

Abstract
Background. The aim was to evaluate the analgesic efficacy and safety of the dexketoprofen/tramadol 25 mg/75 mg fixed-dose combination vs dexketoprofen (25 mg) and tramadol (100 mg) in moderate-to-severe acute pain after total hip arthroplasty.
Methods. This was a randomized, double-blind, parallel-group study in patients experiencing pain of at least moderate intensity on the day after surgery, compared with placebo at first administration to validate the pain model. The study drug was administered orally every 8 h throughout a 5 day period. Rescue medication, metamizole 500 mg, was available during the treatment period. The evaluation of efficacy was based on patient assessments of pain intensity and pain relief. The primary end point was the mean sum of the pain intensity difference values throughout the first 8 h (SPID8).
Results. Overall, 641 patients, mean age 62 (range 29–80) yr, were analysed; mean (sd) values of SPID8 were 247 (157) for dexketoprofen/tramadol, 209 (155) for dexketoprofen, 205 (146) for tramadol, and 151 (159) for placebo. The primary analysis confirmed the superiority of the combination over dexketoprofen 25 mg (P=0.019; 95% confidence interval 6.4–73) and tramadol 100 mg (P=0.012; 95% confidence interval 9.5–76). The single components were superior to placebo (P<0.05), confirming model sensitivity. Most secondary analyses supported the superiority of the combination. The incidence of adverse drug reactions was low and similar among active treatment groups.
Conclusion. The efficacy results confirmed the superiority of dexketoprofen/tramadol over its single components, even at higher doses (tramadol), with a safety profile fully in line with that previously known for these agents in monotherapy.
Clinical trial registration. EudraCT 2012-004548-31 (https://www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2012-004548-31);
ClinicalTrials.gov NCT01902134 (https://www.clinicaltrials.gov/ct2/show/NCT01902134?term=NCT01902134&rank=1).
Keywords: analgesics; arthroplasty, replacement, hip; dexketoprofen trometamol; pain, postoperative; tramadol
Editor's key points.
This study focuses on the efficacy of combining a non-steroidal anti-inflammatory drug with tramadol.
Combining dexketoprofen and tramadol was effective, with a reduction in required tramadol dose.
Less rescue medication was used when dexketoprofen and tramadol were combined.
Adverse events were predictable from the known safety profile, although further studies are needed.
Despite the great variety of analgesics available, acute pain management is still often inadequate.1–3 Possible causes include the subjective nature of pain, incorrect diagnosis, or fear of adverse drug reactions (ADRs).4
Combining different analgesics that act by different mechanisms (multimodal analgesia) to enhance clinical outcome is a common strategy in pain management. Combinations do well in single-dose analgesic studies,5 and there is a strong argument for additivity of drug-specific benefits.6 Some of the potential benefits of combinations include broader spectrum of action, greater efficacy, better compliance, complimentary pharmacokinetic profile, and better efficacy-to-safety ratio.7
Dexketoprofen, the active chiral form of ketoprofen, is effective in acute pain8 in a wide variety of conditions,9 with an onset of analgesic effect within 30 min.10 Tramadol is a widely used opioid of proven efficacy in combination with paracetamol.11 The combination of dexketoprofen/tramadol is expected to result in additive analgesia, thus allowing a decrease in the required doses of the single agents (particularly tramadol), and with the benefit of quick onset (typical of dexketoprofen) and long duration (tramadol) of the analgesic effect. A previous dose-finding study allowed for the selection of dexketoprofen/tramadol 25 mg/75 mg as the optimal combination of doses to be evaluated further.12
The present study was designed to demonstrate the superior efficacy of dexketoprofen/tramadol 25 mg/75 mg over the individual components (tramadol given at a higher dose) in moderate-to-severe acute pain after total hip arthroplasty and to evaluate its safety and tolerability.
Methods
Ethical approval
The study was conducted in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki, was approved by all concerned Ethics committees, and was registered with EudraCT (2012-004548-31) and ClinicalTrials.gov (NCT01902134). Written informed consent was obtained from all patients. It was a multicentre, randomized, double-blind, double-dummy, placebo- and active-controlled, parallel-group, phase III study encompassing a single-dose phase (SDP) and a multiple-dose phase (MDP). It was conducted at 37 study sites in 10 countries (Czech Republic, Germany, Hungary, Latvia, Lithuania, Poland, Serbia, Spain, Taiwan, and Ukraine) between May 2013 and February 2014.
