

Abstract
Autophagy traffics cellular components to the lysosome for degradation. Ral GTPase and the exocyst have been implicated in the regulation of stress‐induced autophagy, but it is unclear whether they are global regulators of this process. Here, we investigate Ral function in different cellular contexts in Drosophila and find that it is required for autophagy during developmentally regulated cell death in salivary glands, but does not affect starvation‐induced autophagy in the fat body. Furthermore, knockdown of exocyst subunits has a similar effect, preventing autophagy in dying cells but not in cells of starved animals. Notch activity is elevated in dying salivary glands, this change in Notch signaling is influenced by Ral, and decreased Notch function influences autophagy. These data indicate that Ral and the exocyst regulate autophagy in a context‐dependent manner, and that in dying salivary glands, Ral mediates autophagy, at least in part, by regulation of Notch.
Keywords: autophagy, cell death, Drosophila, exocyst, Notch, Ral GTPase
Subject Categories: Autophagy & Cell Death, Development & Differentiation
Introduction
Macroautophagy (autophagy) is a catabolic process during which cytoplasmic components, including organelles and long‐lived proteins, are engulfed and trafficked to the lysosomal compartment for degradation 1. Autophagy has been implicated in several diseases, including neurodegeneration and cancer 2. Autophagy plays dual roles to determine cell fate depending on cell context 3. During stress, such as nutrient deprivation or growth factor removal, autophagy promotes cellular homeostasis and survival by recycling cell components for energy production 4. Alternatively, autophagy has been shown to function in developmentally regulated cell death, as in the case of degrading larval salivary glands during Drosophila development 5.
Autophagy is regulated by upstream protein and lipid kinase complexes, and these complexes in turn influence core ubiquitin‐like conjugation pathways that control autophagosome formation around cytoplasmic cargoes 6. The serine threonine kinase Atg1 (Ulk1/2 in mammals) complex is under control of mTOR, and this is a regulatory complex that integrates nutritional status with the requirement for activation of autophagy 7. The Vps34 lipid kinase (class III phosphatidylinositol 3‐kinase in mammals) complex is required for the formation of phosphatidylinositol 3‐phosphate (PI3P) and therefore has been implicated in multiple vesicle trafficking processes, including autophagy, endocytosis, and protein secretion 8, 9, 10.
Ral is a member of the Ras superfamily of small GTPases. Ral has a variety of downstream effectors and has been implicated in several cellular processes, including gene transcription, signal transduction, actin organization, and membrane dynamics 11. Two well‐characterized Ral effectors are the exocyst components, Sec5 and Exo84 12, 13, 14. Through its interactions with these effectors, Ral plays an important role in vesicle trafficking and protein secretion. Recently, autophagy genes have been implicated in both conventional and unconventional protein secretion 15, 16. Importantly, several regulators of autophagy, including Atg6, Vps34, Atg1, and Vps15, have been shown to be required for steroid‐induced secretion of glue proteins from Drosophila salivary glands 16, 17. The requirement of autophagy genes for protein secretion suggests that there may be cross talk between the regulatory factors that control these distinct vesicle trafficking processes.
Ral has been implicated in the regulation of stress‐induced autophagy 18. Interestingly, through physical interaction studies between RalB and Ulk1/Atg1, components of the Vps34 complex, and the Exo84 exocyst subcomplex, a model has been proposed for this large protein complex in the regulation of autophagy 18. This model makes multiple predictions, including that the Exo84 subcomplex of the exocyst functions as a positive regulator of autophagy and that the Sec5 subcomplex of the exocyst functions as a suppressor of autophagy. In addition, Ral may function as a broad regulator of autophagy in multiple cell contexts. However, the function of Ral and exocyst components in the control of autophagy has not been investigated in animals under physiological conditions, and it remains unclear whether these factors are global regulators of autophagy.
