

Abstract
Precise and efficient endocytosis is essential for vesicle recycling during a sustained neurotransmission. The regulation of endocytosis has been extensively studied, but inhibitors have rarely been found. Here, we show that synaptotagmin‐11 (Syt11), a non‐Ca2+‐binding Syt implicated in schizophrenia and Parkinson's disease, inhibits clathrin‐mediated endocytosis (CME) and bulk endocytosis in dorsal root ganglion neurons. The frequency of both types of endocytic event increases in Syt11 knockdown neurons, while the sizes of endocytosed vesicles and the kinetics of individual bulk endocytotic events remain unaffected. Specifically, clathrin‐coated pits and bulk endocytosis‐like structures increase on the plasma membrane in Syt11‐knockdown neurons. Structural–functional analysis reveals distinct domain requirements for Syt11 function in CME and bulk endocytosis. Importantly, Syt11 also inhibits endocytosis in hippocampal neurons, implying a general role of Syt11 in neurons. Taken together, we propose that Syt11 functions to ensure precision in vesicle retrieval, mainly by limiting the sites of membrane invagination at the early stage of endocytosis.
Keywords: bulk endocytosis, clathrin‐mediated endocytosis, exocytosis, synaptotagmin‐11, vesicle recycling
Subject Categories: Membrane & Intracellular Transport, Neuroscience
Introduction
During neurotransmission, precise and efficient exocytosis‐coupled endocytosis is critical for neurons to recapture and reuse vesicle components and to keep the area of plasma membrane constant, especially during a sustained neuronal activity 1, 2, 3. Several modes of endocytosis operate to recycle vesicle proteins and replenish vesicle pools 1, 2, 3, 4, 5. Clathrin‐mediated endocytosis (CME), the best‐characterized pathway, is the predominant route of vesicle retrieval after exocytosis. Elevated neuronal activity also elicits bulk endo‐cytosis when large areas of plasma membrane are internalized 1, 2, 4. To understand the high efficiency of exo‐endocytotic coupling in neurons, the regulation of exocytotic and endocytotic pathways has been extensively studied for the past four decades. Ca2+ has been shown to trigger endocytosis via voltage‐dependent Ca2+ channels and the activation of calmodulin and calcineurin 6, 7, 8, 9, 10. Endocytic machinery and major vesicle proteins, such as synaptotagmin (Syt) 1 11, 12, 13, synaptophysin 14, and the three SNARE proteins such as synaptobrevin 2, SNAP25, and syntaxin 10, 15, 16, 17, 18 have all been shown to promote exo‐endocytosis. However, the mechanisms underlying the precision and fidelity of vesicle retrieval remain elusive.
Recent studies have identified Syt11 as a candidate gene for susceptibility to schizophrenia and a risk locus for Parkinson's disease 19, 20, 21. However, its function in neurons remains unknown. Syt11 belongs to a family of type I membrane proteins with evolutionarily conserved cytoplasmic tandem C2 domains, C2A and C2B 22, 23, 24. Members of the Syt family are well‐characterized Ca2+ sensors for SNARE‐dependent vesicle fusion during neurotransmitter release and hormone secretion 23, 24, 25, 26. Interestingly, Syt1 and Syt4 have also been shown to function in exocytosis‐coupled endocytosis 11, 12, 13, 27. Among the 17 mammalian Syt isoforms, Syt4 and Syt11 are classified as anomalies because they harbor an aspartate‐to‐serine substitution in a Ca2+ coordination site of the C2A domain and do not bind Ca2+ biochemically 22, 24, 28. In rat brain, Syt11 is an abundant isoform at the mRNA level 29, implying an important function in the nervous system. Using membrane capacitance (C m) recording, confocal imaging, and electron microscopy, we demonstrate here that Syt11 specifically inhibits clathrin‐mediated and bulk endocytosis. We propose that Syt11 serves as a clamp for endocytosis to ensure precision in vesicle retrieval.
Results
Syt11 inhibits endocytosis
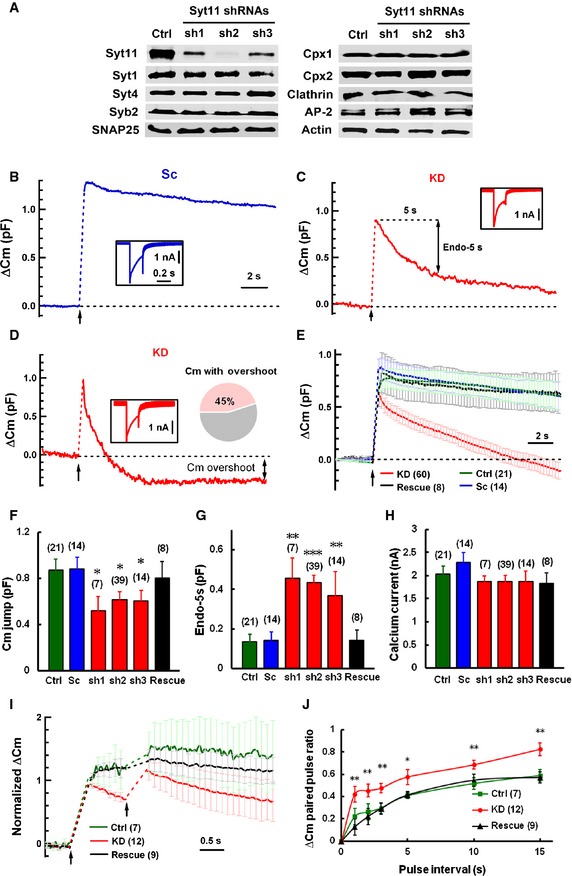
To investigate the function of Syt11 in neurons, especially in exocytosis and endocytosis, we used an shRNA‐based knockdown (KD) approach. Syt11 was efficiently and specifically silenced by three shRNAs (Fig 1A, shSyt11‐1, 73 ± 14% reduction; shSyt11‐2, 85 ± 11%; and shSyt11‐3, 45 ± 14%; n = 4, P < 0.01), while its closest homolog Syt4, the exocytotic proteins Syt1, synaptobrevin 2, SNAP25, and complexins 1 and 2, as well as the endocytic proteins clathrin heavy chain and adaptor protein 2, remained unaffected (Fig 1A, n = 4 for each protein, P > 0.05). Real‐time C m recording was used to monitor exocytosis and endocytosis in the somata of dorsal root ganglion (DRG) neurons (Appendix Fig S1A and B), which release ATP and neuropeptides via clear and dense‐core vesicles 30, 31, 32. In response to a 200‐ms depolarizing pulse from −70 to 0 mV, Syt11 KD (using shSyt11‐2) caused a reduction in the post‐stimulation C m jump, while the subsequent C m decay was greatly accelerated, indicating fast exo‐endocytosis (Fig 1B–G). The Ca2+ currents (Fig 1H) were not affected by Syt11 KD. Furthermore, the basal C m, which reflects the size of the neuronal soma, was similar in KD and control neurons (control: 36.55 ± 3.30 pF, n = 21; KD: 39.29 ± 2.1 pF, n = 58; P = 0.3907). Therefore, the reduced C m jump may reflect impaired exocytosis or faster endocytosis during stimulation. C m overshoot, representing excessive membrane retrieval, was recorded in 45% of KD neurons, but absent from control neurons (Fig 1D and E; Appendix Fig S1C–E), indicating unbalanced endocytosis following exocytosis in the absence of Syt11. Both scrambled shRNA (Sc; Fig 1B and E–H) and untransfected neurons (Ctrl, Fig 1E–H) served as controls in the Syt11 KD experiments. These phenomena also occurred in Syt11 KD neurons with the other two shRNAs (Fig 1E–H) and were completely reversed by expressing an RNAi (shSyt11‐2)‐resistant form of Syt11 (Rescue, Fig 1E–H), indicating that the findings in Syt11 KD neurons were a direct result of Syt11 deletion. Since shSyt11‐2 showed the highest knockdown efficiency among the three shRNAs, we used it in the subsequent experiments. The kinetic analysis of endocytic C m decay revealed two exponential components (time constant (τ), τfast = 1.86 ± 0.26 s, τslow = 6.38 ± 0.57 s, n = 52) in most KD neurons; both components were faster than in control neurons and were reversed by Syt11 rescue (Appendix Fig S2A–D), indicating that at least two modes of endocytosis were accelerated in Syt11 KD neurons. Although Syt4 has been reported to regulate both exocytosis and exo‐endocytosis 27, 33, 34, it failed to rescue the endocytic effects of Syt11 KD, while further reducing the C m jump (Appendix Fig S2E and F).
Figure 1. Syt11 knockdown accelerates exo‐endocytosis and the subsequent vesicle replenishment.
-
AKD efficiency of three different Syt11 shRNAs in DRG neurons. Cultured DRG neurons were infected with control or shRNA‐expressing lentiviruses at DIV 1, and Western blotting for Syt11, Syt1, Syt4, synaptobrevin 2 (Syb2), SNAP25, complexins 1, 2 (Cpx1, Cpx2), clathrin, adaptor protein 2 (AP‐2), and β‐actin was performed at DIV 6–7. n = 4 independent experiments.
-
B, CRepresentative C m traces induced by 200‐ms pulse depolarization (arrows) in DRG neurons. DRG neurons were transfected with plasmids expressing shSyt11‐2 (Syt11 KD, KD) or scrambled shRNA (Sc), and C m recording was performed 5 days after transfection. Endo‐5s represents the C m decay 5 s after stimulation. Insets show Ca2+ currents recorded in the same neurons.
-
DRepresentative C m overshoot recorded from a Syt11 KD neuron. About 45% of the KD neurons showed an excessive membrane retrieval after a 200‐ms depolarizing pulse.
