

Abstract
Aims/Introduction
Circulating cell‐free mitochondrial deoxyribonucleic acid (ccf‐mtDNA) is presumably derived from injured tissues or cells in the body and has been suggested to be potential biomarker in several diseases. The present study explored whether mtDNA could be used as a biomarker to evaluate disease in coronary heart disease (CHD) patients with or without diabetes mellitus (DM).
Materials and Methods
A total of 50 CHD patients with type 2 diabetes, 50 CHD patients without type 2 diabetes, and 50 age‐ and sex‐matched patients without CHD and DM (non‐CHD‐DM) were recruited. Ccf‐mtDNA levels were assessed by measuring the nicotinamide adenine dinucleotide dehydrogenase 1 gene using quantitative real‐time polymerase chain reaction. Receiver operating characteristic curve analysis of plasma mtDNA in CHD with or without DM was also determined. Multivariate logistic regression analyses were carried out to determine the correlation between the mtDNA levels and traditional CHD risk factors.
Results
The plasma ccf‐mtDNA levels were significantly elevated in CHD patients with DM compared with those without and non‐CHD‐DM. The area under the receiver operating characteristic curves of mtDNA in CHD patients with DM vs non‐CHD‐DM was 0.907%. Correlation analyses of the mtDNA levels and traditional CHD risk factors showed that the mtDNA levels were significantly correlated with fasting blood glucose in CHD patients with DM.
Conclusions
Ccf‐mtDNA levels can be used as a biomarker in CHD patients with DM.
Keywords: Circulating cell‐free mitochondrial deoxyribonucleic acid, Coronary heart disease, Diabetes mellitus
Introduction
Diabetes is a major contributor to coronary artery disease (CHD) morbidity and mortality. Patients with diabetes have more than a 200% greater risk of cardiovascular diseases than non‐diabetic individuals1. Although the traditional risk factors for diabetes, namely, high‐density lipoprotein cholesterol, smoking habit, macroalbuminuria, lower estimated glomerular filtration rate, use of diabetes medication and longer duration of diabetes, can explain part of the increasing prevalence of CHD patients, we still lack plasma biomarkers that aid in the evaluation of disease activity.
Recently, circulating cell‐free mitochondrial deoxyribonucleic acid (ccf‐mtDNA) has been used as a potential marker in various diseases. Studies have also shown that ccf‐mtDNA levels are increased in patients or animals with trauma2 and microbial infection3, 4. Furthermore, mtDNA is a damage‐associated molecular pattern5, and can elicit inflammatory response and cause organ injury6, 7. We also found that increased ccf‐mtDNA levels in peripheral blood contribute to cardiovascular risk8, 9. Based on these findings, we compared CHD patients with DM with those without DM and non‐CHD‐DM in order to identify whether ccf‐mtDNA can serve as a biomarker for CHD patients with DM.
Materials and methods
Ethics Statement
The present study protocol was approved by Jingling Hospital's Institutional Review Committee on Human Research, and that it conforms to the provisions of the Declaration of Helsinki (as revised in Edinburgh 2000). All patients provided written informed consent before any study‐related procedure was carried out.
Patient Population
All cases were recruited at the Department of Cardiology of Jinling Hospital (Nanjing, China). A total of 50 CHD patients with DM, 50 CHD patients without DM, and 50 age‐ and sex‐matched patients without CHD and DM were enrolled for the present study. CHD cases were defined as having a history of myocardial infarction, history of angina, positive treadmill test or history of coronary artery bypass graft surgery. Diabetes was defined as taking antidiabetic medication or having a fasting glucose of at least 126 mg/dL or glycated hemoglobin of at least 6.5%. Patients with infection, cancer, other inflammatory diseases, liver disease or renal failure were excluded. Patients without pre‐existing cardiovascular disease and diabetes were selected for the non‐CHD‐DM group. The study protocol was approved by the Regional Committee for Medical Research Ethics of Jinling Hospital, and all patients provided written and oral informed consent.
Plasma Preparation
Whole blood samples were drawn and transferred into ethylenediaminetetraacetic acid‐coated blood collection tubes, and processed within 2 h after venipuncture. Briefly, whole blood was centrifuged at 500 g for 10 min at room temperature, and the supernatant was transferred to a fresh tube followed by centrifugation at 700 g for 5 min at 4°C. Then 240 μL of the supernatant was carefully transferred avoiding any pellets at the bottom of the tubes. The obtained supernatant was further centrifuged at 15 000 g for 10 min at 4°C, and 200 μL of the supernatant was removed to a fresh tube and stored at –80°C for DNA isolation later.
