

Abstract
Background
Non‐small cell lung cancer (NSCLC) harboring kinase‐domain mutations in epidermal growth factor receptors (EGFR) has been observed to be sensitive to ionizing radiation (IR). We explore Rad51‐dependent homologous recombination (HR) DNA repair in regulating radiosensitivity in two NSCLC cell lines with different EGFR mutation status.
Methods
NSCLC cell lines, wild‐type EGFR A549 and mutant EGFR H820 with an in‐frame deletion in exon 19 of EGFR (ΔE746–E750), were cultured. Radiosensitivity was estimated by colony forming assay. Rad51 expression was evaluated by quantitative real time‐polymerase chain reaction and Western‐blot. Lentiviral small hairpin ribonucleic acid‐Rad51 and ΔE746–E750 deletion mutant EGFR were constructed and transfected into cells. Flowcytometry assay was used to analyze DNA double strand breaks, cell cycle alterations, and apoptosis.
Results
A549 had a higher survival factor (SF)2 (0.66 vs. 0.44) and lower α/β value (4.07 vs. 9.01). Compared with the A549 cell, the H820 cell exhibited defective arrest in the S‐phase, a higher rate of G2/M accumulation, early apoptosis, and residual γ‐H2AX. Downregulated Rad51 expression decreased SF2 (0.42 vs. 0.31) and increased the α/β ratio (7.51 vs. 10.5), G2/M accumulation, early apoptosis, and γ‐H2AX in two cell lines. H820 had a low IR‐induced Rad51 expression and nuclear translocation. Exogenous expression of the ΔE746–E750 deletion mutant EGFR caused the A549 cell to become more radiosensitive.
Conclusions
An EGFR mutated NSCLC cell line is sensitive to IR , which is correlated with reduced IR‐induced Rad51 expression and nuclear translocation. The signaling pathway of EGFR maintaining Rad51 protein levels maybe a novel lung cancer therapeutic target to overcome radioresistance.
Keywords: Epidermal growth factor receptor, mutation, non‐small cell lung cancer, Rad51, radiosensitivity
Background
Lung cancer is the most common malignancy and leading cause of mortality worldwide.1, 2 Conventional treatments among advanced non‐small cell lung cancer (NSCLC) patients yield dismal outcomes, with a response rate of ∼30% for cytotoxic chemotherapy and 50∼60% local control for radiotherapy.3 Extensive translational research on NSCLC in search of more effective treatment has led to the recent breakthrough in epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs). Nevertheless, TKIs act totally different among individuals with different EGFR genotypes, raising new challenges for targeted therapy.
The EGFR is a 170‐kDa cell surface receptor that plays a pivotal role in regulating the chain of radiation protective responses. EGFR is frequently overexpressed in 40∼80% of NSCLCs, with poor disease prognosis and reduced survival.4, 5 However, there exists a genetic basis of EGFR underlining the cellular responses to ionizing radiation (IR) in cancer treatment, as patients receiving similar treatment could have different responses to radiotherapy. Recent discoveries have linked somatic mutations in the tyrosine kinase domain (TKD) of the EGFR gene in a subset of NSCLC to a hypersensitivity to IR.6 EGFR mutated patients with locally advanced NSCLC treated with radiotherapy had higher rates of local regional control than EGFR wild‐type (WT) patients.7 Furthermore, Das et al. demonstrate that NSCLC cell lines that harbor somatic activating mutations in TKD of EGFR exhibit a marked sensitivity to IR because of dramatically diminished capacity to resolve radiation‐induced double strand breaks (DSBs) associated with the inefficiency of EGFR nuclear translocation in non homologous end‐joining (NHEJ).8
However, homologous recombination (HR) also contributes to the repair of DSB in mammalian cells and is essential for their viability when cells are exposed to IR.9, 10, 11 Rad51, a key component of DNA homologous repair, can be induced by radiation to express and translocate between cytoplasm and nucleus.12 EGFR may affect the DNA repair process directely or through down‐stream effectors to modulate the intracellular translocation, transcription, and phosphorylation of key proteins/genes involved in the DNA repair process.13 Based on current evidence, an elevated expression of Rad51 confers radioresistance and reduced survival in malignancies such as breast carcinoma and gliomas.14, 15 In lung cancer, a high expression of Rad51 in tumor tissue has been reported as an independent factor for unfavorable prognosis.16 Whether the HR repair protein Rad51 contributes to the absolutely different radioresponse between WT and mutant EGFR in NSCLCs remains unknown.
In this study, we anticipated that somatic activating mutations in the TKD of EGFR interfere with the radioprotective function of EGFR. We intend to explore the difference in radiosensitivity between NSCLC cell lines with and without activating mutations in the TKD of EGFR, and the correlation with their expression levels and subcellular distribution of Rad51 protein.
Materials and methods
Cell culture and treatment
The NSCLC cell line, NCI‐A549, expressing the WT EGFR receptor, was obtained from the State Key Laboratory of Biotherapy and Cancer Center, West China Medical School, Sichuan University. The NCI‐H820 cell containing an in‐frame deletion (ΔE746–E750) in exon 19 of the EGFR was purchased from American Type Culture Collection (Manassas, VA, USA). NSCLC cell lines were cultured at 37°C and 5% CO2 in RPMI 1640 supplemented with 10% fetal bovine serum (Gibco, Mt Waverley, Vic, Australia). Cells were irradiated at different X‐ray doses (0 Gy∼12 Gy) using the digitalized linear accelerator (Elekta Synergy S, Crawley, UK).
Real‐time fluorescent quantitative polymerase chain reaction
Non‐small cell lung cancer cell lines were exposed to different X‐ray doses and messenger ribonucleic acid (mRNA) was extracted from irradiated cells and reverse transcripted to cDNA for subsequent quantitative real‐time fluorescent polymerase chain reaction (PCR) (Invitrogen, Carlsbad, CA, USA). Rad51 was amplified using primers with the sequence 5′‐CAGTGGCTGAGAGGTATGGTCT‐3′ (forward) and 5′‐GGTCTGGTGGTCTGTGTTGAA‐3′ (reverse) in conjunction with a thermal cycling program consisting of 40 cycles of 95°C for three minutes, 95°C for 15 seconds, and 60°C for 15 seconds. The relative RNA expression levels of Rad51 were calculated based on the mean glyceraldehyde 3‐phosphate dehydrogenase expression levels of the representative sample.