Here, we demonstrate that Ral, the Ral guanine nucleotide exchange factor (GEF) Rgl, and components of the exocyst complex are required for proper larval salivary gland degradation. We found that Ral and the exocyst function in salivary gland degradation by regulating autophagy and that Ral may regulate autophagy by influencing Notch levels. By contrast, Ral and the exocyst are not necessary for autophagy in response to nutrient deprivation. These results indicate that Ral and the exocyst regulate autophagy in a context‐dependent manner.
Results
Ral and Rgl are required for salivary gland degradation
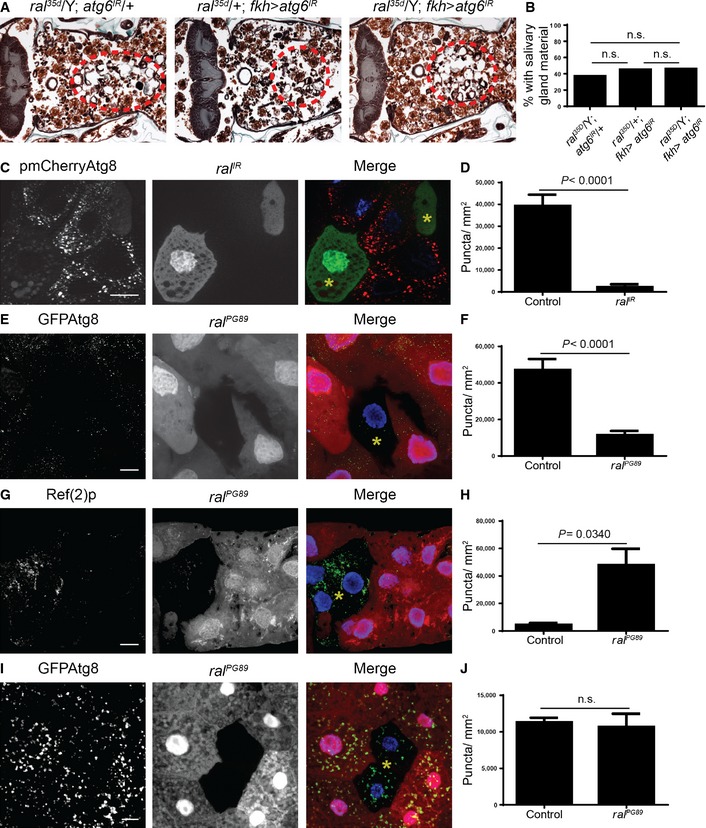
Drosophila larval salivary gland cell death is triggered by a rise in steroid at 12 h after puparium formation, and the glands are completely degraded by 16 h after puparium formation 19. A Ral mutant was identified in a screen of lethal P‐element insertions for persistent larval salivary glands, suggesting that Ral could function in salivary glands during their degradation 20. We tested whether inhibition of Ral in salivary glands would cause a salivary gland degradation defect, and found that knockdown of Ral by expression of a RNAi against ral (ral IR) in salivary glands with the salivary gland‐specific driver fkh‐GAL4 resulted in a degradation defect in 100% of pupae (Figs 1A and B, and EV1). In contrast, 20% of control animals lacking the fkh‐GAL4 driver had persistent salivary gland material at 24 h after puparium formation (Fig 1A and B). Similarly, we found that 90% of pupae with fkh‐GAL4 driving a dominant‐negative Ral, Ral S25N, had a salivary gland degradation defect compared to 15% of control pupae with no fkh‐GAL4 driver (Fig 1C and D). Strong Ral loss‐of‐function mutants are lethal early during development. Therefore, we sought to confirm whether Ral function is necessary for proper salivary gland degradation using weak ral 35d hypomorphic mutants 21. We found that 61% of ral 35d mutant pupae failed to complete salivary gland degradation, whereas most heterozygous control pupae lack salivary gland material at 24 h after puparium formation (Fig 1E and F). Since Ral is a GTPase, we tested whether activation of Ral by its GEF, Rgl, was required for salivary gland degradation. We found that salivary gland‐specific knockdown of Rgl resulted in a similar salivary gland degradation defect phenotype to both Ral knockdown and dominant‐negative Ral expression (Fig 1G and H). Combined, these data indicate that Ral and its upstream activator, Rgl, function in salivary glands during degradation.
