

Abstract
Multiple sclerosis is the most frequent chronic inflammatory disease of the CNS. The entry and survival of pathogenic T cells in the CNS are crucial for the initiation and persistence of autoimmune neuroinflammation. In this respect, contradictory evidence exists on the role of the most potent type of antigen‐presenting cells, dendritic cells. Applying intravital two‐photon microscopy, we demonstrate the gatekeeper function of CNS professional antigen‐presenting CD11c+ cells, which preferentially interact with Th17 cells. IL‐17 expression correlates with expression of GM‐CSF by T cells and with accumulation of CNS CD11c+ cells. These CD11c+ cells are organized in perivascular clusters, targeted by T cells, and strongly express the inflammatory chemokines Ccl5, Cxcl9, and Cxcl10. Our findings demonstrate a fundamental role of CNS CD11c+ cells in the attraction of pathogenic T cells into and their survival within the CNS. Depletion of CD11c+ cells markedly reduced disease severity due to impaired enrichment of pathogenic T cells within the CNS.
Keywords: chemokines, dendritic cells, EAE/MS, Th17, two‐photon microscopy
Subject Categories: Immunology, Neuroscience
Introduction
Multiple sclerosis (MS) is a complex disease of the central nervous system (CNS) involving T cells in its pathology, as also recently evidenced in a genomewide association study (GWAS) (International Multiple Sclerosis Genetics Consortium (IMSGC) et al, 2011; 2013). It has been well established that the clinical severity of the animal disease model, experimental autoimmune encephalomyelitis (EAE), directly correlates with the number and differentiation of CD4+ T‐helper cells inside the target organ (Hofstetter et al, 2007), among which Th17 cells strongly contribute to tissue destruction (Siffrin et al, 2010). Indeed, preliminary data from a phase II clinical study of a blocking antibody against IL‐17 (AIN457) in patients with MS indicate that targeting Th17 cells might be a viable strategy in the treatment of multiple sclerosis (Fernández et al, 2013). Several recent reports indicate that another Th17 cytokine, the granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), represents a major effector molecule in these processes in EAE (Codarri et al, 2011; El‐Behi et al, 2011) and MS (Hartmann et al, 2014; Noster et al, 2014). Since GM‐CSF is crucial for the differentiation and survival of certain DC subsets (Markowicz & Engleman, 1990), the role of DCs in neuroinflammation might be more relevant for disease persistence than previously thought. DCs are professional antigen‐presenting cells, which have a crucial role in the differentiation of T cells and integrate multiple stimuli, most importantly from the innate immune system and invading pathogens, in the decision of whether a pro‐inflammatory or regulatory adaptive immune response is induced. However, DCs are very heterogenic, dependent on their anatomical location and phenotype (Schlitzer & Ginhoux, 2014). It has been reported that DCs are sufficient for promoting an autoreactive response inside the CNS (Greter et al, 2005) and are capable of inducing and amplifying EAE (McMahon et al, 2005; Karman et al, 2006). However, the priming of encephalitogenic T cells has been found to be unaffected or even exaggerated by the absence of DCs in active EAE induced by myelin peptides and strong adjuvants (Isaksson et al, 2012; Yogev et al, 2012). Therefore, a dual role of DCs seems possible, that is, a regulatory role in the context of T‐cell differentiation in the secondary lymphoid organs and a rather pro‐inflammatory role in the CNS. Focusing on CNS DCs, which are a very rare cell population under physiologic conditions, it has been unclear to what extent they contribute to local T‐cell recruitment, activation, and lesion development. In this context, the role of inflammatory chemokines for recruitment and orchestration of tissue inflammation is of significant interest. T cells that express the chemokine receptors CCR2 and CCR5 are more abundant in patients with exacerbated MS (Misu et al, 2001). Furthermore, perivascular T cells in MS and EAE express the chemokine receptor CXCR3. However, the sources of the relevant chemokines remain unclear. Here, we investigate the role of CNS CD11c+ cells by exploiting a transgenic mouse model, CD11c‐DTR/GFP (Jung et al, 2002), which uses the commonly accepted DC marker CD11c to specifically target DCs. This transgenic model allows the conditional depletion of classical DCs after diphtheria toxin (DTX) treatment (Jung et al, 2002). We performed conditional depletion experiments in the effector phase of adoptive transfer EAE, which targets primarily the processes within the CNS. Furthermore, we isolated CNS CD11c+ cells and characterized their chemokine expression profiles, which revealed a unique role of this cell subset for T‐cell migration and recruitment. In addition, on the T‐cell side, we made use of IL‐17 reporter mice, which express the fluorescent protein EGFP, concomitantly to IL‐17 (Esplugues et al, 2011). We utilized in vivo two‐photon laser scanning microscopy (TPLSM) in order to observe the real behavior of DCs and IL‐17‐producing cells at the barrier of, and within, the CNS.
Results
Depletion of CD11c‐GFP+ cells aborts EAE induction by adoptive transfer of encephalitogenic T cells
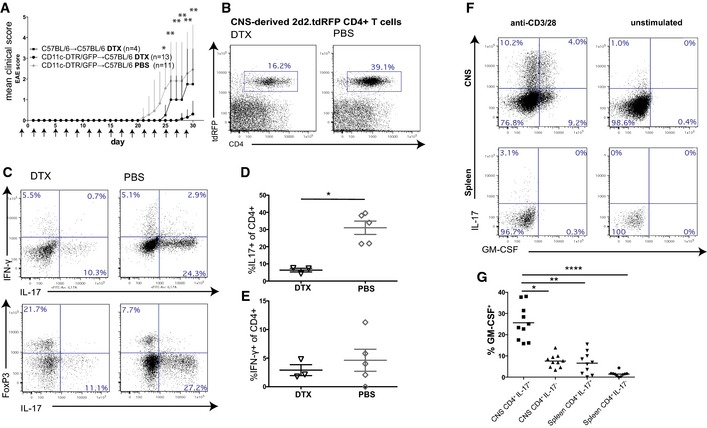
To investigate the role of CD11c‐GFP+ cells in the effector phase of EAE, we used the adoptive transfer EAE model of transgenic myelin‐specific (MOG35‐55‐specific, 2d2) CD4+ T cells, which had been differentiated to Th17 cells in vitro (Siffrin et al, 2010). To analyze the role of CD11c+ cells in the adoptive transfer model, ablation of CD11c+ cells by subcutaneous DTX application was started in chimeric CD11c‐DTR/GFP→C57BL/6 mice before transfer of 2d2.tdRFPxIl‐17‐EGFP Th17 cells and continued every other day during the whole observation period, resulting in reliable CD11c+ cell depletion. Experiments that required CD11c depletion by DTX were performed in chimera to bypass the reported problem of lethality after repetitive DTX injection in the CD11c‐DTR/GFP mice (Probst & van den Broek, 2005; Zaft et al, 2005). Bone marrow chimeric mice were generated as described previously (Siffrin et al, 2009) and yielded a chimerism of > 85% when looking at CD11c‐GFP+ cells of CD11c+ cells and > 93% for CD11c‐DTR‐depleted cells of CD11c+ cells (Appendix Fig S1A and B). Repetitive depletion, even after 11 and more DTX injections, did not result in a loss of depletion efficiency in the CNS or spleen and depleted all relevant subsets of CD11c+ cells (Appendix Fig S1C and D). In adoptive transfer EAE in CD11c+‐depleted recipients, we found a dramatically reduced mean clinical disease score and lower disease incidence (Fig 1A, Appendix Table S1). FACS analysis of mononuclear cells, which were isolated from the CNS of these EAE animals, revealed that the frequencies of transferred Th17 cells were markedly higher in the presence of CD11c+ cells (16.2% in DC‐depleted animals vs. 39.1% in control animals; pooled data for three animals; Fig 1B). In addition, the absence of CD11c+ cells led to the reduction of IL‐17 producers (Fig 1C and D) instead of IFN‐γ producers (Fig 1E). This is supported by similar results in an active EAE model of MOG35‐55/CFA‐immunized CD11c‐DTR/GFP→C57BL/6 mice, which were CD11c+‐depleted after onset of clinical signs to interfere with the effector phase (Appendix Fig S2). Here, a significant reduction of clinical deficits was achieved by CD11c+ cell depletion when started after onset of clinical signs in comparison with non‐CD11c+‐depleted control groups.
Figure 1. Adoptive transfer EAE of encephalitogenic CD4+ T cells into CD11c‐GFP+‐depleted and control mice.