Western blot analysis
Cells were irradiated at different X‐ray doses and the total protein, including nuclear and cytosolic fractions, was prepared for Western blot analysis. An equal amount of lysates from cell lines were subjected to electrophoresis and transferred to polyvinylidene difluoride membranes. Membranes were probed with anti‐delΔE746–A750 EGFR (Cell Signaling Technology, Danvers, MA, USA) and anti‐Rad51 monoclonal mouse monoclonal antibody (Abcam Company, Cambridge, MA, USA). β‐actin protein and lamin B (Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies were used as cytosolic and nuclear fraction markers of Rad51, respectively. Subcellular fractionation was performed using a Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Nanjing, China) according to the manufacturer's recommendations.
Transfection
We constructed two lentivirus vectors which carried the strand of small interfering RNA duplexes targeting Rad51 and scrambled (as a negative control [nc]). The sequences for Rad51 and the negative control were 5′‐UGUAGCAUAUGCUCGAGCG‐3′ (small hairpin [sh]‐Rad51) and 5′‐TTCTCCGAACGTGTCACGT‐3′ (NC).17, 18, 19 The lentivirus were transfected into cells (dilution ratio: 1:20 for A549 cell and 1:30 for H820 cell) and incubated for 72–96 hours before further treatment. The WT EGFR cell line A549 was transfected with delΔE746–E750 EGFR‐pIRES2‐EGFP plasmid by Lipofectamine2000 (Invitrogen), and then screened by G418 (Invitrogen) to get the stable transfection cell line A549‐deletion. Control cells (A549‐nc) were transfected with the empty pIRES2‐EGFP plasmid and similarly sorted. The complementary DNA (cDNA) clone sequence for delΔE746–E750 EGFR is according to the EGFR gene sequences in GenBank retrieval, with the loss of nucleotides (2234–2249) “GGAATTAAGAGAAGC” in exon 19. cDNA synthesis and plasmid construction were fulfilled by Invitrogen.
Clonogenic cell survival assay
Exponentially growing cells were trypsinized and seeded in triplicate 30 mm dishes at various densities commensurate with the dose of radiation. Cells were cultured for 10–14 days, and colonies were fixed with methanol, stained with crystal violet, and manually counted using a microscope. Surviving fraction (SF) values were plotted as a function of X‐ray dose and were determined by the cloning efficiency in the treated cells divided by the plating efficiency (PE) in the untreated control (0 Gy). PE was calculated as the ratio of colonies counted/cells plated in each experiment, each with a standard deviation (SD); the standard error of the mean (SEM) for three experiments were averaged. The SD was calculated using equipartition of variance. The same calculation was also performed for the SF values. Radiosensitivity parameters of α, β, and α/β ratio were derived by fitting to the linear‐quadratic model (Sigmaplot 11.0, SSPS Inc., Chicago, IL, USA). SD and SEM of α, β, and α/β ratio were similarly calculated.
Cell cycle analysis
Twelve hours after various doses of IR, exponentially growing cells were collected and fixed by 70% ethanol overnight, then cell pellets were re‐suspended in 500 μL 1 × phosphate buffered saline (PBS) containing 50 μg/mL propidium iodide (Sigma Aldrich, St Louis, MO, USA) and 20 μg/mL ribonuclease (Sigma) for measuring DNA contents by an FAC Scan flow cytometer (Beckman, Fullerton, CA, USA). Resulting DNA distributions were analyzed by Modfit (Verity Software House, Inc., Topsham, ME, USA) for the proportion of cells in G1, S, and G2‐M phases of the cell cycle. The SD for each measurement based on three replica samples was calculated and averaged over three experiments.
Apoptosis assays (AnnexinV‐fluorescein isothiocyanate/propium iodide)
Apoptotic cells were scored on basis of a double‐labeling for the presence of early and late apoptosis. Thrity‐six hours following radiation, floating and adherent cells were collected and pooled by centrifugation. Cells were then re‐suspended in 500 μL 1 × PBS, sequentially stained with 5 μL AnnexinV‐fluorescein isothiocyanate and 5 μL propidium iodide (PI) in the dark. The samples were quantified using a flow cytometer (Beckman) within an hour.
Flow cytometric analysis of γ‐H2AX
Twenty‐four hours after various irradiation doses, pellets were sequentially fixed in 70% ice‐cold ethanol, incubated at −20°C for up to 24 hours in precooled hybridization buffer (PBS containing 0.5% bovine serum albumin and 0.25% Triton X‐100), hybridized with an Alexa 488‐conjugated monoclonal anti‐γ‐H2AX antibody (Biolegend, San Diego, CA, USA), and diluted at a ratio of 1:50 in hybridization buffer before a flow cytometric test.
Results
Mutant epidermal growth factor receptor (EGFR) non‐small cell lung cancer (NSCLC) cell line H820 exhibited decreased clonogenic survival
Radiosensitivity was assessed by calculating the SF2 (survival fraction at 2 Gy) and α/β ratio (Fig 1a & Table S1). The A549 cell line had an SF2 of 0.66 ± 0.01 and an α/β value of 4.07 ± 0.334 (α = 0.1209 ± 0.015, β = 0.0297 ± 0.003), while the H820 cell line had an SF2 of 0.40 ± 0.02 and an α/β value of 9.01 ± 0.709 (α = 0.3749 ± 0.001, β = 0.0416 ± 0.006). The EGFR‐mutated cell line H820 exhibited high radiosensitivity, which retained a dismal SF (0.037 ± 0.021) at 6 Gy. By contrast, the WT EGFR cell line A549 showed a significant tolerance to radiation with an SF of 0.349 ± 0.016 at 6 Gy. The difference was statistically significant (P < 0.05).
Figure 1.