Figure 1. Ral and Rgl are required for salivary gland degradation.
-
A, BControl animals (+/w; UAS‐ral IR/+, n = 20) and those with salivary gland‐specific knockdown of ral (fkh‐GAL4/w; UAS‐ral IR/+, n = 20) were analyzed by histology for the presence of salivary gland material (red dotted circle) 24 h after puparium formation (A). Quantification of the data is shown in (B).
-
C, DControl animals (+/w; UAS‐ral S25N/+, n = 20) and those with salivary gland‐specific expression of dominant‐negative Ral (fkh‐GAL4/w; UAS‐ral S25N/+, n = 20) were analyzed by histology for the presence of salivary gland material (red dotted circle) 24 h after puparium formation. Quantification of the data is shown in (D).
-
E, FControl animals (ral 35d/+, n = 25) and ral hypomorph mutants (ral 35d/Y, n = 23) were analyzed by histology for the presence of salivary gland material (red dotted circle) 24 h after puparium formation. Quantification of the data is shown in (F).
-
G, HControl animals (+/w; UAS‐rgl IR/+, n = 19) and those with salivary gland‐specific knockdown of rgl (fkh‐GAL4/w; UAS‐rgl IR/+, n = 20) were analyzed by histology for the presence of salivary gland material (red dotted circle) 24 h after puparium formation. Quantification of the data is shown in (H).
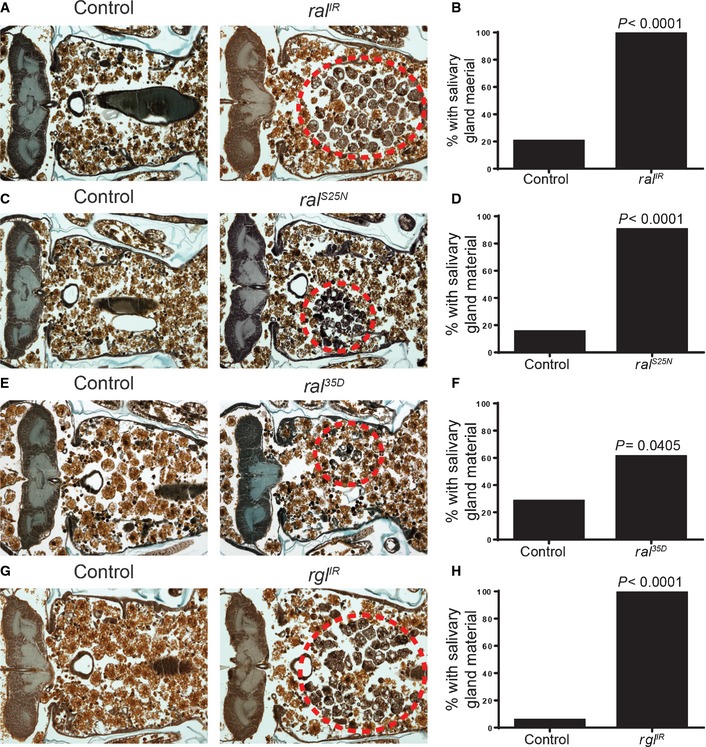
Figure EV1. Description of salivary gland degradation phenotypes observed when Ral is depleted.
- A histological section (left) 24 h after puparium formation of an animal with a strong salivary gland degradation defect (fkh‐GAL4/w; UAS‐ral IR/+). The salivary glands are partially degraded with many cellular fragments remaining. The persistent salivary gland tissue has been highlighted by removal of the surrounding tissues (right).
- A histological section (left) 24 h after puparium formation of an animal with a weak salivary gland degradation defect (ral 35D/Y). The salivary glands are mostly degraded, but a few cellular fragments persist (this time point is 8 h after salivary glands would fully degrade in a wild‐type animal). The remaining salivary gland tissue has been highlighted by removal of the surrounding tissues (right).