-
EAveraged C m traces recorded from Control (Ctrl), Sc, KD (from all three shRNAs, n = 39 for shSyt11‐2, n = 7 for shSyt11‐1, and n = 14 for shSyt11‐3), and rescued (with the shSyt11‐2‐resistant form of Syt11) DRG neurons.
-
F–HStatistics of C m jumps, Endo‐5s, and Ca2+ current recorded from DRG neurons as in (B–E). Data were collected from 4 (Sc, Rescue), 8 (Ctrl), and 16 (KD) independent experiments.
-
INormalized C m changes induced by two pulses of 200‐ms depolarization (arrows) at a 1‐s interval. Data were normalized to the capacitance jump induced by the first depolarization.
-
JPaired‐pulse ratios of ΔC m with different interstimulus intervals as in (I). DRG neurons were stimulated with two 200‐ms pulses at different intervals as indicated, and the paired‐pulse ratio was calculated by normalizing the ΔC m in response to the second stimulation to that induced by the first one.
To determine whether the rapid endocytosis in Syt11 KD neurons could lead to faster vesicle recycling, we performed paired‐pulse stimulation. The paired‐pulse ratios were larger in the KD neurons, especially with short pulse intervals (Fig 1I and J), supporting a faster vesicle replenishment process in the absence of Syt11. We next made C m recordings in DRG neurons with a train of 10 pulses at 1 Hz to investigate whether Syt11 KD also facilitates the sustained exocytosis during tonic transmission. Strikingly, we found that control neurons failed to release after three successive 200‐ms pulses, while the KD neurons showed a stable C m jump with each pulse and maintained the release probability even at the 10th pulse (Appendix Fig S2G). Consistently, when the C m jumps in response to individual pulses were normalized to that induced by the first pulse, they showed increased values in KD neurons (Appendix Fig S2H). Thus, Syt11 KD accelerates exo‐endocytosis and vesicle pool replenishment, which then facilitates the recovery of exocytosis during the sustained neurotransmission.
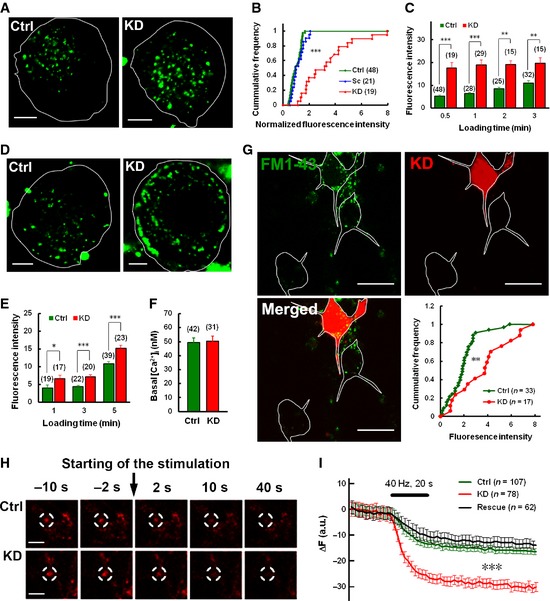
Next, we performed FM1‐43 uptake assays to confirm the endocytic phenotype of Syt11 KD neurons, which took up twice as much FM1‐43 dye as control neurons when stimulated with 100 mM K+ for 30 s (Fig 2A and B). Furthermore, the fluorescence increase was greater at shorter stimulation times as the dye uptake plateaued within 1 min in Syt11 KD neurons (Fig 2C), consistent with the faster endocytosis recorded by C m. Importantly, the accelerated FM1‐43 uptake due to Syt11 KD was not limited to DRG neurons, but was also found in hippocampal neurons (Fig 2G). To investigate whether Syt11 also inhibits endocytosis during synaptic transmission, FM4‐64 was loaded into hippocampal neurons and then unloaded with a train of 800 action potentials at 40 Hz. We found that Syt11 KD increased the dischargeable FM dye in the presynaptic boutons of hippocampal neurons (Fig 2H and I; Appendix Fig S3A), indicating an inhibitory effect of Syt11 on presynaptic endocytosis as well. When the dischargeable FM dye was normalized to the amount taken up, the discharge kinetics was similar in KD and control neurons (Appendix Fig S3B), indicating no effect on exocytosis in KD terminals. These results indicated a general role of Syt11 in neuronal endocytosis. Interestingly, FM1‐43 uptake was also enhanced under resting conditions in Syt11 KD DRG neurons (Fig 2D and E), although the basal intracellular Ca2+ concentration ([Ca2+]i) was not altered (Fig 2F). These findings suggested that Syt11 is involved in both stimulus‐coupled and constitutive endocytosis.
Figure 2. Both stimulus‐coupled endocytosis and constitutive endocytosis are accelerated by Syt11 knockdown.
-
A, BFM1‐43 uptake into DRG neurons stimulated by 100 mM K+ for 30 s. The right‐shifted cumulative frequency in (B) indicates the increased uptake level in KD neurons. Scale bars, 5 μm.
-
CQuantification of KCl‐evoked FM1‐43 uptake with different stimulation times.
-
DRepresentative micrographs showing the increased constitutive FM 1‐43 uptake (for 5 min) in KD neurons. Scale bars, 5 μm.
-
EQuantification of constitutive FM1‐43 uptake with different loading times.
-
FBasal [Ca2+]i in DRG neurons measured with Fura‐2.
-
GFM1‐43 uptake by Syt11 KD (RFP‐positive) and control (RFP‐negative) hippocampal neurons stimulated by 100 mM K+ for 2 min. Quantitative data are shown in the lower‐right panel. Scale bars, 20 μm.
-
HRepresentative images showing the preloaded FM4‐64 fluorescence in presynaptic boutons of hippocampal neurons before and after 800 stimuli at 40 Hz. Scale bars, 5 μm.
-
ITime course of FM4‐64 unloading from control, Syt11 KD, and rescued nerve terminals within the same field of view (six coverslips from at least three biological repeats each).
Syt11‐knockdown‐induced fast endocytosis is dynamin dependent
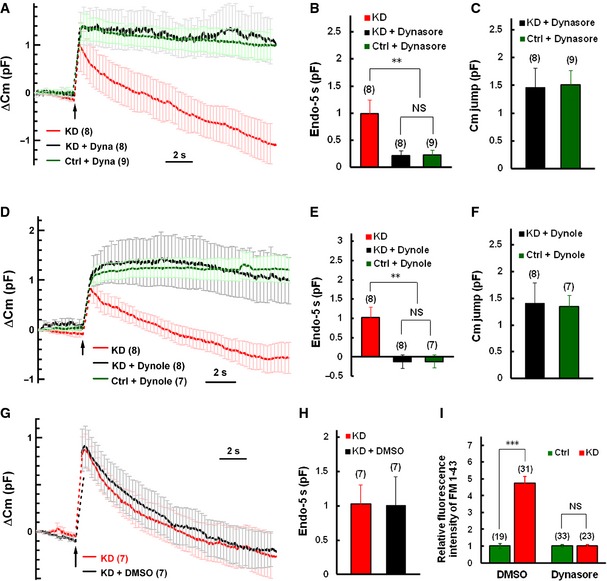
Dynamin plays global and essential roles in endocytosis in mammalian cells 35, 36, 37. We then determined whether the fast endocytosis in Syt11 KD neurons is dynamin dependent. Dynasore, a potent inhibitor of dynamin 38, 39, 40, was included in the intracellular solution and dialyzed into patched DRG neurons. C m traces were recorded 1–3 min and 5–10 min after whole‐cell dialysis. The fast C m decay in KD neurons was completely blocked after 5‐min treatment with 100 μM dynasore (Fig 3A and B). In addition, 10 μM dynole‐34‐2™, another specific inhibitor of dynamin 41, 42, similarly inhibited the fast endocytosis (Fig 3D and E). As a control, 0.1% DMSO had no effect on the C m decay (Fig 3G and H). These results suggested that the fast endocytosis in Syt11 KD neurons is dynamin dependent. We found that after the fast endocytosis was blocked by dynasore or dynole, the C m jumps in Syt11 KD neurons were similar to those in controls (Fig 3C and F). These data indicated that the reduction in the C m jumps in Syt11 KD neurons (Fig 1E and F) is due to faster endocytosis during stimulation, but not a defect in exocytosis.
Figure 3. Syt11‐knockdown‐induced fast endocytosis is dynamin dependent.
-
AAveraged ∆C m traces induced by 200‐ms depolarization in the presence and absence of dynasore. Endocytic inhibition by dynasore (Dyna) was estimated by comparison of C m traces recorded during 1–3 min (KD) and 5–10 min (KD + Dyna and Ctrl + Dyna) after whole‐cell dialysis.
-
B, CEndo‐5s and C m jumps recorded as in (A).
-
DAveraged ∆C m traces induced by 200‐ms depolarization in the presence and absence of dynole‐34‐2™ (Dynole). C m traces and endocytic rate were measured as in (A) except that 10 μM dynole‐34‐2™ with 0.1% DMSO was dialyzed into patched cells.
-
E, FEndo‐5s and C m jumps as in (D).
-
G, HAveraged ∆C m traces and Endo‐5s recorded from KD neurons under control conditions (0.1% DMSO).
-
IKCl‐evoked FM1‐43 uptake in DRG neurons after 10‐min pre‐incubation with 0.1% DMSO (control) or with 100 μM dynasore.