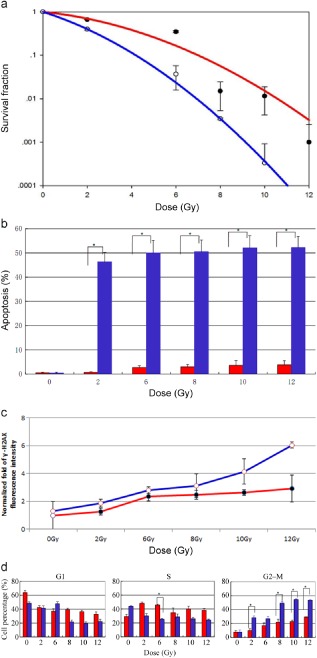
Mutant epidermal growth factor receptor‐expressing cell lines show enhanced sensitivity to radiation. (a) Colonies were counted on the 14th day following radiation, and survival fractions were plotted as a function of dose. The survival curve was obtained by the linear‐quadratic model. Error bars, standard deviation (SD) from mean of triplicate measurements. (b) The apoptosis rate induced by radiation in H820 and A549 cells. Cells were stained with AnnexinV‐ fluorescein isothiocyanate/propium iodide (PI) at 36 hours following radiation; radiation‐induced apoptosis was higher in H820 than in A549 cells. (c) Cells were fixed at 24 hours after irradiation and probed with anti‐phosphorylated histone γ‐H2AX antibody to detect DNA repair capability. H820 cells show significantly more residual γ‐H2AX
, which indicates inefficient DNA repair. Normalized fold fluorescence intensity: the relative values of γ‐H2AX fluorescence intensity divided by unirradiated A549 cells. (d) A549 and H820 cells were stained with PI at 18 hours following radiation. Both cell lines revealed an increase of cells in G2/M with IR treatment. H820 cells show significantly more G2/M arrest and dismal S arrest compared with WT‐expressing cells. Error bars, SD from mean of triple independent measurements. (a) ( ) A549, (
) A549, ( ) H820; (b) (
) H820; (b) ( ) A549, (
) A549, ( ) H820; (c) (
) H820; (c) ( ) A549, (
) A549, ( ) H820; (d) (
) H820; (d) ( ) A549, (
) A549, ( ) H820.
) H820.
Mutant EGFR NSCLC cell line H820 exhibited a higher rate of radiation‐induced apoptosis and diminished DNA repair capacity
Both H820 and A549 cells showed relatively low basal apoptotic fractions (< 1%). After radiation, the apoptosis of H820 cells was significantly higher than that of A549 cells (P < 0.01). As shown in Figure 1b, H820 cells exhibited a high level of apoptosis, which increased to 47% ± 0.03 at 2 Gy and 52.3% ± 0.029 at 12 Gy, while A549 cells were 0.7% ± 0.04 at 2 Gy and slightly increased to 3.8% ± 0.015 at 12 Gy, in a radiation dose‐dependent manner.
The residual γ‐H2AX foci at 24 hours after IR, taken as a surrogate for un‐repaired DSBs, proved to be a reliable predictive factor for radiosensitivity.20, 21, 22 The accumulation of γ‐H2AX were clearly elevated at 24 hours after exposure to IR in two NSCLC cell lines. The mutant EGFR cell line H820 exhibited a strikingly higher rate of γ‐H2AX than A549 (P < 0.01); the retained γ‐H2AX in H820 cells was 1.2∼2‐fold higher than in A549 cells in a dose‐dependent manner (Fig 1c). This indicated that mutant EGFR‐expression was associated with deficient capacity to resolve radiation‐induced DSBs. Moreover, the early apoptosis measurements were taken at a much later time than the γ‐H2AX measurements, thus, the results are not affected because all the cells were present.
Mutant EGFR NSCLC cell line H820 exhibited more radiation induced G2‐M accumulation but less intra‐S arrest
The A549 and H820 cells both revealed an increase of cells in the G2/M phase with IR treatment. This characteristic response was much more apparent in the H820 than in the A549 cells. Compared to the A549 cells, the H820 cells showed 1.5∼3‐fold higher cells in G2/M phase arrest, but 1.5∼1.8‐fold less in the S phase after IR (P < 0.05). It is interesting to note that the A549 cells exhibited radiation induced S‐phase arrest, while the H820 cells failed to do so. (Fig 1d).
Mutant EGFR NSCLC cell line H820 exhibited a low Rad51 expression and nuclear translocation
Evidence has shown that an elevated expression of Rad51 confers radioresistance and reduced survival in malignancies.21, 23 To evaluate the molecular mechanisms of the radiosensitivity phenotype in EGFR‐mutated NSCLCs, we tested the hypothesis that mutant forms of EGFR may be related to the defectiveness of Rad51 expression or subcellular location after irradiation.
We carried out Western‐blot and quantitative real‐time PCR to evaluate Rad51 expression levels. At a protein level, IR increased cellular Rad51 expression levels in a time‐dependent manner, which peaked in 12∼24 hours after IR (Fig 2a). IR‐induced Rad51 protein expression in A549 cells was significantly higher than in H820 cells at various radiation doses, in a dose dependent manner (Fig 2b). A similar trend was comfirmed at the mRNA expression level, as depicted in Figure 2c; Rad51 mRNA expression in A549 cells was significantly higher than in H820, with a magnitude of 2.6∼4.4‐fold at various irradiation doses (P < 0.05). The data indicated an inverse correlation between Rad51 expression and radiosensitivity in the two cell lines.
Figure 2.

Rad51 expression levels in H820 and A549 cells following IR. (a) Rad51 protein expression levels with (10 Gy) or without irradiation at different time points (0, 1, 12, and 24 hours following irradiation). (b) Levels of Rad51 protein in two cells at 12 hours after various doses of radiation. (c) Levels of Rad51 messenger ribonulcleic acid at 12 hours after radiation. Error bars, standard deviation (SD) from mean of triple independent measurements. Normalized fold expression: the relative expression of Rad51 divided by glyceraldehyde 3‐phopshate dehydrogenase; the expression level of Rad51 in A549 cell at 0 Gy was set as 1; *P < 0.05. (d) Levels of Rad51 protein in cytosolic and nuclear fractions with (10 Gy) or without irradiation at different time points (0 and 12 hours following irradiation). Blots were probed for β‐actin and lamin B to identify cytosolic and nuclear fractions, respectively. (c) ( ) A549, (
) A549, ( ) H820.
