

Abstract
In contrast to physiological pain, pathological pain is not dependent on the presence of tissue‐damaging stimuli. One type of pathological pain – neuropathic pain – is often a consequence of nerve injury or of diseases such as diabetes. Neuropathic pain can be agonizing, can persist over long periods and is often resistant to known painkillers. A growing body of evidence shows that many pathological processes within the central nervous system are mediated by complex interactions between neurons and glial cells. In the case of painful peripheral neuropathy, spinal microglia react and undergo a series of changes that directly influence the establishment of neuropathic pain states. After nerve damage, purinergic P2X4 receptors (non‐selective cation channels activated by extracellular adenosine triphosphate) are upregulated in spinal microglia in a manner that depends on the transcription factors interferon regulatory factor 8 and 5, both of which are expressed in microglia after peripheral nerve injury. P2X4 receptor expression on the cell surface of microglia is also regulated at the post‐translational level by signaling from CC chemokine receptor chemotactic cytokine receptor 2. Furthermore, spinal microglia in response to extracellular stimuli results in signal transduction through intracellular signaling cascades, such as mitogen‐activated protein kinases, p38 and extracellular signal‐regulated protein kinase. Importantly, inhibiting the function or expression of these microglial molecules suppresses the aberrant excitability of dorsal horn neurons and neuropathic pain. These findings show that spinal microglia are a central player in mechanisms for neuropathic pain, and might be a potential target for treating the chronic pain state.
Keywords: Microglia, Painful diabetic neuropathy, Purinergic receptors
Introduction
Neuropathic pain is a chronic pain condition that occurs after nerve damage, such as that induced by bone compression in cancer, infection, autoimmune disease, trauma and diabetes1. In addition to spontaneous pain and hyperalgesia (the increased pain perception of noxious stimuli), a troublesome symptom of neuropathic pain is pain hypersensitivity to normally innocuous stimuli, called tactile allodynia. A considerable proportion of diabetic patients describe abnormal hypersensitivity to normally innocuous stimuli2, 3. Allodynia is often resistant to the currently available drugs when administered at doses that do not produce significant side‐effects4, 5, 6. We are now beginning to understand that neuropathic pain is not just a symptom of disease, but is a consequence of disordered functioning of the nervous system7, 8. Unraveling the mechanisms of pain hypersensitivity caused by nerve damage is therefore essential for the development of new therapeutic drugs for neuropathic pain.
The spinal dorsal horn receives sensory information from primary afferent Aδ and C fibers after nociceptive stimuli (Figure 1)9, 10, 11. Aδ fibers have medium diameter myelinated afferents and mediate acute, localized sharp pain sensation. C fibers have small diameter unmyelinated afferents and convey the poorly localized delayed pain. The largest‐diameter, myelinated primary afferent Aβ fibers transmit innocuous mechanical information (i.e., light touch). The terminals of C and Aδ fibers are concentrated in the superficial dorsal horn, and these fibers activate projection neurons and excitatory interneurons (Figure 1). In contrast, the terminals of Aβ fibers are concentrated in the deeper dorsal horn, and mainly target excitatory and inhibitory interneurons (Figure 1) and projection neurons located in the deeper dorsal horn (not shown). Although Aβ fibers polysynaptically link to projection neurons in the superficial dorsal horn, the link is considered to be normally strongly repressed by inhibitory interneurons. Therefore, under normal conditions, Aβ fibers do not activate nociceptive projection neurons and do not cause pain. However, the neuronal networks in the dorsal horn are modulated and modified under pathological conditions, such as peripheral tissue inflammation and peripheral nerve injury (PNI)9, 11. Accumulating evidence from diverse animal models of neuropathic pain suggests that neuropathic pain might involve aberrant excitability in the dorsal horn, resulting from multiple functional alterations after PNI including loss of function of inhibitory interneurons (e.g., diminished activity of these cells or reduced effectiveness of the inhibitory neurotransmitters γ‐aminobutyric acid and glycine)6, 12. Furthermore, a growing body of evidence has shown that PNI‐induced hyperexcitability might not be a consequence merely of changes in neurons, but rather of multiple alterations in glial cells, such as microglia, the immune cells of the CNS13, 14, 15, 16, 17. In the present article, we highlight recent advances that further increase our understanding of the mechanisms underlying neuropathic pain caused by PNI and diabetes, with a specific focus on microglia in the spinal cord.
Figure 1.
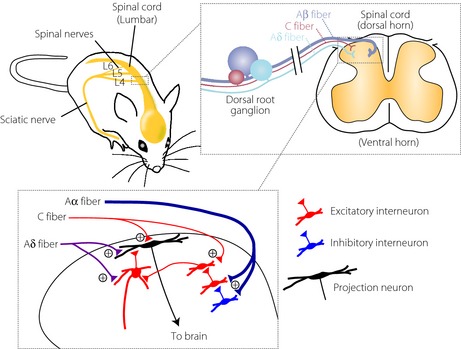
Schematic illustration of primary afferent sensory fibers and neuronal circuits in the dorsal horn. The dorsal root ganglion contains cell bodies of primary afferent neurons that transmit sensory information from the periphery to the spinal dorsal horn. Nociceptive information is mainly mediated by Aδ and C fibers, and innocuous mechanical information is mediated by Aβ fibers. C and Aδ fibers terminate in the superficial dorsal horn, and activate projection neurons and excitatory interneurons. The terminals of Aβ fibers are concentrated in the deeper dorsal horn, and connect to excitatory and inhibitory interneurons.
