

Abstract
Multiple myeloma (MM) is the second most predominant blood malignancy. Proteasome inhibitors like bortezomib have increased life expectancy, but eventually patients develop resistance to therapy. It was proposed that bortezomib acts through the induction of the Unfolded Protein Response (UPR), i.e., accumulation of misfolded proteins causing a lethal stress response. By this theory, increasing the proteasome load by the stimulation of translation may worsen the UPR. Here we evaluated the crosstalk between translation and bortezomib toxicity in both bortezomib sensitive and resistant cells. We found that bortezomib toxicity does not correlate with induction of proapoptotic eIF2α phosphorylation, but rather caused a late reduction in initiation of translation. This effect was accompanied by dephosphorylation of the mTORC1 target 4E-BP1. Infection of myeloma cells with constitutively dephosphorylated 4E-BP1, worsened bortezomib induced cell death. Since mTORC1 inhibitors cause pharmacological inhibition of 4E-BP1 phosphorylation, we tested whether they could act synergistically with bortezomib. We found that both rapamycin, a specific mTORC1 blocker, and PP242 a mTOR antagonist induce the arrest of myeloma cells irrespective of bortezomib sensitivity. Sensitivity to mTOR inhibitors has been associated to the levels of eIF4E/4E-BPs. We found that levels of eIF4E and 4E-BPs are variable among patients, and that 15% of myeloma patients have increased levels of 4E-BP1/2. Primary cells of myeloma retain sensitivity to mTOR inhibition, when plated on stromal cells. We propose that translational load does not contribute to bortezomib-induced death, but rather mTOR targeting may be successful in bortezomib resistant patients, stratified for eIF4E/4EBPs.
Keywords: 4E-BP1, 4E-BP2, ERK, MM.1S, U266, eIF2α, eIF4E
Introduction
Multiple myeloma (MM) is characterized by excess of bone marrow plasma cells, osteolytic bone lesions, immunodeficiency and renal disease.1 Advances in the therapy of MM have led to an increase of survival time. Proteasome inhibitors, such as bortezomib (BZ), have been introduced to treat first relapsed or refractory MM, and more recently newly diagnosed patients. The combination therapy of thalidomide, bortezomib and dexamethasone has increased the time of progression-free survival (PFS) in patients with newly diagnosed multiple myeloma.2 In general, patients respond well to BZ. In spite of this, the disease remains incurable because patients develop resistance to BZ, with average life expectancies of 7–8 years from the diagnosis.3-5 The comprehension of the mechanism of action of BZ, and why myeloma cells are specifically sensitive to it, has therefore attracted the interest of several studies. In agreement with the pleiotropic role of the proteasome in cellular life, many causes have been linked to BZ induced toxicity.6,7 Yet, most of these causes do not fully explain the specific sensitivity of myeloma cells to proteasome inhibitors. Myeloma cells directly interact with bone marrow stromal cells.8 This interaction increases growth, survival, migration, and drug resistance of multiple myeloma cells. BZ reduces the adhesion of myeloma cells to bone marrow stromal cells, therefore reducing their viability.6,8 In addition, genetic and pharmacological evidences suggest that the activation of NFKB is critical for survival of mature B-cells. Activating mutations in the NFKB pathway are common in myeloma patients.9,10 BZ impairs the degradation of IKB, a negative regulator of NFKB, inducing downregulation of growth and anti-apoptotic signaling pathways.11 Other indirect mechanisms to explain BZ toxicity have been proposed.12,13 An attractive model to explain BZ effects is through the Unfolded Protein Response (UPR).14,15 In the UPR, accumulation of misfolded and undegraded proteins in the endoplasmyc reticulum (ER) causes the induction of a three-branched response. Briefly, three ER transmembrane proteins, IRE1 kinase, ATF6 transcription factor and PERK kinase act as sensors of ER stress. The first two branches induce the expression of chaperone proteins and enzymes responsible for ER turnover. The latter branch blocks global translation by phosphorylation of eIF2α and activates translation-driven expression of genes necessary for the stress response.16 If the UPR is not effective, cells are driven to apoptosis.17 This model is attractive because myeloma cells produce large amounts of IgG, which make them potentially susceptible to UPR.18,19 An obvious consequence of this model is that translation inhibitors should reduce the proteasome load, hence BZ-induced toxicity. Evidence for the presence of BZ-induced UPR in myeloma cells has been produced, but without discriminating between sublethal or lethal doses.14 The possibility that the translational load is not contributing to BZ-induced toxicity, but rather contributes to survival, like in most other cancers, has not been addressed in myeloma cells. We decided to analyze how the translational machinery is affected by BZ treatment in myeloma cells, by comparing BZ-resistant cells to BZ-sensitive ones, and analyzing different time courses. We collected data that demonstrate that the reduction of the translational load does not reduce BZ-induced toxicity. Rather, in keeping with other tumor types, the translational machinery is an alternative target to proteasome-induced toxicity.20,21
Results
Bortezomib toxicity is not associated with proapoptotic eIF2α phosphorylation
The treatment of MM cells with BZ is associated with proteasome inhibition, downregulation of growth and activation of apoptotic signaling pathways. We first validated the BZ effect on cell viability in two MM cell lines held as sensitive, MM.1S, or resistant, U266. We treated MM.1S and U266 with increasing BZ concentrations for 24 and 48 h and performed the MTT assay (Fig. 1A). Calculated EC50 at 48 h were 11,93 ± 1,68 nM for MM.1s cells and 16,15 ± 1,81 nM for U266. Within 24 h of treatment, at the concentrations tested, U266 were resistant to BZ at concentrations up to 50 nM, whereas MM.1S had an EC50 of 18.26 nM ± 1,68. We therefore decided to analyze biochemical parameters at times ranging 1–24 h and at 20 nM. The inhibition of proteasome activity leads to polyubiquitinated proteins accumulation. Consistently, 8 h BZ treatment caused, in both MM1.S and U266, accumulation of polyubiquitinated proteins. Further accumulation of polyubiquitinated proteins was observed only in sensitive MM1.S cells (Fig. 1B). In agreement with the MTT data, cleaved caspase 3 and cleaved PARP were observed only in MM1.S cells (Fig. 1C). Thus, proteasome inhibition leads to similar short-term accumulation of poly-Ubiquitinated in both MM1.S and U266 cell. However, only the first undergo an apoptotic response.