) H820.
Research shows that Rad51 protein relocalizes within the nucleus to play its role in DSB repair after IR.24 We examined the levels of Rad51 expression in cytosolic and nuclear fractions of A549 and H820 cells. Results in Figure 2d show that, in response to 10 Gy IR, the nuclear levels of Rad51 protein in the A549 cells increased within 12 hours post irradiation. In striking contrast to WT EGFR‐expressing cells, Rad51 failed to appear in the nucleus of H820 cells with a mutant EGFR phenotype. Moreover, the cytoplasmic levels of Rad51 slightly increased after IR over untreated samples.
Knockdown of Rad51 decreased clonogenic survival
As shown in Figure 3a, the shRNA vector successfully inhibited Rad51 expression with (10 Gy) or without irradiation (0 Gy), compared with the mock (non‐transfected) or NC (transfected with negative control shRNA) samples. After inhibition with shRNA‐Rad51, the SF2 in the A549 and H820 cells decreased to 0.42 ± 0.010 and 0.31 ± 0.019, respectively. Inversely, the α/β ratios were increased to 7.51 ± 0.621 (α = 0.3233 ± 0.004, β = 0.0430 ± 0.015), and 10.5 ± 0.512 (α = 0.4632 ± 0.022, β = 0.0442 ± 0.034), respectively (Fig 3b & Table S2). The effect caused by interfering Rad51 expression was more apparent in the A549 than in the H820 cells.
Figure 3.
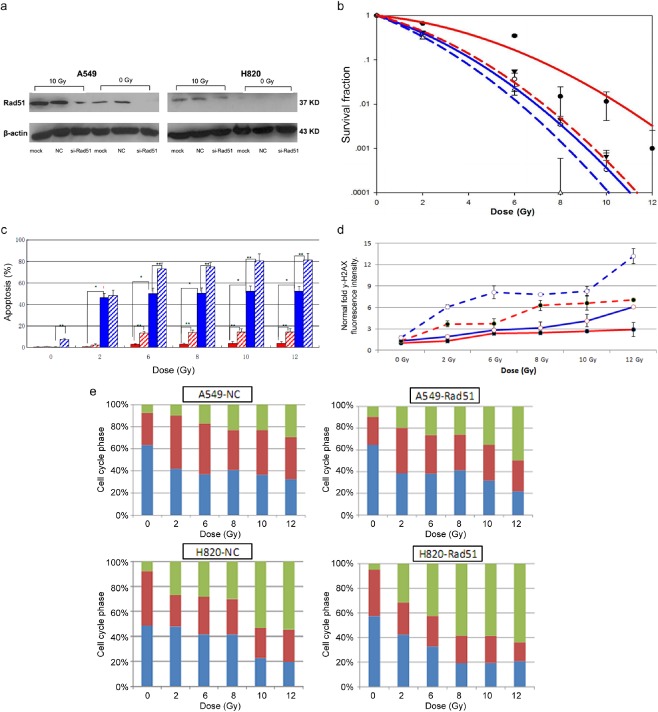
Knockdown of Rad51 sensitizes non‐small cell lung cancer cells to IR. (a) Eighty‐four hours after transfection of the lentivirus, small hairpin ribonucleic acid vector successfully inhibited Rad51 expression with (10 Gy) or without irradiation (0 Gy) in H820 and A549 cells; (b) The survival fractions decreased after downregulation of Rad51; (c) Downregulation of Rad51 increased apoptotic cell fractions in both cell lines. Error bars, SD from mean of triple independent measurements. *P < 0.05; (d) Downregulation of Rad51 induced more residual γ‐H2AX
, indicating shrinkage of DNA repair function. Error bars, SD from mean of triple independent measurements. Normalized fold fluorescence intensity: the relative values of γ‐H2AX fluorescence intensity divided by unirradiated A549 cells. (e) Downregulation of Rad51 increased G2/M phase arrest and decreased S phase fractions. Column, mean values of triple independent experiments. (b) ( ) A549‐nc, (
) A549‐nc, ( ) H820‐nc, (
) H820‐nc, ( ) A549 si‐rad51, (
) A549 si‐rad51, ( ) H820 si‐rad51; (c) (
) H820 si‐rad51; (c) ( ) A549‐NC, (
) A549‐NC, ( ) A549‐Rad51, (
) A549‐Rad51, ( ) H820‐NC, (
) H820‐NC, ( ) H820‐Rad51; (d) (
) H820‐Rad51; (d) ( ) A549‐NC, (
) A549‐NC, ( ) H820‐NC, (
) H820‐NC, ( ) A549‐Rad51, (
) A549‐Rad51, ( ) H820‐Rad51; (e) (
) H820‐Rad51; (e) ( ) G2, (
) G2, ( ) S, (
) S, ( ) G1. Si, small interfering; NC, negative control.
) G1. Si, small interfering; NC, negative control.
Knockdown of Rad51 increased apoptosis and residual γ‐H2AX
Knocking down Rad51 expression resulted in further enhanced apoptosis and residual γ‐H2AX induced by radiation (Fig 3c,d). In A549 cells, the apoptosis rate in the shRNA‐Rad51 group increased nearly 3∼5‐fold more than in the NC group, while the apoptosis rate in H820 cells was much higher, increasing to 82% at 12 Gy (P < 0.05). The inhibition of Rad51 appeared to diminish the resolution of DSBs; the increments of residual γ‐H2AX were 2‐fold in A549 and 0.5–1.5‐fold in H820 cells, respectively, compared with the NC samples.
Knockdown of Rad51 enhanced the G2‐M accumulation and attenuated intra‐S arrest
When transfected with shRNA‐Rad51, radiation induced cell accumulation in the G2/M phase was further increased compared to the control group, with a modest increment of 37.5% ± 0.023 in H820 and 57.7% ± 0.020 in the A549 cells (Fig 3e). By contrast, the data displayed a reduction of cells in the S phase with an average reduction of up to 17% ± 0.033. Although experimental results were reproducible and consistent, the trend of attenuated S phase accumulation did not yield statistical significance (P > 0.05). No distinct change was found in the G1 phase.
