

Abstract
LATS1/2 (large tumor suppressor) kinases and the Angiomotin family proteins are potent inhibitors of the YAP (yes‐associated protein) oncoprotein, but the underlying molecular mechanism is not fully understood. Here, we report for the first time that USP9X is a deubiquitinase of Angiomotin‐like 2 (AMOTL2) and that AMOTL2 mono‐ubiquitination is required for YAP inhibition. USP9X knockdown increased the LATS‐mediated phosphorylation of YAP and decreased the transcriptional output of YAP. Conversely, over‐expression of USP9X reactivated YAP in densely cultured cells. Both genetic and biochemical approaches identified AMOTL2 as a target of USP9X. AMOTL2 was found to be ubiquitinated at K347 and K408, which both reside in the protein's coiled‐coil domain. The AMOTL2 K347/408R mutant, which cannot be ubiquitinated, was impaired in its ability to inhibit YAP. Furthermore, ubiquitinated AMOTL2 can bind to the UBA domain of LATS kinase, and this domain is required for the function of LATS. Our results provide novel insights into the activation mechanisms of core Hippo pathway components.
Keywords: AMOTL2, Hippo pathway, LATS‐YAP, mono‐ubiquitination, USP9X
Subject Categories: Post-translational Modifications, Proteolysis & Proteomics; Signal Transduction
Introduction
Yes‐associated protein (YAP) is a key regulator of stem cell function and has been implicated in cancer development and progression. Acting together with the TEAD (TEA domain) family of transcription factors (among others), YAP turns on the transcriptional programs that drive proliferation, inhibit stem/progenitor cell death, and influence lineage commitment 1, 2, 3, 4. For its crucial role in cell proliferation, the activity of YAP is controlled by diverse upstream inputs. The most classic regulator of YAP is canonical Hippo signaling, which is believed to be activated by cell polarity or cell–cell junction formation. Following the establishment of such upstream signals, several protein complexes (e.g., NF2‐MST1/2–SAV1 and Scribbled–MST1/2–SAV1) activate the LATS kinase 5, 6, which directly phosphorylates and inactivates YAP 7. The LATS kinase phosphorylates YAP on five HxRxxS/T motifs 8. Of those sites, S127 is the principal target, and its phosphorylation causes YAP to be sequestered in the cytoplasm via interaction with 14‐3‐3 proteins 7, 8, 9. YAP is also heavily regulated by the status of the actin cytoskeleton: it is activated by actin polymerization or increased tension on the actin cytoskeleton, but inhibited by actin depolymerization 10, 11, 12. Both LATS‐dependent and ‐independent mechanisms have been implicated in the actin‐mediated regulation of YAP 10, 13, 14. In addition to these mechanical cues, YAP activity is also mediated by intracellular metabolites (e.g., glucose and mevalonate) 15, 16, 17, 18, 19 and extracellular ligands (e.g., LPA, LIF and Wnt) 20, 21, 22. Importantly, many (although not all) of these upstream inputs appear to converge on the LATS kinase. Therefore, gaining a better understanding of the mechanisms that regulate LATS kinase activity is an area of active investigation in the field.
Multiple groups have identified the Angiomotin family proteins as potent suppressors of YAP 23, 24, 25, 26, while some studies have paradoxically suggested that Angiomotins can activate YAP 27, 28. There are three Angiomotin proteins: Angiomotin (including the short p80 and long p130 isoforms), Angiomotin‐like 1 and Angiomotin‐like 2 (AMOTL2). AMOTL2 itself is also a transcriptional target of YAP, highlighting the particular importance of this Angiomotin family member in the negative‐feedback regulation of Hippo signaling 10. Angiomotin proteins can inactivate YAP by at least two mechanisms: First, Angiomotin proteins can sequester YAP in the cytoplasm by direct protein–protein interactions 23, 24. Second, Angiomotin proteins can also activate the LATS kinase 23, 25. Intriguingly, Angiomotin proteins themselves are phosphorylated by LATS, which in turn reinforce their interaction, forming a positive feedback loop 29, 30. Conceptually, Angiomotin proteins might play a key role in the integration of cell–cell junction signals and actin cytoskeleton‐based signals. First, Angiomotin proteins are part of the cell–cell junction complex, and they activate LATS kinase by recruiting it to the junction of the inner cell mass in pre‐implantation embryos 29, 31. Second, Angiomotin proteins bind to F‐actin, by which they are considered to become inactive. Thus, mechanical signals that damage the actin cytoskeleton will free Angiomotin proteins from the non‐functioning compartment. In addition, LATS‐mediated phosphorylation of Angiomotin proteins also loosens their interaction with F‐actin.
While the roles of phosphorylation and dephosphorylation among key components in the Hippo pathway have been extensively studied, the roles of ubiquitination and deubiquitination in this pathway are less well understood. Here, we newly identify the deubiquitinating enzyme, USP9X, as having the ability to deubiquitinate AMOTL2. In addition, we show that AMOTL2 mono‐ubiquitination at K347 and K408 can activate LATS kinase by promoting binding to the UBA domain of LATS. Our studies identify the mono‐ubiquitination and deubiquitination of AMOTL2 as a novel molecular switch in Hippo signaling.
Results
USP9X is required for the activity of YAP in the Hippo pathway
To better understand the mechanism responsible for regulating the LATS–YAP axis, we purified YAP from detached normal human retinal epithelial (RPE) cells by tandem affinity purification (streptavidin followed by S‐tag). Co‐purified proteins were visualized by silver staining and specific bands were identified by mass spectrometry. The list of identified proteins (provided in Appendix Table S1) included USP9X, which was previously reported as a LATS‐binding protein in another comprehensive proteomic screening 32. Functionally, USP9X is well known to act as an oncogene by stabilizing other oncogenes, such as Mcl‐1 33 and ERG 34, although one study conversely suggested that it may act as a tumor suppressor in KRas‐driven pancreatic cancers 35. Because the regulation of the Hippo pathway by ubiquitination and deubiquitination has not been well characterized, we chose to focus on investigating the role of USP9X in Hippo signaling.
We first examined whether USP9X could affect the activity of a YAP‐TEAD reporter. Indeed, USP9X over‐expression increased YAP‐TEAD reporter activity, while expression of a catalytically inactive mutant failed to do so (Fig 1A). Conversely, depletion of USP9X by two independent siRNAs in 293T cells decreased YAP‐TEAD reporter activity (Fig 1B). Next, we examined the impact of USP9X knockdown on YAP target gene expression in two normal human epithelial cell lines: RPE and MCF10A (a normal breast epithelial cell line). Since YAP activity is governed by cell density, we reseeded cells to either sparse or dense conditions 1 day after siRNA transfection. As expected, the levels of CTGF and Cyr61, two well‐established targets of YAP, were decreased in densely cultured cells compared to sparsely cultured cells (Fig 1C and D). Knockdown of USP9X decreased the expressions of CTGF and Cyr61 (at both protein and mRNA levels) in sparsely cultured cells (Fig 1C and D). Furthermore, USP9X knockdown reduced YAP‐TEAD reporter activity in sparsely cultured RPE cells (Fig 1E). Based on these findings, we conclude that USP9X is required for maximal YAP activity.
Figure 1. USP9X is required for YAP activity.
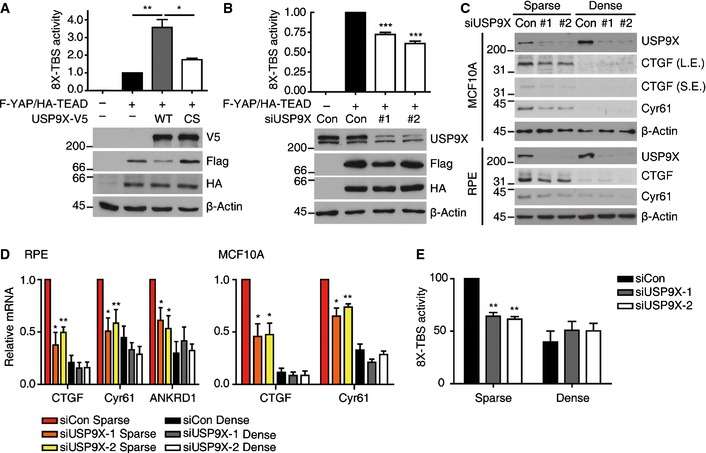
- 293T cells were co‐transfected with CMV‐Renilla, 8X‐TBS (eight repeats of the TEAD‐binding‐sequence) luciferase reporter, Flag‐YAP, HA‐TEAD2 and wild‐type (WT) or catalytically inactive (CS) USP9X‐V5. One day after transfection, luciferase activity was measured and normalized with respect to Renilla activity. The value for Flag‐YAP/HA‐TEAD‐transfected cells (2nd column) was adjusted to 1. Shown below is a representative Western blot showing that the expression levels were comparable between samples (n = 4).