-
AMean clinical scores (±SEM) of C57BL/6→C57BL/6 controls treated with DTX and CD11c‐DTR/GFP→C57BL/6 treated with DTX or PBS during the whole observation period starting from day −1. All animals received in vitro generated 2d2.tdRFP Th17 intravenously on day 0. DC depletion in CD11c‐DTR/GFP→C57BL/6 reduces the encephalitogenicity of adoptively transferred 2d2.tdRFP Th17 cells. Mann–Whitney U‐test was performed on mean clinical scores for DTX vs. PBS‐treated CD11c‐DTR/GFP→C57BL/6 (n = 11 PBS/13 DTX); *P < 0.05, **P < 0.01. See also Appendix Table S1. Pooled data from two independent experiments are shown.
-
BMononuclear cells were isolated on day 30 of animals shown in (A) from the CNS of PBS‐ and DTX‐treated CD11c‐DTR/GFP→C57BL/6 that had been transferred with 2d2.tdRFP Th17 cells. Flow cytometry was performed on surface‐stained cell samples. Cells were pre‐gated on lymphocyte cells (FSC/SSC gate) and PI negative; pooled data of three animals are shown, representative of two independent experiments.
-
CMononuclear cells were isolated from the CNS of PBS‐ and DTX‐treated 2d2.tdRFP Th17‐transferred CD11c‐DTR/GFP→C57BL/6 mice 2–3 days after onset of the disease. Upper panels: Cells were stimulated with plate‐bound anti‐CD3/anti‐CD28, stained for CD4 and cytokines IFN‐γ, IL‐17 (pre‐gated in addition to FSC/SSC on CD45+CD4+). Lower panels: expression of transcription factor FoxP3. Pooled data from three animals are shown, representative for two independent experiments.
-
D, EQuantification of cytokine expression data as shown in (C) for pooled data from two independent experiments. Mann–Whitney U‐test, *P < 0.05.
-
F2d2 Th17 cells were transferred into lymphopenic Rag2−/−cγ−/−. Mononuclear cells were isolated from the spleen and brain at the peak of the disease and analyzed for IL‐17 and GM‐CSF production after stimulation with anti‐CD3/anti‐CD28.
-
GGM‐CSF production of IL‐17 producers versus IL‐17 non‐producers of CNS‐isolated 2d2 Th17 cells (see also F). Each dot/square represents a single animal. One‐way ANOVA and Dunn's multiple comparison test; *P < 0.05, **P < 0.01, ****P < 0.0001.
To exclude a peripheral effect of CD11c+ cell depletion, which has been described in active EAE models where DC depletion led to exacerbation of the disease due to a lack of peripheral FoxP3+ regulatory T‐cell (Treg) induction (Yogev et al, 2012), we also checked lymph nodes and the spleen in our adoptive transfer model. Here, we found similar numbers of transferred cells (Appendix Fig S3A and B), no differences in cytokine production capacity (Appendix Fig S3D and E), and FoxP3 expression by CD4+ T cells (Appendix Fig S3F) in secondary lymphoid organs. This argues against CD11c‐GFP+ cells being relevant in inducing tolerance outside of the brain in this adoptive transfer EAE model. Next, we analyzed the co‐expression of the CD11c+‐relevant T‐cell cytokine GM‐CSF, which we found to be strongly upregulated in IL‐17‐producing Th17 cells in the CNS, but not in the spleen (Fig 1F), indicating that Th17 cells acquire the capacity to produce this cytokine after invasion of the CNS. Furthermore, IL‐17hi 2d2 Th17 cells consistently co‐expressed more frequently GM‐CSF than IL‐17 non‐producers, which shows that these cytokines are selectively co‐expressed within the CNS (Fig 1G).
Pathogenic Th17 cells carry distinct chemokine receptor signatures
To further determine the prerequisites for CD11c‐GFP+ cells and encephalitogenic Th17 cell encounters, we analyzed the chemokine expression pattern of pathogenic T cells by microarray analysis. We compared (i) naïve CD4+CD62Lhi 2d2 T cells before differentiation (Tnaive) and (ii) after repetitive in vitro Th17 differentiation (Th17iv), and (iii) 2d2 Th17 cells, which were isolated from the CNS of these mice at the peak of EAE (Th17eae; see also (Hoppmann et al, 2015)). Additionally, we isolated (iv) CD4+ T cells from the CNS of C57BL/6 mice with active, MOG35‐55‐immunized EAE (CD4eae). Differential gene expression was analyzed using Agilent Whole Mouse Genome Oligo Microarrays 4x44K V2. We concentrated our analysis on chemokine receptors, which showed a distinct activation pattern during EAE development when compared by ratio (Fig 2A). We performed stringent statistical testing on these targets of the microarray, which identified Ccr2, Ccr3, Ccr5, and Cxcr1 to be upregulated in Th17iv cells in comparison with Tnaive, which might indicate their role for homing to the CNS (Fig 2B). For Cxcr1, the expression pattern was confined to the in vitro period, whereas Ccr2 and Ccr5 were strongly expressed in both EAE subtypes (Th17eae and CD4eae). In addition, we identified Ccr8, Cxcr3, and Cxcr4 to be upregulated not in vitro (Th17iv) but in EAE‐derived Th17eae and/or CD4eae, which might indicate a role in their intraparenchymal distribution. Interestingly, the Th17‐associated Ccr6 was not significantly regulated, as the other chemokine receptors involved in the array also did not show relevant regulation in the observed T cells (data not shown).
Figure 2. Regulation of chemokine receptors in T cells at distinct points in and before EAE.
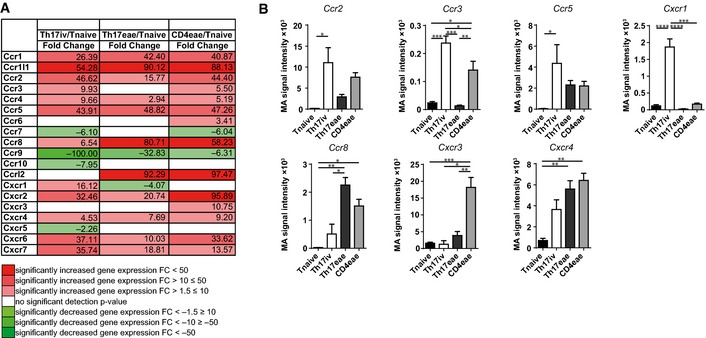
- Expression of chemokine receptors was assessed by microarray analysis of different CD4+ T‐cell populations. Statistical analysis revealed a strong upregulation of most of the chemokine receptors covered in the array for Th17iv/Tnaive (column 2), Th17eae/Tnaive (column 3), and MOG35‐55‐induced EAE‐recovered CD4eae/Tnaive.