DNA Isolation from Plasma
Plasma DNA was isolated from the plasma samples stored at –80°C using the QIAamp DNA Blood Mini Kit (#51104; Qiagen, Valencia, CA, USA) following the manufacturer's instructions. In brief, samples were thawed on ice and then mixed briefly by vortex. Then, the plasma samples were mixed with lysis buffer and proteinase K, and incubated at 56°C for 10 min. At the final step of the procedure, DNA was eluted with 150 μL of nuclease‐free, deionized H2O followed by quantitative real‐time PCR (qPCR) assay.
Primers and qPCR
The total amount of DNA in the sample was measured with spectrophotometry (Nano Drop 2000; Thermo Fischer, Wilmington, DE, USA). The mtDNA primer sequences were derived from human nicotinamide adenine dinucleotide dehydrogenase 1 gene on mtDNA, and were 5′‐ATACCCATGGCCAACCTCCT‐3′ (forward) and 5′‐GGGCCTTTGCGTAGTTGTAT‐3′ (reverse)2, 10; The nuclear DNA primers were derived from human β‐globin gene and were 5′‐GTGCACCTGACTCCTGAGGAGA‐3′ (forward) and 5′‐CCTTGATACCAACCTGCCCAG‐3′ (reverse)11. The mitochondrial and β‐globin DNA concentrations in all of the plasma aliquots were determined by quantitative PCR. The difference in the mitochondrial DNA concentration among the groups was determined statistically. The physical characteristics of the mitochondrial genome were compared with those of the nuclear genome by comparing the mitochondrial DNA concentration and the corresponding β‐globin DNA concentration of the plasma aliquot. The qPCR assays were carried out using a SYBR Green dye‐based kit and the Lightcycle 96 sequence detection system (Roche, Mannheim, Germany).The thermal profile for the qPCR was as follows: 95°C/10 min followed by 40 cycles of 95°C/10 s –58°C/10 s –72°C/10 s.
Analyses of Traditional Risk Factors
We obtained the information of patients' history and of the traditional risk factors, including fasting blood glucose (FBG), creatine kinase, creatine kinase isoenzyme MB, total cholesterol, triglycerides, high‐density lipoprotein cholesterol, low‐density lipoprotein cholesterol, systolic blood pressure, diastolic blood pressure, white blood cells, and neutrophils, urea, creatinine and uric acid. The correlations between the level of ccf‐mtDNA and traditional risk factors were then assessed.
Statistical Analysis
Statistical analysis was carried out using SPSS software, version 18.0 (SPSS Inc., Chicago, IL, USA). All measurements with normal distribution were represented as mean ± standard deviation. For the non‐normal distribution, median (interquartile range) expression was used. Receiver operating characteristic (ROC) curves were established to determine the sensitivity and specificity of the mtDNA for predicting active CHD with DM. Pearson's correlation analysis was carried out to calculate the correlation between the level of mtDNA and clinical features. To evaluate the relative contribution of mtDNA in CHD with DM, a multiple linear regression model was used. P‐values <0.05 were considered statistically significant.
Results
Baseline Characteristics of the Patients
The baseline characteristics of the CHD patients who did and did not have DM are shown in Table 1. The mean age was similar in the subject groups with and without CHD. The ratios of sex in the two groups of patients with or without CHD were also similar (men vs women: 23/27 and 24/26, respectively). The proportion of subjects receiving insulin or oral hypoglycemic agents was significantly higher in the group with DM than in the group without DM (42/50 vs 0/50, P < 0.05). We observed that the patients with a history of DM had significantly higher BMI, FBG, systolic blood pressure, white blood cells and neutrophils than those without; in addition, their serum triglycerides and creatine kinase isoenzyme MB tended to be higher than those of patients without DM, although the difference did not reach a statistical significance. In contrast, total cholesterol, high‐density lipoprotein cholesterol, low‐density lipoprotein cholesterol, creatine kinase, diastolic blood pressure, urea, creatinine and uric acid were not statistically different between the two groups.
Table 1.
