in duplicate. *P < .05 compared to baseline or between linolenic acid vs methyl linolenate. NS = not significant.
GPR40 Knockout Mice Demonstrate Blunted CCK Secretion in Response to Olive Oil Gavage
To determine whether the in vitro effects of GPR40 on fatty acid–induced CCK secretion would be reflected in vivo, circulating CCK levels were measured in GPR40+/+ and GPR40−/− mice after gastric gavage of olive oil, a natural dietary oil rich in oleic acid but also containing a heterogeneous mixture of saturated and unsaturated LCFAs. There was no significant difference in fasting (basal) levels of CCK between GPR40+/+ and GPR40−/− mice (1.0 ± 0.2 pmol/L, n = 7 and 1.0 ± 0.3 pmol/L, n = 6, respectively). However, after a gastric gavage of olive oil, significant genotype (P < .02) and time (P < .0001) effects on plasma CCK levels were present (Figure 7). Compared to fasting levels, plasma CCK was significantly elevated at 30 and 60 minutes post-gavage in both genotypes. At 30 minutes post-gavage, plasma CCK levels rose to 4.2 ± 0.8 pmol/L in the GPR40+/+ mice, and the response in the GPR40−/− mice was slightly lower at 2.8 ± 0.6 pmol/L. At 60 minutes post-gavage, plasma CCK levels continued to rise to 5.4 ± 0.9 pmol/L in the GPR40+/+ mice, which was significantly higher than circulating levels in the GPR40−/− mice (3.1 ± 0.4 pmol/L; P < .05 based on 2-way analysis of variance).
Figure 7.
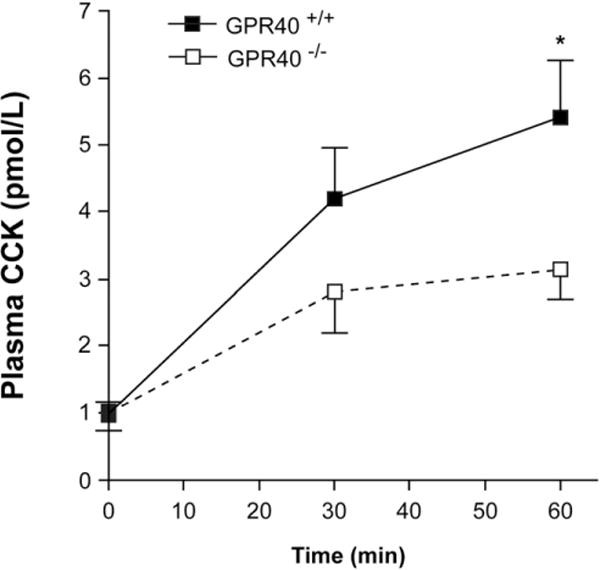
Effect of G-protein–coupled receptor 40 (GPR40) expression on cholecystokinin (CCK) secretion in response to olive oil gavage. Time course of plasma CCK in GPR40+/+ (n = 7) and GPR40−/− mice (n = 6) gavaged with 0.5 mL olive oil. Values are presented as mean ± standard error of mean. Significance was determined by 2-way analysis of variance. *P < .05. Experiment was repeated in duplicate.
Discussion
In the present study, we utilized BAC transgenic mice expressing CCK-promoter driven eGFP to isolate a pure population of intestinal I cells and identify the expression of GPR40 on a gene and protein level. Furthermore, to show that GPR40 is a LCFA receptor directly mediating fatty acid–induced CCK secretion, we obtained in vitro functional data from these isolated cells and demonstrated that targeted deletion of GPR40 markedly reduces [Ca2+]i signaling and CCK secretion in response to LCFAs. This effect is mirrored in vivo using the GPR40 knockout mouse. Taken together, these data strongly support a role for GPR40 in the direct detection of fatty acids leading to CCK secretion from the intestinal I cell.