- Microarray signal intensities of most strongly regulated genes from (A) are shown in detailed statistical analysis for the different T‐cell subgroups. Values are depicted as signal intensity mean ± SEM from three independent experiments. Statistical significance was determined using one‐way ANOVA with post hoc Tukey test for multiple comparisons. P‐values < 0.05 were regarded as statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
CNS dendritic cells grant encephalitogenic T cells access to the CNS
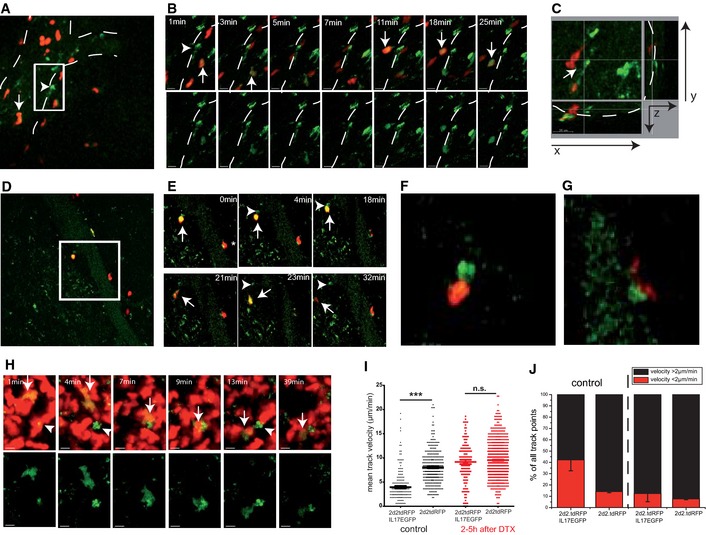
To visualize MOG35‐55‐specific T‐cell receptor transgenic (2d2) Th17 cells, we used genetically encoded constitutively red fluorescent (Rosa26‐tdRFP) and IL‐17 reporter (Il17a‐IRES‐EGFP) IL‐17hi 2d2 Th17 cells. These 2d2 Th17 cells were transferred into CD11c‐DTR/GFP mice, and their interaction with CD11c‐GFP+ cells was monitored at the onset and peak of clinical signs of EAE. Before disease onset, only very few CD11c‐GFP+ cells in CD11c‐DTR/GFP mice could be detected by TPLSM in the CNS. Some of these CD11c‐GFP+ cells were found near the surface of larger venules on the parenchymal side (Appendix Fig S4) and have been described elsewhere as bipolar dendritic cells (Prodinger et al, 2011). At the onset of the clinical disease (days 1–2), vessel‐associated CD11c‐GFP+ cells (Fig 3A) closely interacted with intravascular, rolling IL‐17hi 2d2 Th17 cells (Fig 3B and Video EV1). The elongated perivascular CD11c‐GFP+ cells were closely associated with the vessel walls and made contact with the rolling Th17 cells via filopodium‐like dendrites (Fig 3C). Interestingly, most of the long‐lasting contacts were with Th17 cells actively producing IL‐17. Within the perivascular area, the interaction of CD11c‐GFP+ cells with 2d2 Th17 cells was associated with high EGFP expression as a sign of strong IL‐17 production (Fig 3D), was long‐lasting, and preceded tissue invasion (Fig 3E–G and Video EV2). This holds true not only for the perivascular DCs but also for the few intraparenchymal, round‐shaped CD11c‐GFP+ cells in early inflammatory CNS lesions. As an example, an IL‐17hi 2d2 Th17 cell is shown that migrated directly toward a CD11c‐GFP+ DC and engaged in long‐lasting contact (Fig 3H, Videos EV3 and EV4), which is reminiscent of what has previously been described for the antigen recognition process in secondary lymphoid organs (SLO) (Cahalan & Parker, 2008). The quantitative analysis of the migration pattern of IL‐17hi 2d2 Th17 cells as compared with all 2d2 Th17 cells revealed that the cytokine‐expressing cells were slower in mean velocity but also had a larger number of stopping points (instantaneous velocity) compared to the whole 2d2 Th17 population—despite all of these T cells recognizing the same myelin antigen. This motility pattern was clearly CD11c+‐GFP cell dependent as the depletion of CD11c‐GFP+ cells during T‐cell monitoring led to an increase in the mean velocity of the IL‐17hi cells (Fig 3I) and lower static motility, as shown by reduced (< 2 μm/min) instantaneous velocity (Fig 3J). To exclude any bias by the adoptive transfer EAE model, we performed TPLSM in active EAE of single‐transgenic Il17‐EGFP mice. In active EAE lesions, Il17‐EGFP+ cells showed a perivascular migration pattern with regionalized migration (Appendix Fig S5, Video EV5) in the same way as IL‐17hi 2d2 Th17 cells. Recently, it has been shown ex vivo, based on functional and developmental criteria, that these CNS CD11c‐GFP+ cells are classic DCs (Anandasabapathy et al, 2011) and function as potent antigen‐presenting cells (APCs) (Greter et al, 2005; Bailey et al, 2007). Our in vivo observations by time‐lapse imaging show that these CNS DCs have a crucial role in the interplay of CD11c‐GFP+ cells with IL‐17‐producing Th17 cells.
Figure 3. Preferential interaction of CNS CD11c‐GFP+ cells with IL‐17hi 2d2 Th17 cells at the onset of the disease.
-
AIntravascular IL‐17hi 2d2 Th17 cell (arrow; double positive, green: IL‐17EGFP and red: 2d2.tdRFP) rolling cell toward a CD11c‐GFP+ cell (arrowhead). White dotted lines mark a venous vessel.
-
BTime‐lapse TPLSM (maximal intensity projections) of the boxed area in (A) shows that the perivascular elongated CD11c‐GFP+ cell (arrowhead) enters into contact with IL‐17‐expressing (arrow; double positive, green: IL‐17EGFP and red: 2d2.tdRFP) Th17 cells. Scale bar, 20 μm.
-
CXYZ‐resolved TPLSM depiction reveals that the elongated perivascular CD11c‐GFP+ cell makes contact with the intravascular 2d2 Th17 cells via a filopodium‐like dendrite (xy‐plane, 300 × 300 μm; z‐depth, 70 μm).
-
DStrong interaction of a perivascular IL‐17hi 2d2 Th17 cells with a perivascular CD11c‐GFP+ cell (insert upper left contact).
-
ETime lapse of the insert in (D): Stopping motility of the round‐shaped IL‐17hi 2d2 Th17 cell (arrow) near to a Cd11c‐GFP+ cell (arrowhead) is followed by entry into the CNS parenchyma.
-
F, GMagnified image of interaction of the (F) upper left and (G) lower right perivascular T‐cell–DC contacts as shown in (D) and (E).
-
HTime‐lapse imaging of parenchymal CD11c‐GFP+ cells (arrowhead), targeted migration of an IL‐17hi 2d2 Th17 cell (arrow, double positive; green, IL‐17EGFP; red, 2d2.tdRFP) toward the CD11c‐GFP+ cell (arrowhead). Scale bar, 20 μm.
-
IQuantification of the motility pattern of IL‐17hi 2d2.tdRFP vs. all 2d2.tdRFP in CD11c‐DTR/GFP brainstem lesions before and after (2–5 h) intraperitoneal DTX injection. Evaluation of mean track velocity of the single‐cell tracking (each dot represents a track); pooled data from at least two independent experiments (> 3 imaging areas). For the statistical analysis, the Mann–Whitney U‐test was performed (***P < 0.001; n.s., not significant).
-
JPercentage of stopping cells (red bar, instantaneous velocity < 2 μm/min) of the data set as shown in (I).
CNS CD11c‐GFP+ cells are a mixed population of conventional DCs and monocyte‐derived CD11c+ cells with distinct proportions depending on disease stage
In order to further characterize CNS CD11c+ cells, we isolated and phenotyped mononuclear cells from the CNS of EAE‐affected mice before onset (day 8–9), at the peak (day 13–17) and in the chronic phase of the disease (day 22–27) by flow cytometry. We found that conventional DCs—antigen‐expressing cells defined by expression of CD11c and absence of Ly6C/G—were the most abundant subset in the spleen over the whole course of EAE. The CD11c+Ly6C/G− subset in the spleen consists of approximately equal proportions of CD11b‐expressing cells and CD11b‐negative cells, and CD11c+ Ly6C/Gint (monocyte‐derived CD11c+) cells were a rare subset in the spleen (Appendix Fig S1D).
In the CNS, there is a characteristic change of the subpopulations of CD11c+ cells during the course of EAE. Initially, the conventional DCs (CD11c+Ly6C/G−)—here the CD11b+ subset—were the prevailing population (Fig 4A). At the peak of the disease, this changes with an increasing preponderance of monocyte‐derived CD11c+ cells (CD11c+ CD11b+ Ly6C/Gint), which even increases in the chronic stage of EAE. Looking at absolute numbers, in the CNS, there is an approximately 20‐fold increase in the CNS at the peak (on average 30,347 CD11c+ cells/CNS) and 12‐fold in the chronic phase of the disease (on average 18,065 CD11c+ cells/CNS; Fig 4B) compared with the pre‐onset situation (on average 1,476 CD11c+ cells/CNS). This increase and consecutive contraction of cell numbers is associated with a continuous increase in the proportion of the monocyte‐derived CD11c+ subset (Fig 4C).
Figure 4. Characterization of CD11c+ subpopulations in the CNS in the course of EAE.
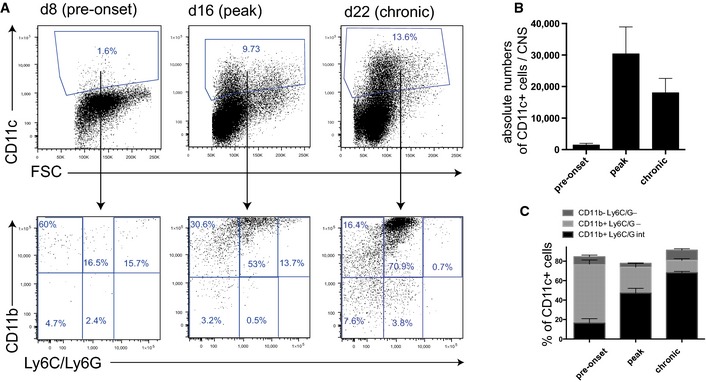
- In the CNS, there was a strong increase in CD11c+ proportions over the different phases of the disease. The composition of CD11c+ subpopulations changed over time, starting with a preponderance of CD11c+Ly6C/G− conventional DCs before onset of EAE. In the peak and chronic phase, there was a continuous increase in the proportion of monocyte‐derived CD11c+ cells.
- In the CNS, the quantification of the absolute numbers shows a strong increase in CD11c+ cells during the course of EAE (mean ± SEM).