- 293T cells were transfected with control or USP9X‐targeting siRNAs. One day after siRNA transfection, the cells were co‐transfected with CMV‐Renilla, 8X‐TBS luciferase, Flag‐YAP and HA‐TEAD2. Reporter activity was quantified as described in (A) (n = 4).
- RPE or MCF10A cells were transfected with the indicated siRNAs for 24 h and then reseeded to either sparse or dense culture conditions. At 48 h after siRNA transfection, the cells were harvested and cell extracts were analyzed by Western blotting for the indicated proteins. L.E., long exposure, S.E., short exposure.
- Cells were treated as described in (C), the indicated mRNAs were analyzed with RT–qPCR, and the results were normalized with respect to the β‐actin mRNA (n = 4).
- RPE cells were transfected with the indicated siRNAs for 24 h, and then co‐transfected with CMV‐Renilla and 8X‐TBS luciferase. One day after the latter transfection, the cells were reseeded to either sparse or dense conditions, and reporter activity was measured at 24 h after reseeding (n = 3).
USP9X regulates YAP phosphorylation and localization through LATS
We next investigated the localization of YAP, as changes in localization form the principal mechanism through which this protein is regulated. In sparsely growing control RPE cells, intense nuclear YAP staining was observed by immunofluorescence (Fig 2A). In USP9X‐depleted cells, in contrast, YAP was diffusely localized throughout the cell (Fig 2A), and the staining intensity of nuclear YAP was significantly decreased (Fig 2B). The reduction of nuclear YAP in USP9X‐depleted cells was also confirmed by cell fractionation experiments (Fig 2C). We also examined whether USP9X over‐expression could reactivate YAP in densely cultured cells. Since USP9X is a large protein, it was technically difficult to establish retrovirus packaging of USP9X for its over‐expression. Therefore, we first transiently transfected USP9X and detected transfected cells by V5 immunostaining. EGFP plasmid was used as a control. In densely growing cells, YAP was diffusely localized throughout the cell, and neither GFP nor catalytically inactive USP9X affected YAP localization (Fig 2D). In contrast, USP9X wild‐type transfected cells showed increased nuclear YAP staining (Fig 2D and E). We also attempted to establish stable RPE clones expressing USP9X WT or CS mutant, which we successfully obtained two independent clones for each construct. When these cells were grown to high density, USP9X WT‐over‐expressing cells showed increased levels of nuclear YAP (Fig 2F) and its target genes (Appendix Fig S1A). As an alternative approach of increasing USP9X expression, we took the advantage of recently developed CRISPR‐SAM (synergistic activation mediator) system 36. We identified three sgRNAs that effectively over‐expressed USP9X. Densely growing cells over‐expressing USP9X by sgRNA also exhibited increased amount of nuclear YAP as determined by cell fractionation (Fig 2G). Accordingly, USP9X over‐expression increased CTGF and Cyr61 protein levels in densely growing cells (Appendix Fig S1B). Importantly, USP9X siRNA transfection reduced CTGF and Cyr61 levels back, excluding the possibility of off‐target effects of sgRNAs (Appendix Fig S1C).
Figure 2. USP9X regulates YAP localization.
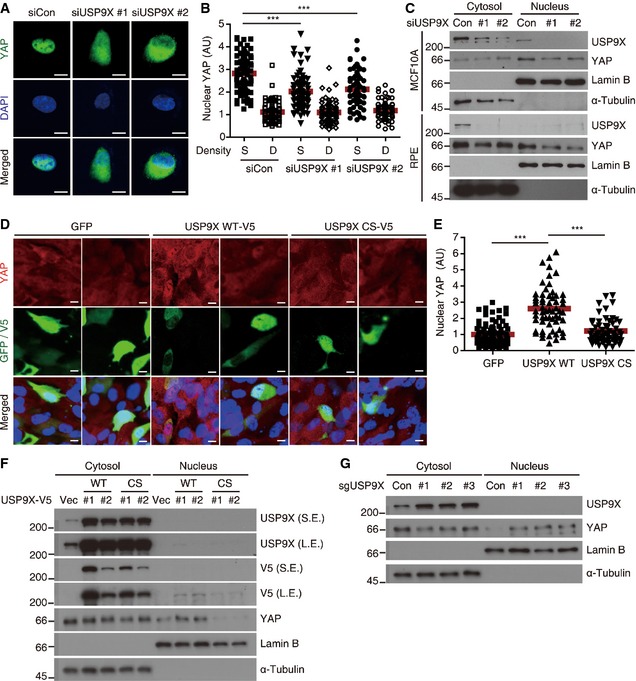
- Representative anti‐YAP immunofluorescence images of sparsely cultured RPE cells transfected with the indicated siRNAs. Green, YAP. Blue, DAPI. Scale bar, 10 μm.
- The nuclear YAP intensities of the cells shown in (A) were quantified using the ImageJ software. At least 70 cells from five or six random fields were analyzed. Quantified values were normalized by adjusting the average of “siControl Dense cells” to 1. We performed two independent experiments and obtained similar results. Data from one representative experiment are presented. The red bar indicates the average value. S, Sparse. D, Dense (***P < 0.001; paired Student's t‐test).
- Sparsely cultured RPE or MCF10A cells were transfected with the indicated siRNAs, fractionated into cytosolic and nuclear extracts, and analyzed by Western blotting for the indicated proteins.
- Representative anti‐YAP immunofluorescence images of densely cultured RPE cells transfected with the indicated plasmids. Green, GFP or V5. Red, YAP. Blue, DAPI. Scale bar, 10 μm.
- The nuclear YAP intensities of transfected cells in (D) were quantified using the ImageJ software. At least 70 cells from five to six random fields were analyzed. Quantified values were normalized by adjusting the average of “EGFP transfected cells” to 1. Similar results were obtained from two independent experiments. Data from one representative experiment are presented. The red bar indicates the average value (***P < 0.001; paired Student's t‐test).
- Densely cultured stable RPE clones integrated with empty vector (Vec), USP9X‐V5 WT or CS mutant were fractionated into cytosolic and nuclear extracts, and analyzed by Western blotting for the indicated proteins.
- Densely cultured RPE cells transduced with CRISPR‐SAM targeting USP9X were fractionated into cytosolic and nuclear extracts, and analyzed by Western blotting for the indicated proteins.
Since YAP S127 phosphorylation is the major determinant of YAP localization, we examined the phosphorylation status of YAP S127 in USP9X knockdown cells. Interestingly, USP9X knockdown markedly increased YAP S127 phosphorylation to a level comparable to that seen in densely cultured cells (Fig 3A). Conversely, over‐expression of USP9X by either stable integration of USP9X WT or CRISPR‐SAM reduced YAP S127 phosphorylation in densely cultured cells (Fig 3B and C). Because LATS1/2 kinases are the major kinases for YAP S127, we next questioned whether LATS1/2 depletion could restore the YAP S127 phosphorylation induced by USP9X knockdown. Due to the technical difficulties inherent in using siRNA to efficiently deplete three proteins, we established stable cell lines expressing USP9X shRNA (using the sequence of siRNA #2). Our results revealed that transfection of USP9X shRNA‐expressing cells with LATS1/2 siRNAs restored YAP S127 phosphorylation (Fig 3D). As an independent approach, we examined the impact of USP9X depletion in RPE cells expressing Flag‐YAP WT or the Flag‐YAP 5SA mutant, which cannot be phosphorylated by LATS. As expected, USP9X depletion decreased YAP target genes in Flag‐YAP WT‐expressing cells, but had no effect in Flag‐YAP 5SA‐expressing cells (Fig 3E and F). Consistent with these observations, cell proliferation was impaired by USP9X knockdown in control cells, whereas YAP 5SA‐expressing cells remained refractory (Fig EV1). Taken together, these results suggest that USP9X promotes YAP activity by restraining LATS kinase.
Figure 3. USP9X regulates YAP phosphorylation through LATS kinase.
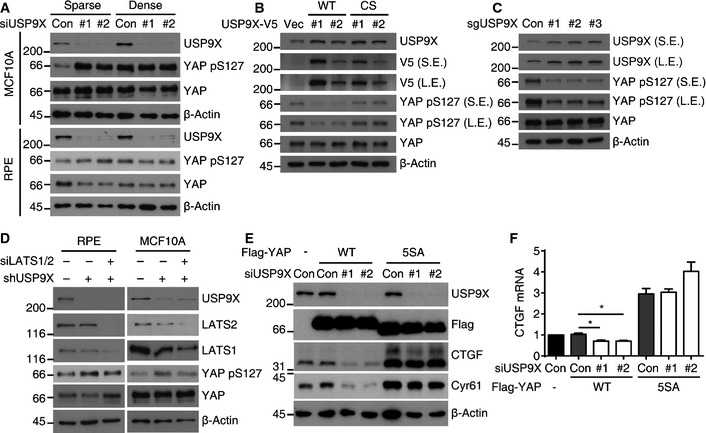
- RPE cells were analyzed by Western blotting for the indicated proteins.