Gastrointestinal hormone secretion in response to luminal nutrients can be mediated directly by the nutrient components themselves or indirectly through paracrine, endocrine, or neuronal factors. Evaluation of isolated enteroendocrine cells is required in order to examine direct stimulatory or inhibitory mechanisms. However, the enteroendocrine cell population makes up <1% of the intestinal mucosa and is divided into 15 known subtypes based on hormone production and secretion.25 Because this population is rare and diffusely scattered, the ability to identify and directly study specific subsets of these cells has been challenging. In the past, unenriched populations of rat intestinal mucosal cells have been studied to interrogate peptone-induced CCK secretion,26,27 but nonenteroendocrine cell types may still exert an indirect effect on the cells. Liddle et al have sorted a pure population of enteroendocrine cells labeled to be CCK-immunopositive, through FACS of monitor peptide-stimulated intestinal mucosal cells,28 but fatty acid secretagogues have not been tested in this population. The advent of fluorescent protein markers and promoter-driven transgenic mice has advanced the ability to separate specific subsets of enteroendocrine cells for study.29–31
By incorporating the CCK-eGFP transgene into GPR40−/− mice, we were able to directly interrogate the significance of this receptor as a direct mediator of LCFA-induced CCK secretion in a pure population of acutely isolated CCK-eGFP cells without confounding paracrine and neuronal influences. CCK-eGFP cells were directly activated, as measured by increasing [Ca2+]i, by LCFAs, linoleic, linolenic, and oleic acids, all of which are established potent ligands for GPR40.10,19,32 Furthermore, CCK secretion is directly stimulated in response to linolenic acid. Targeted deletion of GPR40 markedly reduced both the [Ca2+]i response to linoleic acid and the hormone secretion response to linolenic acid compared to wild-type CCK-eGFP cells. CCK-eGFP wild-type cells were not responsive to methyl linolenate, likely because the C-terminal carboxyl group of the fatty acid is important for appropriate receptor binding and activation, and esterification of the carboxyl group renders the ligand inactive.3,19 Together, with additional evidence of GPR40-specific agonist MEDICA-16 increasing [Ca2+]i in CCK-eGFP cells, these data confirm that LCFAs must bind GPR40 to directly activate hormone secretion from the I cell.
Although there was residual but diminished [Ca2+]i activation of GPR40−/− CCK-eGFP cells to LCFAs, the CCK secretory response to linolenic acid was absent. Therefore, it is unlikely that GPR120, which is also activated by linolenic–acid,7,8 plays a role in direct fatty acid induced CCK secretion in the native I cell. This finding differs from that demonstrated in the murine enteroendocrine cell line STC-1, in which incomplete RNA silencing of GPR120 partially inhibited linolenic acid–stimulated CCK secretion while partial silencing of GPR40 had no effect.8 Unlike intestinal I cells, the STC-1 cell line is a relatively nonspecific transformed cell line that expresses multiple hormones (CCK, secretin, proglucagon-derived peptides, pancreatic polypetide, neurotensin, and peptide YY) and receptors.33,34 Therefore, it is possible that the relative levels of GPR40 and GPR120 expression in STC-1 cells do not necessarily reflect the levels expressed in the native I cell, which may account for the difference in our results. Nonetheless, our results emphasize the importance of studying native cells whenever possible. Further studies on native I cells will be necessary to exclude a potential role of GPR120 on CCK secretion.
Our in vivo findings support what was observed in vitro between GPR40+/+ and GPR40−/− CCK-eGFP cells. Compared to wild-type mice, targeted deletion of GPR40 resulted in significantly reduced plasma CCK levels in response to gastric gavage of olive oil. In humans, intraduodenal infusion of fatty acids of at least 12-carbon length are required to raise plasma CCK levels, inhibit gastric motor function, and suppress appetite.4,35 In rats, duodenal infusion of sodium oleate (C18:1) elevates plasma CCK levels36 and activates rat jejunal vagal afferents through binding of the CCK-A receptor.37 Although we did not use specific fatty acid chain lengths in our in vivo studies, the fat composition of olive oil is primarily oleic acid (C18:1) and other polyunsaturated (linoleic [C18:2], linolenic [C18:3]) and saturated (palmitic [C16:0], stearic [C18:0]) LCFAs of C16–C18 carbon chain length. As described earlier, we have shown that oleic, linoleic, and linolenic acid similarly activate CCK-eGFP cells consistent with that shown by others in GPR40-expressing cell lines.10,19
Although the rise in plasma CCK levels in response to olive oil was significantly less in the GPR40−/− mice compared to GPR40+/+ mice, the response was still greater than fasting CCK levels. Given that CCK secretion is reduced to basal levels in vitro in acutely isolated GPR40−/− CCK-eGFP cells, the in vivo response is mostly likely due to activation of an indirect fatty acid sensing mechanism. One putative mechanism by which intestinal lipid may stimulate CCK secretion independent of GPR40 is through indirect activation by chylomicron component apolipoprotein A-IV (apoA-IV). ApoA-IV is secreted by enterocytes in response to absorption of LCFAs and has been identified as a satiety factor and activator of CCK-responsive vagal afferents and their downstream effects.38–40 The role of apoA-IV on CCK secretion is unclear as an apoA-IV receptor has yet to be identified, and no study has been done looking at apoA-IV on plasma CCK secretion in vivo or in vitro in the STC-1 cell line. However, apoA-IV remains a possible explanation for the residual circulating CCK levels in the GPR40−/− mouse in response to fatty acids that is not observed in vitro in the acutely isolated GPR40−/− CCK-eGFP cells.