- In the CNS, the proportions of the subpopulations show an increasing proportion of monocyte‐derived CD11c+ cells (mean ± SEM).
CNS CD11c‐GFP+ cells express distinct chemokine profiles that converge with pathogenic T‐cell chemokine receptor expression
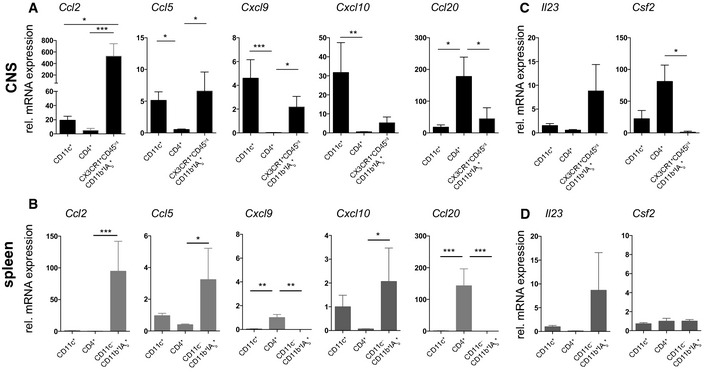
To analyze the chemotactic signature of dendritic cells in autoimmune CNS inflammation, we isolated CD11c‐GFP+ from the CNS of animals at the peak of actively induced EAE (for gating strategy, see Appendix Fig S6). Furthermore, splenic CD11c‐GFP+ cells from these mice were isolated as controls. The CNS non‐CD11c+ fraction consisted mainly of CD4+ T cells and microglia (CD45intCX3CR1‐YFP+CD11b+IAb +) (Bruttger et al, 2015); therefore, we chose these cells as a control population. For spleen samples, we chose instead of CD45intCX3CR1‐YFP+CD11b+IAb + the CD11c−CD11b+IAb + (monocyte/macrophage) population as the reference population. We performed quantitative real‐time PCR (qRT–PCR) of these samples to analyze their chemokine expression pattern. We identified Ccl2 (encoding for MCP‐1) as being expressed in CNS CD11c+ cells and microglia cells (Fig 5A) in EAE, whereas it was expressed at significantly lower levels in the spleen and in general in CD4+ T cells (Fig 5B). We analyzed the expression levels of the T‐cell‐relevant chemokines Ccl5, Cxcl9, and Cxcl10, all of which are important T‐cell‐attracting inflammatory chemokines (Hamann et al, 2008). We found that Ccl5 (encoding for RANTES), Cxcl9, and Cxcl10 were strongly expressed in the CNS CD11c+ cells (Fig 5A). Ccl20 was strongly expressed in the CNS CD4+ cell fraction but not by CNS or splenic CD11c+ cells. In addition, we analyzed the expression of the Th17‐relevant cytokine IL‐23 and the CD11c+‐relevant cytokine GM‐CSF (Fig 5C), which are both essential for EAE induction (Cua et al, 2003; El‐Behi et al, 2011). The expression of Il23 showed a trend toward being more prominent in microglia. Furthermore, we found strong upregulation of Csf2 in CNS CD4+ cells but not in their counterparts in the spleen, which supports our findings of GM‐CSF upregulation in CD4+ Th17 cells (see also Fig 1F and G).
Figure 5. Chemokine expression profiling of CD11c+ cells in the spleen and CNS reveals a key role of CNS CD11c+ cells for inflammatory chemokines.
-
ACD11c+ cells, CD4+ cells, and CX3CR1+CD45intIAb + (microglia) cells were isolated from peak EAE animals from the CNS. Depicted are normalized mRNA expression (to expression in spleen CD11c+ cells) levels as determined by qRT–PCR analysis relative to the housekeeping genes eukaryotic translation elongation factor 1 alpha 1 (Eef1a1) and peptidylprolyl isomerase A (Ppia). CNS CD11c+ cells show gene expression of the inflammatory chemokines Ccl2, Ccl5, Cxcl9, Cxcl10, but not Ccl20.
-
BCD11c+ cells, CD4+ cells, and CD11c−CD11b+IAb + (monocytes/macrophages) were isolated from EAE‐affected mice as described in (A). Relevantly regulated chemokines in other cell populations than CNS CD11c+ cells are shown.
-
C, DQuantitative expression of EAE‐relevant factors Il23 and Csf2 were investigated in (C) the CNS samples as described above and (D) the spleen samples.
Thus, the presence of CD11c+ cells in the CNS in peak EAE lesions is associated with the production of distinct inflammatory chemokines and cytokines that are involved in the pro‐inflammatory cascade in autoimmune neuroinflammation.
CNS CD11c‐GFP+ cells are enriched at the peak of the disease and form clusters in the CNS that are targeted by T cells
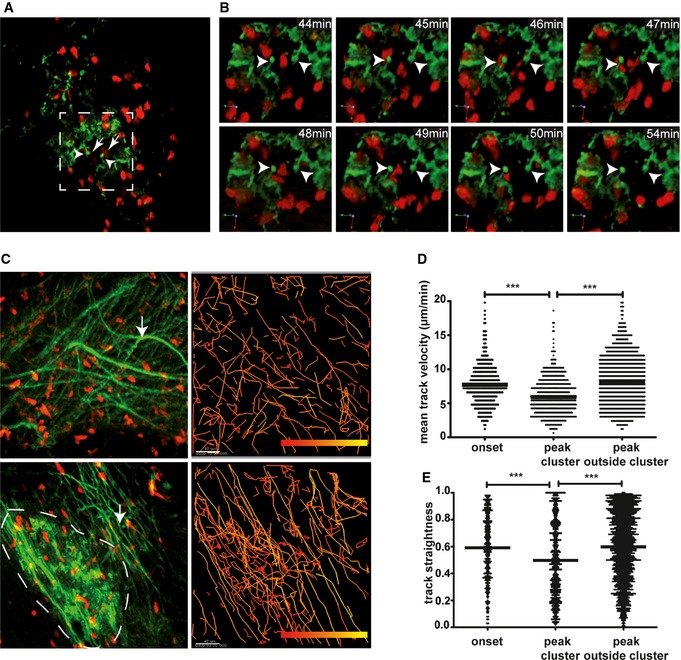
To further characterize the consequences of the T‐cell–CD11c+ cell interaction, we performed TPLSM at the peak of the disease. Here, we found that numerous CD11c‐GFP+ cells were arranged in clusters (Fig 6A) with a mesh of protrusions. These CD11c‐GFP+ cell networks were targeted by 2d2.tdRFP cells (Fig 6B and Video EV6). Whereas at the onset, there were mostly transient, very dynamic interactions between most 2d2.tdRFP T cells and CD11c‐GFP+ cells (with the exception of IL‐17hi cells), at the peak, there was regionalized low‐velocity motility of many of the transferred 2d2.tdRFP cells (Fig 6C, upper panel and Video EV7). In contrast, areas of low DC density were marked by very rapid T‐cell motility, which was a seemingly more random motility (Video EV8). These differences could also be objectified by quantitative tracking analyses (Fig 6D and E). To date, the observed highly regionalized motility pattern of T cells within the CD11c‐GFP+ cell clusters has only been described in lymph node germinal centers (Hauser et al, 2007). Importantly, we did not detect any differences in fiber networks in the CNS inflammatory lesions (Fig 6C), which are highlighted by second harmonic generation (SHG). These SHG signals are generated by the infrared (IR) excitation (1,110 nm) of highly ordered (non‐fluorescent) non‐centrosymmetric fibers, which are mainly collagen fibers outside the CNS, but whose makeup within the CNS remains unclear (Wilson et al, 2009). In our TPLSM of brainstem lesions, we regularly detected these fiber networks in inflammatory CNS lesions as secondary signs of lesion formation (Herz et al, 2011).
Figure 6. Cluster formation of DC within the CNS parenchyma at the peak of the disease.
-
ACD11c‐GFP+ DC cluster formation near venous vessel with protrusions (arrowhead) attracting 2d2.tdRFP Th17 cells (arrow).
-
BThree‐dimensional time‐lapse imaging of the boxed area in (A) shows interaction of 2d2.tdRFP Th17 cells with CD11c‐GFP+ cell protrusions (arrowhead).
-
CLeft column: Visualization of an area without CD11c‐GFP+ cell cluster (upper left) and one with CD11c‐GFP+ cell clusters (lower left). Intercellular matrix fibers (arrows) that are found in inflamed CNS parenchyma are visualized by excitation at 1,110 nm. Right column: Single cell tracking (color‐coded tracks, low velocity: red, high velocity: yellow) of 2d2.tdRFP T cells reveals a confined motility pattern within areas of CD11c‐GFP+ cell clusters (lower right) in comparison to random migration of T cells in areas without CD11c‐GFP+ cell clusters (upper right).