Patient population
The patients were men and women aged 18–80 yr, undergoing standard primary unilateral total hip arthroplasty because of osteoarthritis (excluding osteoarthritis secondary to systemic or metabolic diseases, trauma, or infections) and experiencing pain at rest of at least moderate intensity on the day after surgery. Women participating in the study had to be either of non-childbearing potential or willing to use a highly effective contraceptive method.
Patients with known allergy or contraindication to the study drugs or rescue medication (RM) were excluded from the study, as were patients using and not able to stop analgesics other than those specified in the protocol. Medications whose concomitant use with the study drugs or RM were not advisable or might confound the study results were restricted during pre-specified time frames (related to drug characteristics such as half-life). Restricted medications included monoamine oxidase inhibitors, antiepileptics, antipsychotics, serotonin reuptake inhibitors, tricyclic antidepressants, lithium, methotrexate, antibacterial sulphonamides, and ondansetron. Anticoagulants, thrombolytics, and antiplatelet agents were also restricted, with the exception of standard perioperative thromboprophylaxis and cardioprophylactic use of low doses of aspirin (≤325 mg). Patients with moderate to severe renal dysfunction, severe hepatic or severe cardiac dysfunction, history of gastrointestinal disorders, bleeding disorders, severe asthma, and epilepsy were excluded from participation in the study (according to the investigator's judgement after assessment of the medical history, physical examination, vital signs, ECG, and laboratory safety tests at screening). Patients with a history of drug or alcohol abuse, chronic opioid users (recent use of major opioids or tramadol for more than 1 week), and pregnant or breast-feeding women were also excluded.
Conduct of the study
Patients were enrolled at each site by the investigators according to inclusion and exclusion criteria. The surgical procedure (including the anaesthetic regimen) was performed in accordance with the current site medical practice. Postoperative analgesia consisted of i.v. or i.m. morphine or other short-acting opioids. The day after surgery, 1 or 2 h after cessation of postoperative analgesia (depending on route of administration), and provided that patients were capable of taking oral medications, patients who experienced pain of at least moderate intensity [defined as pain intensity visual analog scale (PI-VAS) ≥40] were eligible to be randomized.
Patients were assigned to one of six possible treatment arms (A, B, C, D, E, or F) through an Interactive Voice/Web Response (IVR/IWR) system after a computer-generated randomization sequence stratified by baseline PI-VAS categories [moderate pain (40–60) and severe pain (>60)] with an imbalanced 1:3:1:3:1:3 ratio, using block size of 12].
The treatment arms determined the drugs to be received during the SDP (relative to the first 8 h after the first study drug administration) and the MDP (encompassing the 12 subsequent doses, each one administered every 8 h) as represented in Table 1.
Table 1.
Treatment arms
| Group assignment | Single-dose phase | Multiple-dose phase |
|---|---|---|
| A | Placebo | Dexketoprofen/tramadol 25 mg/75 mg |
| B | Dexketoprofen/tramadol 25 mg/75 mg | Dexketoprofen/tramadol 25 mg/75 mg |
| C | Placebo | Dexketoprofen 25 mg |
| D | Dexketoprofen 25 mg | Dexketoprofen 25 mg |
| E | Placebo | Tramadol 100 mg |
| F | Tramadol 100 mg | Tramadol 100 mg |
The overall study duration was ∼6 weeks for each patient, including the screening period (within 4 weeks of the randomization day), the treatment period (lasting 5 days), and the end-of-study visit (1 week after the last dose) for final safety follow-up.
Participants, health-care providers, and data collectors involved in the conduct or statistical analysis were unaware of the treatment that participants were receiving. Moreover, double-blind conditions were secured by using a double-dummy design; each study dose consisted of one tablet (containing dexketoprofen/tramadol 25 mg/75 mg, dexketoprofen 25 mg, or placebo) and two capsules (containing either tramadol 50 mg or placebo).
RM (metamizole 500 mg, a maximum of four intakes per day) was available during the entire treatment period, and the use of paracetamol (500 mg) as antipyretic was allowed during the MDP.
The evaluation of efficacy was based on patients' assessments recorded in an electronic diary of the following measures: pain intensity (PI) at rest on a 0–100 visual analog scale (VAS; 0=no pain to 100=worst pain imaginable) at 30 min and 1, 1.5, 2, 3, 4, 6, and 8 h post-dose (SDP) and every 2 h up to 8 h after the morning (seventh) dose on day 3 (MDP); the worst PI whilst moving experienced during the previous 24 h on a 0–100 VAS (assessed 8 h after the corresponding morning dose on days 2 and 3); and pain relief (PAR) on a verbal rating scale (VRS; 0=none to 4=complete) at 30 min and 1, 1.5, 2, 3, 4, 6, and 8 h post-dose (SDP). The amount and the time when RM was used were also recorded.