Ectopic expression of the ΔE746–E750 mutant EGFR sensitizes A549 cells to radiation
It was of interest to test the hypothesis whether ectopic expression of ΔE746–E750 mutant EGFR in WT EGFR genic background would sensitize A549 cells to radiation, like H820 cells. As shown in Figure 4a, A549‐del cells stably expressed ΔE746–E750 mutant EGFR protein, which was confirmed by Western blot.
Figure 4.
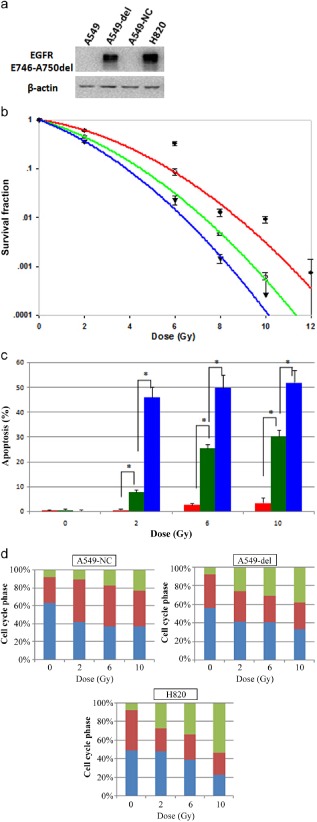
Expression of ΔE746–E750 deletion mutant epidermal growth factor receptor (EGFR) sensitizes cells to radiation. (a) Levels of ΔE746–E750 mutant EGFR protein expression in four cells. (B) expression of ΔE746–E750 deletion mutant EGFR decreased clonogenic survival after IR in A549‐del cells. Error bars, standard deviation (SD) from mean of triplicate measurements. (c) Radiation induced apoptosis was increased in radiosensitive mutant EGFR‐expressing A549‐del cells. Error bars, SD from mean of triple independent measurements. *P < 0.05. (d) A549‐del cells showed significantly more G2/M arrest and dismal S arrest compared with A549‐nc cells. Column, mean values of triple independent experiments. (b) ( ) A549‐nc, (
) A549‐nc, ( ) A549‐del, (
) A549‐del, ( ) H820; (c) (
) H820; (c) ( ) A549‐nc, (
) A549‐nc, ( ) A549‐del, (
) A549‐del, ( ) H820; (d) (
) H820; (d) ( ) G1, (
) G1, ( ) S, (
) S, ( ) G2. NC, negative control.
) G2. NC, negative control.
The effect of radiation on clonogenic cell survival was examined in three different cell lines: A549‐del, A549‐nc, and H820 cells (Fig 4b & Table S2). Ectopic expression of ΔE746–E750 mutant EGFR significantly reduced clonogenic survival after IR in A549‐del cells compared with A549‐nc cells (P < 0.05), with decreased SF2 (0.61 ± 0.014 vs. 0.45 ± 0.029) and increased α/β values (4.02 ± 0.301 vs. 7.27 ± 0.442). Moreover, results in Figure 4c–d show that, in A549‐del cells, the apoptosis rate increased nearly 7.4∼10.2‐fold more than A549‐nc cells after radiation, and the increment of IR‐induced G2/M accumulation was 0.63∼1.57‐fold. Despite the presence of WT EGFR, the A549‐del cell exhibited marked radiosensitivity, a higher rate of G2/M accumulation (P < 0.05), and early apoptosis (P < 0.05), which indicated that the mutant forms of the EGFR probably had a radiosensitizing effect in NSCLCs in an isogenic background.
Ectopic expression of the ΔE746–E750 mutant EGFR A549‐del cells failed to activate ionizing radiation (IR)‐induced Rad51 expression and nuclear translocation
To further comfirm that ΔE746–E750 mutant EGFR affects IR‐induced Rad51 protein expression and nuclear translocation, we examined the levels of Rad51 expression in cytosolic and nuclear fractions of the three cell lines, A549‐nc, A549‐del, and H820 cells at various time points after exposure to IR.
As shown in Figure 5a, Rad51 protein expression was significantly decreased in A549‐del cells with 10 Gy irradiation. A549‐nc cells showed high IR‐induced Rad51 expression within 12 hours, while in A549‐del cells the Rad51 expression level was still low, even after 24 hours (Fig 5b). Furthermore, in A549‐nc cells, Rad51 protein increased in the nucleus within 12 hours following IR treatment and persisted until 24 hours, with a similar trend at the cytoplasmic level (Fig 5b). By constrast, in A549‐del and H820 cells, both of which expressed ΔE746–E750 mutant EGFR, the nuclear levels of Rad51 protein were mainly unchanged even after 24 hours.
Figure 5.
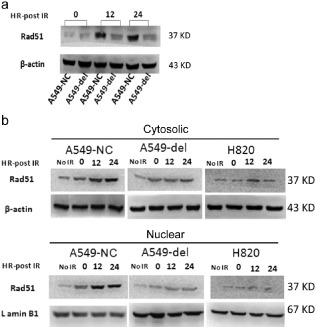
The effects of mutant epidermal growth factor receptor (EGFR) on Rad51 messenger ribonucleic acid, protein expression, and subcellular distribution. (a) Rad51 protein expression in A549‐nc and A549‐del cells with 10 Gy irradiation at different time points (0, 12, and 24 hours following irradiation). (b) Cytosolic and nuclear fractions of Rad51 expression in A549‐nc, A549‐del, and H820 cells with 10 Gy irradiation at different time points (0, 12, and 24 hours following irradiation). Blots were probed for β‐actin and lamin B1 to identify cytosolic and nuclear fractions, respectively. NC, negative control.
Discussion
Although published data show that NSCLC cell lines with mutant EGFR are more radiosensitive than cell lines with WT EGFR, the underlying mechanism has not been fully elucidated. DNA HR mainly occurs in the late S phase, which is a rather radioresistant phase of the cell cycle, and also in the G2 phase, which is not radio‐resistant. No data have directly addressed the role of DNA HR in the radiation response of lung cancer with different EGFR mutation status. We provide evidence that NSCLC cell lines with different EGFR status exhibit distinct radiosensitivity and DSB repair function, both of which are associated with Rad51 expression and subcellular distribution.