-
D, EQuantitative analysis of T‐cell tracking mean velocity (D) and track straightness (E) of 2d2.tdRFP Th17 cells in the onset of the disease with differential distribution at the peak of the disease. Cluster‐associated tracks have significantly lower mean velocities than Th17 cells outside the CD11c‐GFP clusters. One‐way ANOVA and Dunn's multiple comparison test (***P < 0.001) were performed. Pooled data from at least three independent experiments per group are shown.
Discussion
We observed a key function of CNS CD11c+ cells in the entry and persistence of encephalitogenic T cells in an animal model of multiple sclerosis in vivo. In our two‐photon imaging experiments at the onset of the disease, perivascular CD11c+ cells were found to be in contact with intravascular rolling and perivascular IL‐17‐expressing Th17 cells, which consequently invaded the CNS parenchyma. DC depletion reduced and/or delayed EAE induction and IL‐17 production by Th17. Thus, we have proven in vivo that CNS CD11c+ cells in the perivascular space attract encephalitogenic T cells and are a key factor for their function. It has been suggested previously that DCs act as potent APCs (McMahon et al, 2005; Bailey et al, 2007). In addition, IL‐17 seems to be a reliable marker for these pioneering encephalitogenic T cells which showed a characteristic activation motility in the interaction with CNS CD11c+ cells as previously described for the antigen recognition process in secondary lymphoid organs (Cahalan & Parker, 2008). There is debate as to whether EAE and MS are Th1‐ or Th17‐mediated diseases (Stromnes et al, 2008; Steinman, 2010). IL‐23, which is strongly associated with the Th17 phenotype, is the crucial factor for the development of EAE (Cua et al, 2003; Ivanov et al, 2006). Interestingly, it has become clear that Th1‐like (IFN‐γ‐expressing) cells in EAE might develop from originally IL‐17‐expressing T cells. By fate mapping of these cells in transgenic mice, it has been demonstrated that the overwhelming majority of T cells expressing the Th1 cytokine IFN‐γ in the CNS in EAE had previously been IL‐17 producers (“ex‐Th17”). In addition, these IFN‐γ‐expressing ex‐Th17 display distinct features compared to classic Th1 cells (Hirota et al, 2011). We showed that high IL‐17 production is accompanied by high GM‐CSF levels in the CNS. Interestingly, the CNS DC–Th17 interaction did not only have an effect on the Th17 cells; locally reactivated encephalitogenic Th17 cells produced large amounts of GM‐CSF, which is a relevant factor for CD11c+ cell induction. Accordingly, we found prominent CNS CD11c+ cell clusters at the peak of the disease. This is in line with findings of the role of GM‐CSF for EAE induction by Th17 cells (Codarri et al, 2011; El‐Behi et al, 2011) and indicates a crucial role of Th17–DC interplay for the perpetuation of autoimmunity. GM‐CSF blockade during the priming and effector phase in EAE has been shown to reduce clinical signs of active EAE (Codarri et al, 2011; El‐Behi et al, 2011). This effect was independent of Th17 polarization which has been shown to be similar in Csf2 −/− Th17 and wild‐type Th17 cells (El‐Behi et al, 2011). This is indeed a similar finding as we found by depletion of CD11c+ cells. In addition, also GM‐CSF deficient Th1 cells exhibited less encephalitogenicity (Codarri et al, 2011). The latter publication stressed the role of GM‐CSF for the secondary recruitment of inflammatory cells in the effector phase of EAE, which supports the relevance of our findings of an increase in numbers of CD11c+ cells, a continuous proportional increase in (GM‐CSF‐dependent) monocyte‐derived CD11c+ cells, and the formation of CD11c+ clusters as the disease progresses. It has been shown that there is a clear link between chronic CNS inflammation and induction of CD11c+ cells from myeloid and microglia cells in EAE and infectious encephalitis (Fischer & Reichmann, 2001; Ponomarev et al, 2007). For the potential consequence of a lack of this CD11c+ accumulation, Codarri et al also showed that, in their model with actively immunized Csf2 −/− mice, there is a lack of retention or accumulation of CD4+ T cells in the CNS, which again is equivalent to low CNS CD4+ T‐cell numbers in CD11+‐depleted animals in our adoptive transfer EAE.
In fact, active EAE models, which rely on immunization of mice with myelin peptide with strong adjuvants, showed an inverse exacerbating effect of CD11c+ cell/DC depletion on EAE development (Isaksson et al, 2012; Yogev et al, 2012), which could be related to a peripheral role of DCs for Treg induction in the priming phase of those immunologic challenge models. Here, we unraveled the role of CD11c+ cells in the CNS on transferred encephalitogenic T cells and in the effector phase of active EAE, thus focusing on the admission and survival of pathogenic T cells in the CNS. The question of sustained inflammation in the CNS might be more relevant for the human disease, which is usually not elicited by vaccinations (Siffrin et al, 2007) and persists in the absence of any systemic inflammatory challenges. It has been shown that in the CNS, despite the absence of secondary lymphoid organs, adoptive transfer EAE can be induced since local DCs are sufficient for the induction (Greter et al, 2005). High GM‐CSF production by local pathogenic effector T cells might also be relevant for the CD11c+ cell/DC networks, which we describe here for the first time in autoimmune CNS inflammation. We found these CD11c‐GFP+ cell clusters associated with vessels, which might explain the reported finding of CD4+ T cells to migrate preferentially along the CNS vessels (Siffrin et al, 2009). In our chimeric model, CD11c+ depletion is very efficient but does not reach 100%; therefore, we cannot answer whether CD11c+ cells are essential for T‐cell activation/survival within the CNS or if their role can be compensated for in the long term by another population of antigen‐presenting cells. However, CNS CD11c+ cells closely interacted with T cells, which led to a reduction of T‐cell motility and velocity. This prolonged interaction has up to now only been described in SLO and was clearly associated with induction of effector functions (Celli et al, 2007). The entry and/or survival of T cells into and within the CNS, an immune‐specialized organ that has physiologically a very low number of antigen‐presenting cells, might be due to these pathological DC networks. This is supported by findings that DC–T‐cell interactions are apparently responsible for the survival of pro‐inflammatory T cells in long‐standing autoimmune disease, for example, rheumatoid arthritis (Weyand & Goronzy, 2003) and diabetes mellitus (Calderon et al, 2011).
Our analysis of the chemokine expression pattern of immune cells isolated from EAE‐affected mice identified strong production of Ccl5, Cxcl9, and Cxcl10 by CNS CD11c+ cells. According to our data, encephalitogenic T cells have distinct chemokine receptor expression if analyzed before induction and when isolated from EAE‐affected CNS tissue. We found an upregulation of the chemokine receptors Ccr8, Cxcr3, and Cxcr4 in pathogenic T‐helper cells within the CNS in EAE. The CXCR3/CXCL10 interaction has been largely investigated and is commonly associated with autoimmune neuroinflammation (Simpson et al, 2000). CXCL10 (similarly as MCP‐1/CCL2) has been found to be elevated in the cerebrospinal fluid (CSF) of patients with MS (Sørensen et al, 1999; Scarpini et al, 2002) and is already upregulated before clinical signs are visible in EAE (Fife et al, 2001). CXCR3 has been described on the majority of perivascular T cells in MS lesions (Simpson et al, 2000), which makes this chemokine receptor rather a general marker of invading T cells in EAE and MS. More importantly, we identified the chemokine receptor Ccr5 to be highly expressed in encephalitogenic, in vitro repetitively stimulated Th17 cells. Repetitive stimulation of Th17 cells has already been shown to result in an IL‐23‐dependent increase in rather Th1‐associated CCR5 and loss of Th17‐associated CCR6 as typical for the late developmental plasticity of Th17 cells (Lee et al, 2009). This is important also in view of the human disease where there are fewer CCR2+CCR5+ T cells than other invading cells but which were associated with disease exacerbation (Misu et al, 2001). These CCR2+CCR5+ T cells might represent a strongly pathogenic subset with an enrichment of IL‐17+IFN‐γ+ T cells (Sato et al, 2012). These results of the T‐cell analysis correlated strongly with the finding of upregulated Ccl5 as a ligand of Ccr5.
Taken together, the initiation of neuroinflammation is strongly associated with the attraction of pathogenic T cells toward perivascular CD11c+ cells in the CNS. Long‐lasting interaction between CD11c+ cells with IL‐17‐producing Th17 cells can be visualized locally. Consecutive co‐expression of GM‐CSF by these T cells at the onset of the disease eventually results in CNS DC network formation at the peak of the disease, concomitantly with GM‐CSF production by pathogenic T cells. CNS CD11c+ cells at the peak of the disease are strong producers of a distinct inflammatory chemokine pattern. Thus, our findings suggest that DC networks within the CNS are crucial for the perpetuation and may be relevant for chronification of the disease and thus be an attractive target for the treatment of relapsing–remitting disease activity in patients with MS.