Microglia in the Spinal Cord in PNI and Diabetic Animals
Glial cells make up over 70% of the total cell population in the central nervous system (CNS), and are classified into astrocytes, oligodendrocytes and microglia. Microglial cells are known as resident macrophages in the CNS, which derive from primitive macrophages in the yolk sac18. In the adult, microglia are ubiquitously distributed throughout CNS and have small cell bodies bearing branched and motile processes, which might monitor the local environment in the CNS19, 20. Microglia rapidly respond to a wide range of stimuli that threaten physiological homeostasis, including PNI. In a growing body of literature, it is evident that PNI leads to a dramatic activation of microglia in the spinal dorsal horn (Figure 2). This response is commonly observed among various models of neuropathic pain including diabetic neuropathy. The morphological features of microglial activation are a cell body hypertrophy with thickened and retracted processes, an increase in cell number and an increase in the staining level of microglial markers, such as CD11b and ionized calcium‐binding adapter molecule‐1 (Figure 2). Using a model of painful diabetic neuropathy caused by streptozotocin (STZ), we found that microglia in the dorsal horn showed hypertrophic morphology, an increase in cell number and in expression of microglial markers. These changes were also consistently observed in other studies21, 22, 23, 24, 25, 26, and also found in a model of type 2 diabetes, db/db mice27. An interesting observation in STZ‐induced diabetic rats was that microglia activation was more pronounced in the fourth lumbar (L4) segment of the dorsal horn than in the L5 or L6 segments. Similar segmental difference has been observed in the dorsal horn in response to injury to tibial and common peroneal nerves28, 29. Furthermore, activated microglia were observed mainly in the medial side of the dorsal horn, an area that has been shown to receive inputs from these nerves28, and also to be innervated by myelinated fibers29, 30. These lines of evidence lead to the possibility that microglia activation might be related to diabetes‐induced damage and/or aberrant activity of a subpopulation of primary afferent sensory neurons projecting to the L4 dorsal horn rather than to a direct action of glucose on microglia. L4 dorsal root ganglion neurons, whose central fibers project to the L4 dorsal horn, are the principal sensory neurons projecting to the hindpaw31. Because tactile allodynia is consistently observed in the hindpaw of diabetic models, activated spinal microglia could be a mechanistic link to diabetes‐induced tactile allodynia. Clinical evidence also indicates that tactile allodynia in patients with diabetes is observed mainly in distal regions of the body, such as feet and ankles32. Mechanisms underlying the spatial‐specific activation of microglia in the dorsal horn remain to be determined. However, the area in the spinal dorsal horn receiving damaged primary afferent sensory neurons matches that having markedly activated microglia, therefore, microglial activation requires a signal(s) related to ‘nerve injury.’ Indeed, blocking peripheral input from primary afferent fibers by bupivacaine inhibits microglial activation in the dorsal horn after PNI33. In STZ‐treated diabetic mice, Suzuki et al.23 showed that continuous systemic administration of lidocaine inhibited microglial activation in the dorsal horn (although they have proposed a direct action of lidocaine on microglia).
Figure 2.
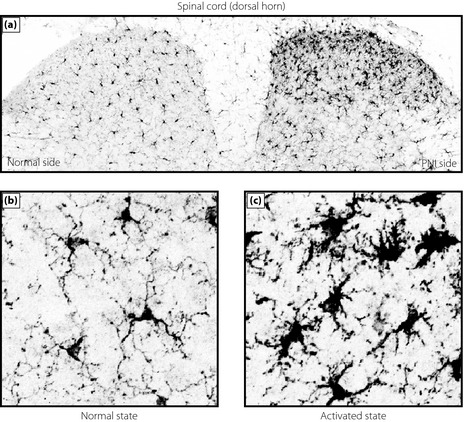
Activation of microglia in the dorsal horn of the spinal cord after peripheral nerve injury. (a) Immunofluorescence of the microglia marker ionized calcium‐binding adapter molecule‐1 in the spinal dorsal horn 7 days after nerve injury. High‐magnified images of (b) normal and (c) activated states of microglia in the contralateral and ipsilateral side, respectively, of the spinal dorsal horn.
A neuronally derived signaling molecule that might be important for microglial activation remains to be determined, but several candidates have been reported. Those include monocyte chemoattractant protein‐1 (MCP‐1 or CCL2) and metalloproteinase‐9 (MMP‐9), whose expressions are markedly increased in dorsal root ganglion neurons after PNI34, 35, 36, 37, 38. Mice lacking chemotactic cytokine receptor 2, which is a receptor for MCP‐1, or MMP‐9‐deficient mice showed a reduction of microglia activation caused by PNI38, 39. Conversely, intrathecal administration of MCP‐1 or MMP‐9 into normal rats produced microglial activation, as well as allodynia37, 38. Substrates of MMP‐9 for microglial activation are unclear, but fractalkine, interleukin‐1β (IL‐1β) and tumor necrosis factor‐α could be potential candidates17.
The role of CCL2 in neuropathic pain might also be associated with its ability to attract and activate monocytes. Zhang et al.39 have shown that bone marrow‐derived cells injected intravenously into lethally irradiated recipient mice were observed in the spinal cord parenchyma ipsilateral to the PNI in a manner that depended on the level of spinal CCL2 expression. Interestingly, bone marrow cells that had migrated into the dorsal horn underwent proliferation, expressed ionized calcium‐binding adapter molecule‐1 and showed microglia‐like morphology. However, the ability of bone marrow‐derived cells to migrate into the parenchyma of the CNS, including the spinal cord remains controversial, as a result of experimental manipulations, such as irradiation (which could influence the blood–spinal cord barrier) and exogenously injected donor cells40. Thus, the extent to which bone marrow‐derived cells contribute to spinal microglia activation after PNI remains unresolved.
It was found that spinal microglia express a receptor for the cytokine interferon‐γ (IFN‐γR), and that stimulating IFN‐γRs in naïve animals by means of intrathecal administration of IFN‐γ produces a hypertrophic morphology and an increase in the microglial number41. Furthermore, IFN‐γR‐knockout mice showed the reduction of morphological and numerical changes of microglia after PNI. Although the source of IFN‐γ remains to be identified, the IFN‐γ/IFN‐γR system is critical in transforming resident spinal microglia into the activated state. Whether IFN‐γ signaling also contributes to microglial activation in diabetic animals remains unknown.