The dynamin dependence of fast endocytosis was also confirmed by FM1‐43 uptake assays. Ten‐minute pre‐incubation with 100 μM dynasore in the extracellular solution, but not with 0.1% DMSO, strongly inhibited the accelerated FM1‐43 uptake in KD cells (Fig 3I). Taken together, the results showed that Syt11 inhibits a dynamin‐dependent endocytosis.
Since Syt11 functions in the endocytic pathway, we then investigated its subcellular localization in DRG neurons. Available antibodies were tested with various protocols, but no specific immunocytochemical signals were detectable. We hence expressed Myc‐tagged Syt11 and found punctate staining in both the somata and axons of DRG neurons (Fig 4A, top left panel). Myc‐Syt11 partly co‐localized with dynamin, the endocytic coat protein clathrin and its adaptor AP‐2, the trans‐Golgi network marker TGN46, the vesicle marker synaptobrevin 2 (VAMP2), and the recycling endosome marker transferrin receptor, but rarely with the lysosome marker LAMP1 (Fig 4). These results support the notion that Syt11 functions in vesicle recycling pathways.
Figure 4. Syt11 localizes to vesicle recycling pathways.
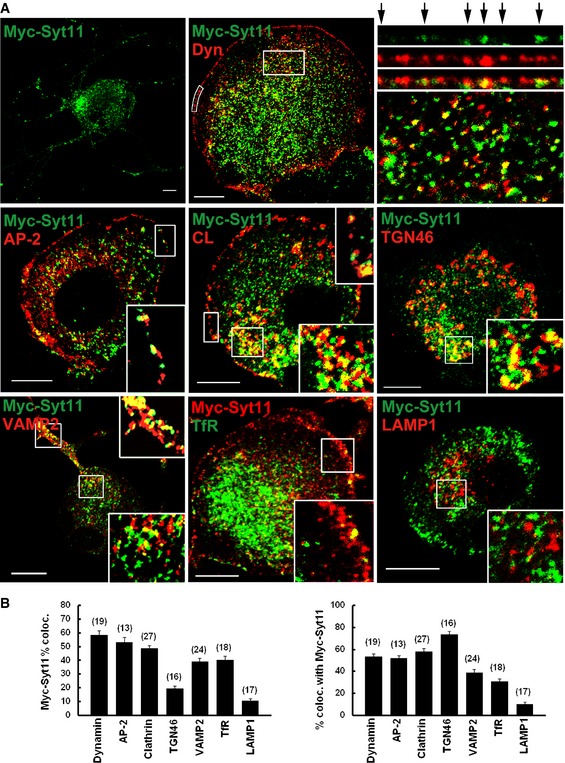
- DRG neurons expressing Myc‐Syt11 were immunostained for Myc‐Syt11, dynamin (Dyn), AP‐2, clathrin (CL), TGN46, synaptobrevin 2 (Syb2), and LAMP1. For dynamin, the box along the plasma membrane was straightened, enlarged, and is shown in the upper right panels with arrows indicating co‐localized puncta. An enlarged inset of the inside of the cell (box) is shown in the lower panel. For the localization of the transferrin receptor, EGFP‐transferrin receptor (TfR) was expressed in DRG neurons. Scale bars, 10 μm.
- Analysis of co‐localization of Myc‐Syt11 with dynamin, AP‐2, clathrin, TGN46, synaptobrevin 2, transferrin receptor, and LAMP1. n = 3 independent experiments.
Syt11 inhibits clathrin‐mediated endocytosis
Clathrin‐mediated endocytosis and bulk endocytosis are both dynamin‐dependent mechanisms of vesicle retrieval during neuronal activity 1, 2, 3, 4. To test whether Syt11 functions in CME, Alexa Fluor‐conjugated transferrin uptake was measured in DRG neurons with Syt11 KD or overexpression. We found that transferrin uptake was enhanced in Syt11 KD neurons, but suppressed in neurons overexpressing Myc‐Syt11 or Syt11 (Fig 5A and B). These results indicated that Syt11 inhibits constitutive CME.
Figure 5. Syt11 inhibits clathrin‐mediated endocytosis.
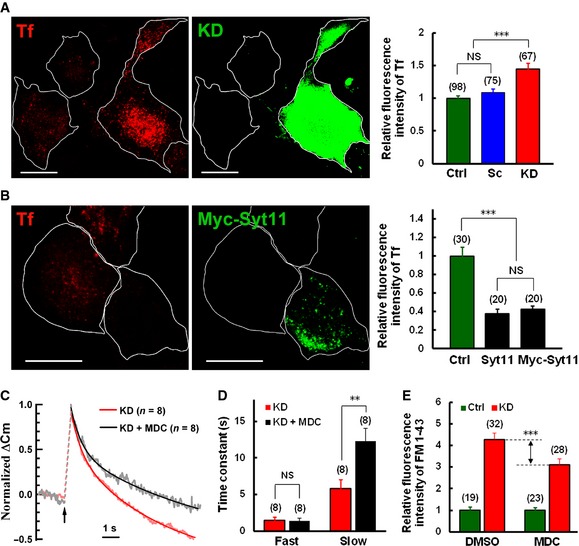
- Transferrin (Tf, red) uptake in Syt11 KD (GFP‐positive) and control neurons. Quantitative data are on the right. Scale bars, 20 μm.
- Tf uptake in Syt11‐overexpressing and control neurons. Quantitative data are on the right. Scale bars, 20 μm.
- Normalized ∆C m induced by 200‐ms depolarization of a Syt11 KD neuron in the presence of 100 μM MDC was fitted to a double‐exponential decay function (solid black and red, fitted curves).
- Fast and slow time constants of endocytosis in Syt11 KD neurons recorded as in (C).
- KCl‐evoked FM1‐43 uptake in DRG neurons after 1‐h pre‐incubation with 0.1% DMSO or 100 μM MDC in the bath solution.
We next determined whether stimulus‐coupled CME is also altered in Syt11 KD neurons. Monodansylcadaverine (MDC), an inhibitor of CME 43, 44, was included in the intracellular solution and dialyzed into patched cells. C m traces were obtained 1–3 min and 5–10 min after whole‐cell dialysis. MDC partly inhibited the accelerated C m decay in Syt11 KD neurons (Fig 5C). When the C m traces were fit to a double‐exponential decay function, two phases of endocytosis were revealed (Fig 5C). The initial fast phase had a time constant of 1.44 ± 0.48 s and was unaffected by MDC (Fig 5D). In contrast, the subsequent slow phase in Syt11 KD cells was significantly inhibited by MDC (Fig 5D, KD, τ = 5.78 ± 1.23 s; KD + MDC, τ = 12.32 ± 1.69 s), suggesting that CME is involved in the slow phase of vesicle retrieval under 200‐ms depolarization. Consistently, FM1‐43 uptake assays also showed a partial block of the accelerated endocytosis in Syt11 KD neurons after pre‐incubation with 100 μM MDC (Fig 5E). To further confirm the function of Syt11 in CME, Pitstop 1, a specific clathrin inhibitor 45, was dialyzed into patched cells during C m recordings. As expected, the Syt11 KD‐induced fast endocytosis was also attenuated by the application of Pitstop 1 (Appendix Fig S4). Although the dynamin and clathrin inhibitors may have off‐target effects 46, 47, our results point to the involvement of Syt11 in stimulus‐coupled CME.
Syt11 inhibits bulk endocytosis
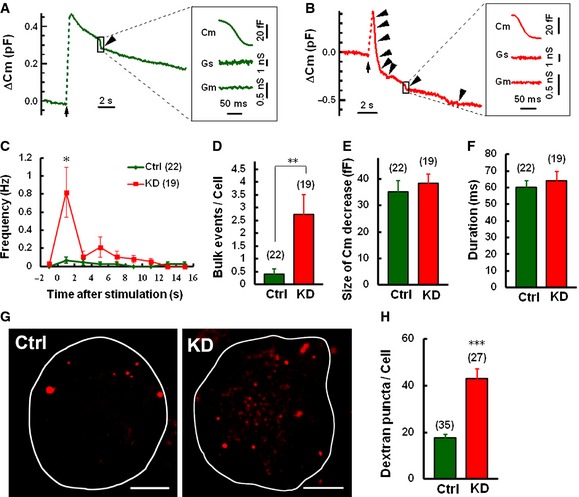
Since the endocytic C m decay revealed that two different kinetic forms of dynamin‐dependent endocytosis were accelerated in Syt11 KD neurons, while the slow mode of endocytosis was reversed by blocking CME (Figs 1, 3 and 5; Appendix Fig S2), we speculated that an additional mode of the exocytosis‐coupled endocytic pathway, which is dependent on dynamin and faster than CME, is modulated by Syt11 expression. Bulk endocytosis provides a high‐capacity pathway for vesicle retrieval during elevated neuronal activity and peaks immediately after stimulation 1, 3, 4, 6. To investigate a possible contribution of bulk endocytosis to the accelerated endocytic activity, we analyzed the C m traces recorded in Syt11 KD and control neurons in detail. Bulk endocytosis was reflected as a brief downward capacitance shift of ~20–500 fF with a decay rate > 50 fF/100 ms, according to the standard protocols 6, 48 (Fig 6A and B). We found that the frequency of bulk endocytotic events was ~5‐fold higher in Syt11 KD neurons with a peak immediately after a 200‐ms stimulus pulse (Fig 6C and D). The size and duration of the brief C m shifts in KD cells were similar to those of controls (Fig 6E and F). Therefore, Syt11 only inhibited the frequency of bulk endocytotic events, leaving the kinetics unchanged. To independently confirm the function of Syt11 in bulk endocytosis, tetramethylrhodamine dextran, a large (40 kDa) fluorescent dextran that is too large to be taken up by small vesicles 49, was used to monitor bulk endocytic events during stimulation. Consistently, the total number of large dextran fluorescent puncta per cell increased dramatically in Syt11 KD DRG neurons (Fig 5G and H), confirming the inhibitory role of Syt11 in bulk endocytosis. Altogether, Syt11 KD accelerates both CME and bulk endocytosis in DRG neurons.