Materials and Methods
Mice
CD11c‐DTR/GFP (Jung et al, 2002), 2d2 (Bettelli et al, 2003), B6.acRFP, Rag2−/−cγ−/−, B6‐CX3CR1‐GFP (Jung et al, 2000; Jolivel et al, 2015), CX3CR1‐Cre‐ERT2 x iDTR x R26‐YFP (Bruttger et al, 2015), and C57BL/6J mice were bred under specifically pathogen‐free (SPF) conditions. IL‐17‐EGFP mice were generated by EE as has been described previously (Esplugues et al, 2011), bred with B6.acRFP and 2d2 mice, and kept in our SPF facility. CX3CR1‐Cre‐ERT2 x iDTR x R26‐YFP mice were induced for microglia‐specific YFP expression by tamoxifen as described previously (Bruttger et al, 2015).
Generation of bone marrow chimeras
Conventional chimeras were generated as described previously (Kursar et al, 2005; Gutcher et al, 2006), with the following modifications: Recipients were wild‐type (WT) C57BL/6 mice; for conventional bone marrow chimera, donor cells were derived from CD11c‐DTR/GFP mice and WT littermate controls. In brief, recipient animals were sublethally irradiated with 1,100 cGy (split dose). Donor animals were sacrificed by neck dissection; and bone marrow cells were isolated by flushing of femur and tibia, dissolved, and MACS®‐depleted of CD90+ T cells. Recipients were reconstituted with 8–15 × 106 donor bone marrow cells ∼8 h after irradiation. Mice were kept on 0.01% enrofloxacin (Bayer HealthCare) in drinking water for about weeks. Engraftment took place over 8 weeks of recovery and was checked by FACS analysis. All animal experiments were conducted according to the German Animal Protection Law.
Experimental autoimmune encephalomyelitis
For adoptive transfer, naïve 2d2 CD4+CD62Lhi cells were cultured as has been previously described (Siffrin et al, 2010) and were injected into B6.CD11c‐DTR/GFP or CD11c‐DTR/GFP→C57BL/6 mice. For active EAE, C57BL/6 mice were immunized subcutaneously with 250 μg of myelin oligodendrocyte glycoprotein (MOG35‐55) peptide (Research Genetics), emulsified in complete Freund's adjuvant (Difco Laboratories), and supplemented with 800 μg of heat‐inactivated M. tuberculosis H37Ra (Difco Laboratories). Mice received 200 ng of pertussis toxin (Sigma Aldrich) intraperitoneally at the time of immunization and after 48 h.
Mice were checked for clinical symptoms daily and these were converted into disease scores as has been described previously (Siffrin et al, 2010). For DC depletion, diphtheria toxin (12 ng/g bodyweight; List laboratories, UK) was diluted in PBS and administered intraperitoneally, starting the day prior to immunization and repeated every 48 h until the end of the experiment. Depletion efficiency was checked on spleen and CNS samples (Appendix Fig S1C and D).
Immune cell isolation
Lethally anesthetized animals were transcardially perfused with ice‐cold PBS. Brain and spinal cord were isolated, cut into small pieces, and diluted in IMDM (Gibco, Germany) substituted with 10 mg/ml Collagenase/Clostridiopeptidase (Sigma, Germany) and 200 U/ml DNase (Roche, Germany). After incubation for 30 min at 37°C under continuous rotation, the CNS tissue samples were put through a mesh (70 μm) and mononuclear cells separated by conventional 40/70% Percoll centrifugation. Spleens were isolated and digested by direct injection of 10 mg/ml Collagenase (Sigma Aldrich, Germany) and subsequent incubation for 30 min at 37°C under continuous rotation. Thereafter, digested spleens and lymph nodes were mechanically disrupted to obtain single‐cell suspensions. Erythrocytes were removed by a washing step with lysis buffer. For qRT–PCR, CD11c‐GFP+ cells were enriched by positive magnetic bead isolation, which yielded a purity of about 80% for CNS and splenic CD11c‐GFP+ cells. In addition, CD11c+ cell subpopulations as described in Fig 4 were isolated by FACS (purity > 90%), which showed similar expression patterns for the investigated chemokines in FACS and MACS isolations.
Intravital imaging
Mice were anesthetized using 1.5% isoflurane (Abbott, Germany) in oxygen/nitrous oxide (2:1) with a facemask. Mice were then tracheotomized and continuously respirated with a Harvard Apparatus Advanced Safety Respirator (Hugo Sachs, Germany). The anesthetized animal was transferred to a custom‐built microscopy table and fixed in a hanging position. The preparation of the imaging field was performed according to adapted protocols for cortical imaging (Göbel & Helmchen, 2007). In brief, the brainstem was exposed by carefully removing musculature above the dorsal neck area and removing dura mater between the first cervical vertebra and occipital skull bone. The head was inclined for access to deeper brainstem regions, and the brainstem was superfused with isotonic Ringer's solution, which was continuously exchanged by a peristaltic pump. A sterile agarose patch (0.5% in 0.9% NaCl solution) was installed on the exposed brain surface to reduce heartbeat and breathing artifacts.
During surgery and microscopy, body temperature was maintained at 35–37°C. We employed near‐infrared (NIR) excitation as described previously (Siffrin et al, 2010). The animal experiments were approved by the appropriate state committees for animal welfare (LUA Rheinland‐Pfalz; G 10‐01‐048) and were performed in accordance with current guidelines and regulations.
Excitation Setup for TPLSM
We employed NIR excitation, that is, two‐photon excitation of the sample at 850 nm, using an automatically tunable Ti:Sa laser (Mai Tai HP, Spectra Physics, USA) which is coupled into a commercially available scan head (TriMScope, LaVision Biotec GmbH, Bielefeld, Germany) via routing mirrors. The Ti:Sa beam is coupled into an upright microscope (BX‐51WI, Olympus, Hamburg, Germany) through the scan–tube lens combination (SL, TL). A dichroic mirror (DM) reflects the excitation beams toward the objective lens (20×, NA 0.95, Olympus Europe, Hamburg, Germany) that focuses them onto the sample at the same spot. XYZ stacks were typically collected within a scan field of 300 × 300 μm at 512 × 512 pixel resolution and a z‐plane distance of 2 μm at a frequency of 400 or 800 Hz. Applied laser powers ranged from 2 to 6 mW at the specimen's surface, which allowed for imaging depths up to 150 μm for EGFP as the fluorophore.
RNA isolation
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Germany) according to the manufacturer's protocol. Isolated RNA was further cleaned off possible genomic DNA by treatment with DNase I (Roche). Quality and integrity of total RNA preparation was confirmed by using a Nanodrop 2000c Spectrophotometer (Thermo Scientific, Germany).
cDNA synthesis was performed by reverse transcription of total RNA using the Superscript III First‐Strand Synthesis System and random hexamer primers (Invitrogen, Germany) following the manufacturer's instructions.
Quantification by real‐time PCR
Amplification primers for real‐time PCR analysis were designed using the Beacon Designer 8 Software (PREMIER Biosoft International, USA) according to the manufacturer's guidelines and subsequently tested for amplification efficiency and specificity. Sequences for primers are listed in Appendix Table S1. Real‐time PCR was performed using iQ SYBR Green supermix (Bio‐Rad Laboratories, Germany) in an CFX Connect™ Real‐Time Detection System (Bio‐Rad). Relative changes in gene expression were determined using the ΔΔC t method (Livak and Schmittgen, 2001) with eukaryotic translation elongation factor 1 alpha (EeF1a1), glyceraldehyde 3‐phosphate dehydrogenase (Gapdh), peptidylprolyl isomerase A (Ppia), and actin‐beta (Actb) as reference genes.