Figure 1. The induction of eIF2α phosphorylation does not correlate with growth arrest in BZ sensitive cells (A) MM.1S cells are more sensitive to BZ than U266 cells. Cells were treated with BZ (5 nM to 50 nM) for 24 and 48 h. MTT assay was performed. Data are presented as percentage of untreated control. Statistical significance was assessed by Student’s t test * P<0,05, **P < 0,01. (B) BZ induces polyubiquitin accumulation in both sensitive and insensitive cells. MM cells were treated with 20 nM BZ for 1, 8 and 24 h. Total protein extracts were analyzed in WB to test for polyubiquitin accumulation. Data were normalized with anti-β-actin. (C) Apoptosis is activated only in sensitive MM.1S cells. MM cells were treated as indicated for 24 h. Total protein extracts were analyzed by WB for anti-caspase 3 and anti PARP antibodies. (D) Constitutive eIF2α phosphorylation is only transiently affected by BZ treatment both in sensitive MM.1S cells and resistant U266 cells. MM cells were treated with 20 nM BZ for indicated times. Thapsigargin (tg) treatment in NIH-3T3 cells was used as a positive control for eIF2α phosphorylation. Data were normalized for the total amount of eIF2α.
It has been proposed that the treatment of MM cells with proteasome inhibitors triggers the Unfoded Protein Response (UPR).14,22,23 In response to UPR, the PERK Kinase is activated by dimerization and phosphorylation. Once activated, PERK phosphorylates eIF2α resulting in translation attenuation.24 Therefore we investigated whether BZ had effects on eIF2α phosphorylation and on protein synthesis. We made these observations: first, the induction of eIF2α phosphorylation by BZ treatment was minimal, and present both in BZ-sensitive MM1.S cells and in BZ-insensitive U266 cells. Second, the basal level of eIF2α phosphorylation of myeloma cells was higher than in fibroblast (Fig. 1D). We conclude that the timing and extent of induction of eIF2α phosphorylation does not associate with BZ-induced death.
4E-BP1 dephosphorylation accompanies and accelerates bortezomib-induced death
Next, we assessed whether translation is affected by proteasome inhibition and if this correlates with induced toxicity. Briefly, the best-characterized pathway converging on translation is driven by mTORC1, which leads to the direct phosphorylation of 4E-BPs, and through S6K1 of rpS6.25 In general, rapid inhibition of mTORC1 by rapamycin or by mTOR blockers leads to the rapid dephosphorylation of both rpS6 and 4E-BP1.We assessed whether the mTORC1 pathway is affected by BZ. Surprisingly, BZ treatment affected phosphorylation of mTORC1 substrates only in BZ-sensitive cells. BZ treatment caused dephosphorylation of 4E-BP1, (Fig. 2A) in MM1.S sensitive cells, but not in U266 resistant cells. Next, we investigated the phosphorylation status of rpS6. The p70 ribosomal S6 kinases, directly regulated by mTOR, phosphorylate rpS6 on Ser-240 and Ser-244.26 The RAS/ERK pathway also regulates rpS6 phosphorylation independent of mTOR through the activation of p90 ribosomal S6K kinases that phosphorylate rpS6 on Ser-235 and Ser-236.27 Our data indicate that while 24 h BZ treatment affects 4E-BP1 phosphorylation, S6 phosphorylation is not compromised by BZ. Thus we hypothesize that mTORC1 activity was still present in BZ-treated cells. We pulled down mTORC1 complex from BZ-treated cells, in conditions of reduced in vivo phosphorylation of 4E-BP1. We found that BZ did not reduce mTORC1 kinase activity, at least in vitro (Fig. 2B). The data shown indicate a clear dephosphorylation of 4E-BP1 that is not accompanied by S6 dephosphorylation (Fig. 2A). The phosphorylation of rpS6 in Ser 235/236 may be explained by activation of the p90 ribosomal S6 Kinase (RSK) downstream of the Ras/ERK signaling cascade.27 Indeed, BZ induces ERK phosphorylation in a dose dependent manner (Supplementary Figure 1A). Since that 4E-BP is dephosphorylated upon BZ treatment in MM.1S, we analyzed whether BZ treatment causes an enrichment of 4E-BP1 bound to eIF4E. We evaluated cap complex assembly of initiation factors in both MM.1S and U266 treated with BZ. Data in Figure 2C show that BZ treatment causes an enrichment of 4E-BP1 bound to eIF4E at 24 h BZ treatment only in MM.1S. In agreement with the observed absence of effects of BZ treatment on 4E-BP1 phosphorylation in U266 cells, BZ did not affect 4E-BP1 binding to eIF4E in the same context (Fig. 2C). Next, we evaluated the relationship between BZ sensitivity and initiation of translation. Polysomal profiles demonstrated that BZ treatment caused an inhibition of initiation in sensitive MM1.S cells, but not in resistant U266 cells (Fig. 3A). To better address the effect of BZ on global translation we measured translational rate by methionine incorporation in MM.1S and U266 cells treated with BZ. Short BZ treatment does not affect global translation while long BZ treatment induces only in MM.1S cells a reduction of translation rate (Fig. 3B). These data suggest that translational load does not increase BZ toxicity, but rather that attenuation of cap dependent translation exacerbates BZ toxicity. Next, we directly tested the hypothesis. MM.1S cells have dephosphorylated 4E-BP1 upon BZ treatment, while 4E-BP1 remains phosphorylated in BZ insensitive U266. We infected U266 cells with retrovirus expressing non-phosphorylable 4E-BP1 (4Ala) or empty control. We evaluated the efficiency of infection by western blotting (Fig. 4A). Next, we observed the viability of infected cells with the MTT assay. We found that 4E-BP1–4Ala infected cells are, even if slightly, more sensitive to 48 h BZ treatment (Fig. 4B). Taken together data demonstrate that the translational load is not accelerating BZ-induced death, and suggest that the block of translation of specific mRNA accelerates BZ toxicity.