Figure 6. Syt11 knockdown increases the frequency of bulk endocytosis.
-
A, BC m traces with bulk endocytosis (arrowheads) in control (A) and KD (B) neurons induced by 200‐ms depolarization. Expanded bulk events are shown on the right.
-
C, DFrequency distribution and bulk endocytic events/cell within 16 s after stimulation.
-
E, FSize and duration of C m decay during bulk endocytosis.
-
G, HRepresentative z‐projected fluorescence images and statistics showing the large dextran uptake in Syt11 KD and control DRG neurons. Neurons were loaded with 50 μM tetramethylrhodamine dextran (40 kDa) by 2‐min exposure to 100 mM KCl. Scale bars, 10 μm.
Syt11 limits the formation of clathrin‐coated pits and bulk endocytosis‐related structures
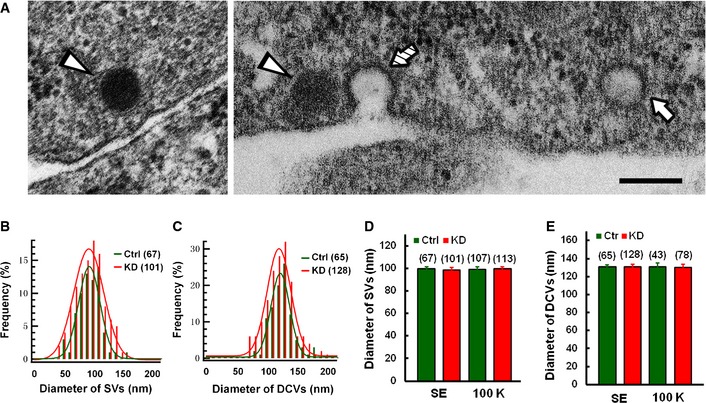
We next performed transmission electron microscopy on single neurons after horseradish peroxide (HRP) uptake to further confirm the nature of Syt11‐accelerated endocytosis and to further examine different stages of CME and bulk endocytosis in Syt11 KD neurons. Clear vesicles and dense‐core vesicles were observed in the somata of DRG neurons (Fig 7A). Since the diameter of the clear vesicles (97.1 ± 22.7 nm) was unaffected in Syt11 KD neurons (Fig 7), and > 90% of the clear vesicles were < 120 nm in diameter, we used 120 nm to distinguish HRP‐labeled small vesicles from bulk endocytosis‐derived endosomes 49, 50. Consistently, Syt11 KD neurons contained more HRP‐labeled small vesicles and endosomes under both resting and stimulated conditions (Fig 8A–D), while their size distributions remained similar to those of controls (Fig 8I and J). The unchanged vesicle/endosome sizes in KD neurons demonstrated that the endocytic processes remain largely intact although they occur at higher frequencies.
Figure 7. Vesicle size remains unaffected in Syt11‐knockdown neurons.
-
ARepresentative electron micrographs showing a small/clear vesicle (SV, arrow), dense‐core vesicles (DCVs, arrowheads), and a clathrin‐coated pit (hatched arrow) in DRG neurons. Scale bar, 200 nm.
-
B, CDiameter distributions of SVs and DCVs (statistics from six control cells and nine KD cells) in DRG neurons. The red and green lines are curves fitted to a Gaussian function.
-
D, EDiameters of SVs and DCVs in Syt11 KD and control neurons with (100 K, statistics from six control cells and nine KD cells) or without 100 mM K+ stimulation (SE, statistics from six cells for each group). Data are presented as mean ± s.e.m. of three independent experiments. ANOVA, P = 0.995 for (D) and P = 0.998 for (E).
Source data are available online for this figure.
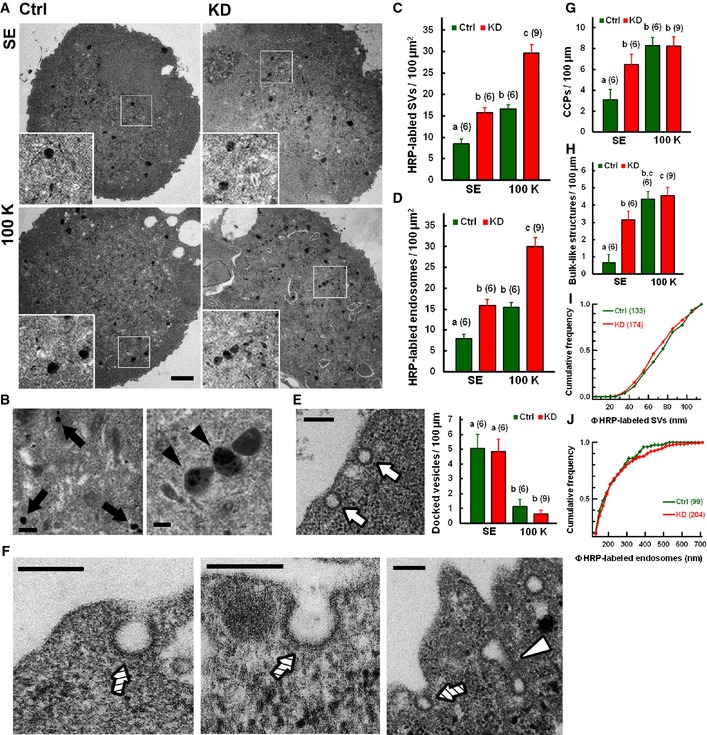
Figure 8. Syt11 inhibits clathrin‐mediated and bulk endocytosis at the early stage of membrane invagination.
-
ARepresentative electron micrographs of HRP uptake from standard (SE) or 100 mM K+ (100 K) bath solution. Scale bar, 2 μm.
-
B–DHRP‐labeled small vesicles (arrows) and endosomes (arrowheads) in DRG neurons. Quantitative data are shown in (C, D). Scale bar, 200 nm.
-
ERepresentative docked vesicles (arrows) in a DRG neuron. Quantitative data are on the right. Scale bar, 200 nm.
-
F–HClathrin‐coated pits (CCPs, hatched arrows) and bulk endocytosis‐related structures of invaginating membrane (open arrowhead) in DRG neurons. Quantitative data are shown in (G, H). Scale bars, 200 nm.
-
I, JCumulative frequencies of diameter distributions of small vesicles and endosomes (statistics from six control cells and nine KD cells). Kolmogorov–Smirnov test, P = 0.304 for (I) and P = 0.553 for (J).
Upon depolarization, docked vesicles diminished while clathrin‐coated pits and bulk endocytosis‐related structures increased on the plasma membrane of control and KD neurons (Fig 8E–H), representing exocytosis and coupled endocytosis. There was no significant difference in the number of docked vesicles before and after stimulation (Fig 8E), confirming that the deletion of Syt11 has no effect on exocytosis.
Since the sizes of endocytic vesicles and endosomes were unaffected in KD neurons, while the frequency of both CME and bulk endocytotic events increased (Figs 5, 6, 7, 8), we reasoned that Syt11 may regulate the initial stage of endocytic pathways. Consistent with this notion, more clathrin‐coated pits and bulk endocytosis‐related structures were observed on the plasma membrane of Syt11 KD neurons (Fig 8F–H). The number of clathrin‐coated pits per 100 μm plasma membrane in KD neurons was double that in control neurons (Ctrl, 3.11 ± 0.98; KD, 6.45 ± 1.03), while bulk endocytosis‐related structures were ~4‐fold higher (Ctrl, 0.67 ± 0.45/100 μm; KD, 3.15 ± 0.49/100 μm) under resting conditions (Fig 8G and H). This was consistent with the enhanced constitutive endocytosis revealed by FM1‐43, transferrin, and HRP uptake. Depolarization with 100 mM K+ further increased the appearance of both structures on the plasma membrane, but there was no difference between Syt11 KD and control neurons (Fig 8G and H), probably due to the saturation of internalization sites.
We next investigated the functional domains of Syt11 in endocytosis and compared the rescue effects of Syt11 harboring deletions of the transmembrane domain, C2A, or C2B, or the KKAA mutant (replacing the two conserved lysine residues with alanine residues in the AP‐2‐binding site) with full‐length Syt11 (Fig 9A). The accelerated uptake of FM4‐64 (all endocytic pathways), transferrin (CME), and large dextran (40 kDa, bulk endocytosis) was completely restored to normal by full‐length Syt11 (Fig 9B–D; Appendix Fig S5A and B), confirming the inhibitory role of Syt11 in both CME and bulk endocytosis. Strikingly, the transmembrane domain and AP‐2‐binding sites were critical for Syt11 to specifically inhibit CME, while C2A was specific for bulk endocytosis (Fig 9B–D; Appendix Fig S5). On the other hand, C2B was required for both pathways. Altogether, these results suggested that Syt11 serves as a clamp for both CME and bulk endocytosis by limiting the sites of membrane invagination. We propose that Syt11, working with many positive regulators, functions to ensure precision during vesicle retrieval.
Figure 9. Functional domains of Syt11 in endocytosis.