- Indicated stable RPE clones in densely cultured conditions were analyzed by Western blotting for the indicated proteins.
- Densely cultured RPE cells transduced with CRISPR‐SAM targeting USP9X were analyzed by Western blotting for the indicated proteins.
- RPE or MCF10A cells expressing the control or USP9X shRNA were transfected with the LATS1/2 siRNA as indicated. Cells were reseeded to low density and harvested after one day.
- RPE cells transduced with Flag‐YAP WT or Flag‐YAP 5SA were transfected with the indicated siRNAs. Cells were reseeded to low density and harvested after one day.
- RT–qPCR of CTGF mRNAs in the cells described in (E) (n = 3). Error bars indicate the SEM (*P < 0.05, paired Student's t‐test).
Figure EV1. USP9X knockdown impairs cell proliferation in a YAP‐dependent manner.
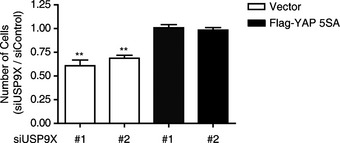
RPE cells were transduced with vector (control) or Flag‐YAP 5SA retroviruses, and then transfected with control or USP9X‐targeting siRNAs. Cells were quantified three days after siRNA transfection and normalized with respect to control siRNA‐transfected cells. USP9X knockdown impaired cell proliferation in control cells, but not in Flag‐YAP 5SA‐expressing cells (n = 4). Error bars indicate the SEM (compared with siControl cells; **P < 0.01, paired Student's t‐test).
AMOTL2 is a substrate of USP9X
To further understand the mechanism through which USP9X regulates LATS kinase, we utilized genetic and biochemical approaches. A number of LATS kinase regulators are known, including NF2, Angiomotin proteins, MST1/2–SAV1 and Rho activity. We co‐depleted each upstream regulator together with USP9X and examined YAP phosphorylation. Among the three Angiomotin family proteins, we chose Angiomotin‐like 2 (AMOTL2) for our analysis, because AMOTL2 is known to be the major isoform expressed in MCF10A cells and its loss‐of‐function phenotypes are well established 24. Co‐depletion of AMOTL2, but not SAV1 or NF2, reduced YAP phosphorylation and increased the expression of its target genes (Fig 4A and B). Using USP9X shRNA‐expressing cells, we further confirmed that an MST1/2 siRNA failed to rescue YAP phosphorylation or target gene expression (Appendix Fig S2). We also confirmed that active LATS (phospho‐LATS S909) was increased by USP9X depletion, and that only AMOTL2 co‐depletion restored it (Fig 4A and Appendix Fig S2A). Notably, co‐depletion of NF2 restored target gene expression but did not affect YAP phosphorylation, suggesting that NF2 might suppress YAP activity independent of YAP phosphorylation. In this regard, it is notable that nuclear NF2 is believed to have tumor‐suppressor function 37. Lastly, we confirmed that RhoA activity was not significantly affected by USP9X knockdown, as judged by a Rhotekin pull‐down assay (Fig EV2). Based on these genetic screening results, we speculate that AMOTL2 could be a potential target of USP9X.
Figure 4. AMOTL2 is downstream of USP9X.
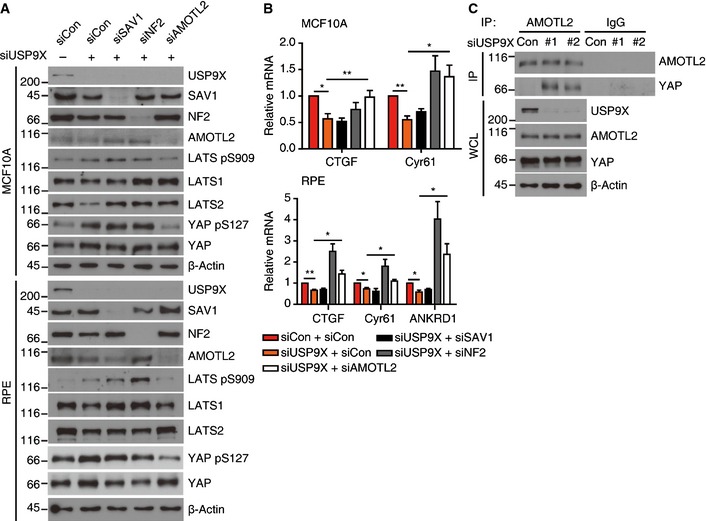
- RPE or MCF10A cells were transfected with the indicated combinations of siRNAs and reseeded to low density, and cell extracts were analyzed by Western blotting for the indicated proteins.
- The cells described in (A) were analyzed by RT–qPCR for the indicated target genes (n = 4). Error bars indicate the SEM (*P < 0.05, **P < 0.01; paired Student's t‐test).
- Sparsely cultured RPE cells transfected with the indicated siRNAs were immunoprecipitated with an anti‐AMOTL2 antibody and analyzed by Western blotting for the indicated proteins. WCL, whole‐cell lysates.
Figure EV2. RhoA activity is not affected by USP9X depletion.
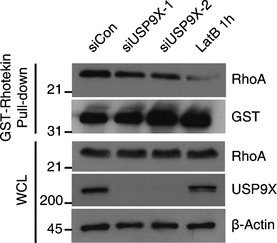
Sparsely cultured RPE cells were transfected with the indicated siRNAs, harvested, lysed and incubated with 5 μg of GST‐Rhotekin pre‐bound to 20 μl of GST beads. After incubation for 1 h at 4°C, the beads were collected and the bound proteins were analyzed by Western blotting.
In parallel, we performed in vivo ubiquitination assays against selected Hippo pathway components (AMOTL2, NF2, MST1, SAV1, LATS1/2, Mob1A, YAP and TEAD4), and tested whether the ubiquitination of each was affected by USP9X knockdown or over‐expression. Interestingly, ubiquitination of AMOTL2 was the only robust result obtained from this screen: knockdown of USP9X increased AMOTL2 ubiquitination (Fig 5A), whereas over‐expression of USP9X WT, but not the catalytically inactive mutant, decreased AMOTL2 ubiquitination (Fig 5B). We also confirmed that immunoprecipitated USP9X could deubiquitinate AMOTL2 in vitro (Appendix Fig S3). Notably, AMOTL2 ubiquitination was increased as cells became confluent (Fig 5C) and also by USP9X knockdown in sparsely cultured cells (Fig 5D). A physical interaction between USP9X and AMOTL2 was demonstrated by co‐immunoprecipitation experiments with tagged proteins in 293T cells (Fig 5E), as well as with endogenous proteins in RPE and MCF10A cells (Fig 5F). Thus, our biochemical screening also indicates that AMOTL2 is a downstream target of USP9X. Consistent with this notion, the interaction between AMOTL2 and YAP was increased by USP9X knockdown (Fig 4C).
Figure 5. AMOTL2 is a substrate of USP9X.
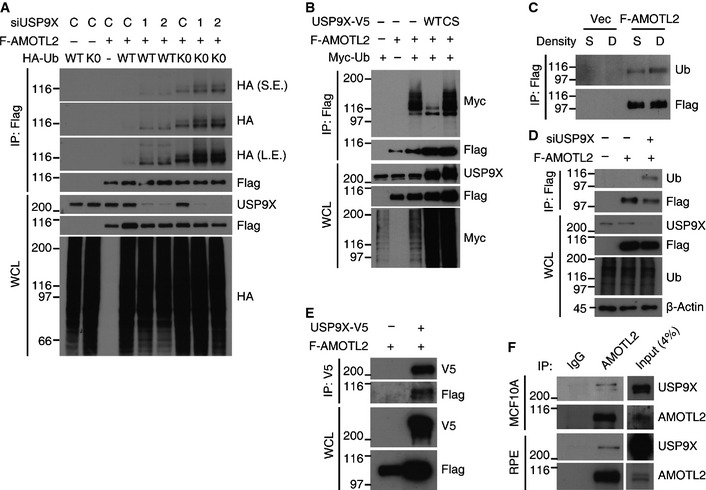
- 293T cells were transfected with the indicated siRNAs, cultured for 24 h, and then transfected with the indicated DNAs. 48 h after siRNA transfection, the cells were harvested and ubiquitination of Flag‐AMOTL2 was examined. C, control siRNA.
- 293T cells were transfected with the indicated combinations of DNAs, cultured for 24 h, and subjected to in vivo ubiquitination assays.
- RPE cells transduced with vector (control) or Flag‐AMOTL2 were seeded under sparse (S) or dense (D) conditions, and an in vivo ubiquitination assay was performed.
- RPE cells transduced with Flag‐AMOTL2 were transfected with a 1:1 mixture of the two USP9X siRNAs. One day after siRNA transfection, the cells were reseeded to the sparse condition and an in vivo ubiquitination assay was performed after one day.
- 293T cells were transfected with the indicated combinations of DNAs, and a co‐immunoprecipitation assay was performed.
- Sparsely cultured RPE or MCF10A cells were immunoprecipitated with an anti‐AMOTL2 antibody.