Ral is required for autophagy in dying salivary gland cells
The requirement for Ral during salivary gland degradation led us to investigate whether Ral functions in previously defined processes that participate in the destruction of this tissue. Salivary gland cell death requires both caspases and autophagy for complete salivary gland degradation 5. Caspases and autophagy act in an additive and parallel manner to control salivary gland cell death. Specifically, decreased function of genes in either pathway alone results in a salivary gland cell fragment phenotype, whereas inhibition of both autophagy and caspases results in an additive phenotype, with a more intact salivary gland tissue fragment phenotype 24 h after puparium formation 5.
We tested whether Ral may function in the same pathway as caspases by expressing the caspase inhibitor, p35. Expression of p35 in the ral 35d/wild‐type heterozygous genetic background resulted in persistence of salivary gland cell fragments in 57% of pupae, and gland tissue fragments in 43% of pupae (Figs 2A and B, and EV2). By contrast, expression of p35 in the hemizygous ral 35d mutant background resulted in persistence of cell fragments in 20% of pupae and gland tissue fragments in 80% of pupae (Figs 2A and B, and EV2). The enhanced gland degradation defect phenotype in the ral 35d mutants expressing p35 indicates that Ral functions in an additive manner with caspases. We further tested the relationship between Ral and caspases by knocking down Ral in salivary glands and assaying for caspase activity by staining for cleaved caspase‐3. At 0 h after puparium formation, well before caspases are activated during salivary gland cell death, both control and ral IR‐expressing cells have little to no cleaved caspase‐3 staining (Fig 2C and E). At 13 h after puparium formation, after caspases have been activated, both control and ral IR‐expressing cells have similarly increased staining for cleaved caspase‐3 (Fig 2D and E). Taken together, these data indicate that Ral is not required for caspase activity in salivary glands and that Ral and caspases function in parallel during salivary gland cell death. In addition, the presence of caspase activity at this stage in salivary glands indicates that altered ral function does not broadly influence development by impairing all of the steroid‐triggered responses that are associated with degradation of this tissue.
Figure 2. Loss of ral does not affect caspase activity during salivary gland degradation.
-
A, Bral hypomorph mutants (ral 35d/Y; UAS‐p35/+, n = 20), animals with salivary gland‐specific expression of p35 (ral 35d/+; UAS‐p35/fkh‐GAL4, n = 21), and ral hypomorph mutants with salivary gland‐specific expression of p35 (ral 35d/Y; UAS‐p35/fkh‐GAL4, n = 20) were analyzed by histology for the presence of salivary gland material (red dotted circles) 24 h after puparium formation. Quantification of the data is shown in (B). Data are represented as means. Statistical significance was determined using a chi‐square test comparing the percentages of gland fragments.
-
C–ESalivary glands dissected from 0 (C) or 13 h (D) after puparium formation control animals (+/w; UAS‐ral IR/+) and those with salivary gland‐specific knockdown of ral (fkh‐GAL4/w; UAS‐ral IR/+) and stained with cleaved caspase‐3 antibody (green) and DAPI (blue). Scale bars represent 20 μm. Quantification of the data is shown in (E). Data are represented as means ± SEM; n ≥ 10. Statistical significance was determined using a Student's t‐test.
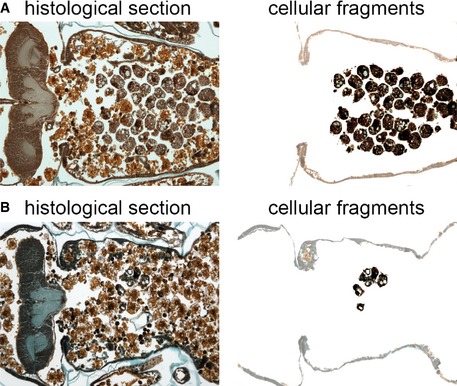
Figure EV2. Description of salivary gland degradation phenotypes.
-
A, BHistological sections (left) 24 h after puparium formation of animals with partially degraded salivary glands resulting in a cellular fragment phenotype (A) ral 35d/Y; UAS‐p35/+, (B) ral 35d/+; UAS‐p35/fkh‐GAL4. The remaining salivary gland tissue has been highlighted by removal of the surrounding tissues (right). Note the diffuse nature of the persistent salivary gland material.