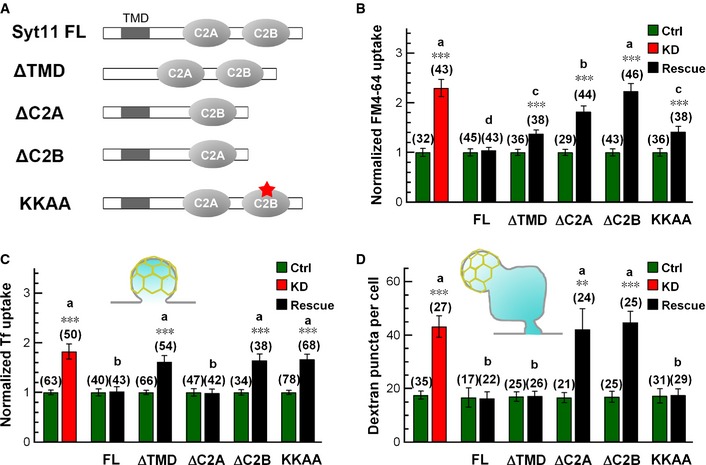
-
ADiagram of the mutant forms of Syt11 used for rescue experiments.
-
B–DStatistics of FM4‐64 (5 min), Tf, and large dextran (40 kDa) uptake into KD neurons with or without rescue by indicated forms of Syt11.
Discussion
Endocytosis is an essential and well‐regulated process in higher eukaryotes 35, 51, 52. Sustained neurotransmission imposes a greater demand on the precision and efficiency of endocytic pathways as vesicles are recycled locally to balance continued exocytosis 1, 2, 3. While numerous players and regulators of endocytosis have been characterized in neurons 3, 13, 14, 15, inhibitors are rare. This work reveals the cellular function of Syt11 as a novel inhibitor in the vesicle retrieval pathways.
Using an RNAi approach, we revealed the function of Syt11 in neuronal endocytosis. Membrane capacitance recording to monitor the somatic exocytosis and endocytosis in DRG neurons showed that Syt11 KD greatly accelerated exo‐endocytosis, and this was often accompanied by an excessive membrane retrieval as evidenced by C m overshoot, indicating an unbalanced coupling of endocytosis to exocytosis (Figs 1B–G and 3A–F). Furthermore, FM1‐43, dextran, and HRP uptake assays demonstrated an enhanced endocytic activity upon membrane depolarization in the absence of Syt11. Interestingly, constitutive endocytosis was accelerated as well (Figs 3D and E, 5A, and 8A–D), suggesting that the Syt11‐mediated inhibition of endocytosis is activity independent. Syt11 did not seem to regulate exocytosis as there was no difference in C m jumps between KD and control neurons when dynamin‐dependent endocytosis was inhibited (Fig 3C and F). Electron microscopic studies also failed to reveal the defects in docked vesicles (Fig 8E). Collectively, Syt11 mainly inhibits endocytosis.
Clathrin‐mediated endocytosis and bulk endocytosis are distinct modes of vesicle retrieval 1, 2, 3, 4. Clathrin‐coated pits are formed by the concerted action of BAR domain‐containing proteins, adaptors, and the clathrin coat to internalize cargos 51, 52, 53. On the other hand, bulk endocytosis takes up large areas of the plasma membrane by an unknown mechanism. We surprisingly discovered that Syt11 inhibited both CME and bulk endocytosis (Figs 5, 6, 7, 8, 9), which contributed to the accelerated endocytosis in Syt11 KD neurons. Specifically, clathrin‐coated pits and bulk endocytosis‐related structures increased on the plasma membrane of Syt11 KD neurons as revealed by electron microscopy (Fig 8F–H). The processes of both pathways seemed to be normal as the sizes of vesicles and endosomes remained unchanged (Fig 8I and J). In addition, C m recordings demonstrated that the kinetics of individual bulk endocytic events was similar in Syt11 KD and control neurons, while their frequency increased in the absence of Syt11 (Fig 6). These findings support the hypothesis that Syt11 limits the internalization sites of both CME and bulk endocytosis during vesicle recycling. Importantly, Syt11 function was not limited to DRG neurons, but also occurred in hippocampal neurons (Fig 2G–I; Appendix Fig S3, soma and synapses). A new study has also revealed that Syt11 negatively regulates particle ingestion by macrophages 54, suggesting that it is a general regulator in several modes of endocytosis.
Synaptotagmins are known to be Ca2+ sensors for SNARE‐dependent vesicle fusion 23, 24, 25, 26. Among them, Syt4 and Syt11 are highly homologous and do not bind Ca2+ biochemically 22, 24, 28. However, the expression of Syt4 failed to reverse the endocytic defect of Syt11 KD (Appendix Fig S2E and F), suggesting that Syt11 is functionally distinct from Syt4. In Syt4‐knockout mice, exo‐endocytosis in posterior pituitary neurons is enhanced since the kiss‐and‐run mode of exocytosis is favored 27. Conversely, the Ca2+ sensor Syt1 has been proposed as a positive regulator of CME by serving as a nucleation site for clathrin‐coated pits through its high‐affinity interactions with AP‐2 and stonin 2 11, 13, 55, 56, 57, 58, 59. Both the transmembrane domain and the AP‐2‐binding site are critical for Syt1 to promote CME 13, 56. Interestingly, they are also required for Syt11 to inhibit CME, but are not necessary for bulk endocytosis. In contrast, either the C2A or the C2B domain of Syt1 facilitates CME 13, but the C2A domain of Syt11 is dispensable for its function in CME. Both C2A and C2B are required for Syt11 to inhibit bulk endocytosis. Our current finding of Syt11 as an inhibitor of both CME and bulk endocytosis suggests that members of the Syt family regulate endocytosis in a highly coordinated and complementary manner to achieve the precision in vesicle retrieval, with Ca2+‐binding Syts functioning as positive regulators, while non‐Ca2+‐binding Syts function as negative regulators. Taken together, we propose that Syt11, working with many positive regulators, functions as a clamp for both CME and bulk endocytosis to ensure the precision during vesicle retrieval. Disruption of this Syt11‐mediated inhibitory mechanism may contribute to certain brain diseases such as Parkinson's disease and schizophrenia.
Materials and Methods
Plasmids
Full‐length rat synaptotagmin‐11 (rSyt11, AF000423) was subcloned into pIRES2‐EGFP (Clontech), pCMV5 (Clontech), pCMV5‐Myc (Clontech), or p3XFLAG‐CMV (Sigma). Full‐length rSyt4 (NM_031693) was subcloned into pIRES2‐EGFP or pCMV5. The nucleotide target sequences GG AAC ATT CAG AAG TGC ATT A (shSyt11‐1), G GCT GAG ATC ACA AAT ATA CG (shSyt11‐2), and G GAC AAA GAT GGT TCT CAT AG (shSyt11‐3) were chosen to silence the expression of Syt11. A random sequence (TTC TCC GAA CGT GTC ACG T) that was predicted to target no genes in human, rat, and mouse cells was chosen as a negative control (Guangzhou RiboBio Co., Ltd). Annealed double‐stranded oligonucleotides encoding the target sequences were inserted into the vector pRNAT‐H1.1‐RFP/GFP or the lentiviral vector pMAGic 4.1 to generate plasmids expressing shRNAs against Syt11. An RNAi (shSyt11‐2)‐resistant form of rSyt11 for the rescue experiments was generated by introducing the following silent mutations: G GCG GAA ATT ACC AAT ATA CG. The K345,346A mutant (KKAA) was generated from the RNAi‐resistant form of Syt11. Both mutants were produced by PCR using the QuikChange site‐directed mutagenesis kit (Stratagene). For truncated forms of Syt11, oligonucleotides encoding the transmembrane domain (residues 3–45), C2A domain (154–281), or C2B domain (288–430) were deleted from the RNAi‐resistant form of Syt11 by PCR using standard procedures to generate ΔTMD‐, ΔC2A‐, and ΔC2B‐expressing plasmids. All constructs were verified by DNA sequencing.
Antibodies
The primary antibodies used for Western blotting and immunofluorescence (IF) studies were anti‐actin (A5316, Sigma), anti‐Syt11 (270003, Synaptic Systems), anti‐Syt1 (105221, Synaptic Systems), anti‐Syt4 (sc‐30095, Santa Cruz), anti‐c‐Myc (sc‐789 and sc‐40, Santa Cruz), anti‐SNAP25 (111011, Synaptic Systems), anti‐complexins 1, 2 (122002, Synaptic Systems), anti‐clathrin heavy chain (610499, BD Transduction Laboratories for Weste rn blot; MA1‐065 (X22), Thermo Scientific for IF), anti‐AP‐2α‐adaptin (610501, BD Transduction Laboratories for Western blot; MA1‐064 (AP6), Thermo Scientific for IF), anti‐synaptobrevin 2 (104211, Synaptic Systems), anti‐LAMP1 (ab25744, Abcam), anti‐dynamin (hudy 1, 28907, Upstate Biotechnology), and anti‐TGN46 (ab2809, Abcam). The highly cross‐adsorbed secondary antibodies for IF staining were all from Invitrogen. They were Alexa Fluor® 488 goat anti‐mouse IgG (H+L) (A11029), Alexa Fluor® 594 goat anti‐mouse IgG (H+L) (A11032), Alexa Fluor® 488 goat anti‐rabbit IgG (H+L) (A11034), and Alexa Fluor® 594 goat anti‐rabbit IgG (H+L) (A11037). The secondary antibodies for Western blotting were IRDye 800CW goat anti‐rabbit IgG (LIC‐926‐32211, LI‐COR Biosciences) and IRDye 680CW goat anti‐mouse IgG (LIC‐926‐32220, LI‐COR Biosciences).