Evaluation of safety was based on the incidence, intensity, seriousness, and causality of treatment-emergent adverse events (AEs) and the frequency of clinically significant changes in physical examination, heart rate (HR), blood pressure (BP), laboratory safety tests (haematology, biochemistry, and urinalysis), and 12-lead ECG.
Outcomes
The primary efficacy outcome was the mean sum of pain intensity differences (SPID) throughout 8 h after the first dose (SPID8 corresponds to the area under the pain intensity difference vs time curve during 8 h post-dose).
Secondary PI-related end points during the SDP and MDP included the following: mean PI-VAS scores, mean SPID at rest, mean percentage of theoretical maximum SPID (% max SPID) at rest, percentage of PI responders (achievement of a mean PI-VAS <40 at rest), and worst pain on movement. The PAR-related end points over SDP included the following: mean PAR-VRS scores, mean total pain relief (TOTPAR), and percentage of TOTPAR responders (achievement of at least 50% of theoretical maximum TOTPAR; ≥50% max TOTPAR). The use of RM was also studied.
Statistics
The null hypothesis of equality between dexketoprofen/tramadol and each single component was tested as co-primary efficacy end points using an analysis of covariance (ancova) and a two-sided overall significance level of 5%. The two covariates were treatment (main effect) and baseline PI category.
The analysis of the primary end point was also carried out for sensitivity purposes in all randomized patients without any imputation and in all patients without major protocol violations.
The PI-VAS, SPID, % max SPID, and TOTPAR were analysed analogously. The PAR-VRS were analysed by Wilcoxon rank-sum test.
The percentage of responders was analysed using a χ2 test. In addition, the percentage of PI responders throughout 8 h post-dose was analysed using a general estimating equations (GEE) analysis.
The number of patients using RM was analysed using a χ2 test. Safety variables were analysed by means of descriptive statistics.
All report outputs were produced using SAS version 9.2 (SAS Institute Inc., Cary, NC, USA) in a secure and validated environment.
For the primary analysis, single missing values were linearly interpolated. A last observation carried forward (LOCF) approach was used for multiple consecutive missing values, unless the reason for a missing value was sleep (reported in the subsequent assessment), in which case this last missing value was replaced by the lowest PI-VAS from the relevant 8 h period.
In order to minimize the impact of RM (or paracetamol as antipyretic during MDP) on the efficacy assessments, the PI and PAR scores recorded for 6 h after RM intake were replaced using the baseline observation carried forward (BOCF) during the SDP13 and the LOCF during the MDP [or worst observation carried forward (WOCF) if the assessment immediately before RM intake was missing].
Sample size calculation
A sample size of 600 patients and a significance level of 0.05 were required for a power higher than 85% to detect the differences in change of SPID8 between dexketoprofen/tramadol and each single component.
A standard deviation of 94 mm h and a between difference of at least 35 mm h was assumed based on data from a previous phase II study (Scartoni S and Nizzardo A, unpublished observations). Assuming an approximate 25% screening failure rate, 800 patients would need to be screened.
Results
A total of 641 patients were randomized to one of six possible treatment arms. The participant flow, with the numbers of participants who were randomly assigned and received the intended treatment and were analysed for the primary outcome, is represented in Fig. 1. All randomized patients were included in the efficacy and safety analysis.
Fig 1.

Study CONSORT flow diagram. Participant flow, with the numbers of participants who were randomly assigned, received intended treatment, and were analysed for the primary outcome. Analysis populations were as follows: the ITT population included all patients randomized; the safety population included all patients randomized who received at least one dose of study treatment; and the PP population included all ITT patients with no major protocol violations. •, received at least one dose; alloc., allocated; DKP, dexketoprofen trometamol 25 mg; DKP/TRAM, dexketoprofen trometamol/tramadol hydrochloride 25 mg/75 mg; ITT, intention to treat; n, number of patients; PP, per protocol; TRAM, tramadol hydrochloride 100 mg.
The mean age of the patients was 62 (range 29–80) yr, with a balanced gender distribution (295 males and 346 females). Baseline pain was moderate (PI-VAS 40–60 mm) in 324 patients (51%) and severe (>60 mm) in 315 (49%). Patient characteristics and baseline data were comparable among different treatment arms (Supplementary material, Table S1).
For the analyses pertinent to the SDP, treatment arms were combined to produce the following four groups: dexketoprofen/tramadol (B=159); dexketoprofen (D=161); tramadol (F=160); and placebo (A+C+E=161). During the MDP, treatment arms including the same active treatment were combined, resulting in the following three groups: dexketoprofen/tramadol (A+B=213); dexketoprofen (C+D=214); and tramadol (E+F=214).