Clinical features of coronary heart disease patients with diabetes mellitus, coronary heart disease patients without diabetes mellitus and non‐coronary heart disease without diabetes mellitus
| CHD | Non‐CHD | P‐value† | P‐value‡ | ||
|---|---|---|---|---|---|
| DM (+) | DM (–) | DM (–) | |||
| Age (years) | 65.83 ± 9.06 | 63.65 ± 11.38 | 59.75 ± 11.45 | P > 0.1 | P > 0.1 |
| Sex (male/female) | 25/25 | 23/27 | 24/26 | ||
| BMI | 26.54 ± 3.12 | 25.29 ± 2.62 | 24.44 ± 2.66 | 0.01 < P < 0.05 | 0.01 < P < 0.05 |
| Fasting blood glucose (mmol/L) | 6.86 ± 4.13 | 4.99 ± 1.54 | 5.08 ± 1.16 | 0.01 < P < 0.05 | P > 0.05 |
| Total cholesterol (mmol/L) | 3.81 ± 0.81 | 3.89 ± 0.84 | 3.93 ± 0.93 | P > 0.1 | P > 0.05 |
| Triglycerides (mmol/L) | 1.61 ± 0.87 | 1.49 ± 0.84 | 1.19 ± 0.61 | P > 0.1 | P < 0.02 |
| HDL cholesterol (mmol/L) | 0.98 ± 0.19 | 1.06 ± 0.23 | 1.12 ± 0.27 | P > 0.1 | P > 0.05 |
| LDL cholesterol (mmol/L) | 2.29 ± 0.71 | 2.32 ± 0.86 | 2.43 ± 0.82 | P > 0.1 | P > 0.05 |
| SBP (mmHg) | 135.68 ± 18.08 | 126.53 ± 16.19 | 127.72 ± 17.35 | P > 0.01 | 0.02 < P < 0.05 |
| DBP (mmHg) | 75.59 ± 9.71 | 76.67 ± 10.06 | 77.85 ± 10.44 | P > 0.1 | P > 0.05 |
| Urea (mmol/L) | 5.89 ± 1.63 | 5.78 ± 1.66 | 5.69 ± 1.60 | P > 0.1 | 0.05 < P < 0.1 |
| Creatinine (μmol/L) | 61.91 ± 16.91 | 68.26 ± 16.53 | 66.30 ± 17.12 | 0.05 < P < 0.1 | 0.05 < P < 0.1 |
| Uric acid (μmol/L) | 300.19 ± 91.55 | 315.23 ± 82.35 | 319.58 ± 89.67 | P > 0.1 | P > 0.1 |
| White blood cells | 7.58 ± 2.62 | 6.39 ± 2.09 | 7.17 ± 2.46 | 0.01 < P < 0.05 | P > 0.05 |
| NEUTR% | 66.74 ± 10.14 | 61.28 ± 10.63 | 62.13 ± 12.84 | 0.01 < P < 0.05 | P > 0.05 |
| CK (μg/L) | 83.05 ± 42.43 | 85.12 ± 58.9 | 114.42 ± 122.34 | P > 0.1 | P > 0.05 |
| CK‐MB (ug/L) | 16.16 ± 8.13 | 14.25 ± 5.96 | 14.82 ± 8.40 | P > 0.1 | P > 0.1 |
†Coronary heart disease (CHD) patients with diabetes mellitus (DM) vs CHD patients without DM. ‡CHD patients with DM vs non‐CHD without DM. BMI, body mass index; CK, creatine kinase; CK‐MB, creatine kinase isoenzyme MB; DBP, diastolic blood pressure; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; NEUTR, neutrophils; SBP, systolic blood pressure.
Ccf‐mtDNA level increased in CHD with DM
We measured the mtDNA levels in the 50 CHD patients with DM, 50 CHD patients without DM and 50 non‐CHD‐DM using qPCR. The results showed that levels of mtDNA were significantly increased in CHD patients with DM compared with the CHD patients without and non‐CHD‐DM. The median plasma ccf‐mtDNA levels in the patients with CHD were approximately twofold higher than those of non‐CHD‐DM the group, whereas patients with CHD and DM were fivefold higher than those of the non‐CHD‐DM group (Figure 1).
Figure 1.
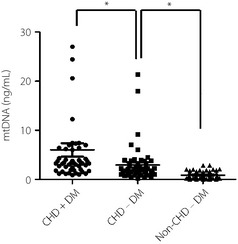
The levels of mitochondrial deoxyribonucleic acid (mtDNA) in coronary heart disease (CHD) patients with or without diabetes mellitus (DM) and non‐CHD‐DM. *P < 0.01. CHD+DM, coronary heart disease patients with type 2 diabetes; CHD‐DM, coronary heart disease patients without type 2 diabetes; non‐CHD‐DM, age‐ and sex‐matched patients without coronary heart disease and diabetes mellitus.