-
CA histological section (left) 24 h after puparium formation of an animal with partially degraded salivary glands resulting in a gland fragment phenotype (ral 35d/Y; UAS‐p35/fkh‐GAL4). The remaining salivary gland tissue has been highlighted by removal of the surrounding tissues (right). Note that the remaining salivary gland tissue retains its basic glandular structure and is less diffuse than in (A) or (B).
We next investigated whether Ral is required for autophagy. We first asked whether the salivary gland degradation phenotype of ral 35d mutants is enhanced by knocking down the autophagy gene, Atg6. We found that 45% of animals expressing Atg6 IR had a persistent cell fragment phenotype. Similarly, 46% of ral 35d mutants expressing Atg6 IR had a cell fragment phenotype; neither group had any persistence of gland tissue fragments (Fig 3A and B). The lack of phenotypic enhancement when Atg6 IR is expressed in the ral 35d background is different from the increased persistence of gland material that we observed in ral 35d mutants expressing p35 (Fig 2A and B), and suggests that Ral and Atg6 may function in the same pathway. Consistent with our histological data suggesting that Ral functions in the autophagic pathway, clonal knockdown of ral function by expression of ral IR in salivary gland cells 14 h after puparium formation lead to a significant decrease in autophagy reporter mCherry‐Atg8a puncta formation when compared to neighboring control cells (Fig 3C and D). Similarly, loss‐of‐function ral PG89 mutant cells possess fewer autophagy reporter GFP‐Atg8a puncta (Fig 3E and F), and possess more autophagy cargo receptor Ref(2)p/p62 puncta than control cells (Fig 3G and H). Combined, these data indicate that ral is required for autophagy in dying salivary gland cells.
Figure 3. ral is required for autophagy in dying salivary glands, but not for starvation‐induced autophagy in fat body.
-
A, Bral hypomorph mutants (ral 35d/Y; UAS‐Atg6 IR/+, n = 27), animals with salivary gland‐specific expression of Atg6 IR (ral 35d/+; UAS‐Atg6 IR/fkh‐GAL4, n = 20), and ral hypomorph mutants with salivary gland‐specific expression of Atg6 IR (ral 35d/Y; UAS‐Atg6 IR/fkh‐GAL4, n = 24) were analyzed by histology for the presence of salivary gland material (red dotted circles) 24 h after puparium formation. Quantification of the data is shown in (B).
-
C, DSalivary glands dissected from 14 h after puparium formation animals expressing mCherry‐Atg8a in all cells, and ral IR specifically in GFP‐marked clone cells (yw, hsflp/w; pmCherry‐Atg8a/UAS‐ral IR; act < FRT, cd2, FRT > GAL4, UAS‐GFP/+) analyzed for mCherry‐Atg8a puncta. GFP‐marked clone ral IR‐expressing cells contain a yellow asterisk. Quantification of the data is shown in (D).
-
E, FSalivary glands dissected from 14 h after puparium formation animals that have ral loss of function (ral PG89) specifically in non‐RFP cells, and that express GFP‐Atg8a in all cells (yw, hs‐Flp, FRT19A, Ubi‐RFP/ral PG89, FRT19A; pGFP‐Atg8a) analyzed for GFP‐Atg8a puncta. ral PG89 mutant cells are marked with a yellow asterisk. Quantification of the data is shown in (F).
-
G, HSalivary glands dissected from 14 h after puparium formation animals that have ral loss of function (ral PG89) specifically in non‐RFP cells (yw, hs‐Flp, FRT19A, Ubi‐RFP/ral PG89, FRT19A) and are stained for Ref(2)p (green) and DAPI (blue). ral PG89 mutant cells are marked with a yellow asterisk. Quantification of the data is shown in (H).