Overall, 93 (14.5%) patients had major protocol deviations during the study. Most common major protocol deviations were related to use of restricted or prohibited medication (34) or study procedures (34), with a potential impact on both SDP and MDP.
Single-dose phase
For the primary end point, SPID8, the highest mean (sd) value was reported in the dexketoprofen/tramadol group [247 (157)]; values reported by dexketoprofen and tramadol groups were very similar, [209 (155)] and [205 (146)], respectively, and the lowest value was reported in the placebo group [151 (159)]. The combination was significantly better than dexketoprofen [P=0.019; 95% confidence interval (CI) 6.4–73] and tramadol (P=0.012; 95% CI 9.5–76). Active treatments were superior to placebo (P<0.05), thus confirming model sensitivity. The analysis in all randomized patients without any imputation confirmed these results. In the analysis of all patients without major protocol violations (n=548), the differences between dexketoprofen/tramadol and both single components did not reach statistical significance at the 5% level (Table 2).
Table 2.
Statistical analysis of SPID8 at rest (analysis of covariance). DKP, dexketoprofen trometamol 25 mg; DKP/TRAM, dexketoprofen trometamol/tramadol hydrochloride 25 mg/75 mg; se, standard error; SPID8, sum of the pain intensity differences throughout 8 h after the first dose; TRAM, tramadol hydrochloride 100 mg
| Population |
Point estimate (se) (Treatment A) | Point estimate (se) (Treatment B) | Estimated difference (se) (Treatment A−B) | 95% Confidence interval | P-value | |
|---|---|---|---|---|---|---|
| Treatment A | Treatment B | |||||
| All randomized patients | ||||||
| DKP/TRAM | DKP | 248 (12) | 208 (12) | 39 (17) | 6.4–73 | 0.019 |
| DKP/TRAM | TRAM | 248 (12) | 205 (12) | 43 (17) | 9.5–76 | 0.012 |
| DKP | Placebo | 208 (12) | 152 (12) | 57 (17) | 24–90 | <0.001 |
| TRAM | Placebo | 205 (12) | 152 (12) | 54 (17) | 21–87 | 0.002 |
| All randomized patients with no imputations | ||||||
| DKP/TRAM | DKP | 243 (11) | 207 (11) | 36 (16) | 4.7–68 | 0.024 |
| DKP/TRAM | TRAM | 243 (11) | 208 (11) | 35 (16) | 3.7–67 | 0.029 |
| DKP | Placebo | 207 (11) | 170 (11) | 37 (16) | 5.0–68 | 0.023 |
| TRAM | Placebo | 208 (11) | 170 (11) | 38 (16) | 6.1–69 | 0.020 |
| All patients with no major protocol violations | ||||||
| DKP/TRAM | DKP | 251 (13) | 222 (13) | 29 (18) | −6.0 to 64 | 0.104 |
| DKP/TRAM | TRAM | 251 (13) | 217 (13) | 33 (18) | −2.5 to 69 | 0.068 |
| DKP | Placebo | 222 (13) | 156 (13) | 66 (18) | 30–101 | <0.001 |
| TRAM | Placebo | 217 (13) | 156 (13) | 61 (18) | 25–97 | <0.001 |
Analysis of PI-VAS scores at rest (Fig. 2 and Supplementary material, Table S2), mean SPID at rest (Table 3), and mean % max SPID at rest (Supplementary material, Table S3) during the SDP confirmed in general the superiority of dexketoprofen/tramadol over dexketoprofen and tramadol from the 3 and 4 h time point onwards.
Fig 2.

Observed PI-VAS at rest for the single-dose phase (first 8 h) by treatment (all randomized patients). DKP, dexketoprofen trometamol 25 mg; DKP/TRAM, dexketoprofen trometamol/tramadol hydrochloride 25 mg/75 mg; PI, pain intensity; PI-VAS, pain intensity visual analog scale; TRAM, tramadol hydrochloride 100 mg; VAS, visual analog scale.
Table 3.