The role of toll‐like receptors, a family of type I transmembrane signaling proteins that recognize pathogen‐associated molecular patterns, has also been reported in PNI models of neuropathic pain. Mice lacking either toll‐like receptor 2, 3 or 4 show impaired microglial activation in the dorsal horn after PNI42, 43, 44.
Spinal Microglia are Crucial for Neuropathic Pain
Activated microglia show dramatic changes in the expression of various genes, including cell‐surface receptors for neurotransmission (e.g., purinergic receptors) and intracellular signaling molecules (e.g., mitogen‐activated protein kinases [MAPKs]) and bioactive diffusible factors (e.g., pro‐inflammatory cytokines and neurotrophic factors).
Purinergic Receptors
In 1972, Burnstock45 proposed new roles of nucleotides as neurotransmitters, even though it was primarily recognized that intracellular adenosine triphosphate (ATP) is the source of free energy to maintain life, and nucleotides are key molecules within cells. In 1993, the first receptors for nucleotides, called P2 purinoceptors, were cloned46, 47. Afterwards, numerous subtypes of these receptors were also cloned, and subsequently scientists began to gradually accept the ‘purinergic system.’ Now purinergic P2 receptors are divided into two families, ionotropic receptors (P2X) and metabotropic receptors (P2Y). P2X receptors (of which there are seven types, P2X1–P2X7) contain intrinsic pores that open on binding of extracellular ATP48. P2Y receptors (of which there are eight types, P2Y1, 2, 4, 6, 11, 12, 13 and 14) are coupled to heteromeric G‐proteins49. Nucleotides are considered to be released or leaked from glial cells as well as neurons, and thus purinergic signaling plays an important role in cell‐to‐cell communications under physiological and pathophysiological conditions50.
It was found that expression of P2X4Rs was upregulated exclusively in microglia after PNI, and that PNI‐induced tactile allodynia was reversed by pharmacological blockade of P2X4Rs in the spinal cord51. It was shown that PNI‐induced pain hypersensitivity depends on ongoing purinergic signaling through microglial P2X4Rs. A marked reduction in neuropathic pain in both mice knocked down and knocked out of P2X4R further demonstrated the necessity of P2X4Rs51, 52, 53. Intrathecal delivery of P2X4R‐stimulated microglia caused normal rats to produce allodynia, indicating the sufficiency of P2X4R14, 51. These findings on the necessity and sufficiency of microglial P2X4Rs provided the first definitive evidence for a causal role of spinal microglia in neuropathic pain. This was supported by the findings in studies using minocycline, a tetracycline antibiotic that has an inhibitory effect on microglial activation. These studies consistently showed that minocycline attenuates both microglial activation and mechanical allodynia in models of neuropathic pain caused by PNI and diabetes24, 54, 55, 56.
The mechanisms by which microglia are crucial for producing neuropathic pain must involve signaling from activated microglia to dorsal horn neurons. It was shown that activation of microglial P2X4Rs stimulates the synthesis and release of brain‐derived neurotrophic factor (BDNF)52, 57, and that BDNF then causes an alteration of transmembrane anion gradient in a subpopulation of dorsal horn lamina I neurons presumably through the downregulation of the neuronal chloride transporter, KCC2, which in turn renders γ‐aminobutyric acid and glycine effects depolarizing, rather than hyperpolarizing, in these neurons (Figure 3). Furthermore, Keller et al.58 have shown that local spinal administration of ATP‐stimulated microglia changes the phenotype of in vivo spinal lamina I output neurons, such that they relay innocuous mechanical input, which could account for allodynia. Thus, P2X4R‐stimulated microglia release BDNF as a crucial factor to signal to lamina I neurons, causing aberrant nociceptive output that contributes to neuropathic pain (Figure 3)8. Interestingly, a decrease in KCC2 expression in the spinal cord has also been reported in STZ‐induced diabetic rats24. The decrease was suppressed by treatment with minocycline. Furthermore, blocking the BDNF action in STZ‐injected rats by tropomyosin‐related kinase B/fragment, crystallizable domain was found to induce moderate effects on mechanical hyperalgesia, although BDNF levels were not increased in STZ‐diabetic rats24. Although P2X4R expression in spinal microglia under diabetic conditions remains unknown, microglial regulation of KCC2 could be a mechanism of diabetes‐induced neuropathic pain.
Figure 3.
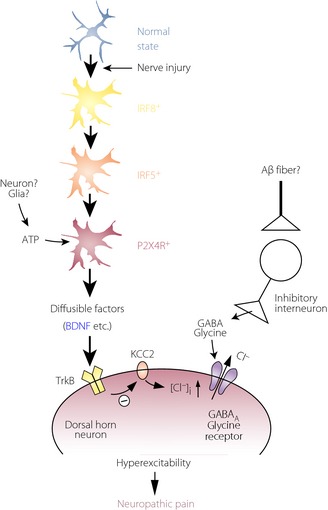
Schematic illustration for shifting spinal microglia toward a P2X4 receptor (P2XR4)‐expressing reactive state through an interferon regulatory factor 8 (IRF8)–IRF5 transcriptional axis after nerve injury, and a potential mechanism by which P2X4+ microglia cause hyperexcitability in dorsal horn neurons and neuropathic pain. After nerve injury, activated microglia show increased expression of IRF8, which in turn leads to induction of IRF5 expression. IRF5 then induces P2X4R expression by directly binding to the promoter region of the P2rx4 gene. P2X4R is activated by extracellular adenosine triphosphate (ATP; which could be presumably released from neurons or glial cells) and, in turn, release bioactive diffusible factors, such as brain‐derived neurotrophic factor (BDNF). BDNF downregulates the potassium‐chloride transporter, KCC2, through tropomyosin‐related kinase B, causes an increase in intracellular [Cl−], and leads to the collapse of the transmembrane anion gradient in dorsal horn neurons, which in turn induces depolarization of these neurons after stimulation by γ‐aminobutyric acid and glycine (which might be released in response to stimulation of Aβ fiber). The resultant hyperexcitability in the dorsal horn pain network induced by microglial factors could be responsible for neuropathic pain. GABA, γ‐aminobutyric acid.