AMOTL2 is mono‐ubiquitinated at K347 and K408
As our results suggested that ubiquitinated AMOTL2 is the more active form, we sought to identify the ubiquitination site(s) and prove the functionality of this modification. Of note, AMOTL2 ubiquitination seems to be independent of protein stability control, as ubiquitination was readily detected in the absence of proteasome inhibitor treatment. Interestingly, our molecular weight analysis revealed that mono‐ubiquitinated AMOTL2 was by far the major ubiquitinated species, although a small portion of poly‐ubiquitinated AMOTL2 was also detected upon longer exposure (Fig 5A). Importantly, AMOTL2 was equally ubiquitinated with K0 ubiquitin mutant (which carries no lysine residues, so that it cannot be polymerized), confirming that AMOTL2 is mono‐ubiquitinated (Fig 5A, lanes 7–9).
Domain mapping experiments showed AMOTL2 to be majorly ubiquitinated within the central coiled‐coil domain (Fig 6A). Since a previously published proteomic study found that AMOTL2 K408, which resides in the coiled‐coil domain, is ubiquitinated in cells 38, we mutated this site to generate the AMOTL2 K408R mutant. This mutant was less ubiquitinated than the WT control, and showed decreased YAP binding in RPE and MCF10A cells (Fig EV3). Since residual ubiquitination was still observed in the K408R mutant, we suspected that additional site(s) should exist. Thus, we isolated the AMOTL2 coiled‐coil domain from 293T cells and analyzed it by mass spectrometry. Our results validated the ubiquitination of K408, and also newly identified ubiquitination at K347 (Fig 6B). The AMOTL2 K347 and K408 double mutant showed complete abolition of ubiquitination (Fig 6C and D) and had a significantly decreased interaction with YAP in RPE and MCF10A cells (Fig 6D). Next, we hoped to determine how AMOTL2‐YAP interaction might affect AMOTL2 ubiquitination. Thus, we generated AMOTL2 Y213A mutant which fails to interact with YAP (Fig 6E). Strikingly, the AMOTL2 Y213A mutant also failed to be ubiquitinated (Fig 6E). This result indicates that AMOTL2‐YAP interaction is required for AMOTL2 ubiquitination.
Figure 6. AMOTL2 is ubiquitinated at K347 and K408.
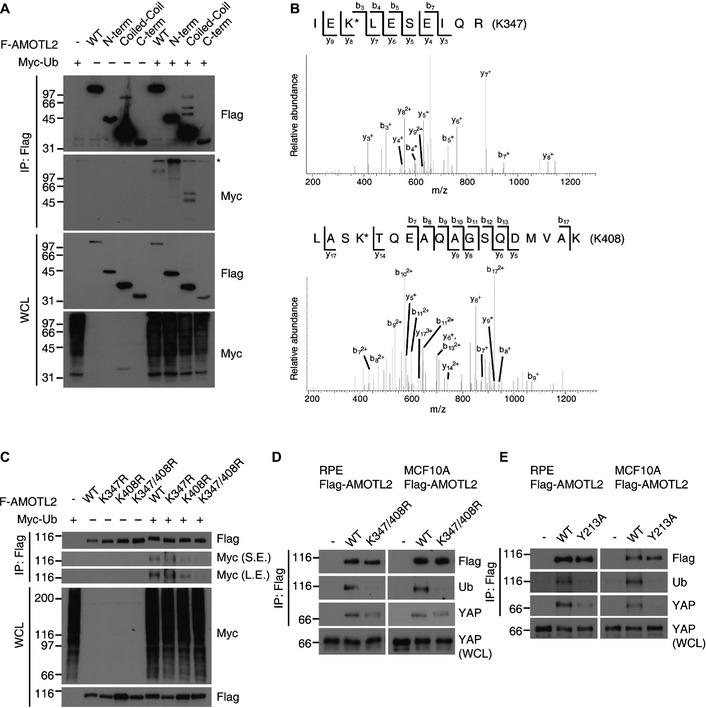
- Flag‐AMOTL2 constructs were designed to express regions spanning from the N‐terminus to the coiled‐coil domain (AA 1–306), the coiled‐coil domain (AA 307–581), and the C‐terminal portion (AA 582–781). These constructs were co‐transfected into 293T cells with Myc‐Ub, and an in vivo ubiquitination assay was conducted. *Non‐specific band.
- The SBP‐AMOTL2 coiled‐coil domain was expressed in 293T cells, subjected to tandem affinity purification, fractionated by SDS–PAGE and stained with Coomassie blue. Bands corresponding to the AMOTL2 coiled‐coil domain were excised and analyzed by mass spectrometry for identification of ubiquitinated sites (K‐G‐G linkages). Shown are MS/MS spectra recoded using an LTQ‐Orbitrap mass spectrometer for the doubly charged peptide, IEK*LESEIQR (MH+ = 1358.72, z = 2+ ; upper), and the triply charged peptide, LASK*TQEAQAGSQDMVAK (MH+ = 1992.96, z = 3+ ; lower). These peptides contain the K347 and K408 ubiquitination sites, respectively (marked by asterisks). Fragmented ions are annotated according to the nomenclature for peptide fragmentation in mass spectrometry.
- The indicated Flag‐AMOTL2 WT or mutant proteins were subjected to an in vivo ubiquitination assay in 293T cells.
- RPE or MCF10A cells were transduced with Flag‐AMOTL2 WT or Flag‐AMOTL2 K347/408R, and immunoprecipitated with an anti‐Flag antibody. Immunoprecipitated and WCL samples were analyzed by Western blotting for the indicated proteins.
- RPE or MCF10A cells were transduced with Flag‐AMOTL2 WT or Flag‐AMOTL2 Y213A, and processed as in (D).
Figure EV3. Analysis of the AMOTL2 K408R mutant.
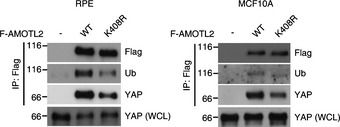
RPE or MCF10A cells were transduced with Flag‐AMOTL2 WT or the K408R mutant and immunoprecipitated with an anti‐Flag antibody. Immunoprecipitated and WCL samples were analyzed by Western blotting for the indicated proteins.
Ubiquitination of AMOTL2 at K347 and K408 is required for its function
To examine if ubiquitination of AMOTL2 is required for its tumor‐suppressor function, we performed complementation experiments with shRNA‐resistant AMOTL2 WT or K347/408R mutant. As expected, AMOTL2 knockdown decreased YAP S127 phosphorylation and increased levels of putative YAP target genes, whereas AMOTL2 WT reintroduction reverted them. However, the AMOTL2 K347/408R mutant failed to show such effects (Fig 7A). YAP activation or AMOTL2 depletion is known to trigger epithelial‐to‐mesenchymal transition. Thus, we checked the expression of epithelial and mesenchymal markers. AMOTL2 knockdown decreased epithelial marker (E‐cadherin), and increased several mesenchymal markers including N‐cadherin. Again, while AMOTL2 WT reintroduction reverted these changes, AMOTL2 K347/408R mutant failed to do so (Fig 7B). Accordingly, AMOTL2 WT rescued the increased migration of AMOTL2‐depleted cells in transwell migration assay, but AMOTL2 K347/408R mutant was ineffective (Fig 7C). Finally, we performed soft agar assay to check for tumorigenic potential of each cell line. AMOTL2‐depleted cells showed marked induction of colony‐forming ability, which was only rescued by AMOTL2 WT (Fig 7D and E). Taken altogether, these results show that mono‐ubiquitination of AMOTL2 is required for its function.
Figure 7. AMOTL2 ubiquitination is required for its function.
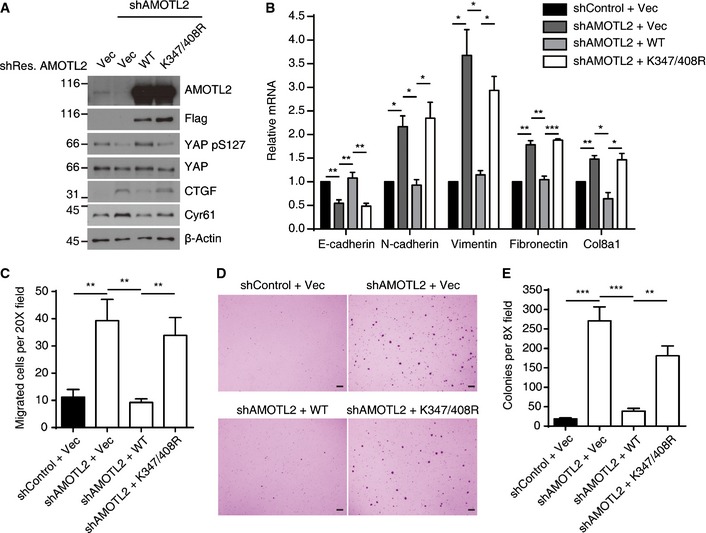
- MCF10A cells expressing shRNA against AMOTL2 were complemented with shRNA‐resistant Flag‐AMOTL2 WT or the K347/408R mutant. The indicated cells were harvested and analyzed by Western blotting for the indicated proteins.