ROC Curve Analysis of Plasma mtDNA
To determine whether plasma levels of mtDNA could serve as a biomarker to predict CHD with DM, we tested the ability to discriminate CHD patients with DM from the CHD patients without DM and non‐CHD‐DM. The concentration of mtDNA in the plasma was used to generate a ROC curve to evaluate the predictive capability of mtDNA for CHD patients with DM. The results showed that the area under the ROC curves of mtDNA in CHD patients with DM vs without DM was 0.675%, whereas that in CHD patients with DM vs non‐CHD‐DM was 0.907% (Table 2). These results show that mtDNA can be used to discriminate CHD patients with DM from non‐CHD‐DM.
Table 2.
Receiver operating characteristic curve analysis to assess the mitochondrial deoxyribonucleic acid ability to discriminate coronary heart disease patients with diabetes mellitus from without diabetes mellitus and non‐coronary heart disease without diabetes mellitus
| AUC | SE | Asymptomatic significance | 95% CI | Sensitivity | Specificity | Optimal concentration (ng/mL) | |
|---|---|---|---|---|---|---|---|
| CHD with DM (n = 50) vs CHD without DM (n = 50) | 0.676 | 0.075 | 0.03 | 0.528–0.823 | 0.761 | 0.593 | 2.64 |
| CHD with DM (n = 50) vs non‐CHD‐DM (n = 50) | 0.907 | 0.04 | 0.000 | 0.828–0.986 | 0.96 | 0.556 | 1.003 |
Optimal concentration refers to the concentration of mitochondrial deoxyribonucleic acid that can result in an optimal specificity and sensitivity if it is used as a cut‐off value for the positivity of coronary heart disease (CHD) with diabetes mellitus (DM). AUC, area under the receiver operating characteristic curves; CI, confidence interval; non‐CHD‐DM, non‐coronary heart disease without diabetes mellitus; SE, standard error.
Correlation Between the Level of Plasma ccf‐mtDNA and Traditional Risk Factors
We further examined the relationship between plasma mtDNA concentrations in CHD patients with and without DM, and the traditional risk factors using Pearson's correlation analysis. The results showed a significant correlation between ccf‐mtDNA levels and blood glucose in CHD patients with DM (Figure 2). There was no significant correlation between plasma levels of ccf‐mtDNA and other traditional risk factors. Pearson's correlation analysis was also carried out between the levels of the mtDNA and other clinical parameters of CHD patients with DM; however, no correlations were observed (Table 3). In multivariate linear regression analysis, mtDNA was associated with FBG (β = –0.107, P < 0.001; adjusted R 2 = 0.272), whereas it was not correlated with other parameters (Table 4).
Figure 2.
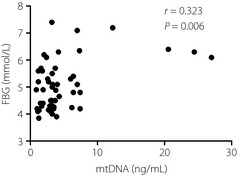
Plasma levels of mitochondrial deoxyribonucleic acid (mtDNA) correlate with the levels of fasting blood glucose (FBG; r = 0.323, P < 0.05) in coronary heart disease patients with diabetes mellitus.
Table 3.
Pearson correlation analysis between the levels of plasma mtDNA and the clinical characteristics of CHD with DM
| Variable | R/ P | mtDNA |
|---|---|---|
| BMI | R | 0.186 |
| P value | 0.43 | |
| FBG | R | 0.323 |
| P value | 0.006 | |
| Total cholesterol | R | −0.072 |
| P value | 0.4 | |
| Triglycerides | R | −0.161 |
| P value | 0.5 | |
| SBP | R | −0.041 |
| P value | 0.6 | |
| DBP | R | 0.081 |
| P value | 0.3 | |
| White Blood Cell | R | 0.019 |
| P value | 0.9 | |
| Neutrophil | R | 0.164 |
| P value | 0.4 | |
| ALT | R | −0.088 |
| P value | 0.6 | |
| AST | R | 0.181 |
| P value | 0.3 | |
| CK | R | 0.192 |
| P value | 0.3 | |
| CK‐MB | R | 0.017 |
| P value | 0.9 | |
| SBP | R | 0.139 |
| P value | 0.5 | |
| DBP | R | 0.172 |
| P value | 0.3 | |
| HDL | R | −0.1005 |
| P value | 0.6 | |
| LDL | R | −0.289 |
| P value | 0.1 |
R, Pearson correlation coefficient. A total of 50 samples described in the methods were used in the analysis.