EGFR mutant NSCLCs are radiosensitive and deficient in IR‐induced Rad51 expression and relocation
Our findings indicate that, compared to EGFR WT A549 cells, H820 and A549‐del cells that expressed the ΔE746–E750 mutant forms of EGFR are 1.5 times more radiosensitive in correlation with a higher level of apoptosis and DSBs induced by radiation. This suggests a causal link between ΔE746–E750 mutations in EGFR and intrinsic radiosensitivity. The difference in radiation sensitivity between EGFR wild and mutant NSCLC has been proven by earlier research to be p53‐independent.8 We further show that the H820 cell failed to activate IR‐induced Rad51 expression and nuclear translocation. Stable ectopic expression of ΔE746–E750 mutant EGFR in the WT EGFR NSCLC cell perfectly recapitulates the deficiencies in survival and IR‐induced Rad51 nuclear translocation. This was consistent with earlier data which reported that Rad51‐overexpressing mammalian cells exhibit an increased resistance to IR with decreased DNA damage induced apoptosis.11, 25 However, emerging data also suggest that Ras‐extracellular signal‐related kinases 1/2 and phosphatidylinositol‐4,5‐bisphosphate 3‐kinase‐protein kinase B pathways might contribute to modulating radiosensitivity with Rad51 in EGFR‐mutated NSCLCs.26, 27 These findings further underscore that NSCLCs with EGFR mutation are hypersensitive in relation to aberrant signal transduction that can lead to defective Rad51 mediated DNA repair.
Our study suggested that the different characteristics of radiation induced cell cycle arrest may also account for the disparity in radiosensitivity between mutant and WT EGFR expressing NSCLC cell lines. The G2‐M arrest was greater, but the intra‐S phase arrest was depleted in mutant EGFR expressing H820 cells compared to the WT A549 cells in response to IR. It was further proven that ectopic expression of the ΔE746–E750 mutant EGFR in WT EGFR A549 cells leads to increased radiation sensitivity with an increased G2/M phase accumulation and a decrease in S phase arrest. These results support the findings of Das et al.8 IR induced DNA damage leads to arrest at the G1, intra‐S, and/or the G2‐M transition points of the cell cycle, protecting cells from unchecked DNA synthesis and defective DNA repair to improve cell survival.28, 29, 30 Inability to block DNA synthesis in response to radiation promotes cells to transit through radiation induced cell cycle checkpoints with amplified DNA damage, resulting in chromosomal catastrophe and apoptosis. As a result, mutant EGFR‐expressing cell lines lacking intra‐S phase arrest may develop decreased clonogenic survival when exposed to radiation compared to WT EGFR NSCLCs. Our results support previous reports that the radiation induced block in the DNA synthesis in WT EGFR A549 cells could persist 18 hours following radiation, but was absent in EGFR mutant cells.6 The depleted radiation‐induced intra‐S phase arrest and increased G2/M arrest in the H820 NSCLC cell line could provide a basis for choosing optimal cycle‐specific cytotoxic drugs for chemoradiation of NSCLCs harbouring EGFR activating mutation of exon‐19 deletion.
Knockdown of Rad51 expression sensitizing NSCLC cells to IR
With the use of specific shRNA to downregulate Rad51 expression, our study further confirmed that Rad51 participates in regulating the radiation response of NSCLC cells through mediating cell cycle transition, inducing apoptosis and interfering DNA repair. After downregulating Rad51 expression, the radiation sensitivity of both the A549 and H820 cell lines were enhanced, as well as increased radiation‐induced apoptosis, G2‐M phase arrest, and γ‐H2AX residual.
Interestingly, the radiation sensitizing effect of Rad51 shRNA is greater in the A549 than in the H820 cell line (radiosensitivity enhancement ratio: 3.44 vs. 1.49), suggesting that suppression of Rad51 is an effective way to overcome the resistance of WT EGFR‐expressing NSCLCs during radiation treatment. However, although the knockdown of Rad51 did reduce the magnitude of radiosensitivity difference between the mutant and WT EGFR cell lines, it did not eliminate the difference. The SF2 was 0.31 ± 0.019 for EGFR mutant and 0.42 ± 0.010 for WT NSCLC cell lines, even after the Rad51 knockdown, indicating that apart from the HR DNA repair pathway, any other mechanisms could be involved in regulating the radiation response associated with EGFR, such as recently reported by Soni et al., that if NHEJ is blocked there is backup NHEJ.31
The suppression of Rad51 protein actually increased radiation‐induced cell degradation with apoptosis and G2 phase arrest. Rad51 has an inhibitory effect on P53 transactivator activity; targeting Rad51 may abort the inhibitory effect on P53 and, thus, lead to increased apoptosis.9, 32 The increased apoptosis in Rad51‐knockdown H820 cells (harboring mutated P53) should be p53 independent. Increased DSBs can activate the ataxia telangiectasia mutated (ATM)/ataxia telangiectasia and Rad3 related (ATR)/checkpoint kinase (Chk)1/2 pathways resulting in cell G2/M cycle arrest; the depletion of Rad 51 apparently delays G2/M recovery, which accounts for why targeting Rad51 involved an increased accumulation of cells in the G2/M phase.33, 34 Despite increased G2/M arrest in H820 cells, the low expression of Rad51 still fails to repair DSB, and eventually leads to reduced cell survival.