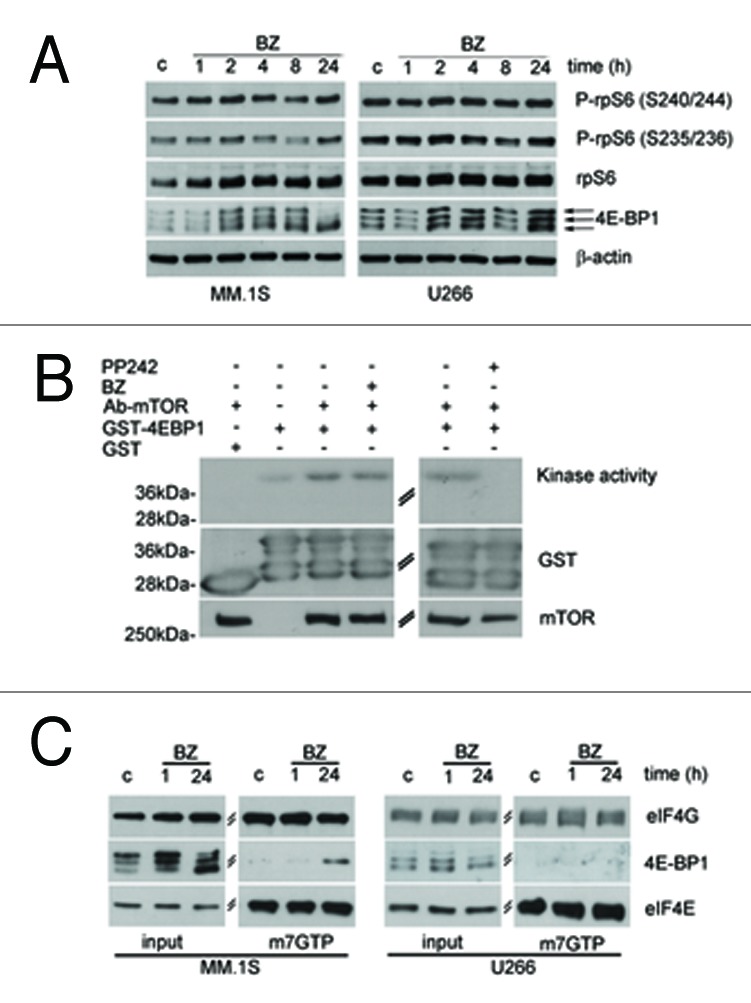
Figure 2. BZ induced toxicity is accompanied by 4E-BP1 dephosphorylation (A) 4E-BP1 phosphorylation is affected by BZ only in sensitive MM.1S cells. MM cell lines were treated with 20 nM of BZ for indicated times. The samples were subjected to SDS-PAGE and western blotting to analyze 4E-BP1 isoforms and S6 phosphorylation. (B) mTORC1 activity is not reduced by BZ in vitro. MM.1S cells were exposed to 20 nM BZ for 24 h. mTOR, immunoprecipitated with anti-mTOR antibody, was analyzed for kinase activity with γ-32P ATP. GST-4E-BP1 was used as substrate and GST as negative control of kinase assay (left panel) Immunoprecipitated mTOR from MM.1S cellular extracts was preincubated with 1,5 μM PP242 for 30 min prior to the kinase reaction. PP242 is a negative control of mTOR Kinase activity (right panel). (C) BZ treatment induces 4E-BP1 binding to eIF4E in MM.1S treated with BZ for 24 h. MM cells were treated as indicated and total proteins were incubated with 7-Methyl GTP-Sepharose beads. Input is 10% of the purification. Cap binding proteins were analyzed by WB with anti 4E-BP1 and eIF4G. eIF4E shows equal amount of purified proteins.

Figure 3. BZ treatment induces attenuation of cap dependent translation in MM.1S cells. (A) Polysome profile of MM.1S and U266 treated with BZ 20 nM for 24 h. BZ treated MM.1S cells have reduced polysomal peaks and augmented 80S. BZ does not affect polysome distribution in U266 cells. (B) MM cells were treated with 20 nM BZ for indicated times and pulsed with 35-S Methionine. Methionine incorporation was normalized on proteins. Cycloheximide (CHX) is a negative control of translational elongation. Statistical significance was assessed by Student’s t test **P < 0,01.

Figure 4. U266 cells expressing a not phosphorylable 4E-BP1(4Ala) show increased BZ sensitivity (A) U266 cells expressing empty vector and HA-4E-BP1(4Ala) were immunoblotted with anti-HA and anti-4E-BP1 antibodies. The HA-4E-BP1(4Ala) construct is overexpressed. (B) The transduced cells were treated with increasing concentrations of BZ for 48 h. MTT assay was performed. The viability of untreated cells was set as 100%. Statistical significance was assessed by Student’s t test * P < 0,05, **P < 0,01.
mTOR inhibition delays MM growth independently from bortezomib resistance
The data obtained suggest that the pharmacological inhibition of 4E-BP1 may act synergistically to BZ and/or be of value in treatment of MM. The most widely mTORC1 inhibitor is rapamycin; however, rapamycin treatment causes activation of a pro-survival feedback loop by ERK and Akt kinase activation.28,29 As previously shown, ERK is activated also by BZ treatment (Figure S1A). We examined the effect of ERK inhibition on MM viability, in conditions of BZ treatment. Data show that ERK inhibition in MM1.S cells does not worsen BZ-mediated toxicity (Figure S1B), thus providing a rational for further analysis of mTOR inhibitors in myeloma cells. Next, we analyzed the effect of either mTORC1 inactivation by rapamycin or mTOR pharmacological blockade by PP242, singly or in combination with BZ. Briefly, we found that both i) mTORC1 inhibition by either rapamycin or PP242 decreased the survival of MM cells, ii) the effect of PP242 and rapamycin was independent from the one of BZ (Fig. 5A). Early trials with rapalogues have shown a limited response in myeloma cells.30,31 Thus, we examined the effects of PP242 on the growth of primary myeloma cells derived from patients. Since the interaction between stromal cells and myeloma cells is critical to their survival, the effect of mTORC blockers was examined on cocultured tumor and stromal cells. It must be emphasized that primary myeloma cells in culture do not proliferate vigorously. Of five patients cells tested, only one was found to partly respond to PP242, in conditions of BZ resistance (Fig. 5B). These data suggest that a subset of patients may respond to mTORC inhibition.