- mRNAs isolated from cells in (A) were analyzed by RT–qPCR against indicated EMT marker genes (n = 4). Error bars indicate the SEM (*P < 0.05, **P < 0.01, ***P < 0.001; paired Student's t‐test).
- Transwell migration assay was performed using cells as in (A) (n = 8). We analyzed 5 random fields for each sample. Error bars indicate the SEM (**P < 0.01, paired Student's t‐test).
- Representative images of soft agar assay results using cells as in (A). Scale bar, 1 mm.
- Soft agar assay was performed using cells as in (A) (n = 8). Error bars indicate the SEM (**P < 0.01, *** P < 0.001, paired Student's t‐test).
Ubiquitinated AMOTL2 can bind to the LATS UBA domain
Next, we examined how AMOTL2 ubiquitination might confer its function. The LATS1/2 kinases have evolutionarily well‐conserved UBA domains at their N‐termini (Fig 8A). Such domains are known to bind ubiquitinated proteins, but no previous study has examined the ligands or functional significance of the LATS UBA domain. Prompted by our above findings, we postulated that ubiquitinated AMOTL2 could be a ligand of the LATS UBA domain. Indeed, we found that the AMOTL2 coiled‐coil domain interacted with LATS2 WT, but not with the LATS2 ΔUBA mutant (Fig 8B). Furthermore, the AMOTL2 K347/408R mutant showed reduced interaction with LATS2 (Fig 8C). Using full‐length AMOTL2, we confirmed that ΔUBA mutation of LATS2 reduces the interaction with AMOTL2 (Fig EV4). Next, to examine whether the LATS UBA domain is required for the AMOTL2‐induced activation of LATS kinase, we immunoprecipitated HA‐LATS2 WT or ΔUBA mutant proteins from 293T cell extracts co‐expressing AMOTL2, and subjected the immune complexes to a kinase assay using His‐YAP as the substrate. Our results revealed that LATS2 WT was robustly activated by AMOTL2 (lanes 2 and 4, Fig 8D), whereas LATS2 ΔUBA was only weakly affected (lanes 3 and 5, Fig 8D). Both LATS2 WT and the LATS2 ΔUBA mutant showed marginal activity toward YAP in the absence of AMOTL2 over‐expression (lanes 2 and 3, Fig 8D), confirming that AMOTL2 contributes to the activation of LATS. K0 Ub mutant activated LATS2 to a level comparable to WT Ub, confirming that mono‐ubiquitination of AMOTL2 plays a functional role (Appendix Fig S4). We also performed immune complex kinase assays using AMOTL2 WT or the AMOTL2 K347/408R mutant. Our results revealed that although AMOTL2 WT effectively increased LATS activity, the AMOTL2 K347/408R mutant was less potent in doing so (Fig 8E). To further examine the functional importance of the LATS UBA domain, we employed our previously established Lats1 −/− ; Lats2 fl/fl ; SV40 immortalized mouse embryonic fibroblasts 13. As expected, ablation of Lats2 by Cre infection abolished Yap phosphorylation and markedly induced the expression of CTGF and Cyr61 (lane 2, Fig 8F). Also consistent with our expectations, reconstitution with LATS2 WT reverted these changes (lane 3, Fig 8F). Strikingly, however, reconstitution with the LATS2 ΔUBA mutant almost completely failed to restore Yap phosphorylation and reduce the target gene expression (lane 4, Fig 8F). Based on these results, we propose that ubiquitination of AMOTL2 at K347 and K408 contributes to activation of the LATS kinase by supporting the binding of AMOTL2 to the LATS UBA domain.
Figure 8. Ubiquitinated AMOTL2 can bind to the LATS UBA domain.
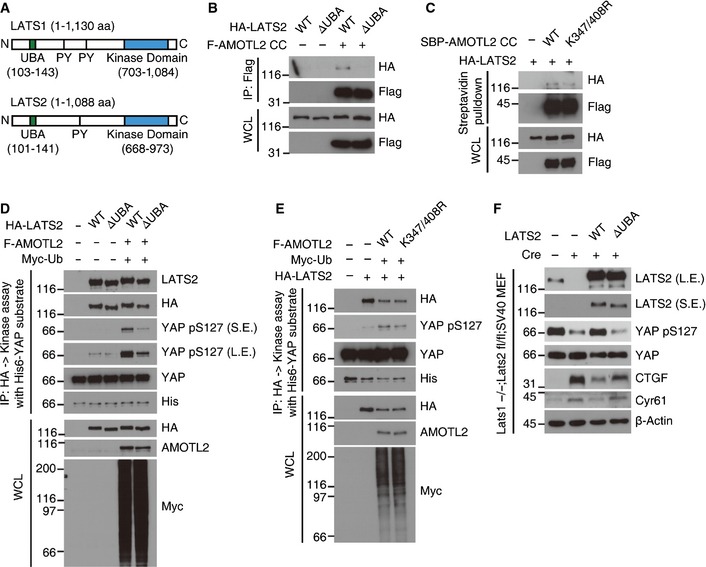
-
ASchematics of human LATS1 and LATS2. PY, PY motif.
-
B, C293T cells were transfected with the indicated DNAs and immunoprecipitated with an anti‐Flag antibody or pulled down with streptavidin‐binding beads.
-
D, E293T cells were transfected as indicated and immunoprecipitated with an anti‐HA antibody. LATS2 immune complexes were subjected to a kinase assay with His‐YAP and cold ATP.
-
FLats1 −/− ; Lats2 fl/fl ; SV40 mouse embryonic fibroblasts complemented with vector, LATS2 WT or ΔUBA mutant were infected with empty (control) or Zeocin‐Cre retroviruses and selected with 400 μg/ml Zeocin for 4 days. Harvested cells were analyzed by Western blotting for the indicated proteins.
Figure EV4. Interaction between full‐length AMOTL2 and LATS2 WT or ΔUBA mutant.
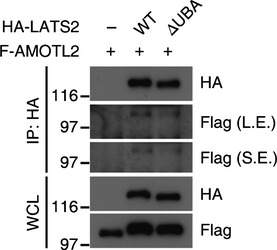
293T cells were transfected with the indicated combinations of DNAs and subjected to co‐immunoprecipitation.
Discussion
Here, we identify AMOTL2 mono‐ubiquitination, which is regulated by the deubiquitinating enzyme, USP9X, as a new regulatory layer of the Hippo pathway. Our studies with the K347/408R mutant clearly show that ubiquitination of these residues is required for the activity of AMOTL2. We also propose a working model in which ubiquitinated AMOTL2 might function (at least in part) by acting as a ligand of the LATS UBA domain (Fig 9). In cells experiencing low cell–cell contact and strong actin tension, AMOTL2 should be inactive to allow YAP‐mediated transcription. In this context, AMOTL2 is deubiquitinated by USP9X activity and probably by down‐regulation of a yet‐unidentified E3 ligase. As cells become confluent, AMOTL2 undergoes ubiquitination and binds to the UBA domain of LATS, thereby activating LATS and subsequently inhibiting YAP. In confluent cells, ubiquitination of AMOTL2 requires E3 ligase activity and (likely) functional down‐regulation of USP9X.
Figure 9. Model for the role of AMOTL2 ubiquitination in the Hippo pathway.
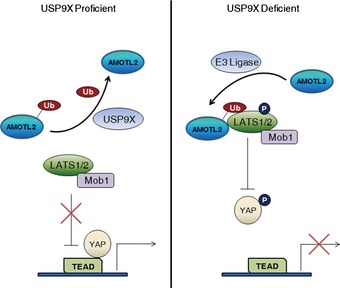
See Discussion for further details.
Our current results raise several immediate issues. First, the E3 ligase of AMOTL2 remains to be identified. This E3 ligase would be expected to have a tumor‐suppressor function. Of note, a previous study showed that the Itch E3 ligase activates Angiomotin by ubiquitinating its coiled‐coil domain 39. Although the target lysine residue identified in the previous study was not conserved in AMOTL2, it is conceivable that ubiquitination of Angiomotin proteins in the coiled‐coil domain promotes their activity in general.
Second, we need to understand how USP9X itself is regulated. Interestingly, we noticed an evolutionarily well‐conserved LATS consensus motif in USP9X (Appendix Fig S5), prompting us to speculate that USP9X activity could be inhibited by LATS‐mediated phosphorylation. In this regard, it is notable that a previous study detected USP9X‐LATS2 interaction only in the okadaic acid‐treated condition, wherein LATS2 is considered to be active 32. It is also important to note that we initially co‐purified USP9X as a YAP‐associated complex in detached cells, which activates LATS2 as well 14. Although USP9X was co‐precipitated with YAP (Fig EV5), we believe that this interaction would be an indirect consequence arising from the USP9X–LATS2–AMOTL2–YAP complex. Further investigation of USP9X as a LATS substrate is required to resolve this issue. From an evolutionary perspective, USP9X is conserved in Drosophila as the fat facets (faf) protein, but homologs of Angiomotin proteins are not found in the fly. Notably, the LATS target site is not conserved in faf. We thus propose that as the Angiomotin proteins emerged during evolution and became incorporated into Hippo signaling, faf may have coevolved to serve the feedback regulation by LATS kinase.