Cell culture and transfection
The use and care of animals was approved and directed by the Animal Care and Use Committee of Peking University. All chemicals were from Sigma unless otherwise stated. Isolation of dorsal root ganglion (DRG) neurons was performed as previously described 31. Briefly, adult Wistar rats (male, ~60 g) were used and DRGs were isolated in ice‐cold L15 medium (Gibco). The ganglia were treated with trypsin (0.3 mg/ml) and collagenase (1 mg/ml) for 40 min at 37°C. Cells were dissociated, collected, and transfected with the Neon™ 100 μl transfection system MPK10096 (Invitrogen). Cells were then plated on poly‐L‐lysine‐coated coverslips and maintained in Dulbecco's modified Eagle's medium (DMEM)–F12 (Gibco) supplemented with 10% FBS (Gibco). For gene silencing experiments, 24 h after transfection, the culture medium was replaced by Neurobasal‐A (Gibco) supplemented with 2% B27 (Gibco), 0.5 mM l‐glutamine (Gibco), 10 ng/ml nerve growth factor, and 5 μM cytosine arabinoside. Experiments were performed at 1–2 days in vitro (DIV) for overexpressing neurons and DIV 5–6 for KD neurons. For KD efficiency, shRNA‐carrying and control lentiviruses (Shanghai Sunbio Medical Biotech Co., Ltd.) were used to infect DRG neurons at DIV 1, and Western blotting was performed on DIV 6–7.
For hippocampal cultures 60, hippocampi were dissected from Wistar rats at postnatal day 1 and treated with 0.25% trypsin at 37°C for 12 min. Cells were plated on polyethyleneimine‐coated glass coverslips in 35‐mm dishes and maintained in DMEM (Gibco) supplemented with 10% FBS. After 3 h, the medium was replaced by Neurobasal (Gibco) supplemented with 2% B27, 0.5 mM l‐glutamine, and 5 μM cytosine arabinoside. Cultures at 5 DIV were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Transfected neurons were cultured until 13–15 DIV for FM uptake assays.
HEK293A cells were cultured in DMEM supplemented with 10% FBS. For gene silencing experiments, shRNAs and target gene‐expressing plasmids were delivered using VigoFect (Vigorous Biotechnology Beijing Co.). Western blotting analysis was performed 3 days after transfection.
Gel electrophoresis and Western blotting
Cells were washed with phosphate‐buffered saline (PBS) and homogenized on ice with lysate buffer [20 mM Hepes at pH 7.4, 100 mM KCl, 2 mM EDTA, 1% NP‐40, 1 mM PMSF, and 2% proteinase inhibitor (539134, Calbiochem)]. The homogenates were centrifuged at 16,000 g for 15 min at 4°C, and the supernatants were collected and boiled in SDS–PAGE buffer. Proteins were electrophoresed and transferred to nitrocellulose filter membranes. Each membrane was blocked by incubation for 1 h with PBS containing 0.1% Tween‐20 (v/v) and 5% non‐fat dried milk (w/v). After washing with 0.1% Tween‐20 containing PBS (PBST), the blots were incubated with primary antibodies at 4°C overnight in PBST containing 2% bovine serum albumin (BSA). Secondary antibodies were then applied at room temperature for 1 h. Blots were scanned with an Odyssey infrared imaging system (LI‐COR Biosciences) and quantified with ImageJ (National Institutes of Health, USA).
Immunofluorescence
Cells were washed three times with PBS, fixed in 4% paraformaldehyde for 20 min, and permeabilized with 0.3% Triton X‐100 in PBS containing 2% BSA for 5 min at room temperature. After blocking for 1 h with 2% BSA in PBS, cells were incubated for 1 h with primary antibodies, washed three times with blocking solution, and then incubated for 1 h with secondary antibodies. After three washes in blocking solution and one wash in PBS, cells were mounted on slides with 50% glycerol. A z‐series of 1‐μm optical sections was scanned through the 63× oil‐immersion lens of a Zeiss 710 inverted confocal microscope. Images were processed with the Zeiss LSM Image Browser (version 3.0) and Adobe Photoshop (Adobe Systems Inc.). Co‐localization analysis was performed with default M1 and M2 coefficients in ImageJ.
Membrane capacitance recording
Membrane capacitance (C m) was measured under the whole‐cell configuration using an EPC10/2 amplifier controlled by Pulse software (HEKA Elektronik) as described previously 30, 31. The membrane potential was clamped at −70 mV, and pipette resistance was between 3 and 4 MΩ. The external solution contained (in mM) 150 NaCl, 5 KCl, 2.5 CaCl2, 1 MgCl2, 10 H‐HEPES, and 10 D‐glucose, pH 7.4. In 100 mM K+ external solutions, the NaCl concentration was reduced to maintain the same ionic strength. The intracellular pipette solution contained (in mM) 153 CsCl, 1 MgCl2, 10 H‐HEPES, and 4 Mg‐ATP, pH 7.2. Dynasore at 100 μM or dynole‐34‐2™ at 10 μM with 0.1% DMSO was included in the intracellular pipette solution to block dynamin‐dependent endocytosis, while 100 μM MDC with 0.1% DMSO or 30 μM Pitstop 1 was used to block clathrin‐mediated endocytosis. DMSO (0.1%) served as the control for dynasore, dynole‐34‐2™, and MDC. Endocytic inhibition was estimated by comparison of C m traces recorded during 1–3 min and 5–10 min after whole‐cell dialysis, except that C m traces from the same batches of DRG neurons with or without Pitstop 1 were used for comparison due to the faster inhibitory effect of this drug. All recordings were performed at room temperature (22–25°C). Igor software (Wavemetrics) was used for all offline data analysis, and series conductance (G s) and membrane conductance (G m) were used to monitor the seal condition in patch‐clamp recordings. In cultured DRG neurons, depolarization‐induced C m jumps were all Ca2+ dependent. Bulk endocytosis, which was reflected as a brief downward capacitance shift, was measured as previously described 48. Capacitance traces were low‐pass‐filtered at 30 Hz, and a downward capacitance shift was identified when the size of the capacitance decay was > 20 fF and the decay rate was > 50 fF/100 ms, with unchanged G s and G m.
FM uptake
FM1‐43 uptake was assessed as described previously 61. Cells were washed 3 times with standard extracellular bath solution and then incubated with 10 μM FM1‐43 or 20 μM FM4‐64 in standard or 100 mM K+‐containing external solution at room temperature. Unbound dye was washed out with standard bath solution immediately after incubation. A z‐series of 1‐μm optical sections was scanned through the 42× oil‐immersion lens of a Zeiss 710 inverted confocal microscope. Identical settings were applied to all samples in each experiment. The total FM1‐43 signal in the cell body was measured, and FM fluorescence intensity (total signal divided by the area of the cell body) was calculated with ImageJ. For the loading and unloading of FM4‐64 in the terminals of hippocampal neurons, 20 μM FM4‐64 was loaded with 800 action potentials at 40 Hz and then washed out immediately. AP‐5 (50 μM) and CNQX (10 μM) were included in the external solutions to block glutamate signaling during stimulation. Dye unloading was achieved using the same stimuli, and fluorescence changes at individual boutons were visualized at 543‐nm excitation and > 560‐nm emission. Images were acquired for 20 s before the stimulation to establish a stable baseline. Data were collected and analyzed offline using ImageJ and Adobe Photoshop. To analyze image stacks, boutons that showed fluorescence changes (ΔF) > 5 arbitrary units (A.U.) during field stimulation were initially selected and marked with 2‐μm‐diameter circular selection areas. Among the marked boutons, those that showed ΔF > 5 A.U. during the 10 s before stimulation were discarded. Then, the fluorescence intensity values during the first 5 s were averaged and used for normalization. The normalized fluorescence traces were averaged from 107 control, 78 KD, and 62 rescued boutons imaged from 6 coverslips each in at least three biological repeats.
Ca2+ imaging
Cytosolic Ca2+ was measured with the Ca2+ indicator Fura‐2 AM (Invitrogen) as previously described 31. Briefly, DRG neurons were loaded with 1 μM Fura‐2 AM for 5 min at 37°C and then washed 3 times with normal external solution at room temperature. [Ca2+]i was measured by epifluorescence imaging using an Olympus IX‐70 inverted microscope equipped with a monochromator‐based system (TILL Photonics). X‐chart software (HEKA Elektronik) was used to collect imaging data at 1 Hz. [Ca2+]i was calculated from the ratio (R) of the fluorescent signals excited at 340 nm and 380 nm with the following equation: [Ca2+]i = K d × (R − R min)/(R max − R), where K d, R min, and R max are constants obtained from in vitro calibration.
Transferrin uptake
The transferrin (Tf) uptake assay was modified from that previously described 62. Cells were washed with serum‐free DMEM containing 20 mM Hepes, pH 7.4, and 1 mg/ml BSA, before being serum‐starved in the same medium for 45 min at 37°C. They were then incubated for 30 min at 37°C with serum‐free medium containing 25 μg/ml human Tf conjugated to Alexa Fluor 594 (Invitrogen). The unbound Tf was washed out with ice‐cold PBS containing 0.3 mM CaCl2 and 0.3 mM MgCl2. The cells were then fixed in ice‐cold 4% formaldehyde and processed for confocal microscopy. For Tf uptake into neurons expressing Syt11 or Myc‐Syt11, immunofluorescence staining with antibody against c‐Myc was performed after fixation. A z‐series of 1‐μm optical sections was scanned through the 42× oil‐immersion lens of a Zeiss 710 inverted confocal microscope. Identical settings were applied to all samples in each experiment. The total Tf signal in the cell body was measured, and Tf fluorescence intensity was calculated with ImageJ.