In summary, this study presents evidence that native I cells express receptors for GPR40 and that GPR40 is the primary receptor directly mediating LCFA-induced CCK secretion from these cells. The roles of GPR120 and potentially apoA-IV as minor or indirect mediators of fatty acid induced CCK secretion remain to be evaluated.
Supplementary Material
Acknowledgments
Funding
Supported by the Veterinary Scientists Training Program, University of California, Davis School of Veterinary Medicine (APL), National Institutes of Health DK41004 (HER), and National Institute of Diabetes, Digestive, and Kidney Diseases Intramural funding.
Abbreviations
- apoA-IV
apolipoprotein A-IV
- BAC
bacterial artificial chromosome
- CCK
cholecystokinin
- eGFP
enhanced green fluorescent protein
- FACS
fluorescence-activated cell sorting
- FFA
free fatty acid
- FI
fluorescence intensity
- GPR
G-protein–coupled receptor
- LCFA
long-chain fatty acid
- PBS
phosphate-buffered saline
- RT-PCR
reverse transcription polymerase chain reaction
Footnotes
Supplementary Material
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi: 10.1053/j.gastro.2010.10.012.
Conflicts of interest
The authors disclose no conflicts.
References
- 1.Raybould HE, Lloyd KC. Integration of postprandial function in the proximal gastrointestinal tract. Role of CCK and sensory pathways. Ann N Y Acad Sci. 1994;713:143–156. doi: 10.1111/j.1749-6632.1994.tb44061.x. [DOI] [PubMed] [Google Scholar]
- 2.Ivy AC, Oldberg E. A hormone mechanism for gall-bladder contraction and evacuation. Am J Physiol. 1928;86:559–613. [Google Scholar]
- 3.McLaughlin JT, Lomax RB, Hall L, et al. Fatty acids stimulate cholecystokinin secretion via an acyl chain length-specific, Ca2+-dependent mechanism in the enteroendocrine cell line STC-1. J Physiol. 1998;513(Pt 1):11–18. doi: 10.1111/j.1469-7793.1998.011by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLaughlin J, Grazia Luca M, Jones MN, et al. Fatty acid chain length determines cholecystokinin secretion and effect on human gastric motility. Gastroenterology. 1999;116:46–53. doi: 10.1016/s0016-5085(99)70227-1. [DOI] [PubMed] [Google Scholar]
- 5.Guimbaud R, Moreau JA, Bouisson M, et al. Intraduodenal free fatty acids rather than triglycerides are responsible for the release of CCK in humans. Pancreas. 1997;14:76–82. doi: 10.1097/00006676-199701000-00012. [DOI] [PubMed] [Google Scholar]
- 6.Covington DK, Briscoe CA, Brown AJ, et al. The G-protein-coupled receptor 40 family (GPR40-GPR43) and its role in nutrient sensing. Biochem Soc Trans. 2006;34:770–773. doi: 10.1042/BST0340770. [DOI] [PubMed] [Google Scholar]
- 7.Hirasawa A, Tsumaya K, Awaji T, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. 2005;11:90–94. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka T, Katsuma S, Adachi T, et al. Free fatty acids induce cholecystokinin secretion through GPR120. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:523–527. doi: 10.1007/s00210-007-0200-8. [DOI] [PubMed] [Google Scholar]
- 9.McLaughlin J. Long-chain fatty acid sensing in the gastrointestinal tract. Biochem Soc Trans. 2007;35:1199–1202. doi: 10.1042/BST0351199. [DOI] [PubMed] [Google Scholar]
- 10.Briscoe CP, Tadayyon M, Andrews JL, et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem. 2003;278:11303–11311. doi: 10.1074/jbc.M211495200. [DOI] [PubMed] [Google Scholar]