Figure 5. mTOR inhibition impairs MM growth independently from BZ and in the presence of stromal cells (A) Growth inhibition induced by BZ alone and in combination with mTOR inhibitors. MM.1S and U266 were treated for 24 and 48 h with increasing concentrations of BZ alone and in the presence of rapamycin (10 nM) or PP242 (500 nM). MTT assay was subsequently performed. Data are presented as percentage of the control, in which cells were treated with 0,02% (v/v) DMSO. (B) HS-5 stromal cells were plated and incubated O/N. Isolated primary MM cells from a patient were cultured on HS-5 stromal cells layer. The co-culture was treated with 2 nM BZ, 250 nM, 500 nM of PP242. Two nM BZ was combined with 250 nM PP242 and 500 nM PP242. The co-culture was incubated for 24, 48 and 72 h. MTT assay was performed. Data are presented as percentage of control treated with 0,02% (v/v) DMSO. Statistical significance was assessed by Student’s t test * P < 0,05, **P < 0,01.
Variable levels of eIF4E and 4E-BP1/2 in myeloma patients
Determinants of sensitivity to mTORC1 inhibition are mutations in PI3K/RAS pathways or altered levels of 4E-BP1/2 vs. eIF4E.32-34 Specifically, higher levels of eIF4E vs 4E-BP1 confer rapamycin insensitivity and viceversa. We analyzed in myeloma patients (n = 122) the relative levels of 4E-BP1 and 4E-BP2 vs. eIF4E: Data show that approximately 15% of patients have higher relative levels of 4E-BP1 and 4E-BP2 (Fig. 6A–B).

Figure 6. Differential ratios of eIF4E/4E-BP mRNAs in MM patients (A) Transcriptional levels of 4E-BP1, 4E-BP2 and EIF4E were evaluated in 122 Multiple Myeloma patients at the onset of disease. The 4E-BP1/EIF4E, 4E-BP2/EIF4E ratio were calculated for each patient. The average ratio was imposed as threshold. In both graphs the red dots represent the patients who have 4E-BP1/EIF4E or 4E-BP2/EIF4E ratio higher than the threshold. (B) The chart indicates that 39 patients have 4E-BP1 levels higher than eIF4E, 49 patients have 4E-BP2 levels higher than EIF4E and 18 patients have both 4E-BP1, 4E-BP2 mRNA levels higher than EIF4E.
Discussion
On the basis of available literature, we hypothesized that BZ induced an UPR response in myeloma cells, allowing us to identify eIF2α phosphorylation and uORF mRNAs critical for survival and resistance of cells.14,19,23 To address the problem, we set up conditions that allowed us to discriminate between BZ-induced toxicity and survival. In these conditions, we did not find evidence for eIFα phosphorylation in BZ-induced lethality. This result is in apparent contrast with previous reports that analyzed the UPR in MM cells.23,35 However, here we focused on BZ concentrations able to induce cell death, thus ruling out a direct relationship between eIF2α phosphorylation and toxicity. Several groups have tried to explain the molecular bases of different individual responsiveness to bortezomib, exploiting human MM lines characterized by differential sensitivity.36,37 Nevertheless, the molecular basis of sensitivity or resistance to BZ among patients, remain largely unknown. We note that, MM1S cells have only 2-fold higher sensitivity than U266 to BZ. In the case of other drugs, such as for instance rapamycin, sensitive cells show about 100 fold difference vs. resistant cells in term of response to the drug.32 Sensitivity/resistance to BZ seems more a clinical concept than a genetically driven clear-cut difference. This said, U266 cells are considered by all means a good paradigm of BZ-resistant myeloma cells, whereas MM1.S are considered sensitive. We found that, as in other tumor models, myeloma cells exhibit a sensitivity to translational inhibition, and a prosurvival activity of the mTORC1-eIF4E axis. Similarly to other tumor cell types, inhibition of mTORC1 may be beneficial to therapy.20 Mechanistic evidence demonstrates that the cytostatic effects of rapamycin, an highly specific mTORC1 inhibitor, is due to inhibition of eIF4F formation through dephosphorylation of the eIF4E repressor 4E-BPs.38 Indeed, either decrease of eIF4E or increase of 4E-BP1 can bypass mTORC1 inhibition in vivo. Conversely, eIF4E increase or 4E-BP1 downregulation overcome rapamycin inhibition.34 We found that when BZ represses 4E-BP phosphorylation, in vivo, overexpression of not phosphorylable 4E-BP1 worsens BZ toxicity. We also found an unexpected effect of ERK inhibition on BZ-treated cells. Rapamycin induces a feedback loop that activates ERK and can lead to resistance in other cancer cell types.28 However, in MM ERK inhibition does not increase BZ-induced toxicity, but reduces it. One unexpected observation is that BZ induces 4E-BP1 dephoshorylation, but not rpS6 dephoshorylation as rapamycin does. However, it is intriguing that kinase activity assays suggest that mTOR activity is not affected directly by BZ treatment. Thus, the dephosphorylation of 4E-BP1 may be due to specific phosphatase activities stimulated by BZ or other mechanisms such as mTOR and 4E-BP1 delocalization. A recent work suggests specificity of rapamycin sensitivity for substrate choice for mTORC1 kinase,39 possibly suggesting that BZ may act similarly. Alternatively, BZ may induce variations in steady-state levels of adaptors regulating mTORC1 specificity, in vivo. Future work is needed. We show that both mTORC1 and mTOR inhibition are effective in myeloma cell lines. Moreover, as shown in our and another study, the mTOR inhibitor PP242 has demonstrated efficacy against primary MM cells.40 The major effect of PP242 on tumor cells is the inhibition of cell proliferation.41 Primary myeloma cells grow poorly outside their bone marrow microenvironment. In spite of different conditions of culture (various cytokine combinations, various stroma substrates), primary MM cells cultured in vitro display a decline in growth and proliferation within three days of culture.42 This aspect limits the measurement of drug sensitivity in primary myeloma cells, especially for cytostatic agents like for PP242. Therefore, the inhibitory effect of PP242, in vitro in our conditions, is underestimated. Which would be the best way to employ mTORC1 inhibitors? In this sense, in agreement with a recent study, we found that blockade of mTOR is not synergistic with BZ treatment.43 Thus, mTOR inhibition may be useful in patients resistant to BZ. Data from this and other works would however suggest that patients that would benefit most from mTOR inhibition are without RAS mutations,32,44and have high levels of 4E-BP, low eIF4E.33,34 These patients represent a subset of the whole MM population: it might be thus mandatory to identify them, before treatment. As a final remark, recent genome sequencing has unveiled new somatic mutations in myeloma cancer cells. Among them, it is curious to note that several of them are on factors associated with translational control, and that at least 50% patients have one mutation in one gene involved in protein synthesis.45 These data, together with our observations, may suggest that the translational machinery will be an attractive target for therapy in myeloma cells.