-
I, JFat bodies dissected from 4 h starved animals that have ral loss of function (ral PG89) specifically in non‐RFP cells, and that express GFP‐Atg8a in all cells (yw, hs‐Flp, FRT19A, Ubi‐RFP/ral PG89, FRT19A; pGFP‐Atg8a) analyzed for GFP‐Atg8a puncta. ral PG89 mutant cells are marked with a yellow asterisk. Quantification of the data is shown in (J).
Several factors have been identified that regulate autophagy in a dying cell context‐specific manner 22, 23, 24. Since RalB was shown to be required for resource‐deprivation‐triggered autophagy in mammalian cell lines 18, this prompted us to ask whether Ral is required for starvation‐triggered autophagy in vivo. We tested this by assaying for GFP‐Atg8a puncta formation in fat bodies of starved animals with loss‐of‐function ral PG89 mutant cells. In contrast to what we observed in dying salivary gland cells, fat bodies of starved larvae had similar levels of GFP‐Atg8a puncta in both cells lacking ral PG89 function and their neighboring control cells (Fig 3I and J). This indicates that Ral functions in a context‐specific manner to regulate autophagy, as it is required for autophagy in dying salivary gland cells, but it appears to be expendable for starvation‐induced autophagy in the fat body.
The exocyst is required for autophagy associated with cell death, not starvation‐induced autophagy
Ral has several effector proteins, the best characterized are RALBP (or Rlip) which is involved in endocytosis, and the exocyst subunits, Sec5 and Exo84 25. The exocyst is an evolutionarily conserved octameric complex involved in the tethering of secretory vesicles to the plasma membrane 26. Previous studies in mammalian cell lines suggest that Ral regulates autophagy through its interactions with Sec5 and Exo84 18, 27. Since our results indicate that Ral regulates autophagy in a context‐dependent manner, we wondered whether the exocyst could be regulating autophagy in a similar way. We tested whether knockdown of exocyst subunits could affect glue peptide exocytosis from salivary glands, and identified RNAi strains against sec5, sec3, sec8, and exo84 that inhibited secretion (Fig EV3), indicating that these RNAis are functional. In addition, knockdown of ral in the salivary gland led to a similar secretion defect (Fig EV3).
Figure EV3. Ral and the exocyst are required for protein secretion in salivary glands.
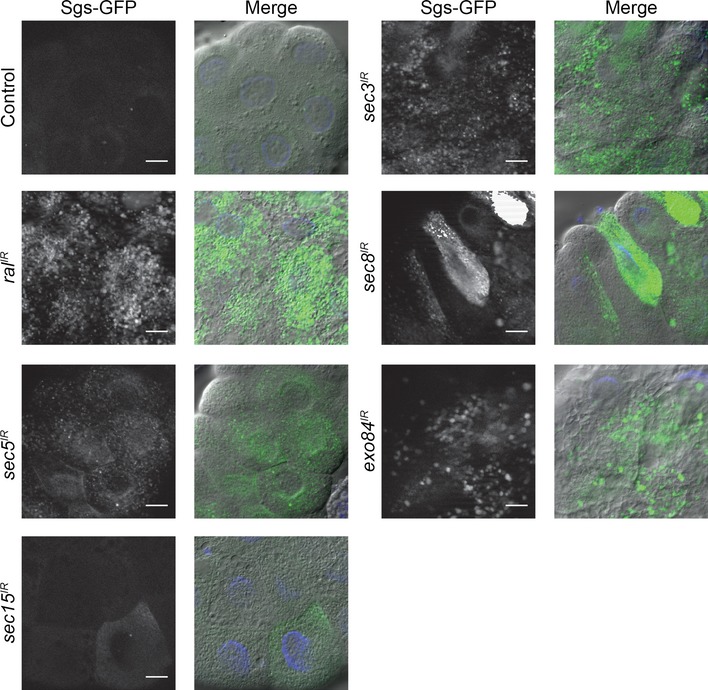
At the end of larval development, the salivary gland cells secrete a large amount of glue proteins in response to steroid signaling, and the glue is extruded at pupariation (0 h after puparium formation). Exocytosis of the glue proteins can be monitored in vivo using a transgenic fusion of the secreted glue protein, Sgs3, and GFP. Here, salivary glands dissected from 6 h after puparium formation control animals (fkh‐GAL4/+; SgsΔ3‐GFP/+), and those with salivary gland‐specific knockdown of ral (fkh‐GAL4/w; SgsΔ3‐GFP/+; UAS‐ral IR/+), sec5 (fkh‐GAL4/w; SgsΔ3‐GFP/+; UAS‐sec5 IR/+), sec15 (fkh‐GAL4/w; SgsΔ3‐GFP/+; UAS‐sec15 IR/+), sec3 (fkh‐GAL4/w; SgsΔ3‐GFP/UAS‐sec3 IR), sec8 (fkh‐GAL4/w; SgsΔ3‐GFP/UAS‐sec8 IR), exo84 (fkh‐GAL4/w; SgsΔ3‐GFP/UAS‐exo84 IR) were analyzed for the presence of SgsΔ3‐GFP. The merged images are GFP (green), Hoescht (blue), and DIC. Scale bars represent 20 μm.