Statistical analysis
All data were analyzed using PRISM5 (GraphPad software). Data are presented as mean ± SEM from four independent experiments. Statistical analysis of the data was conducted using a nonparametric test (Mann–Whitney U or one‐way analysis of variance [ANOVA] and Dunn's multiple comparison test) or a parametric test (one‐way ANOVA followed by post‐hoc Tukey test) (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Microarray samples and array construction
We used collected CD4 magnetic bead‐enriched CD4+ T cells at different time points of adoptive transfer and active EAE. Four different cell populations were analyzed (from three independent experiments each): (i) naïve (CD4+CD62Lhi), myelin‐specific (2d2) T cells before differentiation (Tnaive) and (ii) after repetitive in vitro Th17 differentiation (Th17iv). Adoptive transfer EAE was induced by injecting these in vitro primed, (2d2) Th17 cells into lymphopenic Rag−/− mice. At the peak of EAE, 2d2 Th17 cells (iii) were isolated from the CNS of these mice (Th17eae). All procedures necessary for microarray measurements, starting at RNA isolation until first data analysis steps, were carried out at Miltenyi Biotec (Bergisch Gladbach, Germany). RNA was isolated using standard RNA extraction protocols (NucleoSpin® RNA II). Quality was checked via the Agilent 2100 Bioanalyzer platform (Agilent Technologies). In total, 100 ng of each total RNA sample was used for the linear T7‐based amplification step. To produce Cy3‐labeled cRNA, the RNA samples were amplified and labeled using the Agilent Low Input Quick Amp Labelling Kit (Agilent Technologies) following the manufacturer's protocol. Hybridization to Agilent Whole Mouse Genome Oligo Microarrays 4x44K V2 was performed according to Agilent Gene Expression Hybridization Kit protocol (Agilent Technologies). Fluorescence signals of the hybridized Agilent Microarrays were detected using Agilent's Microarray Scanner System (Agilent Technologies).
Microarray data analysis
The Agilent Feature Extraction Software (FES) was used to read out and process the microarray image files. The raw and normalized data are available online at the NCBI GEO database (Edgar et al, 2002), accession number GSE57098 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE57098). For determination of differential gene expression, FES‐derived output data files were further analyzed using the Rosetta Resolverâ gene expression data analysis system (Rosetta Biosoftware) and gene expression ratios were calculated by dividing sample signal intensity through control signal intensity. For the detection of differentially expressed genes, P‐values were calculated based on signal intensity variances using an error‐model‐based hypothesis test (Weng et al, 2006).
Author contributions
FZ and VS conceived and initiated the study and evaluated data. MP, VS, JOV, JuB, and JW performed the EAEs. MP, VS, and TB performed the immune cell isolation and FACS analysis. NH was responsible for the microarray samples and analysis. NH, MP, and VS collected and analyzed the real‐time PCR data. VS, JéB, RG, and PB conducted the intravital imaging. SJ, EE, RAF, and NY provided mice and analyzed EAE data. MP, VS, and FZ wrote and revised the manuscript. All authors were involved in discussions and critical commentary of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Video EV1
Video EV2
Video EV3
Video EV4
Video EV5
Video EV6
Video EV7
Video EV8
Review Process File
Acknowledgements
This work was supported by grants from the German Research Council (DFG) and to F.Z./T.B. (CRC‐TR‐128, B4) and to V.S. (CRC‐TR‐128, B9). Furthermore, we thank Christin Liefländer, Andreas Zymny, and Heike Ehrengard for expert technical assistance and Darragh O'Neill for proofreading the manuscript.
The EMBO Journal (2016) 35: 89–101
References
- Anandasabapathy N, Victora GD, Meredith M, Feder R, Dong B, Kluger C, Yao K, Dustin ML, Nussenzweig MC, Steinman RM, Liu K (2011) Flt3L controls the development of radiosensitive dendritic cells in the meninges and choroid plexus of the steady‐state mouse brain. J Exp Med 208: 1695–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey SL, Schreiner B, McMahon EJ, Miller SD (2007) CNS myeloid DCs presenting endogenous myelin peptides “preferentially” polarize CD4+ T(H)‐17 cells in relapsing EAE. Nat Immunol 8: 172–180 [DOI] [PubMed] [Google Scholar]
- Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK (2003) Myelin oligodendrocyte glycoprotein‐specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 197: 1073–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruttger J, Karram K, Wörtge S, Regen T, Marini F, Hoppmann N, Klein M, Blank T, Yona S, Wolf Y, Mack M, Pinteaux E, Müller W, Zipp F, Binder H, Bopp T, Prinz M, Jung S, Waisman A (2015) Genetic cell ablation reveals clusters of local self‐renewing microglia in the mammalian central nervous system. Immunity 43: 92–106 [DOI] [PubMed] [Google Scholar]
- Cahalan MD, Parker I (2008) Choreography of cell motility and interaction dynamics imaged by two‐photon microscopy in lymphoid organs. Annu Rev Immunol 26: 585–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon B, Carrero JA, Miller MJ, Unanue ER (2011) Cellular and molecular events in the localization of diabetogenic T cells to islets of Langerhans. Proc Natl Acad Sci USA 108: 1561–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli S, Lemaître F, Bousso P (2007) Real‐time manipulation of T cell‐dendritic cell interactions in vivo reveals the importance of prolonged contacts for CD4+ T cell activation. Immunity 27: 625–634 [DOI] [PubMed] [Google Scholar]
- Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B (2011) RORγt drives production of the cytokine GM‐CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 12: 560–567 [DOI] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD (2003) Interleukin‐23 rather than interleukin‐12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421: 744–748 [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang G‐X, Dittel BN, Rostami A (2011) The encephalitogenicity of T(H)17 cells is dependent on IL‐1‐ and IL‐23‐induced production of the cytokine GM‐CSF. Nat Immunol 12: 568–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, O'Connor W Jr, Rongvaux A, Van Rooijen N, Haberman AM, Iwakura Y, Kuchroo VK, Kolls JK, Bluestone JA, Herold KC, Flavell RA (2011) Control of TH17 cells occurs in the small intestine. Nature 475: 514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández Ó, Arnal‐García C, Arroyo‐González R, Brieva L, Calles‐Hernández MC, Casanova‐Estruch B, Comabella M, de las Heras V, García‐Merino JA, Hernández‐Pérez MA, Izquierdo G, Matas E, Meca‐Lallana JE, del M, Mendibe‐Bilbao M, Muñoz‐García D, Olascoaga J, Oreja‐Guevara C, Prieto JM, Ramió‐Torrentà L et al (2013) Review of the novelties presented at the 28th Congress of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) (III). Rev Neurol 57: 317–329 [PubMed] [Google Scholar]
- Fife BT, Kennedy KJ, Paniagua MC, Lukacs NW, Kunkel SL, Luster AD, Karpus WJ (2001) CXCL10 (IFN‐gamma‐inducible protein‐10) control of encephalitogenic CD4+ T cell accumulation in the central nervous system during experimental autoimmune encephalomyelitis. J Immunol 166: 7617–7624 [DOI] [PubMed] [Google Scholar]
- Fischer HG, Reichmann G (2001) Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J Immunol 166: 2717–2726 [DOI] [PubMed] [Google Scholar]
- Göbel W, Helmchen F (2007) New angles on neuronal dendrites in vivo. J Neurophysiol 98: 3770–3779 [DOI] [PubMed] [Google Scholar]
- Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B (2005) Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med 11: 328–334 [DOI] [PubMed] [Google Scholar]
- Gutcher I, Urich E, Wolter K, Prinz M, Becher B (2006) Interleukin 18‐independent engagement of interleukin 18 receptor‐alpha is required for autoimmune inflammation. Nat Immunol 7: 946–953 [DOI] [PubMed] [Google Scholar]
- Hamann I, Zipp F, Infante‐Duarte C (2008) Therapeutic targeting of chemokine signaling in Multiple Sclerosis. J Neurol Sci 274: 31–38 [DOI] [PubMed] [Google Scholar]
- Hartmann FJ, Khademi M, Aram J, Ammann S, Kockum I, Constantinescu C, Gran B, Piehl F, Olsson T, Codarri L, Becher B (2014) Multiple sclerosis‐associated IL2RA polymorphism controls GM‐CSF production in human TH cells. Nat Commun 5: 5056 [DOI] [PubMed] [Google Scholar]
- Hauser AE, Junt T, Mempel TR, Sneddon MW, Kleinstein SH, Henrickson SE, von Andrian UH, Shlomchik MJ, Haberman AM (2007) Definition of germinal‐center B cell migration in vivo reveals predominant intrazonal circulation patterns. Immunity 26: 655–667 [DOI] [PubMed] [Google Scholar]