Materials and Methods
Cell culture and proliferation assay
The MM cell lines MM.1S and U266 cells were kindly provided by Dr Tonon. Cells were cultured in RPM1 1640 (Euroclone) supplemented with 10% FBS (fetal bovine serum; Gibco) 1% glutamine and a commercial antibiotic mix (Gibco). Cell proliferation was measured by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide]. Cells were plated in 96 well plate at a density of 20,000 cells per well. After treatment MTT solution was added and incubated for 3 h. The reaction product was quantified reading the absorbance at 570 nm using a microplate reader (Biorad).
Primary myeloma specimens and proliferation assay
Bone marrow (BM) samples for molecular studies were obtained during standard diagnostic procedures. Written informed consent was obtained from each patient. Plasma cells were purified from mononuclear BM cells obtained by Ficoll-Hypaque density gradient centrifugation using anti-CD138 micro beads on an AutoMacs Magnetic Cell Separator (MACS system, Miltenyi Biotec, Auburn, CA). The purity of positively selected plasma cells was assessed by flow cytometry and was ≥ 90% in all cases. For co-colture experiment HS-5 stromal cells were plated O/N in 96 well plates at a density of 500 cells per well. Primary Myeloma cells were plated at density of 10.000 cells/well on a layer of HS-5 stromal cells. Drugs were added at the concentration indicated and compared with DMSO treated controls. Cultures were then incubated for 24, 48 and 72 h in a 37 °C incubator with 5% of CO2. MTT assay was performed. The experiment was done in triplicate measurement.
Drugs and reagents
The following antibodies were used: rabbit polyclonal anti-rpS6, anti-phospho-rpS6 (Ser235/236), anti-phospho-rpS6 (Ser240/244), anti-4E-BP1, anti-p44/42 MAPK (ERK1/2), anti-phospho-p44/42 MAPK-ERK 1/2 (Thr202/Tyr204), anti-eIF2α, anti-phospho-eIF2α (Ser51), anti-mTOR, (Cell signaling); mouse monoclonals anti-β actin (sigma), anti caspase -3 (Alexis), anti-PARP-1 (Millipore), anti-HA (Covance). Retrovirus pBABE-puro and 4E-BP1 4Ala were a gift of Dr. N. Sonenberg. Proteasome inhibitor bortezomib (BZ) was from Millennium Pharmaceuticals, Cambrige MA, mTOR inhibitor PP242, rapamycin and cycloheximide were from Sigma.
Western blot
MM cells were lysed with buffer containing 10 mM NaCl, 10 mM MgCl2, 10 mM TRIS-HCl (pH 7.3), 1% Triton X-100, 1% sodium deoxycholate, 1 mM DTT, 5 mM NaF, 2 mM Na3VO4, 40 units/ml RNasin® (Promega, Milan, Italy) and protease inhibitor cocktail. The whole cell extract was clarified at 4 °C at 15,000 g for 10 min. The amount of recovered protein was quantified by the bicinchoninic acid (BCA) protein assay. Extracts were resolved on SDS-PAGE, transferred to Immobilon-P membranes (Millipore), and probed with appropriate antibodies. Equal amount of proteins was analyzed.
Kinase assay
Proteins from MM.1S cells were extracted in lysis buffer (50 mM Tris-HCL pH 7.4, 150 mM NaCl, 1 mM EDTA, 5 mM EGTA, 20 mM β-glycerolphosphate and protease inhibitor cocktail) by freezing and thawing and clarified by centrifugation. Protein concentration was quantified by BCA. One mg of total extract protein was incubated with anti-mTOR antibody (1:100) for 2 h at 4οC in constant rotation. Immunoprecipitation was performed with protein A for 30 min. Beads were washed one time with high-salt wash buffer (100 mM TRIS-HCl pH 7.4, 500 mM LiCl) and three times with kinase buffer (10 mM Hepes, 50 mM β-glycerolphosphate, 150 mM NaCl). The beads were resuspended in kinase buffer. The kinase assay was performed by adding 10 μg GST-4E-BP1 recombinant protein or GST alone, 10 mM MnCl2 and 4 μCi of γ-32P-ATP. The reaction was run at 30οC for 1 h and terminated by adding one volume of sample buffer. Samples were boiled 5 min, separated by SDS PAGE, transferred to Immobilon-P membranes (Millipore), and probed with anti-GST, anti-mTOR. Autoradiography was performed for one hour at room temperature.
m7GTP Cap column pull-down
Cells were collected by scraping, washed three times with cold PBS and pelleted by centrifugation. Cell pellets were resuspended in lysis buffer (50 mM TRIS-HCl at pH 7.4, 100 mM KCl, 1 mM EDTA, 1 mM EGTA, 20 mM NaF, 0,5 mM Na3VO4, 1% Triton X-100, and protease inhibitors), and incubated for 30 min at 4 °C. Cell debris were removed by centrifugation at 10,000 g for 10 min at 4 °C, and protein concentration was determined using the BCA (bicinchoninic acid) protein assay (Thermo scientific). Extract (300 μg) was incubated with 30 μL of m7GTP-agarose resin (GE Healthcare) for 1 h at 4 °C. The resin was washed three times with 1 mL of lysis buffer, boiled for 6 min in Laemmli buffer, and proteins were resolved by SDS-PAGE.