The upregulation of P2X4R expression in microglia is therefore a key process in neuropathic pain. Several studies have identified molecules that upregulate P2X4R expression in microglia59, 60, 61. We have recently identified interferon regulatory factor‐8 (IRF8) as a crucial transcription factor for P2X4R upregulation62. IRF8 is a member of the IRF family (IRF1–9), and is expressed in immune cells, such as lymphocytes and dendritic cells63. The role of IRFs in the CNS was entirely unknown, but Masuda et al.62 recently showed that within the spinal cord, IRF8 expression is selectively upregulated in microglia after PNI. It was also found that IRF8‐deficient mice show a reduction of PNI‐induced tactile allodynia. Furthermore, suppressing upregulated expression of spinal IRF8 caused a significant recovery of the PNI‐induced allodynia. These results show that microglial IRF8 is necessary for the development and maintenance of tactile allodynia after PNI. In in vitro studies, it was shown that IRF8 promotes the expression of genes associated with reactive states of microglia including P2X4R. Furthermore, the PNI‐induced P2X4R upregulation was prevented in IRF8‐deficient mice. More recently, we identified IRF5 as a target of IRF864. IRF5 expression was also induced selectively in spinal microglia after PNI. IRF5‐deficient mice showed substantial resistance to pain hypersensitivity. Interestingly, IRF5 expression in activated microglia led to an induction of P2X4R expression by directly binding to the promoter region of the P2rx4 gene. Consistent with this, IRF5‐deficient mice did not upregulate spinal P2X4R after PNI. Thus, an IRF8–IRF5 transcriptional axis could contribute to shifting spinal microglia toward a P2X4R‐expressing reactive state after PNI (Figure 3)65, 66.
Other transcription factors regulating the states of microglia have also been reported. For example, the E26‐transformation‐specific family transcription factor and master regulator of myeloid development, PU.1, is expressed in the microglia of adult CNS tissue67. PU.1 has been shown to control function and phenotype of human brain microglia68. Furthermore, expression of spinal cord PU.1 is markedly increased after PNI69. Likewise, Runt‐related transcription factor 1 has been reported to regulate the activity state of microglia70, and its expression in the spinal microglia is also upregulated after PNI70. These microglial transcription factors might play a part in altering microglial phenotype and modulating pain signaling. However, their roles in the pathogenesis of neuropathic pain remain unknown and thus require further investigation, which could provide new insights into the molecular mechanisms underlying microglial activation and neuropathic pain.
To detect extracellular ATP, P2X4Rs are necessary to be expressed on the cell surface of microglia. Surprisingly, a large amount of P2X4R protein within microglia (and macrophages) localizes predominantly to intracellular lysosomal compartments71, and P2X4R protein remains stable within the proteolytic environment of lysosomes. How P2X4R protein is recruited to the cell surface of microglia remains elusive, but recent studies have shown that trafficking of P2X4R protein to the cell surface occurs when microglia are stimulated by a toll‐like receptor 4 agonist, lipopolysaccharide72, 73, or the Ca2+ ionophore, ionomycin71. Furthermore, the chemokine CCL2 increased P2X4R protein levels on the cell surface (without changing total cellular expression) through chemotactic cytokine receptor 274. Notably, CCL2 changed the distribution of lysosomes with P2X4R protein within microglial cells and induced the release of a lysosomal enzyme74. Thus, CCL2 might promote the expression of P2X4R protein on the cell surface of microglia through exocytosis of P2X4R‐containing lysosomes. A recent study using single‐molecule imaging to track P2X4Rs in the processes of microglia showed that lateral mobility of P2X4Rs is enhanced in activated microglia by the p38, a member of the MAPKs, pathway that selectively regulates slowly mobile P2X4Rs75. These results showed that microglial P2X4Rs are dynamically regulated on the cell surface. Thus, post‐translational regulation to enhance P2X4R expression and mobility on the cell surfaces of microglia might render cells hyperresponsive to extracellular ATP, which might be important in neuropathic pain.
MAPKs
In response to activation of cell‐surface receptors on spinal microglia by extracellular ligands, a variety of cellular responses occur through activation of intracellular signaling cascades. p38 was activated in spinal microglia after PNI, and contributes to neuropathic pain76, 77. p38 activation in spinal microglia has been shown in different animal models of neuropathic pain78, 79, 80, 81, including diabetic models23, 26. Furthermore, pharmacological inhibition of p38 activity by repeated injection or by infusion of p38 inhibitors attenuated tactile allodynia caused by PNI76, 77, 82, 83 and by diabetes21, 84. Thus, activation of this kinase is necessary for the pathogenesis of neuropathic pain. It remains unclear how nerve damage activates p38 in spinal microglia. A blockade of the sciatic nerve using bupivacaine before PNI prevented p38 activation in spinal microglia85, raising the possibility that activity in the peripheral nerve is required for microglial p38 activation. It was found that cathepsin S rapidly induces p38 phosphorylation in spinal microglia after intrathecal injection, an effect that is dependent on fractalkine and its receptor signaling86, 87. In addition, the purinergic P2Y12R might also be involved in p38 activation in spinal microglia88.