Statistical analysis of SPID at rest during 2, 4, 6, 24, and 48 h (analysis of covariance; all randomized patients). DKP, dexketoprofen trometamol 25 mg; DKP/TRAM, dexketoprofen trometamol/tramadol hydrochloride 25 mg/75 mg; se, standard error; SPIDn, sum of the pain intensity differences during ‘n’ hours after the first dose; TRAM, tramadol hydrochloride 100 mg
| Time points |
Point estimate (se) (Treatment A) | Point estimate (se) (Treatment B) | Estimated treatment difference (se) (Treatment A−B) | 95% Confidence interval | P-value | |
|---|---|---|---|---|---|---|
| Treatment A | Treatment B | |||||
| SPID2 at rest | ||||||
| DKP/TRAM | DKP | 52 (2.7) | 44 (2.7) | 7.9 (3.8) | 0.4–16 | 0.038 |
| DKP/TRAM | TRAM | 52 (2.7) | 47 (2.7) | 5.7 (3.8) | −1.8 to 13 | 0.136 |
| DKP | Placebo | 44 (2.7) | 40 (2.7) | 4.1 (3.8) | −3.4 to 12 | 0.282 |
| TRAM | Placebo | 47 (2.7) | 6.4 (3.8) | 6.4 (3.8) | −1.2 to 14 | 0.097 |
| SPID4 at rest | ||||||
| DKP/TRAM | DKP | 124 (5.8) | 103 (5.8) | 22 (8.2) | 5.9–38 | 0.008 |
| DKP/TRAM | TRAM | 124 (5.8) | 105 (5.8) | 19 (8.2) | 3.2–35 | 0.019 |
| DKP | Placebo | 103 (5.8) | 85 (5.8) | 18 (8.2) | 1.7–34 | 0.030 |
| TRAM | Placebo | 105 (5.8) | 85 (5.8) | 20 (8.2) | 4.4–36 | 0.013 |
| SPID6 at rest | ||||||
| DKP/TRAM | DKP | 191 (8.9) | 158 (8.8) | 33 (13) | 8.1–57 | 0.009 |
| DKP/TRAM | TRAM | 191 (8.9) | 159 (8.8) | 32 (13) | 7.5–57 | 0.011 |
| DKP | Placebo | 158 (8.8) | 120 (8.8) | 38 (13) | 14–63 | 0.002 |
| TRAM | Placebo | 159 (8.8) | 120 (8.8) | 39 (13) | 14–63 | 0.002 |
| SPID24 at rest | ||||||
| DKP/TRAM | DKP | 929 (34) | 752 (34) | 177 (48) | 82–271 | <0.001 |
| DKP/TRAM | TRAM | 929 (34) | 827 (34) | 102 (48) | 8.1–197 | 0.033 |
| SPID48 at rest | ||||||
| DKP/TRAM | DKP | 1949 (65) | 1674 (65) | 274 (92) | 93–455 | 0.003 |
| DKP/TRAM | TRAM | 1949 (65) | 1771 (65) | 178 (92) | −2.8 to 359 | 0.054 |
In the dexketoprofen/tramadol group, 77% were PI responders (mean PI-VAS <40) at the 8 h time point of the SDP, in comparison to 67% in the dexketoprofen group (P=0.055), 66% in the tramadol group (P=0.029), and 50% for placebo. Both single components had a significantly higher response than placebo (P<0.05 for both comparisons). The results of the GEE analysis showed the superiority of the combination over both dexketoprofen (P=0.012, 95% CI 1.1–2.5) and tramadol (P=0.039, 95% CI 1.0–2.3), and confirmed the superiority of dexketoprofen and tramadol over placebo (P=0.001, 95% CI 1.3–2.7 and P<0.001, 95% CI 1.4–3.0, respectively).
In terms of mean TOTPAR and percentage of TOTPAR responders (at least 50% max TOTPAR) at the end of the SDP, the higher values were observed in the dexketoprofen/tramadol group, but the differences did not reach statistical significance (Supplementary material, Table S4).
Multiple-dose phase
Analysis of PI-VAS scores at rest (Fig. 3 and Supplementary material, Table S2), mean SPID at rest (Table 3), and mean % max SPID at rest (Supplementary material, Table S3) during the MDP confirmed in general the superiority of dexketoprofen/tramadol over dexketoprofen and tramadol throughout 48 h (for mean SPID, the superiority of the combination over tramadol approached statistical significance).
Fig 3.
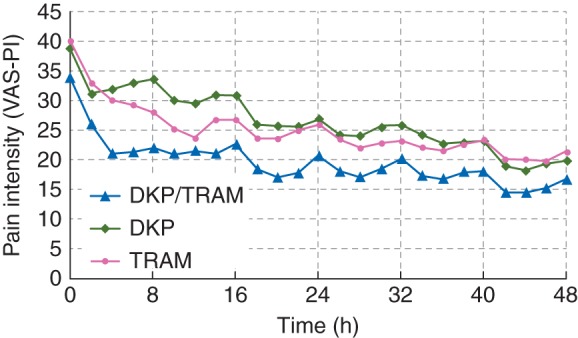
Observed PI-VAS at rest during the first 48 h of the multiple-dose phase by treatment (all randomized patients). DKP, dexketoprofen trometamol 25 mg; DKP/TRAM, dexketoprofen trometamol/tramadol hydrochloride 25 mg/75 mg; PI, pain intensity; PI-VAS, pain intensity visual analog scale; TRAM, tramadol hydrochloride 100 mg; VAS, visual analog scale.