Figure EV5. Interaction between USP9X and YAP.
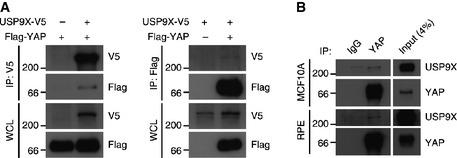
- 293T cells were transfected with the indicated combinations of DNAs and subjected to co‐immunoprecipitation with the indicated antibodies.
- Densely cultured RPE or MCF10A cells were lysed and subjected to co‐immunoprecipitation with the indicated antibodies.
Finally, our results give insights into the molecular mechanism of AMOTL2 regulation. So far, AMOTL2‐YAP interaction 23, 26, AMOTL2 phosphorylation by LATS 29, 30, and AMOTL2 mono‐ubiquitination (our study) have been found to regulate AMOTL2 function. Intriguingly, these regulatory mechanisms seem to cooperate in a positive feed‐forward manner. In the future, a systematic study using multiple AMOTL2 mutants would allow us to dissect the precise mechanism of AMOTL2 regulation.
Our findings are closely linked with previous reports on the functions of USP9X. The crosstalk between Hippo signaling and TGF‐β–Smad signaling has been well characterized 40, 41. YAP determines the localization of Smad, such that TGF‐β signaling becomes dampened when upstream Hippo signaling is active. USP9X also plays an essential role in TGF‐β–Smad signaling by deubiquitinating Smad4, which is required for assembly of the Smad2/3/4 complex 42. Given our new finding that USP9X promotes the nuclear localization of YAP, it is tempting to propose that USP9X acts as an orchestrator of Hippo–TGF‐β crosstalk. Furthermore, USP9X is over‐expressed in epithelial cancers, generally functions as an oncogene 43, 44, enhances the self‐renewal of mouse neural stem cells in vitro 45, and is needed in the brain for proper neural development 46. In the future, it would be interesting to examine whether YAP mediates these phenotypes.
Numerous regulators of YAP have been identified through the years. Although many of them converge on the LATS kinase, the molecular mechanism responsible for regulating LATS is poorly understood. In addition, the Hippo pathway is known to be regulated by phosphorylation and dephosphorylation of key components, but this is the first study to reveal the importance of mono‐ubiquitination and deubiquitination of these Hippo pathway components. In light of this, our findings offer significant insights into a new regulatory mechanism responsible for mediating the Hippo pathway, and suggest many interesting new questions that warrant future investigation.
Materials and Methods
Cell culture
RPE, MCF10A and 293T cells were purchased from ATCC. MCF10A cells were cultured in DMEM/F12 supplemented with 5% horse serum (Invitrogen), 20 ng/ml EGF (Peprotech), 10 μg/ml insulin (Sigma), 0.5 μg/ml hydrocortisone (Sigma), 100 ng/ml cholera toxin (Sigma) and penicillin/streptomycin (Invitrogen). 293T and RPE cells were cultured in DMEM or DMEM/F12 supplemented with 10% FBS and penicillin/streptomycin. Immortalized Lats1/2 mouse embryonic fibroblasts were described previously 13 and were cultured in DMEM supplemented with 10% FBS and penicillin/streptomycin. For generation of stable RPE clones, USP9X‐V5 transfected cells were selected with 5 μg/ml of blasticidin for 2 weeks and surviving clones were picked using cloning discs (Sigma).
Transfection of siRNAs
The siRNAs were synthesized by Samchully Pharm, and siRNA duplexes were annealed to a concentration of 20 μM. For transfection, the Lipofectamine RNAiMAX reagent (Invitrogen) was used according to the manufacturer's instructions. Where indicated, cells were reseeded to either sparse (3.4 × 103 cells/cm2) or dense (105 cells/cm2) culture conditions 24 h post‐transfection, and subjected to analysis 1 day thereafter. The siRNA‐targeting sequences are presented in Appendix Table S2.
Reporter assay
293T cells were cultured in 6‐well dishes and transfected with 0.25 μg Flag‐YAP, 0.25 μg HA‐TEAD2, 1 μg 8X‐TBS luciferase, 15 ng CMV‐Renilla and 1 μg USP9X‐V5 using polyethylenimine (PEI). RPE cells were cultured in 6‐well dishes and transfected with 2 μg 8X‐TBS luciferase and 10 ng CMV‐Renilla using FuGENE HD (Promega). Reporter activities were measured using a Dual Luciferase Kit (Promega) according to the manufacturer's guides.
RNA isolation and analysis
Total RNAs were isolated using the Ribo‐EX reagent (GeneALL) according to the manufacturer's instructions. Briefly, 2 μg RNA was heated at 70°C for 5 min and then reverse‐transcribed using an oligo‐dT (18dT) primer for 2 h at 37°C. The resulting cDNAs were diluted with 1 volume of water and analyzed by quantitative polymerase chain reaction (qPCR) using a Bio‐Rad Connect machine. The mRNA levels of target genes were normalized with respect to that of β‐actin. The sequences of the utilized primers are listed in Appendix Table S2.
Immunofluorescence and image acquisition
Anti‐YAP immunostaining was performed as previously described 13. Images were collected with an LSM 510 microscope using a C‐Apochromat 40×/1.2 W and C‐Apochromat 63×/1.2 W objectives, and the LSM 510 NLO acquisition software (all from Zeiss). The ImageJ software was used for image processing and signal quantification.
Co‐immunoprecipitation
Harvested cells were lysed with 0.5% Triton X‐100 buffer [50 mM Tris–Cl (pH 7.5), 150 mM NaCl, 1 mM EDTA and 0.5% Triton X‐100]. Cell extracts (1 mg in 1 ml) were incubated overnight at 4°C with 1–2 μg of the indicated antibodies. The extracts were incubated with 20 μl of protein A/G agarose beads (GenDepot) for 1 h, and then the beads were washed three times with lysis buffer (Triton X‐100 reduced to 0.1%) and boiled with Laemmli buffer.
In vivo ubiquitination assay
For assays in 293T cells, the cells were transfected with Flag‐AMOTL2 and Myc‐ubiquitin or HA‐ubiquitin at a 1:4 ratio. For assays in RPE or MCF10A cells, the cells were transduced with a Flag‐AMOTL2 retrovirus. Harvested cells were lysed with RIPA buffer [50 mM Tris–Cl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 0.5% deoxycholate, 1% NP‐40 and 0.1% SDS]. Cell extracts (1 mg of protein in 1 ml) were incubated at 4°C overnight with 2 μg of anti‐Flag antibody. The extracts were then incubated with 20 μl of protein A/G agarose beads for 1 h, and then the beads were washed five times with RIPA buffer and boiled with Laemmli buffer.
In vitro deubiquitination assay
293T cells transfected with either vector (control), USP9X‐V5 WT or USP9X‐V5 CS mutant were lysed with 0.5% Triton X‐100 lysis buffer, followed by immunoprecipitation with an anti‐V5 antibody. Beads were washed three times with lysis buffer and once with reaction buffer (50 mM HEPES (pH 7.5), 100 mM NaCl, 5% glycerol, 5 mM MgCl2, and 1 mM DTT). 15 μl of ubiquitinated AMOTL2 was added to the beads as a substrate and incubated overnight at 30°C. Reaction was terminated by adding Laemmli buffer. To prepare ubiquitinated AMOTL2, SBP‐AMOTL2 coiled‐coil and Myc‐Ub were co‐transfected to 293T cells. SBP‐AMOTL2 was pulled down with streptavidin‐binding beads, and eluted with 2 mM biotin dissolved in deubiquitination reaction buffer. Eluted proteins were aliquoted and stored at −70°C until use.
CRISPR‐SAM
CRISPR‐SAM stable cell lines were generated according to a previously described method 36. Related constructs were purchased from Addgene. For selection, we used 1 μg/ml of blasticidin, 25 μg/ml of hygromycin and 100 μg/ml of zeocin. sgRNA sequences targeting USP9X were taken from the same paper 36, and are provided in Appendix Table S2.
LATS kinase assay
Harvested cells were lysed with 0.5% Triton X‐100 buffer and the resulting lysate (1 mg of protein in a 1 ml volume) was incubated with 2 μg of anti‐HA antibody and processed for immunoprecipitation. Protein A/G agarose beads were washed three times with lysis buffer and once with LATS kinase assay buffer [25 mM HEPES (pH 7.4), 50 mM NaCl, 10 mM MgCl2, and 1 mM dithiothreitol (DTT)]. The immunoprecipitated HA‐LATS2 was reacted for 30 min at 30°C with 1 μg of recombinant His‐YAP (purified from BL21 Rosetta strain) and 200 μM of cold ATP. The reaction was stopped by boiling with an equal volume of Laemmli buffer.