- Herz J, Paterka M, Niesner RA, Brandt AU, Siffrin V, Leuenberger T, Birkenstock J, Mossakowski A, Glumm R, Zipp F, Radbruch H (2011) In vivo imaging of lymphocytes in the CNS reveals different behaviour of naïve T cells in health and autoimmunity. J Neuroinflammation 8: 131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B (2011) Fate mapping of IL‐17‐producing T cells in inflammatory responses. Nat Immunol 12: 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofstetter HH, Toyka KV, Tary‐Lehmann M, Lehmann PV (2007) Kinetics and organ distribution of IL‐17‐producing CD4 cells in proteolipid protein 139‐151 peptide‐induced experimental autoimmune encephalomyelitis of SJL mice. J Immunol 178: 1372–1378 [DOI] [PubMed] [Google Scholar]
- Hoppmann N, Graetz C, Paterka M, Poisa‐Beiro L, Larochelle C, Hasan M, Lill CM, Zipp F, Siffrin V (2015) New candidates for CD4 T cell pathogenicity in experimental neuroinflammation and multiple sclerosis. Brain 138: 902–917 [DOI] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics Consortium (IMSGC) , Wellcome Trust Case Control Consortium , Sawcer S, Hellenthal G, Pirinen M, Spencer CCA, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, Edkins S, Gray E, Booth DR, Potter SC, Goris A, Band G, Oturai AB, Strange A et al (2011) Genetic risk and a primary role for cell‐mediated immune mechanisms in multiple sclerosis. Nature 476: 214–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Multiple Sclerosis Genetics Consortium (IMSGC) , Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, Cotsapas C, Shah TS, Spencer C, Booth D, Goris A, Oturai A, Saarela J, Fontaine B, Hemmer B, Martin C, Zipp F, D'Alfonso S, Martinelli‐Boneschi F, Taylor B et al (2013) Analysis of immune‐related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 45: 1353–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaksson M, Lundgren BA, Ahlgren KM, Kämpe O, Lobell A (2012) Conditional DC depletion does not affect priming of encephalitogenic Th cells in EAE. Eur J Immunol 42: 2555–2563 [DOI] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL‐17+ T helper cells. Cell 126: 1121–1133 [DOI] [PubMed] [Google Scholar]
- Jolivel V, Bicker F, Biname F, Ploen R, Keller S, Gollan R, Jurek B, Birkenstock J, Poisa‐Beiro L, Bruttger J, Opitz V, Thal SC, Waisman A, Bauerle T, Schafer MK, Zipp F, Schmidt MH (2015) Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol 129: 279–295 [DOI] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20: 4106–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Unutmaz D, Wong P, Sano G‐I, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA (2002) In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell‐associated antigens. Immunity 17: 211–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karman J, Chu HH, Co DO, Seroogy CM, Sandor M, Fabry Z (2006) Dendritic cells amplify T cell‐mediated immune responses in the central nervous system. J Immunol 177: 7750–7760 [DOI] [PubMed] [Google Scholar]
- Kursar M, Höpken UE, Koch M, Köhler A, Lipp M, Kaufmann SHE, Mittrücker H‐W (2005) Differential requirements for the chemokine receptor CCR7 in T cell activation during Listeria monocytogenes infection. J Exp Med 201: 1447–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT (2009) Late developmental plasticity in the T helper 17 lineage. Immunity 30: 92–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2‐ΔΔCT method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Markowicz S, Engleman EG (1990) Granulocyte‐macrophage colony‐stimulating factor promotes differentiation and survival of human peripheral blood dendritic cells in vitro. J Clin Invest 85: 955–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD (2005) Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med 11: 335–339 [DOI] [PubMed] [Google Scholar]
- Misu T, Onodera H, Fujihara K, Matsushima K, Yoshie O, Okita N, Takase S, Itoyama Y (2001) Chemokine receptor expression on T cells in blood and cerebrospinal fluid at relapse and remission of multiple sclerosis: imbalance of Th1/Th2‐associated chemokine signaling. J Neuroimmunol 114: 207–212 [DOI] [PubMed] [Google Scholar]
- Noster R, Riedel R, Mashreghi M‐F, Radbruch H, Harms L, Haftmann C, Chang H‐D, Radbruch A, Zielinski CE (2014) IL‐17 and GM‐CSF expression are antagonistically regulated by human T helper cells. Sci Transl Med 6: 241ra80 [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Shriver LP, Maresz K, Pedras‐Vasconcelos J, Verthelyi D, Dittel BN (2007) GM‐CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol 178: 39–48 [DOI] [PubMed] [Google Scholar]
- Probst HC, van den Broek M (2005) Priming of CTLs by lymphocytic choriomeningitis virus depends on dendritic cells. J Immunol 174: 3920–3924 [DOI] [PubMed] [Google Scholar]
- Prodinger C, Bunse J, Krüger M, Schiefenhövel F, Brandt C, Laman JD, Greter M, Immig K, Heppner F, Becher B, Bechmann I (2011) CD11c‐expressing cells reside in the juxtavascular parenchyma and extend processes into the glia limitans of the mouse nervous system. Acta Neuropathol 121: 445–458 [DOI] [PubMed] [Google Scholar]
- Sato W, Tomita A, Ichikawa D, Lin Y, Kishida H, Miyake S, Ogawa M, Okamoto T, Murata M, Kuroiwa Y, Aranami T, Yamamura T (2012) CCR2(+)CCR5(+) T cells produce matrix metalloproteinase‐9 and osteopontin in the pathogenesis of multiple sclerosis. J Immunol 189: 5057–5065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpini E, Galimberti D, Baron P, Clerici R, Ronzoni M, Conti G, Scarlato G (2002) IP‐10 and MCP‐1 levels in CSF and serum from multiple sclerosis patients with different clinical subtypes of the disease. J Neurol Sci 195: 41–46 [DOI] [PubMed] [Google Scholar]
- Schlitzer A, Ginhoux F (2014) Organization of the mouse and human DC network. Curr Opin Immunol 26: 90–99 [DOI] [PubMed] [Google Scholar]
- Siffrin V, Brandt AU, Herz J, Zipp F (2007) New insights into adaptive immunity in chronic neuroinflammation. Adv Immunol 96: 1–40 [DOI] [PubMed] [Google Scholar]
- Siffrin V, Brandt AU, Radbruch H, Herz J, Boldakowa N, Leuenberger T, Werr J, Hahner A, Schulze‐Topphoff U, Nitsch R, Zipp F (2009) Differential immune cell dynamics in the CNS cause CD4+ T cell compartmentalization. Brain 132: 1247–1258 [DOI] [PubMed] [Google Scholar]
- Siffrin V, Radbruch H, Glumm R, Niesner R, Paterka M, Herz J, Leuenberger T, Lehmann SM, Luenstedt S, Rinnenthal JL, Laube G, Luche H, Lehnardt S, Fehling H‐J, Griesbeck O, Zipp F (2010) In vivo imaging of partially reversible th17 cell‐induced neuronal dysfunction in the course of encephalomyelitis. Immunity 33: 424–436 [DOI] [PubMed] [Google Scholar]
- Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN (2000) Expression of the interferon‐gamma‐inducible chemokines IP‐10 and Mig and their receptor, CXCR3, in multiple sclerosis lesions. Neuropathol Appl Neurobiol 26: 133–142 [DOI] [PubMed] [Google Scholar]
- Sørensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, Qin S, Rottman J, Sellebjerg F, Strieter RM, Frederiksen JL, Ransohoff RM (1999) Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest 103: 807–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L (2010) Mixed results with modulation of TH‐17 cells in human autoimmune diseases. Nat Immunol 11: 41–44 [DOI] [PubMed] [Google Scholar]
- Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM (2008) Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med 14: 337–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng L, Dai H, Zhan Y, He Y, Stepaniants SB, Bassett DE (2006) Rosetta error model for gene expression analysis. Bioinformatics 22: 1111–1121 [DOI] [PubMed] [Google Scholar]
- Weyand CM, Goronzy JJ (2003) Ectopic germinal center formation in rheumatoid synovitis. Ann N Y Acad Sci 987: 140–149 [DOI] [PubMed] [Google Scholar]
- Wilson EH, Harris TH, Mrass P, John B, Tait ED, Wu GF, Pepper M, Wherry EJ, Dzierzinski F, Roos D, Haydon PG, Laufer TM, Weninger W, Hunter CA (2009) Behavior of parasite‐specific effector CD8+ T cells in the brain and visualization of a kinesis‐associated system of reticular fibers. Immunity 30: 300–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogev N, Frommer F, Lukas D, Kautz‐Neu K, Karram K, Ielo D, von Stebut E, Probst H‐C, van den Broek M, Riethmacher D, Birnberg T, Blank T, Reizis B, Korn T, Wiendl H, Jung S, Prinz M, Kurschus FC, Waisman A (2012) Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of pd‐1 receptor(+) regulatory T cells. Immunity 37: 264–275 [DOI] [PubMed] [Google Scholar]
- Zaft T, Sapoznikov A, Krauthgamer R, Littman DR, Jung S (2005) CD11chigh dendritic cell ablation impairs lymphopenia‐driven proliferation of naive and memory CD8+ T cells. J Immunol 175: 6428–6435 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Video EV1
Video EV2
Video EV3
Video EV4
Video EV5
Video EV6
Video EV7
Video EV8
Review Process File