The use of RM, defined as the percentages of patients using RM during 24 and 48 h and overall during the MDP, were lower in the dexketoprofen/tramadol group (7.0, 8.9, and 11%, respectively) than in the dexketoprofen group (17, 24, and 29%, respectively) and in the tramadol group (19, 22, and 23%, respectively). These differences were statistically significant throughout all time points (all P≤0.001).
Mean (sd) scores for worst pain whilst moving during the previous 24 h (days 2 and 3) were slightly lower in the dexketoprofen/tramadol group (29 [21] and 24 [19], respectively) than in the dexketoprofen group (34 [24] and 28 [22], respectively) and the tramadol group (33 [22] and 27 [20], respectively).
Safety results
Of the 641 patients randomized and dosed, 27 (4.2%) patients experienced a total of 39 ADRs during the active treatment, of which 16 were mild, 18 were moderate, and five were severe. The most frequent ADRs (≥1% amongst the treatment group) were nausea (0.9% patients; six events) and vomiting (0.6% patients; four events).
The dexketoprofen/tramadol group presented a lower incidence of ADRs (2.8% of patients) in comparison with the dexketoprofen group (4.7% of patients) and the tramadol group (5.1% of patients).
Two patients reported a total of five serious ADRs. One patient in the dexketoprofen group experienced duodenal ulcer. Another patient in the dexketoprofen/tramadol group experienced periorbital oedema, face oedema, laryngeal oedema, and haematuria. These events were resolved on the same day (haematuria resolved within 2 days).
There were no marked differences between treatment groups in terms of safety outcomes, including HR, BP, physical examination, 12-lead ECG, or laboratory safety parameters.
Discussion
The present study was conducted in a model of moderate-to-severe acute nociceptive somatic pain (major orthopaedic surgery) regarded as clinically relevant by regulators4 and prescribing physicians.14
The design was primarily aimed at testing the superior efficacy of dexketoprofen/tramadol vs the single agents during the SDP (primary end point), whereas the MDP was intended to confirm the sustained efficacy of the combination (during the first 48 h of the MDP) and to establish its safety during the repeated administration (up to day 5). It was deemed that evaluations of efficacy beyond this point would not provide the same degree of assay sensitivity because of the inherent increased variability and because postoperative pain usually decreases with time owing to the normal course of healing, and pain control may become less distinguishable.15,16
The surgical technique and anaesthetic regimen were not standardized in the study because local standards for surgical technique and anaesthesia represent the best patient management available at each site. Potential surgical confounders were reduced by selecting only patients undergoing primary (first time) unilateral total hip arthroplasty because of primary osteoarthritis. Other important factors, such as the postoperative analgesic care, were standardized, and only those patients experiencing a certain level of pain intensity on the day after surgery were included in the study.
Given that early mobilization is crucial after hip arthroplasty, pain on movement was assessed on days 2 and 3 by means of the ‘worst pain whilst moving experienced during the previous 24 h’. The limited value of these results is acknowledged but, owing to the study characteristics, pain on movement could not have been measured on day 1 (patients could have received different treatments during this period). Moreover, other procedures to measure pain on movement, such as pain evoked by a standardized manoeuvre, might be more precise because a recall effect is avoided, but they are hardly feasible in such a well-sampled study considering the variability in strategies for early mobilization after total hip arthroplasty.
The results of the present study confirmed that dexketoprofen/tramadol 25 mg/75 mg oral fixed-dose combination was able to provide analgesic efficacy greater than was achieved with each component in monotherapy, one of them administered at a higher dose than in the combination (tramadol 100 mg). The efficacy results were consistent after single- and multiple-dose administration, thus supporting the selection of the doses and the posology (dosing) proposed according to the previous phase II dose-finding study.12 The fact that the sensitivity analysis of the primary end point including all patients without major protocol violations did not reach statistical significance was possibly related to the reduction in the overall number of patients.
Regarding safety, results showed no increase of ADRs with the fixed-dose combination over the single agents. Most of the serious ADRs reported in the dexketoprofen/tramadol-treated patients are already described in dexketoprofen Summary of Product Characteristics [and also associated with other non-steroidal anti-inflammatory drugs (NSAIDs)] with a low frequency, as follows: laryngeal oedema (rare=0.01–0.1%) and face and periorbital oedema (angioedema, facial oedema; very rare/isolated reports ≤0.01%). Angioneurotic oedema is also associated with tramadol (rare=0.01–0.1%). The serious ADR haematuria could plausibly be attributed to other concomitant treatments (rivaroxaban) received by the patient.