- 11.Salehi A, Flodgren E, Nilsson NE, et al. Free fatty acid receptor 1 (FFA(1)R/GPR40) and its involvement in fatty-acid-stimulated insulin secretion. Cell Tissue Res. 2005;322:207–215. doi: 10.1007/s00441-005-0017-z. [DOI] [PubMed] [Google Scholar]
- 12.Itoh Y, Hinuma S. GPR40, a free fatty acid receptor on pancreatic beta cells, regulates insulin secretion. Hepatol Res. 2005;33:171–173. doi: 10.1016/j.hepres.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 13.Edfalk S, Steneberg P, Edlund H. Gpr40 is expressed in enteroendocrine cells and mediates FFA stimulation of incretin secretion. Diabetes. 2008;57:2280–2287. doi: 10.2337/db08-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gong S, Zheng C, Doughty ML, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- 15.Kebede M, Alquier T, Latour MG, et al. The fatty-acid receptor GPR40 plays a role in insulin secretion in vivo after high-fat feeding. Diabetes. 2008;57:2432–2437. doi: 10.2337/db08-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Latour MG, Alquier T, Oseid E, et al. GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes. 2007;56:1087–1094. doi: 10.2337/db06-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pang Z, Wu N, Zhang X, et al. GPR40 is partially required for insulin secretion following activation of beta3-adrenergic receptors. Mol Cell Endocrinol. 2010;325:18–25. doi: 10.1016/j.mce.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 18.Lan H, Hoos LM, Liu L, et al. Lack of FFAR1/GPR40 does not protect mice from high-fat diet-induced metabolic disease. Diabetes. 2008;57:2999–3006. doi: 10.2337/db08-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itoh Y, Kawamata Y, Harada M, et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422:173–176. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- 20.Leverkoehne I, Gruber AD. The murine mCLCA3 (alias gob-5) protein is located in the mucin granule membranes of intestinal, respiratory, and uterine goblet cells. J Histochem Cytochem. 2002;50:829–838. doi: 10.1177/002215540205000609. [DOI] [PubMed] [Google Scholar]
- 21.Shapiro H, Shachar S, Sekler I, et al. Role of GPR40 in fatty acid action on the beta cell line INS-1E. Biochem Biophys Res Commun. 2005;335:97–104. doi: 10.1016/j.bbrc.2005.07.042. [DOI] [PubMed] [Google Scholar]
- 22.Suh HN, Huong HT, Song CH, et al. Linoleic acid stimulates gluconeogenesis via Ca2+/PLC, cPLA2, and PPAR pathways through GPR40 in primary cultured chicken hepatocytes. Am J Physiol Cell Physiol. 2008;295:C1518–C1527. doi: 10.1152/ajpcell.00368.2008. [DOI] [PubMed] [Google Scholar]
- 23.Hara T, Hirasawa A, Sun Q, et al. Novel selective ligands for free fatty acid receptors GPR120 and GPR40. Naunyn Schmiedebergs Arch Pharmacol. 2009;380:247–255. doi: 10.1007/s00210-009-0425-9. [DOI] [PubMed] [Google Scholar]
- 24.Hu H, He LY, Gong Z, et al. A novel class of antagonists for the FFAs receptor GPR40. Biochem Biophys Res Commun. 2009;390:557–563. doi: 10.1016/j.bbrc.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 25.Rindi G, Leiter AB, Kopin AS, et al. The “normal” endocrine cell of the gut: changing concepts and new evidences. Ann N Y Acad Sci. 2004;1014:1–12. doi: 10.1196/annals.1294.001. [DOI] [PubMed] [Google Scholar]
- 26.Sharara AI, Bouras EP, Misukonis MA, et al. Evidence for indirect dietary regulation of cholecystokinin release in rats. Am J Physiol. 1993;265:G107–G112. doi: 10.1152/ajpgi.1993.265.1.G107. [DOI] [PubMed] [Google Scholar]