Furthermore, it is notable that knockdown of Rad51 reduced IR‐induced intra‐S arrests in both mutant and WT EGFR cell lines, which confirms the regulation of Rad51 on IR‐induced S phase arrest in NSCLC, regardless of EGFR mutation status. This might be caused by suppressed Rad51 interfering with the intra‐S phase checkpoint response axis, based on the ATM–Chk2–cell division cycle (Cdc) 25A–cyclin dependent kinase 2–Cdc45 pathway.35 However, the difference in S phase arrest after knockdown of Rad51 is quite remarkable between the two cell lines. The WT EGFR A549 cells still exhibit a higher S phase fraction after radiation than the non‐irradiated cells, even with knockdown of Rad51 expression. This effect was absent in mutant EGFR cells marked by a further depleted S phase fraction. This effect may be a result of other DNA repair (NHEJ), which is still functional in WT EGFR NSCLCs, but defected in EGFR mutant NSCLC cell lines.8 In addition, the mutated P53 in H820 cell may add to greater intra‐S fraction reduction because of the aberrant S/G2 checkpoint function.36
Rad51 and radiation fractionation
Linear–quadratic (LQ) formalism is the most prevalent model used to predict the radiosensitivity and radiation fractionation of cells in clinical applications, which estimates the parameters' biologically based constraints on α and β.37 A higher α/β ratio is usually a result of a higher α. In the DSB model, α is a measure of both DSB induction and their repair kinetics, while β is a measure of the probability of fragment loss. It has been considered that cancers with a higher α/β ratio respond well to hyperfractionation radiotherapy, while in those with a lower α/β ratio, hypofractionation should be recommended. In this study, the α/β ratio differed between A549 and H820 cell lines. A549 cells had a lower α/β (4.07 ± 0.334) value than H820 cells (9.01 ± 0.709). After downregulating Rad51 expression, the α/β value increased (7.51 ± 0.621 for A549, 10.5 ± 0.512 for H820). Our data imply that EGFR WT NSCLC cells might benefit from hypofractionation, while for EGFR mutant NSCLC or both genotypes with downregulating Rad51, hyperfraction radiotherapy is more appropriate. However, all results were obtained in vitro which might differ from the true clinical situation because of uncontrollable circumstances of the body (e.g. proliferative, oxygen and nutritional states).38
Several implications for future research could be derived from our study: (i) the upstream signaling pathway for mutant EGFR in downregulating Rad51 expression; (ii) mechanisms of mutant EGFR mediating depletion of intra‐S phase arrest and the role of Rad51 in regulating radiation‐induced cell cycle distribution; and (iii) Rad51 could provide a radiosensitivity predictive factor and an effective target in NSCLC. Moreover, as the small molecular TKIs were reported to radiosensitize NSCLC by decreasing the expression of Rad51, our data could provide a clue for choosing fractionation in NSCLC patients receiving concurrent TKI and radiotherapy.26, 27 Further in vitro and clinical research will help to identify the prognostic and predictive role of Rad51 in NSCLC with different EGFR expression status.
Conclusions
Rad51 contributes to differences in radiosensitivity between mutant and WT EGFR NSCLCs. Strategies that specifically target Rad51 may represent novel and effective therapeutic modalities in NSCLC, especially in radiation resistant WT EGFR lung cancer cells.
Disclosure
No authors report any conflict of interest.
Supporting information
Table S1 The surviving fractions at different X‐ray doses (a) and radiosensitivity parameters based on fitting to L‐Q model (b).
Table S2 The radiosensitivity parameters of A549 and H820 cells after transfection.
Acknowledgments
This study was supported in part by grants from the Science and Technology Support Projects of Sichuan, China (No. 03SG022‐008), and the National Basic Research Program of China (No.2011CB935800).
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62: 10–29. [DOI] [PubMed] [Google Scholar]
- 2. Yang P, Allen MS, Aubry MC et al Clinical features of 5,628 primary lung cancer patients: Experience at Mayo Clinnic from 1997 to 2003. Chest 2005; 128: 452–462. [DOI] [PubMed] [Google Scholar]
- 3. Maemondo M, Inoue A, Kobayashi K et al Gefitinib or chemotherapy for non‐small‐cell lung cancer with mutated EGFR. N Engl J Med 2010; 362: 2380–2388. [DOI] [PubMed] [Google Scholar]
- 4. Ang KK, Bonner JA, Curran WJ, Harari PM, Langer CJ. The Expanding Role of EGFR‐targeted Therapies in NSCLC and Head and Neck Cancer. American Academy of CME, Houston: 2006. [Google Scholar]
- 5. Raymond E, Faivre S, Armand JP. Epidermal growth factor receptor tyrosine kinase as a target for anticancer therapy. Drugs 2000; 60 (Suppl. 1): 15–23. [DOI] [PubMed] [Google Scholar]
- 6. Das AK, Sato M, Story MD et al Non‐small‐cell lung cancers with kinase domain mutations in the epidermal growth factor receptor are sensitive to ionizing radiation. Cancer Res 2006; 66: 9601–9608. [DOI] [PubMed] [Google Scholar]
- 7. Mak RH, Doran E, Muzikansky A et al Outcomes after combined modality therapy for EGFR‐mutant and wild‐type locally advanced NSCLC. Oncologist 2011; 16: 886–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Das AK, Chen BP, Story MD et al Somatic mutations in the tyrosine kinase domain of epidermal growth factor receptor (EGFR) abrogate EGFR‐mediated radioprotection in non‐small cell lung carcinoma. Cancer Res 2007; 67: 5267–5274. [DOI] [PubMed] [Google Scholar]
- 9. Henning W, Stürzbecher HW. Homologous recombination and cell cycle checkpoints: Rad51 in tumour progression and therapy resistance. Toxicology 2003; 193: 91–109. [DOI] [PubMed] [Google Scholar]