Table 4.
Multivariate linear regression analysis model including mitochondrial deoxyribonucleic acid and systolic blood pressure
| Variable | β | P‐value | Adjusted R 2 | |
|---|---|---|---|---|
| mtDNA | Constant | –13.974 | 0.156 | |
| FBG | 2.231 | 0.033 | ||
| SBP | 0.049 | NS |
FBG, fasting blood glucose; SBP, systolic blood pressure; mtDNA, mitochondrial deoxyribonucleic acid; NS, not significant.
Discussion
To our knowledge, this is the first study showing an increase of ccf‐mtDNA in CHD without DM compared with non‐CHD‐DM, and a consistent increase of ccf‐mtDNA in CHD with DM compared with those without. When 1.003 ng/mL is used as a cut‐off value of mtDNA plasma concentration for CHD patients with DM positivity, an optimal specificity and sensitivity of 96.0 and 55.6%, respectively, can be obtained according to the ROC analysis.
Our data are consistent with previous studies showing that ccf‐mtDNA levels are elevated in a variety of diseases, such as myocardial infarction8, major trauma2, sepsis12, malignancy13, 14 and intensive care unit conditions15. The present study also showed that mtDNA can be used to discriminate CHD patients with DM from non‐CHD‐DM, although the ability of mtDNA to discriminate CHD patients with DM from those without DM was decreased, suggesting mtDNA is a biomarker for CHD with DM. We found that CHD patients with or without DM had extraordinarily high levels of circulating mtDNA, whereas no one in the normal control group did. As can be seen, these patients were special in some parameters as listed in Table 1, such as higher body mass index, triglycerides and systolic blood pressure compared with normal controls, these might cause the different levels of mtDNA. Previous studies have shown that mtDNA can induce inflammatory responses by activating neutrophils (PMN) through TLR92. These findings suggest that ccf‐mtDNA might represent an important pathogenic determinant that contributes to a systemic inflammatory response16. In the present study, we also found that white blood cells and neutrophils in CHD with DM were increased relative to CHD without DM, suggesting inflammatory responses could contribute to the elevation of plasma ccf‐mtDNA. Furthermore, our data showed that mtDNA was positively correlated with FBG in CHD with DM. It is well known that chronic hyperglycemia induces overproduction of reactive oxygen species17, which have a very short half‐life, and react rapidly with DNA, protein and lipids, thereby resulting in oxidative damage18. Mitochondrial dysfunction is induced by high glucose and free fatty acids in various type cells19, 20. Mitochondria are the major site of reactive oxygen species production within the cell21. Overproduction of reactive oxygen species will change mitochondrial morphology21 and mtDNA replication22. Furthermore, the majority of plasma DNA is derived from apoptotic or necrotic cells23. Thus, we speculated that the increased mtDNA in the plasma of CHD patients with DM might be associated with damaged cells, which could directly release mtDNA into the circulation, or they might act on other cell types in the body that secrete the cc‐mtDNA into circulation. Coronary microvascular disease and endothelial dysfunction is the pathogenesis of CHD, we speculated that the injured endothelial cell‐derived mtDNA might be the main origin in CHD patients. Further effort is required to test this hypothesis.
However, the present study had some limitations. First, the levels of plasma ccf‐mtDNA could be influenced by age and pre‐existing diseases24. Second, the number of cases was small, and large‐scale prospective studies are warranted to evaluate the diagnosis contribution of plasma ccf‐mtDNA on clinical outcomes.
Disclosure
The authors declare no conflict of interest.
Acknowledgment
The study was supported by grants from the National Natural Science Foundation of China (81400238).