Polysomal profiles
Cells (40 × 106) were collected and washed with cold PBS (phosphate saline buffer) with 10 μg/ml cycloheximide. Cells were resuspended in lysis buffer composed by 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 30 mM MgCl2, 0.1% NP-40, 100 μg/ml cycloheximide, 40 U/ml RNasin® (Promega), protease inhibitor cocktail (Sigma, St. Louis, MO). Lysed cells were left for 20 min at 4 °C, and lysates were then clarified by centrifugating at 18,000 g for 5 min at 4 °C. Supernatants were collected, and RNA concentration was quantified by reading Abs254. The equivalent of 10 absorbance units at 254 nm were layered on to a 15–50% sucrose gradient in 50 mM Tris/acetate (pH 7.5), 50 mM NH4Cl, 12 mM MgCl2 and 1 mM DTT and centrifuged at 4 °C in a SW41Ti Beckman rotor for 3:30 h at 39000 rev./min. Samples were analyzed with BioLogic LP (BioRad) by reading Abs254.
Methionine labeling
One million cells were incubated at 37 οC with methionine-free medium and pulsed with 33 μCi of Promix 35S-labeled methionine (PerkinElmer Life Sciences) for 1 h. Cells were lysed in 50μl of RIPA buffer without SDS (10 mM Tris-HCL, pH 7.5, 1% Na-deoxycholate, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, protease inhibitor cocktail from Sigma). The lysate was cleared by centrifugation. Extracts of 10 μl were TCA-precipitated on glass microfiber filters (Whatman) and counted. Obtained values were normalized by sample protein content, quantified using the BCA (bicinchoninic acid) protein assay (Thermo scientific). Each sample was analyzed in triplicate and results are expressed as a means ± sd.
Retroviral infections
Retroviral constructs, pBABE empty vector and pBABE -HA-4E-BP1 (4Ala) were transfected into amphotropic phoenix 293T packaging cells. After 48 h virus containing medium was collected. 106 cells were plated in 24-well plate and incubated with virus supernatant and 8 mg/ml polybrene. Cells were spun at 1100 g for 2 h and then incubated for 3 h at 37 οC, 5% CO2. Transduced cells were selected with puromycin 3 mg/ml for one week and further analyzed by immunoblot.
Gene expression profiling
Total RNA from CD138 positive cells was obtained from each sample by the RNeasy® kit (Qiagen, Valencia, CA) extraction procedure. To measure concentration and purity of RNA, a NanoDrop ND-1000 spectrophotometer was used (NanoDrop Technologies), purity of the extracted RNA was based on the 260/280 and the 260/230 O.D. ratios, as calculated and displayed by the NanoDrop spectrophotometer. Moreover, disposable RNA chips (Agilent RNA 6000 Nano LabChip kit) were used to determine the concentration and purity/integrity of RNA samples using Agilent 2100 Bioanalyzer. Samples with at least 30 ng/uL RNA were labeled for gene expression profiling, using the Affymetrix Two-cycles Gene Chip microarray system (Affymetrix, Santa Clara, CA). cDNA synthesis, biotin-labeled target synthesis, HG U133 Plus 2.0 GeneChip arrays hybridization, staining, and scanning were performed according to the standard protocol supplied by Affymetrix. Microarray data were used to identify gene expression profile of 4E-BP1, 4E-BP2 and eIF4E in BM samples obtained from 122 newly diagnosed MM patients.
Supplementary Material
Glossary
Abbreviations:
- MM
multiple myeloma
- UPR
unfolded protein response
- BZ
bortezomib
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Aknowledgements
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC; Investigator Grant and Special Program Molecular Clinical Oncology, 5 per mille no. 9965); Association for International Cancer Research, UK. We thank N. Sonenberg for vectors and G. Tonon for Myeloma cell lines.