- 10. Buchhop S, Gibson MK, Wang XW, Wagner P, Stürzbecher HW, Harris CC. Interaction of p53 with the human Rad51 protein. Nucleic Acids Res 1997; 25: 3868–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vispe S, Cazaux C, Lesca C, Defais M. Overexpression of Rad51 protein stimulates homologous recombination and increases resistance of mammalian cells to ionizing radiation. Nucleic Acids Res 1998; 26: 2859–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gildemeister OS, Sage JM, Knight KL. Cellular redistribution of Rad51 in response to DNA damage: Novel role for Rad51C. J Biol Chem 2009; 284: 31945–31952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meyn RE, Munshi A, Haymach JV, Milas L, Ang KK. Receptor signaling as a regulatory mechanism of DNA repair. Radiother Oncol 2009; 92: 316–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maacke H, Opitz S, Jost K et al Over‐expression of wild‐type Rad51 correlates with histological grading of invasive ductal breast cancer. Int J Cancer 2000; 88: 907–913. [DOI] [PubMed] [Google Scholar]
- 15. Raderschall E, Stout K, Freier S, Suckow V, Schweiger S, Haaf T et al Elevated levels of Rad51 recombination protein in tumor cells. Cancer Res 2002; 62: 219–225. [PubMed] [Google Scholar]
- 16. Qiao GB, Wu YL, Yang XN et al High‐level expression of Rad51 is an independent prognostic marker of survival in non‐small‐cell lung cancer patients. Br J Cancer 2005; 93: 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ko JC, Ciou SC, Jhan JY et al Roles of MKK1/2‐ERK1/2 and phosphoinositide 3‐kinase‐AKT signaling pathways in erlotinib‐induced Rad51 suppression and cytotoxicity in human non‐small cell lung cancer cells. Mol Cancer Res 2009; 7: 1378–1389. [DOI] [PubMed] [Google Scholar]
- 18. Ko JC, Hong JH, Wang LH, Lin YW. The role of repair protein Rad51 in synergistic cytotoxicity and mutagenicity induced by epidermal growth factor receptor inhibitor (Gefitinib, IressaR) and benzo[a]pyrene in human lung cancer. Exp Cell Res 2008; 314: 1881–1891. [DOI] [PubMed] [Google Scholar]
- 19. Su YJ, Tsai MS, Kuo YH et al Role of Rad51 down‐regulation and extracellular signal‐regulated kinases 1 and 2 inactivation in emodin and mitomycin C‐induced synergistic cytotoxicity in human non‐small‐cell lung cancer cells. Mol Pharmacol 2010; 77: 633–643. [DOI] [PubMed] [Google Scholar]
- 20. Gagou ME, Zuazua‐Villar P, Meuth M. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol Biol Cell 2010; 21: 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sak A, Stueben G, Groneberg M, Böcker W, Stuschke M. Targeting of Rad51‐dependent homologous recombination: Implications for the radiation sensitivity of human lung cancer cell lines. Br J Cancer 2005; 92: 1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Menegakis A, Yaromina A, Eicheler W et al Prediction of clonogenic cell survival curves based on the number of residual DNA double strand breaks measured by gammaH2AX staining. Int J Radiat Biol 2009; 85: 1032–1041. [DOI] [PubMed] [Google Scholar]
- 23. Taki T, Ohnishi T, Yamamoto A et al Antisense inhibition of the RAD51 enhances radiosensitivity. Biochem Biophys Res Commun 1996; 223: 434–438. [DOI] [PubMed] [Google Scholar]
- 24. Tarsounas M, Davies AA, West SC. RAD51 localization and activation following DNA damage. Philos Trans R Soc Lond B Biol Sci 2004; 359: 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raderschall E, Bazarov A, Cao J et al Formation of higher‐order nuclear Rad51 structures is functionally linked to p21 expression and protection from DNA damage induced apoptosis. J Cell Sci 2002; 115: 153–164. [DOI] [PubMed] [Google Scholar]
- 26. Chinnaiyan P, Huang S, Vallabhaneni G et al Mechanisms of enhanced radiation response following epidermal growth factor receptor signaling inhibition by erlotinib (Tarceva). Cancer Res 2005; 65: 3328–3335. [DOI] [PubMed] [Google Scholar]
- 27. Tanaka T, Munshi A, Brooks C, Liu J, Hobbs ML, Meyn RE. Gefitinib radiosensitizes non‐small cell lung cancer cells by suppressing cellular DNA repair capacity. Clin Cancer Res 2008; 14: 1266–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003; 3: 421–429. [DOI] [PubMed] [Google Scholar]
- 29. Falck J, Petrini JH, Williams BR, Lukas J, Bartek J. The DNA damage‐dependent intra‐S phase checkpoint is regulated by parallel pathways. Nat Genet 2002; 30: 290–294. [DOI] [PubMed] [Google Scholar]
- 30. Kastan MB, Bartek J. Cell‐cycle checkpoints and cancer. Nature 2004; 432: 316–323. [DOI] [PubMed] [Google Scholar]
- 31. Soni A, Siemann M, Grabos M, Murmann T, Pantelias GE, Iliakis G. Requirement for Parp‐1 and DNA ligases 1 or 3 but not of Xrcc1 in chromosomal translocation formation by backup end joining. Nucleic Acids Res 2014; 42: 6380–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marmorstein LY, Ouchi T, Aaronson SA. The BRCA2 gene product functionally interacts with p53 and RAD51. Proc Natl Acad Sci U S A 1998; 95: 13869–13874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen T, Stephens PA, Middleton FK, Curtin N. Targeting the S and G2 checkpoint to treat cancer. Drug Discov Today 2012; 17: 194–202. [DOI] [PubMed] [Google Scholar]
- 34. Huang M, Miao ZH, Zhu H, Cai YJ, Lu W, Ding J. Chk1 and Chk2 are differentially involved in homologous recombination repair and cell cycle arrest in response to DNA double‐strand breaks induced by captothecins. Mol Cancer Ther 2008; 7: 1440–1449. [DOI] [PubMed] [Google Scholar]
- 35. Bartek J, Lukas J. Mammalian G1‐ and S‐phase checkpoints in response to DNA damage. Curr Opin Cell Biol 2001; 13: 738–747. [DOI] [PubMed] [Google Scholar]
- 36. Somyajit K, Basavaraju S, Scully R, Nagaraju G. ATM‐ and ATR‐mediated phosphorylation of XRCC3 regulates DNA double‐strand break‐induced checkpoint activation and repair. Mol Cell Biol 2013; 33: 1830–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guerrero M, Stewart RD, Wang JZ, Li XA. Equivalence of the linear–quadratic and two‐lesion kinetic models. Phys Med Biol 2002; 47: 3197–3209. [DOI] [PubMed] [Google Scholar]
- 38. Mu X, Löfroth PO, Karlsson M, Zackrisson B. The effect of fraction time in intensity modulated radiotherapy: theoretical and experimental evaluation of an optimisation problem. Radiother Oncol 2003; 68: 181–187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 The surviving fractions at different X‐ray doses (a) and radiosensitivity parameters based on fitting to L‐Q model (b).
Table S2 The radiosensitivity parameters of A549 and H820 cells after transfection.