Dextran uptake
Tetramethylrhodamine dextran (40 kDa) uptake was assessed using a previously described method 49 with minor modifications. Cells were washed three times with standard extracellular bath solution and then incubated for 2 min at 37°C with 50 μM dextran in 100 mM K+‐containing external solution. Unbound dye was washed out with standard bath solution immediately after incubation. Z‐series of 1‐μm optical sections was scanned through the 42× oil‐immersion lens of a Zeiss 710 inverted confocal microscope. The consecutive optical sections were z‐projected, and the total numbers of dextran fluorescent puncta per cell were counted. Images were processed with ImageJ and Adobe Photoshop.
HRP uptake and electron microscopy of single DRG neurons
Horseradish peroxidase (HRP) uptake and electron microscopy of single DRG neurons were performed according to the previously described methods 49, 50, 63 with small modifications. Neurons were transfected with Syt11 shRNA‐expressing plasmids and plated on gridded dishes (Corning Inc.). Cells were observed under a fluorescence microscope 5–6 days after transfection, and transfected and control cells were identified. For HRP uptake, cells were washed three times with standard external solution and then incubated with standard or 100 mM K+ external solution supplemented with 10 mg/ml HRP for 2 min. They were then fixed in 2.5% glutaraldehyde and 2% paraformaldehyde in PBS for at least 30 min at room temperature. After washing with 100 mM Tris (pH 7.4), the cells were exposed to 0.1% diaminobenzidine and 0.2% H2O2 in 100 mM Tris for 4–5 min and then washed with 100 mM Tris (pH 7.4). The cells were then post‐fixed for 30 min with 1% osmium tetroxide, washed, dehydrated through an ethanol series, and embedded in Epon and polymerized at 60°C for 36 h. The embedded samples were separated from the dishes by dipping in liquid nitrogen and hot water. Cells of interest were isolated and mounted on pre‐polymerized Epon blocks. Ultra‐thin sections (~80 nm) were cut parallel to the cell monolayer, collected on single‐slot formvar‐coated copper grids, and stained with 2% uranyl acetate for 30 min and 0.5% lead citrate for 15 min. Samples were imaged at 120 KV in a Tecnai G2 20 200KV transmission electron microscope. Docked vesicles were identified as those without any measurable distance from the plasma membrane. Vesicle diameters and numbers of HRP‐labeled vesicles and endosomes were measured manually with Zeiss LSM Image Browser (version 3.0).
Statistical analysis
All experiments were replicated biologically for at least three times. Data are shown as mean ± s.e.m. Statistical comparisons were made with the two‐tailed unpaired Student's t‐test, Kolmogorov–Smirnov test, or one‐way ANOVA as indicated. No statistical method was used to predetermine sample sizes, but our sample sizes are similar to those generally used in the field. No randomization or exclusion of samples was used. Normality of the data was tested with the Shapiro–Wilk test, and the equality of variance was determined with Levene's test. All tests were conducted using the Statistical Package for the Social Sciences, version 13.0. Significant differences were accepted at P < 0.05. Numbers of cells analyzed are indicated in the figures unless otherwise indicated in the figure legends.
Author contributions
CXZ and CW conceived the study. CW, YW, MH, ZC, QW, and RH performed the experiments and analyses. WH contributed unpublished reagents. CXZ, CW, WH, and ZZ wrote the manuscript with input from the other authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
This study was supported by grants from the National Basic Research Program of China (2007CB512100 and 2012CB518006), the National Natural Science Foundation of China (30770521, 30970660, 31171026, 31100597, 30911120491, 30830043, 31330024, 31400708, and 31471085), the National Key Technology R&D Program (SQ2011SF11B01041), Beijing Natural Science Foundation Program and Scientific Research Key Program of Beijing Municipal Commission of Education (KZ201510025023), Beijing Institute for Brain Disorders (PXM2013_014226_07_000087), the Natural Science Foundation of Heilongjiang Province of China (C201453), and the A*STAR Biomedical Research Council (WH). CW was supported in part by a Postdoctoral Fellowship of the Peking‐Tsinghua Center for Life Sciences. We thank Drs. Pietro De Camilli (Department of Cell Biology and Howard Hughes Medical Institute, Yale University School of Medicine), Chen Zhang (College of Life Sciences, Peking University), Xuelin Lou (School of Medicine and Public Health, University of Wisconsin), Jianyuan Sun (Institute of Biophysics, Chinese Academy of Sciences), and Bo Zhang (Institute of Molecular Medicine, Peking University; Stanford University) for discussion and comments; Yingchun Hu (Core Facilities of the College of Life Sciences, Peking University) and Junlin Teng (College of Life Sciences, Peking University) for assistance with electron microscopy; Zhengxing Wu (College of Life Science & Technology, Huazhong University of Science and Technology) for plasmids; and IC Bruce (Institute of Molecular Medicine, Peking University) for reading the manuscript.
EMBO Reports (2016) 17: 47–63
References
- 1. Smith SM, Renden R, von Gersdorff H (2008) Synaptic vesicle endocytosis: fast and slow modes of membrane retrieval. Trends Neurosci 31: 559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dittman J, Ryan TA (2009) Molecular circuitry of endocytosis at nerve terminals. Annu Rev Cell Dev Biol 25: 133–160 [DOI] [PubMed] [Google Scholar]
- 3. Saheki Y, De Camilli P (2012) Synaptic vesicle endocytosis. Cold Spring Harb Perspect Biol 4: a005645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Clayton EL, Cousin MA (2009) The molecular physiology of activity‐dependent bulk endocytosis of synaptic vesicles. J Neurochem 111: 901–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Watanabe S, Rost BR, Camacho‐Perez M, Davis MW, Sohl‐Kielczynski B, Rosenmund C, Jorgensen EM (2013) Ultrafast endocytosis at mouse hippocampal synapses. Nature 504: 242–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu XS, McNeil BD, Xu JH, Fan JM, Xue L, Melicoff E, Adachi R, Bai L, Wu LG (2009) Ca2+ and calmodulin initiate all forms of endocytosis during depolarization at a nerve terminal. Nat Neurosci 12: 1003–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun T, Wu XS, Xu JH, McNeil BD, Pang ZP, Yang WJ, Bai L, Qadri S, Molkentin JD, Yue DT et al (2010) The role of calcium/calmodulin‐activated calcineurin in rapid and slow endocytosis at central synapses. J Neurosci 30: 11838–11847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yamashita T, Eguchi K, Saitoh N, von Gersdorff H, Takahashi T (2010) Developmental shift to a mechanism of synaptic vesicle endocytosis requiring nanodomain Ca2+ . Nat Neurosci 13: 838–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xue L, Zhang Z, McNeil BD, Luo FJ, Wu XS, Sheng JS, Shin WC, Wu LG (2012) Voltage‐dependent calcium channels at the plasma membrane, but not vesicular channels, couple exocytosis to endocytosis. Cell Rep 1: 632–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu JH, Luo FJ, Zhang Z, Xue L, Wu XS, Chiang HC, Shin W, Wu LG (2013) SNARE proteins synaptobrevin, SNAP‐25, and syntaxin are involved in rapid and slow endocytosis at synapses. Cell Rep 3: 1414–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang JZ, Davletov BA, Sudhof TC, Anderson RGW (1994) Synaptotagmin‐I is a high‐affinity receptor for Clathrin‐Ap‐2 – implications for membrane recycling. Cell 78: 751–760 [DOI] [PubMed] [Google Scholar]
- 12. Poskanzer KE, Marek KW, Sweeney ST, Davis GW (2003) Synaptotagmin I is necessary for compensatory synaptic vesicle endocytosis in vivo . Nature 426: 559–563 [DOI] [PubMed] [Google Scholar]
- 13. Yao J, Kwon SE, Gaffaney JD, Dunning FM, Chapman ER (2012) Uncoupling the roles of synaptotagmin I during endo‐ and exocytosis of synaptic vesicles. Nat Neurosci 15: 243–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kwon SE, Chapman ER (2011) Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron 70: 847–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Deak F, Schoch S, Liu XR, Sudhof TC, Kavalali ET (2004) Synaptobrevin is essential for fast synaptic‐vesicle endocytosis. Nat Cell Biol 6: 1102–1108 [DOI] [PubMed] [Google Scholar]
- 16. Hosoi N, Holt M, Sakaba T (2009) Calcium dependence of Exo‐ and endocytotic coupling at a glutamatergic synapse. Neuron 63: 216–229 [DOI] [PubMed] [Google Scholar]
- 17. Zhang Z, Wang DS, Sun T, Xu JH, Chiang HC, Shin W, Wu LG (2013) The SNARE proteins SNAP25 and synaptobrevin are involved in endocytosis at hippocampal synapses. J Neurosci 33: 9169–9175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu LG, Hamid E, Shin W, Chiang HC (2014) Exocytosis and endocytosis: modes, functions, and coupling mechanisms. Annu Rev Physiol 76: 301–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huynh DP, Scoles DR, Nguyen D, Pulst SM (2003) The autosomal recessive juvenile Parkinson disease gene product, parkin, interacts with and ubiquitinates synaptotagmin XI. Hum Mol Genet 12: 2587–2597 [DOI] [PubMed] [Google Scholar]