- 27.Nishi T, Hara H, Hira T, et al. Dietary protein peptic hydrolysates stimulate cholecystokinin release via direct sensing by rat intestinal mucosal cells. Exp Biol Med (Maywood) 2001;226:1031–1036. doi: 10.1177/153537020122601110. [DOI] [PubMed] [Google Scholar]
- 28.Liddle RA, Misukonis MA, Pacy L, et al. Cholecystokinin cells purified by fluorescence-activated cell sorting respond to monitor peptide with an increase in intracellular calcium. Proc Natl Acad Sci U S A. 1992;89:5147–5151. doi: 10.1073/pnas.89.11.5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Samuel BS, Shaito A, Motoike T, et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci U S A. 2008;105:16767–16772. doi: 10.1073/pnas.0808567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reimann F, Habib AM, Tolhurst G, et al. Glucose sensing in L cells: a primary cell study. Cell Metab. 2008;8:532–539. doi: 10.1016/j.cmet.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parker HE, Habib AM, Rogers GJ, et al. Nutrient-dependent secretion of glucose-dependent insulinotropic polypeptide from primary murine K cells. Diabetologia. 2009;52:289–298. doi: 10.1007/s00125-008-1202-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Briscoe CP, Peat AJ, McKeown SC, et al. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: identification of agonist and antagonist small molecules. Br J Pharmacol. 2006;148:619–628. doi: 10.1038/sj.bjp.0706770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Geraedts MC, Troost FJ, Saris WH. Peptide-YY is released by the intestinal cell line STC-1. J Food Sci. 2009;74:H79–H82. doi: 10.1111/j.1750-3841.2009.01074.x. [DOI] [PubMed] [Google Scholar]
- 34.Rindi G, Grant SG, Yiangou Y, et al. Development of neuroendocrine tumors in the gastrointestinal tract of transgenic mice. Heterogeneity of hormone expression. Am J Pathol. 1990;136:1349–1363. [PMC free article] [PubMed] [Google Scholar]
- 35.Feltrin KL, Little TJ, Meyer JH, et al. Effects of intraduodenal fatty acids on appetite, antropyloroduodenal motility, and plasma CCK and GLP-1 in humans vary with their chain length. Am J Physiol Regul Integr Comp Physiol. 2004;287:R524–R533. doi: 10.1152/ajpregu.00039.2004. [DOI] [PubMed] [Google Scholar]
- 36.Lewis LD, Williams JA. Regulation of cholecystokinin secretion by food, hormones, and neural pathways in the rat. Am J Physiol. 1990;258:G512–G518. doi: 10.1152/ajpgi.1990.258.4.G512. [DOI] [PubMed] [Google Scholar]
- 37.Lal S, Kirkup AJ, Brunsden AM, et al. Vagal afferent responses to fatty acids of different chain length in the rat. Am J Physiol Gastrointest Liver Physiol. 2001;281:G907–G915. doi: 10.1152/ajpgi.2001.281.4.G907. [DOI] [PubMed] [Google Scholar]
- 38.Fujimoto K, Cardelli JA, Tso P. Increased apolipoprotein A-IV in rat mesenteric lymph after lipid meal acts as a physiological signal for satiation. Am J Physiol. 1992;262:G1002–G1006. doi: 10.1152/ajpgi.1992.262.6.G1002. [DOI] [PubMed] [Google Scholar]
- 39.Glatzle J, Darcel N, Rechs AJ, et al. Apolipoprotein A-IV stimulates duodenal vagal afferent activity to inhibit gastric motility via a CCK1 pathway. Am J Physiol Regul Integr Comp Physiol. 2004;287:R354–R359. doi: 10.1152/ajpregu.00705.2003. [DOI] [PubMed] [Google Scholar]
- 40.Kalogeris TJ, Monroe F, Demichele SJ, et al. Intestinal synthesis and lymphatic secretion of apolipoprotein A-IV vary with chain length of intestinally infused fatty acids in rats. J Nutr. 1996;126:2720–2729. doi: 10.1093/jn/126.11.2720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.