References
- 1.Kyle RA, Rajkumar SV. . Multiple myeloma. N Engl J Med 2004; 351:1860 - 73; http://dx.doi.org/ 10.1056/NEJMra041875; PMID: 15509819 [DOI] [PubMed] [Google Scholar]
- 2.Cavo M, Pantani L, Petrucci MT, Patriarca F, Zamagni E, Donnarumma D, et al. Bortezomib-thalidomide-dexamethasone is superior to thalidomide-dexamethasone as consolidation therapy after autologous hematopoietic stem cell transplantation in patients with newly diagnosed multiple myeloma. Blood 2012; 120:9-19; PMID: 22498745; DOI: http://dx.doi.org/23897961 10.1182/blood-2012-02-408898; http://dx.doi.org/ 10.1182/blood-2012-02-408898. [DOI] [PubMed] [Google Scholar]
- 3.Munshi NC, Anderson KC. New strategies in the treatment of multiple myeloma. Clin Cancer Res 2013; 19:3337-44; PMID: 23515406; DOI: http://dx.doi.org/23897961 10.1158/1078-0432.CCR-12-1881; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sonneveld P, Goldschmidt H, Rosiñol L, Bladé J, Lahuerta JJ, Cavo M, Tacchetti P, Zamagni E, Attal M, Lokhorst HM, et al. . Bortezomib-based versus nonbortezomib-based induction treatment before autologous stem-cell transplantation in patients with previously untreated multiple myeloma: a meta-analysis of phase III randomized, controlled trials. J Clin Oncol 2013; 31:3279 - 87; http://dx.doi.org/ 10.1200/JCO.2012.48.4626; PMID: 23897961 [DOI] [PubMed] [Google Scholar]
- 5.Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Zeldenrust SR, Dingli D, Russell SJ, Lust JA, et al. . Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008; 111:2516 - 20; http://dx.doi.org/ 10.1182/blood-2007-10-116129; PMID: 17975015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Fanourakis G, Gu X, Bailey C, Joseph M, Libermann TA, Treon SP, et al. . Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci USA 2002; 99:14374 - 9; http://dx.doi.org/ 10.1073/pnas.202445099; PMID: 12391322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mujtaba T, Dou QP. . Advances in the understanding of mechanisms and therapeutic use of bortezomib. Discov Med 2011; 12:471 - 80; PMID: 22204764 [PMC free article] [PubMed] [Google Scholar]
- 8.Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. . Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer 2007; 7:585 - 98; http://dx.doi.org/ 10.1038/nrc2189; PMID: 17646864 [DOI] [PubMed] [Google Scholar]
- 9.Demchenko YN, Glebov OK, Zingone A, Keats JJ, Bergsagel PL, Kuehl WM. . Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood 2010; 115:3541 - 52; http://dx.doi.org/ 10.1182/blood-2009-09-243535;; PMID: 20053756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, et al. . Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007; 12:131 - 44; http://dx.doi.org/ 10.1016/j.ccr.2007.07.003; PMID: 17692805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Panwalkar A, Verstovsek S, Giles F. . Nuclear factor-kappaB modulation as a therapeutic approach in hematologic malignancies. Cancer 2004; 100:1578 - 89; http://dx.doi.org/ 10.1002/cncr.20182; PMID: 15073843 [DOI] [PubMed] [Google Scholar]
- 12.Roccaro AM, Hideshima T, Raje N, Kumar S, Ishitsuka K, Yasui H, Shiraishi N, Ribatti D, Nico B, Vacca A, et al. . Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res 2006; 66:184 - 91; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-1195; PMID: 16397231 [DOI] [PubMed] [Google Scholar]
- 13.Shin DH, Chun YS, Lee DS, Huang LE, Park JW. Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood 2008; 111:3131-6; PMID: 18174379; DOI: http://dx.doi.org/16507771 10.1182/blood-2007-11-120576; http://dx.doi.org/ 10.1182/blood-2007-11-120576. [DOI] [PubMed] [Google Scholar]
- 14.Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr., Lee KP, Boise LH. . Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006; 107:4907 - 16; http://dx.doi.org/ 10.1182/blood-2005-08-3531;; PMID: 16507771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Dunner K Jr., Huang P, Abbruzzese JL, McConkey DJ. . Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res 2005; 65:11658 - 66; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-2370; PMID: 16357177 [DOI] [PubMed] [Google Scholar]
- 16.Hetz C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 2012; 13:89-102; PMID: 22251901; DOI: http://dx.doi.org/9531536 10.1038/nrm3270; http://dx.doi.org/ 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 17.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. . CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 1998; 12:982 - 95; http://dx.doi.org/ 10.1101/gad.12.7.982; PMID: 9531536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meister S, Schubert U, Neubert K, Herrmann K, Burger R, Gramatzki M, Hahn S, Schreiber S, Wilhelm S, Herrmann M, et al. . Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res 2007; 67:1783 - 92; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2258; PMID: 17308121 [DOI] [PubMed] [Google Scholar]
- 19.Vincenz L, Jager R, O'Dwyer M, Samali A. Endoplasmic reticulum stress and the unfolded protein response: Targeting the achilles heel of multiple myeloma. Mol Cancer Ther 2013; 12:831-43; PMID: 23729400; DOI: http://dx.doi.org/23241507 10.1158/1535-7163.MCT-12-0782; http://dx.doi.org/ 10.1158/1535-7163.MCT-12-0782. [DOI] [PubMed] [Google Scholar]
- 20.Ramirez-Fort MK, Case EC, Rosen AC, Cerci FB, Wu S, Lacouture ME. Rash to the mTOR inhibitor everolimus: Systematic review and meta-analysis. Am J Clin Oncol 2012; •••: http://dx.doi.org/ 10.1097/COC.0b013e318277d62f;PMID: 23241507 [DOI] [PubMed]
- 21.Kim A, Park S, Lee JE, Jang WS, Lee SJ, Kang HJ, et al. The dual PI3K and mTOR inhibitor NVP-BEZ235 exhibits anti-proliferative activity and overcomes bortezomib resistance in mantle cell lymphoma cells. Leuk Res 2012; 36:912-20; PMID: 22560334; DOI: http://dx.doi.org/12902539 10.1016/j.leukres.2012.02.010; http://dx.doi.org/ 10.1016/j.leukres.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 22.Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH. . Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci USA 2003; 100:9946 - 51; http://dx.doi.org/ 10.1073/pnas.1334037100; PMID: 12902539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ling SC, Lau EK, Al-Shabeeb A, Nikolic A, Catalano A, Iland H, et al. Response of myeloma to the proteasome inhibitor bortezomib is correlated with the unfolded protein response regulator XBP-1. Haematologica 2012; 97:64-72; PMID: 21993678; DOI: http://dx.doi.org/15659334 10.3324/haematol.2011.043331; http://dx.doi.org/ 10.3324/haematol.2011.043331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Proud CG. . eIF2 and the control of cell physiology. Semin Cell Dev Biol 2005; 16:3 - 12; http://dx.doi.org/ 10.1016/j.semcdb.2004.11.004; PMID: 15659334 [DOI] [PubMed] [Google Scholar]