Nevertheless, safety results should be considered cautiously because the study, being primarily intended to assess the short-term analgesic efficacy of dexketoprofen/tramadol, lacks the power fully to assess specific risks, for instance those associated with the use of NSAIDs, such as cardiovascular, renal, and gastrointestinal bleeding. Moreover, the study population was selected according to the current restrictions for the single components because all precautions for use applicable for the single agents remain applicable to the combination. In particular, non-selective NSAIDs should be used with caution in the postoperative setting because the antiplatelet effect may result in an increase of perioperative blood loss.17,18 Likewise, clinical trial and epidemiological data suggest that use of some NSAIDs may be associated with an increased risk of arterial thrombotic events.19 Consequently, patients with risk factors for cardiovascular disease (such as uncontrolled hypertension, congestive heart failure, established ischaemic heart disease, peripheral arterial disease, or cerebrovascular disease) should be treated with NSAIDs only after careful consideration.
In conclusion, the study results provided robust evidence of the efficacy of dexketoprofen/tramadol fixed-dose combination in the management of moderate-to-severe acute pain, with a safety profile fully in line with that previously known for the single agents in monotherapy.
Authors' contributions
Study design: H.J.M., R.A.M., M.B., B.P.V., S.C., M.P.C., A.N.
Patient recruitment and data collection: A.B., O.G., B.F., N.P., S.P., M.M., L.B., L.S., V.Z., M.L.A.
Data analysis: H.J.M., R.A.M., M.B., B.P.V., S.C., A.N.
Preparation of the manuscript: H.J.M., R.A.M., B.P.V., S.C.
All authors contributed to the development of interim and final drafts, and read and approved the final manuscript.
Supplementary material
Supplementary material is available at British Journal of Anaesthesia online.
Declaration of interest
The study sponsor (Menarini Group) contributed to the study design, data analysis, and manuscript preparation. H.J.M. and R.A.M. have been paid consultants for Menarini Group and have been members of speakers' bureaux, but received no direct funding for this work. B.P.V., S.C., A.N., M.P.C., and M.B. are employees of Menarini Group. The other authors report no conflict of interest.
Funding
Menarini Group.
Supplementary Material
Acknowledgements
The authors acknowledge the contribution to the conduct of the study from the following participating investigators, who contributed to the patient recruitment and data collection: Richard Chaloupka (Fakultni nemocnice Brno, Brno, Czech Republic); Jiri Sedivy (Nemocnice Jihlava, p.o., Jihlava, Czech Republic); Jan Deniger (Oblastni nemocnice Kladno, Kladno, Czech Republic); Jiri Novacek (Oblastni nemocnice Mlada Boleslav a.s., Mlada Boleslav, Czech Republic); Patrick Mouret (Klinikum Frankfurt Höchst GmbH, Frankfurt am Main, Germany); Tibor Mintál (PTE KK Trauma Központ-Balesetsebészeti és Kézsebészeti, Pécs, Hungary); Uldis Argalis (Liepaja Regional Hospital, Liepaja, Latvia); Arunas Gelmanas (Hospital of Lithuanian University of Health Sciences Kaunas, Kaunas, Lithuania); Juozas Belickas (Kaunas Clinical Hospital, Kaunas, Lithuania); Pawel Bazela (SPS ZOZ Szpital Miejski, Elbla˛g, Poland); Andrzej Pozowski (Wojewodzki Szpital Specjalistyczny, Wrocław, Poland); Marek Synder (Medical University of Lodz, Łódź, Poland); Marko Bumbasirevic (Clinical Center of Serbia, Belgrade, Serbia); Vladan Stevanovic (Institute for Orthopedic Surgery Banjica, Belgrade, Serbia); Branko Ristic (Clinical Center Kragujevac, Kragujevac, Serbia); Antonio Montes Pérez (Parc de Salut MAR, Barcelona, Spain); Luis Torres Morera (Hospital Universitario Puerta del Mar, Cádiz, Spain); Horng-Chaung Hsu (China Medical University Hospital, Taichung, Taiwan); Fang-Tsai Lee (Kuang Tien General Hospital, Taichung, Taiwan); and Volodymyr Filipenko (Instytut patologii khrebta ta suglobiv im. prof. M.I. Sytenka NAMN Ukraine, Kharkiv, Ukraine).