In contrast, the other member of MAPKs extracellular signal‐regulated protein kinase (ERK) in the spinal cord was found in dorsal horn neurons soon after nerve damage, then predominantly in microglia for the next several days and in astrocytes after 3 weeks89. In contrast, the sequential activation of spinal ERK was not observed in the spinal cord of a model of diabetic neuropathic pain90, in which ERK activation consistently occurred in microglia at least for 4 weeks. Furthermore, inhibition of ERK phosphorylation in the dorsal horn produced a striking alleviation of existing, long‐term tactile allodynia of diabetic rats. Thus, activated microglia could be a crucial component of PNI‐ and diabetes‐induced tactile allodynia, mediated, in part, by the ERK signaling pathway. However, Xu et al.27 have shown in db/db mice that ERK activation occurs in neurons and astrocytes in the spinal cord, and that inhibition of ERK also attenuated mechanical allodynia. Thus, the type of cells that ERK is activated in might be different among models of diabetes.
Potential candidates for intermediary molecules involved in MAPK‐dependent pain modulation are thought to be pro‐inflammatory cytokines, such as IL‐1β, IL‐6 and tumor necrosis factor‐α13, 14, 15, 91, 92. Inhibiting spinal p38 suppresses IL‐1β upregulation in the spinal cord of PNI animals93. C‐Fiber‐evoked responses, N‐methyl‐D‐aspartate receptor‐mediated responses, and wind‐up in wide‐dynamic‐range dorsal horn neurons are enhanced by IL‐1β94, 95. IL‐1β has also been reported to decrease γ‐aminobutyric acid A receptor‐mediated currents96. A powerful role of these cytokines in excitatory or inhibitory synaptic transmission and on neuronal activity in the superficial dorsal horn neurons has been shown97, 98. Also, the inhibition of microglial ERK decreases the hyperresponsiveness of dorsal horn neurons in spinal cord‐injured rats through a reduction of prostaglandin E2 production99.
Conclusion
We have primarily focused on the role of microglia in neuropathic pain caused by PNI and diabetes. A model of mechanisms underlying microglia‐mediated neuropathic pain modulation in the dorsal horn is presented in Figure 3. Importantly, pharmacological, molecular and genetic manipulations of the function or expression of these microglial molecules have substantially influenced pain behaviors and hyperexcitability of the dorsal horn pain pathway. Therefore, spinal microglia critically contribute to pathologically enhanced pain processing in the dorsal horn, and microglial molecules might be promising targets for treating neuropathic pain. In addition to microglia, recent studies have also identified astrocyte‐specific molecules, and have shown a critical role of spinal astrocytes in neuropathic pain27, 38, 100, 101, 102, 103. Interestingly, it was found that oral administration of gabapentin attenuated activation of microglia and astrocytes in STZ‐induced diabetic rats21, raising the possibility that gabapentin could exert its anti‐allodynic actions partially through alterations of activation of glial cells in the spinal cord. It is expected that increased understanding of the functions of microglial molecules will provide us with exciting insights into pain mechanisms and clues to develop new therapeutic agents for the management of neuropathic pain.
Disclosure
The author declares no conflict of interest.
J Diabetes Investig 2016; 7: 17–26
References
- 1. Baron R. Mechanisms of disease: neuropathic pain–a clinical perspective. Nat Clin Pract Neurol 2006; 2: 95–106. [DOI] [PubMed] [Google Scholar]
- 2. Bastyr EJ 3rd, Price KL, Bril V. Development and validity testing of the neuropathy total symptom score‐6: questionnaire for the study of sensory symptoms of diabetic peripheral neuropathy. Clin Ther 2005; 27: 1278–1294. [DOI] [PubMed] [Google Scholar]
- 3. Vinik AI, Suwanwalaikorn S, Stansberry KB, et al Quantitative measurement of cutaneous perception in diabetic neuropathy. Muscle Nerve 1995; 18: 574–584. [DOI] [PubMed] [Google Scholar]
- 4. Calcutt NA. Experimental models of painful diabetic neuropathy. J Neurol Sci 2004; 220: 137–139. [DOI] [PubMed] [Google Scholar]
- 5. Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 1999; 353: 1959–1964. [DOI] [PubMed] [Google Scholar]
- 6. Scholz J, Woolf CJ. Can we conquer pain? Nat Neurosci 2002; 5: 1062–1067. [DOI] [PubMed] [Google Scholar]
- 7. Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci 2009; 32: 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beggs S, Trang T, Salter MW. P2X4R+ microglia drive neuropathic pain. Nat Neurosci 2012; 15: 1068–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Braz J, Solorzano C, Wang X, et al Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron 2014; 82: 522–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci 2010; 11: 823–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Prescott SA, Ma Q, De Koninck Y. Normal and abnormal coding of somatosensory stimuli causing pain. Nat Neurosci 2014; 17: 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science 2000; 288: 1765–1769. [DOI] [PubMed] [Google Scholar]
- 13. Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci 2001; 24: 450–455. [DOI] [PubMed] [Google Scholar]
- 14. Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci 2005; 28: 101–107. [DOI] [PubMed] [Google Scholar]
- 15. Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci 2005; 6: 521–532. [DOI] [PubMed] [Google Scholar]
- 16. Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci 2007; 10: 1361–1368. [DOI] [PubMed] [Google Scholar]
- 17. Suter MR, Wen YR, Decosterd I, et al Do glial cells control pain? Neuron Glia Biol 2007; 3: 255–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ginhoux F, Greter M, Leboeuf M, et al Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010; 330: 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davalos D, Grutzendler J, Yang G, et al ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 2005; 8: 752–758. [DOI] [PubMed] [Google Scholar]