- 25.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 2009; 10:307-18; PMID: 19339977; DOI: http://dx.doi.org/17360704 10.1038/nrm2672; http://dx.doi.org/ 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 26.Meyuhas O. Physiological roles of ribosomal protein S6: One of its kind. Int Rev Cell Mol Biol 2008; 268:1-37; PMID: 18703402; DOI: http://dx.doi.org/17360704 10.1016/S1937-6448(08)00801-0; http://dx.doi.org/ 10.1016/S1937-6448(08)00801-0. [DOI] [PubMed] [Google Scholar]
- 27.Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N, Blenis J. . RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem 2007; 282:14056 - 64; http://dx.doi.org/ 10.1074/jbc.M700906200; PMID: 17360704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, et al. . Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest 2008; 118:3065 - 74; http://dx.doi.org/ 10.1172/JCI34739;; PMID: 18725988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. . Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene 2007; 26:1932 - 40; http://dx.doi.org/ 10.1038/sj.onc.1209990; PMID: 17001314 [DOI] [PubMed] [Google Scholar]
- 30.Ghobrial IM, Weller E, Vij R, Munshi NC, Banwait R, Bagshaw M, et al. Weekly bortezomib in combination with temsirolimus in relapsed or relapsed and refractory multiple myeloma: A multicentre, phase 1/2, open-label, dose-escalation study. Lancet Oncol 2011; 12:263-72; PMID: 21345726; DOI: http://dx.doi.org/22216185 10.1016/S1470-2045(11)70028-6; http://dx.doi.org/ 10.1016/S1470-2045(11)70028-6. [DOI] [PubMed] [Google Scholar]
- 31.Farag SS, Zhang S, Jansak BS, Wang X, Kraut E, Chan K, et al. Phase II trial of temsirolimus in patients with relapsed or refractory multiple myeloma. Leuk Res 2009; 33:1475-80; PMID: 19261329; DOI: http://dx.doi.org/22216185 10.1016/j.leukres.2009.01.039; http://dx.doi.org/ 10.1016/j.leukres.2009.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Di Nicolantonio F, Arena S, Tabernero J, Grosso S, Molinari F, Macarulla T, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest 2010; 120:2858-66; PMID: 20664172; DOI: http://dx.doi.org/22216185 10.1172/JCI37539; http://dx.doi.org/ 10.1172/JCI37539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alain T, Morita M, Fonseca BD, Yanagiya A, Siddiqui N, Bhat M, et al. eIF4E/4E-BP ratio predicts the efficacy of mTOR targeted therapies. Cancer Res 2012; 72:6468-76; PMID: 23100465; DOI: http://dx.doi.org/22216185 10.1158/0008-5472.CAN-12-2395; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-2395. [DOI] [PubMed] [Google Scholar]
- 34.Grosso S, Pesce E, Brina D, Beugnet A, Loreni F, Biffo S. . Sensitivity of global translation to mTOR inhibition in REN cells depends on the equilibrium between eIF4E and 4E-BP1. PLoS ONE 2011; 6:e29136; http://dx.doi.org/ 10.1371/journal.pone.0029136; PMID: 22216185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schewe DM, Aguirre-Ghiso JA. Inhibition of eIF2alpha dephosphorylation maximizes bortezomib efficiency and eliminates quiescent multiple myeloma cells surviving proteasome inhibitor therapy. Cancer Res 2009; 69:1545-52; PMID: 19190324; DOI: http://dx.doi.org/15314020 10.1158/0008-5472.CAN-08-3858; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campanella A, Santambrogio P, Fontana F, Frenquelli M, Cenci S, Marcatti M, et al. Iron increases the susceptibility of multiple myeloma cells to bortezomib. Haematologica 2013; 98:971-9; PMID: 23242599; DOI: http://dx.doi.org/15314020 10.3324/haematol.2012.074872; http://dx.doi.org/ 10.3324/haematol.2012.074872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bianchi G, Oliva L, Cascio P, Pengo N, Fontana F, Cerruti F, et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009; 113:3040-9; PMID: 19164601; DOI: http://dx.doi.org/15314020 10.1182/blood-2008-08-172734; http://dx.doi.org/ 10.1182/blood-2008-08-172734. [DOI] [PubMed] [Google Scholar]
- 38.Hay N, Sonenberg N. . Upstream and downstream of mTOR. Genes Dev 2004; 18:1926 - 45; http://dx.doi.org/ 10.1101/gad.1212704; PMID: 15314020 [DOI] [PubMed] [Google Scholar]
- 39.Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, Ottina K, et al. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science 2013; 341:1236566; PMID: 23888043; DOI: http://dx.doi.org/22045983 10.1126/science.1236566; http://dx.doi.org/ 10.1126/science.1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoang B, Frost P, Shi Y, Belanger E, Benavides A, Pezeshkpour G, et al. Targeting TORC2 in multiple myeloma with a new mTOR kinase inhibitor. Blood 2010; 116:4560-8; PMID: 20686120; DOI: http://dx.doi.org/22045983 10.1182/blood-2010-05-285726; http://dx.doi.org/ 10.1182/blood-2010-05-285726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol 2009; 7:e38; PMID: 19209957; DOI: http://dx.doi.org/22045983 10.1371/journal.pbio.1000038; http://dx.doi.org/ 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zlei M, Egert S, Wider D, Ihorst G, Wäsch R, Engelhardt M. . Characterization of in vitro growth of multiple myeloma cells. Exp Hematol 2007; 35:1550 - 61; http://dx.doi.org/ 10.1016/j.exphem.2007.06.016 [DOI] [PubMed] [Google Scholar]
- 43.Maiso P, Liu Y, Morgan B, Azab AK, Ren P, Martin MB, Zhang Y, Liu Y, Sacco A, Ngo H, et al. . Defining the role of TORC1/2 in multiple myeloma. Blood 2011; 118:6860 - 70; http://dx.doi.org/ 10.1182/blood-2011-03-342394; PMID: 22045983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steinbrunn T, Stühmer T, Gattenlöhner S, Rosenwald A, Mottok A, Unzicker C, Einsele H, Chatterjee M, Bargou RC. . Mutated RAS and constitutively activated Akt delineate distinct oncogenic pathways, which independently contribute to multiple myeloma cell survival. Blood 2011; 117:1998 - 2004; http://dx.doi.org/ 10.1182/blood-2010-05-284422; PMID: 21149634 [DOI] [PubMed] [Google Scholar]
- 45.Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, et al. . Initial genome sequencing and analysis of multiple myeloma. Nature 2011; 471:467 - 72; http://dx.doi.org/ 10.1038/nature09837; PMID: 21430775 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.