References
- 1.Curatolo M, Sveticic G. Drug combinations in pain treatment: a review of the published evidence and a method for finding the optimal combination. Best Pract Res Clin Anaesthesiol 2002; 16: 507–19 [DOI] [PubMed] [Google Scholar]
- 2.Kehlet H, Wilmore DW. Multimodal strategies to improve surgical outcome. Am J Surg 2002; 183: 630–41 [DOI] [PubMed] [Google Scholar]
- 3.Hartrick CT. Multimodal post-operative pain management. Am J Health Syst Pharm 2004; 61(Suppl 1): S4–10 [DOI] [PubMed] [Google Scholar]
- 4.European Medicines Agency. Note for guidance on clinical investigation of medicinal products for treatment of nociceptive pain (CPMP/EWP/612/00). Available from http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003525.pdf (accessed 15 April 2015)
- 5.Moore RA, Derry S, McQuay HJ, Wiffen PJ. Single dose oral analgesics for acute postoperative pain in adults. Cochrane Database Syst Rev 2011; 7: CD008659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore RA, Derry CJ, Derry S, Straube S, McQuay HJ. A conservative method of testing whether combination analgesics produce additive or synergistic effects using evidence from acute pain and migraine. Eur J Pain 2012; 16: 585–91 [DOI] [PubMed] [Google Scholar]
- 7.Raffa RB, Tallarida RJ, Taylor R, Jr, Pergolizzi JV., Jr Fixed-dose combinations for emerging treatment of pain. Expert Opin Pharmacother 2012; 13: 1261–70 [DOI] [PubMed] [Google Scholar]
- 8.Barden J, Derry S, McQuay HJ, Moore RA. Single dose oral ketoprofen and dexketoprofen for acute post-operative pain in adults. Cochrane Database Syst Rev 2009; 7: CD007355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore RA, Barden J. Systematic review of dexketoprofen in acute and chronic pain. BMC Clin Pharmacol 2008; 8: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGurk M, Robinson P, Rajayogeswaran V, et al. Clinical comparison of dexketoprofen trometamol, ketoprofen, and placebo in post-operative dental pain. J Clin Pharmacol 1998; 38: 46S–54S [PubMed] [Google Scholar]
- 11.Sawaddiruk P. Tramadol hydrochloride/acetaminophen combination for the relief of acute pain. Drugs Today (Barc) 2011; 47: 763–72 [DOI] [PubMed] [Google Scholar]
- 12.Moore RA, Gay-Escoda C, Figueiredo R, et al. Dexketoprofen/tramadol: randomised double-blind trial and confirmation of empirical theory of combination analgesics in acute pain. J Headache Pain 2015; 16: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore RA, Edwards JE, McQuay HJ. Acute pain: individual patient meta-analysis shows the impact of different ways of analysing and presenting results. Pain 2005; 116: 322–31 [DOI] [PubMed] [Google Scholar]
- 14.Singla NK, Desjardins PJ, Chang PD. A comparison of the clinical and experimental characteristics of four acute surgical pain models: dental extraction, bunionectomy, joint replacement, and soft tissue surgery. Pain 2014; 155: 441–56 [DOI] [PubMed] [Google Scholar]
- 15.McQuay HJ, Bullingham RE, Moore RA, et al. Zomepirac, dihydrocodeine and placebo compared in postoperative pain after day-case surgery. The relationship between the effects of single and multiple doses. Br J Anaesth 1985; 57: 412–9 [DOI] [PubMed] [Google Scholar]
- 16.Ridgway D. Analgesics for acute pain: meeting the United States Food and Drug Administration's requirements for proof of efficacy. Clin J Pain 2004; 20: 123–32 [DOI] [PubMed] [Google Scholar]
- 17.Reicin A, Brown J, Jove M, et al. Efficacy of single-dose and multidose rofecoxib in the treatment of post-orthopedic surgery pain. Am J Orthop 2001; 30: 40–8 [PubMed] [Google Scholar]
- 18.Chan VW, Clark AJ, Davis JC, Wolf RS, Kellstein D, Jayawardene S. The postoperative analgesic efficacy and tolerability of lumiracoxib compared with placebo and naproxen after total knee or hip arthroplasty. Acta Anaesthesiol Scand 2005; 49: 1491–500 [DOI] [PubMed] [Google Scholar]
- 19.Shau WY, Chen HC, Chen ST, et al. Risk of new acute myocardial infarction hospitalization associated with use of oral and parenteral non-steroidal anti-inflammation drugs (NSAIDs): a case-crossover study of Taiwan's National Health Insurance claims database and review of current evidence. BMC Cardiovasc Disord 2012; 12: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.