- 20. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005; 308: 1314–1318. [DOI] [PubMed] [Google Scholar]
- 21. Wodarski R, Clark AK, Grist J, et al Gabapentin reverses microglial activation in the spinal cord of streptozotocin‐induced diabetic rats. Eur J Pain 2009; 13: 807–811. [DOI] [PubMed] [Google Scholar]
- 22. Toth CC, Jedrzejewski NM, Ellis CL, et al Cannabinoid‐mediated modulation of neuropathic pain and microglial accumulation in a model of murine type I diabetic peripheral neuropathic pain. Mol Pain 2010; 6: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suzuki N, Hasegawa‐Moriyama M, Takahashi Y, et al Lidocaine attenuates the development of diabetic‐induced tactile allodynia by inhibiting microglial activation. Anesth Analg 2011; 113: 941–946. [DOI] [PubMed] [Google Scholar]
- 24. Morgado C, Pereira‐Terra P, Cruz CD, et al Minocycline completely reverses mechanical hyperalgesia in diabetic rats through microglia‐induced changes in the expression of the potassium chloride co‐transporter 2 (KCC2) at the spinal cord. Diabetes Obes Metab 2011; 13: 150–159. [DOI] [PubMed] [Google Scholar]
- 25. Zychowska M, Rojewska E, Kreiner G, et al Minocycline influences the anti‐inflammatory interleukins and enhances the effectiveness of morphine under mice diabetic neuropathy. J Neuroimmunol 2013; 262: 35–45. [DOI] [PubMed] [Google Scholar]
- 26. Cheng KI, Wang HC, Chuang YT, et al Persistent mechanical allodynia positively correlates with an increase in activated microglia and increased P‐p38 mitogen‐activated protein kinase activation in streptozotocin‐induced diabetic rats. Eur J Pain 2014; 18: 162–173. [DOI] [PubMed] [Google Scholar]
- 27. Xu X, Chen H, Ling BY, et al Extracellular signal‐regulated protein kinase activation in spinal cord contributes to pain hypersensitivity in a mouse model of type 2 diabetes. Neurosci Bull 2014; 30: 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Molander C, Grant G. Laminar distribution and somatotopic organization of primary afferent fibers from hindlimb nerves in the dorsal horn. A study by transganglionic transport of horseradish peroxidase in the rat. Neuroscience 1986; 19: 297–312. [DOI] [PubMed] [Google Scholar]
- 29. Beggs S, Salter MW. Neuropathic pain: symptoms, models, and mechanisms. Drug Dev Res 2006; 67: 289–301. [Google Scholar]
- 30. Robertson B, Grant G. Immunocytochemical evidence for the localization of the GM1 ganglioside in carbonic anhydrase‐containing and RT 97‐immunoreactive rat primary sensory neurons. J Neurocytol 1989; 18: 77–86. [DOI] [PubMed] [Google Scholar]
- 31. Thornton PD, Gerke MB, Plenderleith MB. Histochemical localisation of a galactose‐containing glycoconjugate expressed by sensory neurones innervating different peripheral tissues in the rat. J Peripher Nerv Syst 2005; 10: 47–57. [DOI] [PubMed] [Google Scholar]
- 32. Vinik A. CLINICAL REVIEW: use of antiepileptic drugs in the treatment of chronic painful diabetic neuropathy. J Clin Endocrinol Metab 2005; 90: 4936–4945. [DOI] [PubMed] [Google Scholar]
- 33. Suter MR, Berta T, Gao YJ, et al Large A‐fiber activity is required for microglial proliferation and p38 MAPK activation in the spinal cord: different effects of resiniferatoxin and bupivacaine on spinal microglial changes after spared nerve injury. Mol Pain 2009; 5: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tanaka T, Minami M, Nakagawa T, et al Enhanced production of monocyte chemoattractant protein‐1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci Res 2004; 48: 463–469. [DOI] [PubMed] [Google Scholar]
- 35. White FA, Jung H, Miller RJ. Chemokines and the pathophysiology of neuropathic pain. Proc Natl Acad Sci USA 2007; 104: 20151–20158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang J, De Koninck Y. Spatial and temporal relationship between monocyte chemoattractant protein‐1 expression and spinal glial activation following peripheral nerve injury. J Neurochem 2006; 97: 772–783. [DOI] [PubMed] [Google Scholar]
- 37. Thacker MA, Clark AK, Bishop T, et al CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur J Pain 2009; 13: 263–272. [DOI] [PubMed] [Google Scholar]
- 38. Kawasaki Y, Xu ZZ, Wang X, et al Distinct roles of matrix metalloproteases in the early‐ and late‐phase development of neuropathic pain. Nat Med 2008; 14: 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang J, Shi XQ, Echeverry S, et al Expression of CCR2 in both resident and bone marrow‐derived microglia plays a critical role in neuropathic pain. J Neurosci 2007; 27: 12396–12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ajami B, Bennett JL, Krieger C, et al Local self‐renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 2007; 10: 1538–1543. [DOI] [PubMed] [Google Scholar]
- 41. Tsuda M, Masuda T, Kitano J, et al IFN‐gamma receptor signaling mediates spinal microglia activation driving neuropathic pain. Proc Natl Acad Sci USA 2009; 106: 8032–8037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tanga FY, Nutile‐McMenemy N, DeLeo JA. The CNS role of Toll‐like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci USA 2005; 102: 5856–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim D, Kim MA, Cho IH, et al A critical role of toll‐like receptor 2 in nerve injury‐induced spinal cord glial cell activation and pain hypersensitivity. J Biol Chem 2007; 282: 14975–14983. [DOI] [PubMed] [Google Scholar]
- 44. Obata K, Katsura H, Miyoshi K, et al Toll‐like receptor 3 contributes to spinal glial activation and tactile allodynia after nerve injury. J Neurochem 2008; 105: 2249–2259. [DOI] [PubMed] [Google Scholar]
- 45. Burnstock G. Purinergic nerves. Pharmacol Rev 1972; 24: 509–581. [PubMed] [Google Scholar]