- 20. Inoue S, Imamura A, Okazaki Y, Yokota H, Arai M, Hayashi N, Furukawa A, Itokawa M, Oishi M (2007) Synaptotagmin XI as a candidate gene for susceptibility to schizophrenia. Am J Med Genet B 144B: 332–340 [DOI] [PubMed] [Google Scholar]
- 21. Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, Saad M, Simon‐Sanchez J, Schulte C, Lesage S, Sveinbjornsdottir S et al (2011) Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta‐analysis of genome‐wide association studies. Lancet 377: 641–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. von Poser C, Ichtchenko K, Shao XG, Rizo J, Sudhof TC (1997) The evolutionary pressure to inactivate – A subclass of synaptotagmins with an amino acid substitution that abolishes Ca2+ binding. J Biol Chem 272: 14314–14319 [DOI] [PubMed] [Google Scholar]
- 23. Gustavsson N, Han WP (2009) Calcium‐sensing beyond neurotransmitters: functions of synaptotagmins in neuroendocrine and endocrine secretion. Biosci Rep 29: 245–259 [DOI] [PubMed] [Google Scholar]
- 24. Pang ZPP, Sudhof TC (2010) Cell biology of Ca2+‐triggered exocytosis. Curr Opin Cell Biol 22: 496–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chapman ER (2002) Synaptotagmin: a Ca2+ sensor that triggers exocytosis? Nat Rev Mol Cell Bio 3: 498–508 [DOI] [PubMed] [Google Scholar]
- 26. Sudhof TC, Rothman JE (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323: 474–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang ZJ, Bhalla A, Dean C, Chapman ER, Jackson MB (2009) Synaptotagmin IV: a multifunctional regulator of peptidergic nerve terminals. Nat Neurosci 12: 163–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dai H, Shin OH, Machius M, Tomchick DR, Sudhof TC, Rizo J (2004) Structural basis for the evolutionary inactivation of Ca2+ binding to synaptotagmin 4. Nat Struct Mol Biol 11: 844–849 [DOI] [PubMed] [Google Scholar]
- 29. Mittelsteadt T, Seifert G, Alvarez‐Baron E, Steinhauser C, Becker AJ, Schoch S (2009) Differential mRNA expression patterns of the synaptotagmin gene family in the rodent brain. J Comp Neurol 512: 514–528 [DOI] [PubMed] [Google Scholar]
- 30. Huang LYM, Neher E (1996) Ca2+‐dependent exocytosis in the somata of dorsal root ganglion neurons. Neuron 17: 135–145 [DOI] [PubMed] [Google Scholar]
- 31. Zhang C, Zhou Z (2002) Ca2+‐independent but voltage‐dependent secretion in mammalian dorsal root ganglion neurons. Nat Neurosci 5: 425–430 [DOI] [PubMed] [Google Scholar]
- 32. Zhang X, Chen Y, Wang C, Huang LYM (2007) Neuronal somatic ATP release triggers neuron‐satellite glial cell communication in dorsal root ganglia. Proc Natl Acad Sci USA 104: 9864–9869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dean C, Liu H, Dunning FM, Chang PY, Jackson MB, Chapman ER (2009) Synaptotagmin‐IV modulates synaptic function and long‐term potentiation by regulating BDNF release. Nat Neurosci 12: 767–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Johnson SL, Franz C, Kuhn S, Furness DN, Ruttiger L, Munkner S, Rivolta MN, Seward EP, Herschman HR, Engel J et al (2010) Synaptotagmin IV determines the linear Ca2+ dependence of vesicle fusion at auditory ribbon synapses. Nat Neurosci 13: 45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Doherty GJ, McMahon HT (2009) Mechanisms of endocytosis. Annu Rev Biochem 78: 857–902 [DOI] [PubMed] [Google Scholar]
- 36. Schmid SL, Frolov VA (2011) Dynamin: functional design of a membrane fission catalyst. Annu Rev Cell Dev Biol 27: 79–105 [DOI] [PubMed] [Google Scholar]
- 37. Ferguson SM, De Camilli P (2012) Dynamin, a membrane‐remodelling GTPase. Nat Rev Mol Cell Biol 13: 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T (2006) Dynasore, a cell‐permeable inhibitor of dynamin. Dev Cell 10: 839–850 [DOI] [PubMed] [Google Scholar]
- 39. Newton AJ, Kirchhausen T, Murthy VN (2006) Inhibition of dynamin completely blocks compensatory synaptic vesicle endocytosis. Proc Natl Acad Sci USA 103: 17955–17960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kirchhausen T, Macia E, Pelish HE (2008) Use of dynasore, the small molecule inhibitor of dynamin, in the regulation of endocytosis. Method Enzymol 438: 77–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hill TA, Gordon CP, McGeachie AB, Venn‐Brown B, Odell LR, Chau N, Quan A, Mariana A, Sakoff JA, Chircop M et al (2009) Inhibition of dynamin mediated endocytosis by the dynoles‐synthesis and functional activity of a family of indoles. J Med Chem 52: 3762–3773 [DOI] [PubMed] [Google Scholar]
- 42. Richard JP, Leikina E, Langen R, Henne WM, Popova M, Balla T, McMahon HT, Kozlov MM, Chernomordik LV (2011) Intracellular curvature‐generating proteins in cell‐to‐cell fusion. Biochem J 440: 185–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schutze S, Machleidt T, Adam D, Schwandner R, Wiegmann K, Kruse ML, Heinrich M, Wickel M, Kronke M (1999) Inhibition of receptor internalization by monodansylcadaverine selectively blocks p55 tumor necrosis factor receptor death domain signaling. J Biol Chem 274: 10203–10212 [DOI] [PubMed] [Google Scholar]
- 44. Chen CL, Hou WH, Liu IH, Hsiao G, Huang SS, Huang JS (2009) Inhibitors of clathrin‐dependent endocytosis enhance TGFbeta signaling and responses. J Cell Sci 122: 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, MacGregor KA, Tomlin N, Pechstein A, Chau N et al (2011) Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell 146: 471–484 [DOI] [PubMed] [Google Scholar]
- 46. Park RJ, Shen HY, Liu LJ, Liu XR, Ferguson SM, De Camilli P (2013) Dynamin triple knockout cells reveal off target effects of commonly used dynamin inhibitors. J Cell Sci 126: 5305–5312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Munafo DB, Colombo MI (2001) A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci 114: 3619–3629 [DOI] [PubMed] [Google Scholar]
- 48. Lou XL, Paradise S, Ferguson SM, De Camilli P (2008) Selective saturation of slow endocytosis at a giant glutamatergic central synapse lacking dynamin 1. Proc Natl Acad Sci USA 105: 17555–17560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Clayton EL, Sue N, Smillie KJ, O'Leary T, Bache N, Cheung G, Cole AR, Wyllie DJ, Sutherland C, Robinson PJ et al (2010) Dynamin I phosphorylation by GSK3 controls activity‐dependent bulk endocytosis of synaptic vesicles. Nat Neurosci 13: 845–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Di Paolo G, Sankaranarayanan S, Wenk MR, Daniell L, Perucco E, Caldarone BJ, Flavell R, Picciotto MR, Ryan TA, Cremona O et al (2002) Decreased synaptic vesicle recycling efficiency and cognitive deficits in amphiphysin 1 knockout mice. Neuron 33: 789–804 [DOI] [PubMed] [Google Scholar]
- 51. Grant BD, Donaldson JG (2009) Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol 10: 597–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sigismund S, Confalonieri S, Ciliberto A, Polo S, Scita G, Di Fiore PP (2012) Endocytosis and signaling: cell logistics shape the eukaryotic cell plan. Physiol Rev 92: 273–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McMahon HT, Boucrot E (2011) Molecular mechanism and physiological functions of clathrin‐mediated endocytosis. Nat Rev Mol Cell Biol 12: 517–533 [DOI] [PubMed] [Google Scholar]
- 54. Duque GA, Fukuda M, Descoteaux A (2013) Synaptotagmin XI regulates phagocytosis and cytokine secretion in macrophages. J Immunol 190: 1737–1745 [DOI] [PubMed] [Google Scholar]
- 55. Haucke V, Wenk MR, Chapman ER, Farsad K, De Camilli P (2000) Dual interaction of synaptotagmin with mu 2‐and alpha‐adaptin facilitates clathrin‐coated pit nucleation. EMBO J 19: 6011–6019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jarousse N, Kelly RB (2001) The AP2 binding site of synaptotagmin 1 is not an internalization signal but a regulator of endocytosis. J Cell Biol 154: 857–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kononenko NL, Diril MK, Puchkov D, Kintscher M, Koo SJ, Pfuhl G, Winter Y, Wienisch M, Klingauf J, Breustedt J et al (2013) Compromised fidelity of endocytic synaptic vesicle protein sorting in the absence of stonin 2. Proc Natl Acad Sci USA 110: E526–E535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Walther K, Krauss M, Diril MK, Lemke S, Ricotta D, Honing S, Kaiser S, Haucke V (2001) Human stoned B interacts with AP‐2 and synaptotagmin and facilitates clathrin‐coated vesicle uncoating. EMBO Rep 2: 634–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Martina JA, Bonangelino CJ, Aguilar RC, Bonifacino JS (2001) Stonin 2: an adaptor‐like protein that interacts with components of the endocytic machinery. J Cell Biol 153: 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kaech S, Banker G (2006) Culturing hippocampal neurons. Nat Protoc 1: 2406–2415 [DOI] [PubMed] [Google Scholar]
- 61. Zhang C, Xiong W, Zheng H, Wang LC, Lu B, Zhou Z (2004) Calcium‐ and dynamin‐independent endocytosis in dorsal root ganglion neurons. Neuron 42: 225–236 [DOI] [PubMed] [Google Scholar]
- 62. Engqvist‐Goldstein AE, Zhang CX, Carreno S, Barroso C, Heuser JE, Drubin DG (2004) RNAi‐mediated Hip1R silencing results in stable association between the endocytic machinery and the actin assembly machinery. Mol Biol Cell 15: 1666–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. de Wit H, Walter AM, Milosevic I, Gulyas‐Kovacs A, Riedel D, Sorensen JB, Verhage M (2009) Synaptotagmin‐1 docks secretory vesicles to syntaxin‐1/SNAP‐25 acceptor complexes. Cell 138: 935–946 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8