J Diabetes Investig 2016; 7: 109–114
References
- 1. Grundy SM, Howard B, Smith S Jr, et al Prevention Conference VI: Diabetes and Cardiovascular Disease: executive summary: conference proceeding for healthcare professionals from a special writing group of the American Heart Association. Circulation 2002; 105: 2231–2239. [DOI] [PubMed] [Google Scholar]
- 2. Zhang Q, Raoof M, Chen Y, et al Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464: 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cossarizza A, Pinti M, Nasi M, et al Increased plasma levels of extracellular mitochondrial DNA during HIV infection: a new role for mitochondrial damage‐associated molecular patterns during inflammation. Mitochondrion 2011; 11: 750–755. [DOI] [PubMed] [Google Scholar]
- 4. Sursal T, Stearns‐Kurosawa DJ, Itagaki K, et al Plasma bacterial and mitochondrial DNA distinguish bacterial sepsis from sterile systemic inflammatory response syndrome and quantify inflammatory tissue injury in nonhuman primates. Shock 2013; 39: 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krysko DV, Agostinis P, Krysko O, et al Emerging role of damage‐associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 2011; 32: 157–164. [DOI] [PubMed] [Google Scholar]
- 6. Nakahira K, Haspel JA, Rathinam VA, et al Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 2011; 12: 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collins LV, Hajizadeh S, Holme E, et al Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol 2004; 75: 995–1000. [DOI] [PubMed] [Google Scholar]
- 8. Bliksoen M, Mariero LH, Ohm IK, et al Increased circulating mitochondrial DNA after myocardial infarction. Int J Cardiol 2012; 158: 132–134. [DOI] [PubMed] [Google Scholar]
- 9. Andreassi MG. Coronary atherosclerosis and somatic mutations: an overview of the contributive factors for oxidative DNA damage. Mutat Res 2003; 543: 67–86. [DOI] [PubMed] [Google Scholar]
- 10. McGill MR, Sharpe MR, Williams CD, et al The mechanism underlying acetaminophen‐induced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest 2012; 122: 1574–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moreira VG, Prieto B, Rodriguez JS, et al Usefulness of cell‐free plasma DNA, procalcitonin and C‐reactive protein as markers of infection in febrile patients. Ann Clin Biochem 2010; 47(Pt 3): 253–258. [DOI] [PubMed] [Google Scholar]
- 12. Chou CC, Fang HY, Chen YL, et al Plasma nuclear DNA and mitochondrial DNA as prognostic markers in corrosive injury patients. Dig Surg 2008; 25: 300–304. [DOI] [PubMed] [Google Scholar]
- 13. Yu M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci 2011; 89: 65–71. [DOI] [PubMed] [Google Scholar]
- 14. Ellinger J, Muller SC, Wernert N, et al Mitochondrial DNA in serum of patients with prostate cancer: a predictor of biochemical recurrence after prostatectomy. BJU Int 2008; 102: 628–632. [DOI] [PubMed] [Google Scholar]
- 15. Nakahira K, Kyung SY, Rogers AJ, et al Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med 2013; 10: e1001577; discussion e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Escames G, Lopez LC, Garcia JA, et al Mitochondrial DNA and inflammatory diseases. Hum Genet 2012; 131: 161–173. [DOI] [PubMed] [Google Scholar]
- 17. Garcia‐Ramirez M, Francisco G, Garcia‐Arumi E, et al Mitochondrial DNA oxidation and manganese superoxide dismutase activity in peripheral blood mononuclear cells from type 2 diabetic patients. Diabetes Metab 2008; 34: 117–124. [DOI] [PubMed] [Google Scholar]
- 18. Schrauwen P, Schrauwen‐Hinderling V, Hoeks J, et al Mitochondrial dysfunction and lipotoxicity. Biochim Biophys Acta 2010; 1801: 266–271. [DOI] [PubMed] [Google Scholar]
- 19. Gao CL, Zhu C, Zhao YP, et al Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3‐L1 adipocytes. Mol Cell Endocrinol 2010; 320: 25–33. [DOI] [PubMed] [Google Scholar]
- 20. Liu J, Chen Z, Zhang Y, et al Rhein protects pancreatic beta‐cells from dynamin‐related protein‐1‐mediated mitochondrial fission and cell apoptosis under hyperglycemia. Diabetes 2013; 62: 3927–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Green K, Brand MD, Murphy MP. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes 2004; 53(Suppl. 1): S110–S118. [DOI] [PubMed] [Google Scholar]
- 22. Baughman JM, Mootha VK. Buffering mitochondrial DNA variation. Nat Genet 2006; 38: 1232–1233. [DOI] [PubMed] [Google Scholar]
- 23. Jahr S, Hentze H, Englisch S, et al DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res 2001; 61: 1659–1665. [PubMed] [Google Scholar]
- 24. Jylhava J, Nevalainen T, Marttila S, et al Characterization of the role of distinct plasma cell‐free DNA species in age‐associated inflammation and frailty. Aging Cell 2013; 12: 388–397. [DOI] [PubMed] [Google Scholar]