- 46. Lustig KD, Shiau AK, Brake AJ, et al Expression cloning of an ATP receptor from mouse neuroblastoma cells. Proc Natl Acad Sci USA 1993; 90: 5113–5117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Webb TE, Simon J, Krishek BJ, et al Cloning and functional expression of a brain G‐protein‐coupled ATP receptor. FEBS Lett 1993; 324: 219–225. [DOI] [PubMed] [Google Scholar]
- 48. Khakh BS, North RA. P2X receptors as cell‐surface ATP sensors in health and disease. Nature 2006; 442: 527–532. [DOI] [PubMed] [Google Scholar]
- 49. Abbracchio MP, Burnstock G, Boeynaems JM, et al International Union of Pharmacology LVIII: update on the P2Y G protein‐coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev 2006; 58: 281–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Burnstock G. Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov 2008; 7: 575–590. [DOI] [PubMed] [Google Scholar]
- 51. Tsuda M, Shigemoto‐Mogami Y, Koizumi S, et al P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 2003; 424: 778–783. [DOI] [PubMed] [Google Scholar]
- 52. Ulmann L, Hatcher JP, Hughes JP, et al Up‐regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci 2008; 28: 11263–11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tsuda M, Kuboyama K, Inoue T, et al Behavioral phenotypes of mice lacking purinergic P2X4 receptors in acute and chronic pain assays. Mol Pain 2009; 5: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther 2003; 306: 624–630. [DOI] [PubMed] [Google Scholar]
- 55. Narita M, Yoshida T, Nakajima M, et al Direct evidence for spinal cord microglia in the development of a neuropathic pain‐like state in mice. J Neurochem 2006; 97: 1337–1348. [DOI] [PubMed] [Google Scholar]
- 56. Pabreja K, Dua K, Sharma S, et al Minocycline attenuates the development of diabetic neuropathic pain: possible anti‐inflammatory and anti‐oxidant mechanisms. Eur J Pharmacol 2011; 661: 15–21. [DOI] [PubMed] [Google Scholar]
- 57. Trang T, Beggs S, Wan X, et al P2X4‐receptor‐mediated synthesis and release of brain‐derived neurotrophic factor in microglia is dependent on calcium and p38‐mitogen‐activated protein kinase activation. J Neurosci 2009; 29: 3518–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Keller AF, Beggs S, Salter MW, et al Transformation of the output of spinal lamina I neurons after nerve injury and microglia stimulation underlying neuropathic pain. Mol Pain 2007; 3: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tsuda M, Toyomitsu E, Komatsu T, et al Fibronectin/integrin system is involved in P2X(4) receptor upregulation in the spinal cord and neuropathic pain after nerve injury. Glia 2008; 56: 579–585. [DOI] [PubMed] [Google Scholar]
- 60. Tsuda M, Tozaki‐Saitoh H, Masuda T, et al Lyn tyrosine kinase is required for P2X(4) receptor upregulation and neuropathic pain after peripheral nerve injury. Glia 2008; 56: 50–58. [DOI] [PubMed] [Google Scholar]
- 61. Tsuda M, Toyomitsu E, Kometani M, et al Mechanisms underlying fibronectin‐induced upregulation of P2XR expression in microglia: distinct roles of PI3K‐Akt and MEK‐ERK signaling pathways. J Cell Mol Med 2009; 13: 3251–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Masuda T, Tsuda M, Yoshinaga R, et al IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Rep 2012; 1: 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tamura T, Yanai H, Savitsky D, et al The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol 2008; 26: 535–584. [DOI] [PubMed] [Google Scholar]
- 64. Masuda T, Iwamoto S, Yoshinaga R, et al Transcription factor IRF5 drives P2X4R+‐reactive microglia gating neuropathic pain. Nat Commun 2014; 5: 3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tsuda M, Masuda T, Tozaki‐Saitoh H, et al Microglial regulation of neuropathic pain. J Pharmacol Sci 2013; 121: 89–94. [DOI] [PubMed] [Google Scholar]
- 66. Tsuda M, Masuda T, Tozaki‐Saitoh H, et al P2X4 receptors and neuropathic pain. Front Cell Neurosci 2013; 7: 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Horiuchi M, Wakayama K, Itoh A, et al Interferon regulatory factor 8/interferon consensus sequence binding protein is a critical transcription factor for the physiological phenotype of microglia. J Neuroinflammation 2012; 9: 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Smith AM, Gibbons HM, Oldfield RL, et al The transcription factor PU.1 is critical for viability and function of human brain microglia. Glia 2013; 61: 929–942. [DOI] [PubMed] [Google Scholar]
- 69. Imai S, Ikegami D, Yamashita A, et al Epigenetic transcriptional activation of monocyte chemotactic protein 3 contributes to long‐lasting neuropathic pain. Brain 2013; 136: 828–843. [DOI] [PubMed] [Google Scholar]
- 70. Zusso M, Methot L, Lo R, et al Regulation of postnatal forebrain amoeboid microglial cell proliferation and development by the transcription factor Runx1. J Neurosci 2012; 32: 11285–11298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Qureshi OS, Paramasivam A, Yu JC, et al Regulation of P2X4 receptors by lysosomal targeting, glycan protection and exocytosis. J Cell Sci 2007; 120: 3838–3849. [DOI] [PubMed] [Google Scholar]
- 72. Boumechache M, Masin M, Edwardson JM, et al Analysis of assembly and trafficking of native P2X4 and P2X7 receptor complexes in rodent immune cells. J Biol Chem 2009; 284: 13446–13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Toulme E, Garcia A, Samways D, et al P2X4 receptors in activated C8‐B4 cells of cerebellar microglial origin. J Gen Physiol 2010; 135: 333–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Toyomitsu E, Tsuda M, Yamashita T, et al CCL2 promotes P2X4 receptor trafficking to the cell surface of microglia. Purinergic Signal 2012; 8: 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Toulme E, Khakh BS. Imaging P2X4 receptor lateral mobility in microglia regulation by calcium and p38 MAPK. J Biol Chem 2012; 287: 14734–14748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jin SX, Zhuang ZY, Woolf CJ, et al p38 mitogen‐activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci 2003; 23: 4017–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Tsuda M, Mizokoshi A, Shigemoto‐Mogami Y, et al Activation of p38 mitogen‐activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia 2004; 45: 89–95. [DOI] [PubMed] [Google Scholar]