Mass spectrometry for identification of YAP‐binding proteins and the ubiquitination sites of AMOTL2
RPE cells were transduced with SBP alone (streptavidin‐binding protein/S‐tag) or SBP‐YAP retroviruses and selected with 1 μg/ml of puromycin for establishment of stable cell lines. Control and SBP‐YAP‐expressing cells were trypsinized and suspended for 1 h. Harvested cells were lysed with 0.5% Triton X‐100 buffer followed by tandem affinity purification, and the obtained samples were resolved by SDS–PAGE and subjected to silver staining. To identify the ubiquitination sites on AMOTL2, SBP‐AMOTL2 coiled‐coil domain was transfected to 293T cells in 25 100‐mm dishes. Harvested cells were lysed with 0.5% Triton X‐100 buffer and subjected to tandem affinity purification. Briefly, cell lysates were divided into multiple tubes (3 mg of proteins in a 1 ml volume per tube), incubated with streptavidin‐binding beads (20 μl per tube) and incubated at 4°C overnight. The beads were washed three times with lysis buffer, and AMOTL2 was eluted with lysis buffer containing 2 mM biotin. The eluted samples were collected in a single tube and concentrated with 20 μl of S protein binding beads. After three washes with lysis buffer, the beads were boiled with Laemmli buffer, and the samples were fractionated by SDS–PAGE and stained with Coomassie blue. For mass spectrometric analysis, the stained protein bands were excised and subjected to in‐gel tryptic digestion, as previously described 47. The digested peptides were injected into a reverse‐phase Magic 18aq column (15 cm × 75 μm, 5 μm, 200Å) on an Eksigent MDLC system at a flow rate of 300 nl/min. Peptides were eluted with a linear gradient of 5–40% acetonitrile in 0.1% formic acid. The HPLC system was coupled to an LTQ XL‐Orbitrap mass spectrometer (Thermo Scientific). The source ionization parameters were as follows: spray voltage, 1.9 kV; and capillary temperature, 250°C. Survey full‐scan MS spectra (300–2,000 m/z) were acquired with a resolution of 100,000 for precursor selection and charge‐state determination. The MS/MS spectra of the 10 most intense ions from the MS scan were acquired with the following options: isolation width, 2 m/z; normalized collision energy, 35%; and dynamic exclusion duration, 30 s. The raw data were searched using the SEQUEST algorithm in Proteome Discoverer 1.4 (Thermo Scientific). The human UniProt database released in 2013.07 was used with the following search parameters: full tryptic peptide cleavage specificity; two missed cleavages; static modification of carbamidomethylation at cysteine (+ 57.021 Da); dynamic modifications of oxidation at methionine (+ 15.995 Da); and ubiquitination at lysine (+ 114.043 Da).
Nuclear‐cytoplasmic fractionation
Harvested cells were resuspended in hypotonic buffer [10 mM HEPES (pH 7.8), 10 mM KCl, 0.1 mM EDTA, 0.1 mM ethylene glycol tetraacetic acid (EGTA), 1 mM DTT] and incubated on ice for 20 min. Triton X‐100 was added to a final concentration of 0.2% and the samples were briefly vortexed for 10 s to break the plasma membrane. Nuclei were pelleted by centrifugation for 1 min, and the supernatant was saved as the cytoplasmic fraction. The nuclear pellet was washed with hypotonic buffer and PBS, and then extracted with hypertonic buffer [20 mM HEPES (pH 7.8), 0.5 mM EDTA, 0.5 mM EGTA, 1 mM DTT, 420 mM NaCl] for 30 min on ice with frequent vortexing. Nuclear debris was removed by centrifugation for 10 min. The obtained cytoplasmic and nuclear lysates were boiled with Laemmli buffer and resolved by SDS–PAGE.
Rhotekin pull‐down assay
The GST‐Rhotekin‐encoding plasmid was purchased from Addgene, and the recombinant GST‐Rhotekin protein was affinity purified from BL21 strain. Briefly, cells were lysed with 0.5% Triton X‐100 lysis buffer without EDTA, and 1 mg of cell lysate in 0.8 ml was incubated at 4°C for 2 h with 5 μg of GST‐Rhotekin bound to 20 μl of GST beads. The beads were washed three times with lysis buffer and boiled with Laemmli buffer.
Transwell migration assay
MCF10A stable cell lines were starved overnight in MCF10A growth medium containing 2% serum and no EGF. After 24 h, the starved cells were trypsinized, 1 × 105 cells were added to the top chambers of the transwell and MCF10A growth medium was added to the bottom chambers of the transwell. After incubation for 24 h, the migratory cells were fixed with methanol and mounted with DAPI.
Soft agar assay
A total of 1 ml of 0.5% bottom agar in MCF10A growth medium was solidified in 6‐well plates. 1 × 104 cells were added to 0.4% top agar in MCF10A growth medium and layered onto bottom agar. A total of 2 ml of MCF10A growth medium was added on top to prevent drying and was replenished every 4 days. After 21 days, colonies were stained with 0.1% crystal violet followed by washing with distilled water. Stained colonies were examined under a dissecting microscope.
Antibodies
Antibodies against the following proteins were used in our Western blot analyses: V5 (Life Technologies, R960‐25), Flag (WAKO, 012‐22384), HA (Covance, MMS‐101P), β‐actin (Sigma‐Aldrich, A5316), USP9X (Abnova, H00008239‐M01), CTGF (Santa Cruz Biotechnology, sc‐14939), Cyr61 (Santa Cruz Biotechnology, sc‐13100), YAP pS127 (Cell Signaling Technology, #4911), YAP (Cell Signaling Technology, #4912), LATS pS909 (Cell Signaling Technology #9157), LATS1 (Bethyl Laboratories, A300‐477A), LATS2 (Cell Signaling Technology, #5888), SAV1 9, NF2 (Santa Cruz Biotechnology, sc‐331), AMOTL2 (Santa Cruz Biotechnology, sc‐82501), Myc (Santa Cruz Biotechnology, sc‐40), ubiquitin (BD Pharmingen, 550944), His (Santa Cruz Biotechnology, sc‐803), lamin B (Santa Cruz Biotechnology, sc‐6217), α‐tubulin (Calbiochem, CP06), MST1 (Cell Signaling Technology, #3682), MST2 (Cell Signaling Technology, #3952), RhoA (Santa Cruz Biotechnology, sc‐418) and GST (Santa Cruz Biotechnology, sc‐138). For immunofluorescence analyses, an anti‐YAP antibody from Novus Biologicals (H00010413‐M01) and an anti‐V5 antibody from Sigma‐Aldrich (V8137) were used. Rabbit anti‐GFP serum was a kind gift from Eunjoon Kim. For the immunoprecipitation studies, we used the same antibodies described for the Western blot and immunofluorescence analyses.
Statistical analysis
Graphs were drawn using the GraphPad Prism software. Statistical analyses were performed using the paired Student's t‐test (two‐tailed) with a 95% confidence interval.
Author contributions
MijK and MinK conceived the project, performed the experiments and analyzed the data. D‐SL supervised the project and wrote the manuscript, together with MijK and MinK. S‐JP and CL performed the mass spectrometric analyses.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgments
We thank Dr. Stephen Woods for providing the mouse USP9X constructs and Dr. Eunjoon Kim for rabbit anti‐GFP polyclonal serum. This work was funded by a grant from the National Creative Research Program (20120001228). CL acknowledges support received from the KIST intramural program.