- 78. Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci 2006; 26: 4308–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Piao ZG, Cho IH, Park CK, et al Activation of glia and microglial p38 MAPK in medullary dorsal horn contributes to tactile hypersensitivity following trigeminal sensory nerve injury. Pain 2006; 121: 219–231. [DOI] [PubMed] [Google Scholar]
- 80. Ito T, Ohtori S, Inoue G, et al Glial phosphorylated p38 MAP kinase mediates pain in a rat model of lumbar disc herniation and induces motor dysfunction in a rat model of lumbar spinal canal stenosis. Spine 2007; 32: 159–167. [DOI] [PubMed] [Google Scholar]
- 81. Xu JT, Xin WJ, Wei XH, et al p38 activation in uninjured primary afferent neurons and in spinal microglia contributes to the development of neuropathic pain induced by selective motor fiber injury. Exp Neurol 2007; 204: 355–365. [DOI] [PubMed] [Google Scholar]
- 82. Schafers M, Svensson CI, Sommer C, et al Tumor necrosis factor‐alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci 2003; 23: 2517–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Terayama R, Omura S, Fujisawa N, et al Activation of microglia and p38 mitogen‐activated protein kinase in the dorsal column nucleus contributes to tactile allodynia following peripheral nerve injury. Neuroscience 2008; 153: 1245–1255. [DOI] [PubMed] [Google Scholar]
- 84. Daulhac L, Mallet C, Courteix C, et al Diabetes‐induced mechanical hyperalgesia involves spinal mitogen‐activated protein kinase activation in neurons and microglia via N‐methyl‐D‐aspartate‐dependent mechanisms. Mol Pharmacol 2006; 70: 1246–1254. [DOI] [PubMed] [Google Scholar]
- 85. Wen YR, Suter MR, Kawasaki Y, et al Nerve conduction blockade in the sciatic nerve prevents but does not reverse the activation of p38 mitogen‐activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology 2007; 107: 312–321. [DOI] [PubMed] [Google Scholar]
- 86. Clark AK, Yip PK, Grist J, et al Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci USA 2007; 104: 10655–10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Clark AK, Yip PK, Malcangio M. The liberation of fractalkine in the dorsal horn requires microglial cathepsin S. J Neurosci 2009; 29: 6945–6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kobayashi K, Yamanaka H, Fukuoka T, et al P2Y12 receptor upregulation in activated microglia is a gateway of p38 signaling and neuropathic pain. J Neurosci 2008; 28: 2892–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhuang ZY, Gerner P, Woolf CJ, et al ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain 2005; 114: 149–159. [DOI] [PubMed] [Google Scholar]
- 90. Tsuda M, Ueno H, Kataoka A, et al Activation of dorsal horn microglia contributes to diabetes‐induced tactile allodynia via extracellular signal‐regulated protein kinase signaling. Glia 2008; 56: 378–386. [DOI] [PubMed] [Google Scholar]
- 91. DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain 2001; 90: 1–6. [DOI] [PubMed] [Google Scholar]
- 92. Inoue K. The function of microglia through purinergic receptors: neuropathic pain and cytokine release. Pharmacol Ther 2006; 109: 210–226. [DOI] [PubMed] [Google Scholar]
- 93. Ji RR, Suter MR. p38 MAPK, microglial signaling, and neuropathic pain. Mol Pain 2007; 3: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Reeve AJ, Patel S, Fox A, et al Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur J Pain 2000; 4: 247–257. [DOI] [PubMed] [Google Scholar]
- 95. Viviani B, Bartesaghi S, Gardoni F, et al Interleukin‐1beta enhances NMDA receptor‐mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci 2003; 23: 8692–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wang S, Cheng Q, Malik S, et al Interleukin‐1beta inhibits gamma‐aminobutyric acid type A (GABA(A)) receptor current in cultured hippocampal neurons. J Pharmacol Exp Ther 2000; 292: 497–504. [PubMed] [Google Scholar]
- 97. Ikeda H, Tsuda M, Inoue K, et al Long‐term potentiation of neuronal excitation by neuron‐glia interactions in the rat spinal dorsal horn. Eur J Neurosci 2007; 25: 1297–1306. [DOI] [PubMed] [Google Scholar]
- 98. Kawasaki Y, Zhang L, Cheng JK, et al Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin‐1beta, interleukin‐6, and tumor necrosis factor‐alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci 2008; 28: 5189–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhao P, Waxman SG, Hains BC. Extracellular signal‐regulated kinase‐regulated microglia‐neuron signaling by prostaglandin E2 contributes to pain after spinal cord injury. J Neurosci 2007; 27: 2357–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ji RR, Kawasaki Y, Zhuang ZY, et al Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron Glia Biol 2006; 2: 259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhuang ZY, Wen YR, Zhang DR, et al A peptide c‐Jun N‐terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci 2006; 26: 3551–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Katsura H, Obata K, Miyoshi K, et al Transforming growth factor‐activated kinase 1 induced in spinal astrocytes contributes to mechanical hypersensitivity after nerve injury. Glia 2008; 56: 723–733. [DOI] [PubMed] [Google Scholar]
- 103. Tsuda M, Kohro Y, Yano T, et al JAK‐STAT3 pathway regulates spinal astrocyte proliferation and neuropathic pain maintenance in rats. Brain 2011; 134: 1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]