EMBO Reports (2016) 17: 64–78
References
- 1. Moroishi T, Hansen CG, Guan KL (2015) The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer 15: 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Piccolo S, Dupont S, Cordenonsi M (2014) The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev 94: 1287–1312 [DOI] [PubMed] [Google Scholar]
- 3. Mo JS, Park HW, Guan KL (2014) The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep 15: 642–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim M, Kim T, Johnson RL, Lim DS (2015) Transcriptional co‐repressor function of the hippo pathway transducers YAP and TAZ. Cell Rep 11: 270–282 [DOI] [PubMed] [Google Scholar]
- 5. Yin F, Yu JZ, Zheng YG, Chen Q, Zhang NL, Pan DJ (2013) Spatial organization of hippo signaling at the plasma membrane mediated by the tumor suppressor merlin/NF2. Cell 154: 1342–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A et al (2011) The hippo transducer TAZ confers cancer stem cell‐related traits on breast cancer cells. Cell 147: 759–772 [DOI] [PubMed] [Google Scholar]
- 7. Dong JX, Feldmann G, Huang JB, Wu S, Zhang NL, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan DJ (2007) Elucidation of a universal size‐control mechanism in Drosophila and mammals. Cell 130: 1120–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L et al (2007) Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev 21: 2747–2761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee JH, Kim TS, Yang TH, Koo BK, Oh SP, Lee KP, Oh HJ, Lee SH, Kong YY, Kim JM et al (2008) A crucial role of WW45 in developing epithelial tissues in the mouse. EMBO J 27: 1231–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S et al (2011) Role of YAP/TAZ in mechanotransduction. Nature 474: 179–183 [DOI] [PubMed] [Google Scholar]
- 11. Wada KI, Itoga K, Okano T, Yonemura S, Sasaki H (2011) Hippo pathway regulation by cell morphology and stress fibers. Development 138: 3907–3914 [DOI] [PubMed] [Google Scholar]
- 12. Sansores‐Garcia L, Bossuyt W, Wada KI, Yonemura S, Tao CY, Sasaki H, Halder G (2011) Modulating F‐actin organization induces organ growth by affecting the Hippo pathway. EMBO J 30: 2325–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim M, Lee S, Kuninaka S, Saya H, Lee H, Lim DS (2013) cAMP/PKA signalling reinforces the LATS‐YAP pathway to fully suppress YAP in response to actin cytoskeletal changes. EMBO J 32: 1543–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL (2012) Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev 26: 54–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. DeRan M, Yang J, Shen CH, Peters EC, Fitamant J, Chan P, Hsieh M, Zhu S, Asara JM, Zheng B et al (2014) Energy stress regulates hippo‐YAP signaling involving AMPK‐mediated regulation of angiomotin‐like 1 protein. Cell Rep 9: 495–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Z, Wu Y, Wang H, Zhang Y, Mei L, Fang X, Zhang X, Zhang F, Chen H, Liu Y et al (2014) Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc Natl Acad Sci USA 111: E89–E98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S et al (2014) Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol 16: 357–366 [DOI] [PubMed] [Google Scholar]
- 18. Wang W, Xiao ZD, Li X, Aziz KE, Gan B, Johnson RL, Chen J (2015) AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol 17: 490–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mo JS, Meng Z, Kim YC, Park HW, Hansen CG, Kim S, Lim DS, Guan KL (2015) Cellular energy stress induces AMPK‐mediated regulation of YAP and the Hippo pathway. Nat Cell Biol 17: 500–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H et al (2012) Regulation of the Hippo‐YAP pathway by G‐protein‐coupled receptor signaling. Cell 150: 780–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen DH, Sun YT, Wei YK, Zhang PJ, Rezaeian AH, Teruya‐Feldstein J, Gupta S, Liang H, Lin HK, Hung MC et al (2012) LIFR is a breast cancer metastasis suppressor upstream of the Hippo‐YAP pathway and a prognostic marker. Nat Med 18: 1511–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, Bresolin S, Frasson C, Basso G, Guzzardo V et al (2014) YAP/TAZ incorporation in the beta‐catenin destruction complex orchestrates the Wnt response. Cell 158: 157–170 [DOI] [PubMed] [Google Scholar]
- 23. Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei QY, Guan KL (2011) Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Gene Dev 25: 51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chan SW, Lim CJ, Chong YF, Pobbati AV, Huang CX, Hong WJ (2011) Hippo Pathway‐independent Restriction of TAZ and YAP by Angiomotin. J Biol Chem 286: 7018–7026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paramasivam M, Sarkeshik A, Yates JR, Fernandes MJG, McCollum D (2011) Angiomotin family proteins are novel activators of the LATS2 kinase tumor suppressor. Mol Biol Cell 22: 3725–3733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang WQ, Huang J, Chen JJ (2011) Angiomotin‐like proteins associate with and negatively regulate YAP1. J Biol Chem 286: 4364–4370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yi CL, Troutman S, Fera D, Stemmer‐Rachamimov A, Avila JL, Christian N, Persson NL, Shimono A, Speicher DW, Marmorstein R et al (2011) A tight junction‐associated merlin‐angiomotin complex mediates Merlin's regulation of mitogenic signaling and tumor suppressive functions. Cancer Cell 19: 527–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yi CL, Shen ZW, Stemmer‐Rachamimov A, Dawany N, Troutman S, Showe LC, Liu Q, Shimono A, Sudol M, Holmgren L et al (2013) The p130 isoform of angiomotin is required for yap‐mediated hepatic epithelial cell proliferation and tumorigenesis. Sci Signal 6: ra77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hirate Y, Hirahara S, Inoue K, Suzuki A, Alarcon VB, Akimoto K, Hirai T, Hara T, Adachi M, Chida K et al (2013) Polarity‐dependent distribution of angiomotin localizes Hippo signaling in preimplantation embryos. Curr Biol 23: 1181–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chan SW, Lim CJ, Guo F, Tan I, Leung T, Hong W (2013) Actin‐binding and cell proliferation activities of angiomotin family members are regulated by Hippo pathway‐mediated phosphorylation. J Biol Chem 288: 37296–37307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wells CD, Fawcett JP, Traweger A, Yamanaka Y, Goudreault M, Elder K, Kulkarni S, Gish G, Virag C, Lim C et al (2006) A Rich1/Amot complex regulates the Cdc42 GTPase and apical‐polarity proteins in epithelial cells. Cell 125: 535–548 [DOI] [PubMed] [Google Scholar]
- 32. Couzens AL, Knight JD, Kean MJ, Teo G, Weiss A, Dunham WH, Lin ZY, Bagshaw RD, Sicheri F, Pawson T et al (2013) Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase‐phosphatase interactions. Sci Signal 6: rs15 [DOI] [PubMed] [Google Scholar]
- 33. Schwickart M, Huang XD, Lill JR, Liu JF, Ferrando R, French DM, Maecker H, O'Rourke K, Bazan F, Eastham‐Anderson J et al (2010) Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463: 103–107 [DOI] [PubMed] [Google Scholar]
- 34. Wang S, Kollipara RK, Srivastava N, Li R, Ravindranathan P, Hernandez E, Freeman E, Humphries CG, Kapur P, Lotan Y et al (2014) Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer. Proc Natl Acad Sci USA 111: 4251–4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perez‐Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL, Silverstein KAT, Grutzmann R, Aust D, Rummele P et al (2012) The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 486: 266–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H et al (2015) Genome‐scale transcriptional activation by an engineered CRISPR‐Cas9 complex. Nature 517: 583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li W, You LR, Cooper J, Schiavon G, Pepe‐Caprio A, Zhou L, Ishii R, Giovannini M, Hanemann CO, Long SB et al (2010) Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell 140: 477–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ et al (2011) Systematic and quantitative assessment of the ubiquitin‐modified proteome. Mol Cell 44: 325–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Adler JJ, Johnson DE, Heller BL, Bringman LR, Ranahan WP, Conwell MD, Sun Y, Hudmon A, Wells CD (2013) Serum deprivation inhibits the transcriptional co‐activator YAP and cell growth via phosphorylation of the 130‐kDa isoform of Angiomotin by the LATS1/2 protein kinases. Proc Natl Acad Sci USA 110: 17368–17373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Varelas X, Samavarchi‐Tehrani P, Narimatsu M, Weiss A, Cockburn K, Larsen BG, Rossant J, Wrana JL (2010) The Crumbs complex couples cell density sensing to Hippo‐dependent control of the TGF‐beta‐SMAD pathway. Dev Cell 19: 831–844 [DOI] [PubMed] [Google Scholar]
- 41. Varelas X, Wrana JL (2012) Coordinating developmental signaling: novel roles for the Hippo pathway. Trends Cell Biol 22: 88–96 [DOI] [PubMed] [Google Scholar]
- 42. Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, Martello G, Stinchfield MJ, Soligo S, Morsut L et al (2009) FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell 136: 123–135 [DOI] [PubMed] [Google Scholar]
- 43. Peng J, Hu Q, Liu W, He X, Cui L, Chen X, Yang M, Liu H, Wei W, Liu S et al (2013) USP9X expression correlates with tumor progression and poor prognosis in esophageal squamous cell carcinoma. Diagn Pathol 8: 177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luise C, Capra M, Donzelli M, Mazzarol G, Jodice MG, Nuciforo P, Viale G, Di Fiore PP, Confalonieri S (2011) An atlas of altered expression of deubiquitinating enzymes in human cancer. PLoS ONE 6: e15891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jolly LA, Taylor V, Wood SA (2009) USP9X enhances the polarity and self‐renewal of embryonic stem cell‐derived neural progenitors. Mol Biol Cell 20: 2015–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stegeman S, Jolly LA, Premarathne S, Gecz J, Richards LJ, Mackay‐Sim A, Wood SA (2013) Loss of Usp9x disrupts cortical architecture, hippocampal development and TGFbeta‐mediated axonogenesis. PLoS ONE 8: e68287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kabir MH, Suh EJ, Lee C (2012) Comparative phosphoproteome analysis reveals more ERK activation in MDA‐MB‐231 than in MCF‐7. Int J Mass Spectrom 309: 1–12 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File