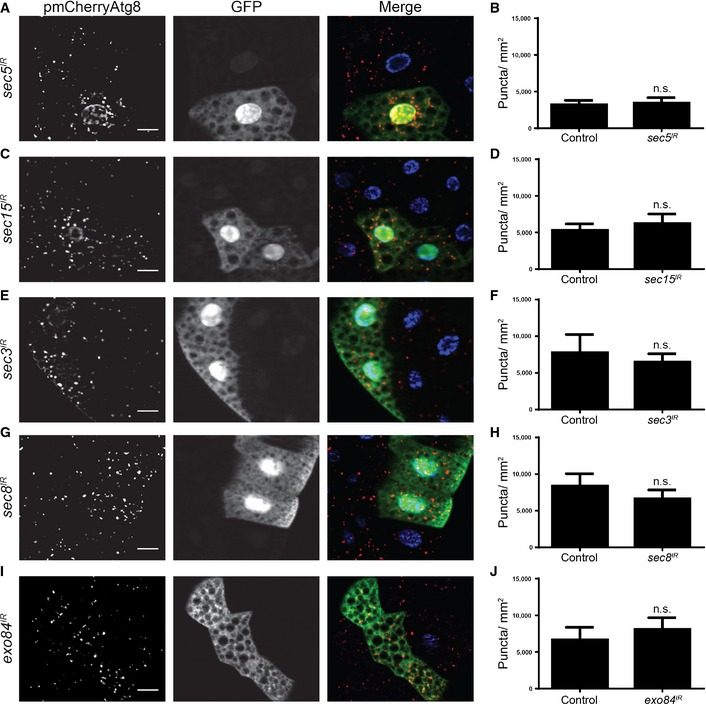
We next asked whether the exocyst could regulate starvation‐induced autophagy in the fly. We tested this by clonally knocking down several exocyst subunits in fat body cells, starving these animals, and then assaying for mCherry‐Atg8a puncta formation. Similar to our results with ral loss of function (Fig 3), we did not observe a difference in mCherry‐Atg8a puncta formation between control cells and the various exocyst subunit knockdown cells (Fig 4). Furthermore, when we clonally knocked down the exocyst subunits and checked for mCherry‐Atg8a puncta formation in the fat bodies of feeding animals, we observed no difference (Fig EV4). Taken together, these results suggest that similarly to Ral, the exocyst is not required for starvation‐induced autophagy in the fly.
Figure 4. The exocyst is not required for starvation‐induced autophagy in fat body.
-
A–JFat bodies dissected from 4‐h starved animals expressing mCherry‐Atg8a in all cells, and (A) sec5 IR, (C) sec15 IR, (E) sec3 IR, (G) sec8 IR, (I) exo84 IR specifically in GFP‐marked clone cells (yw, hsflp/w; pmCherry‐Atg8a/+; act < FRT, cd2, FRT > GAL4, UAS‐GFP/+) analyzed for mCherry‐Atg8a puncta. Quantification of the data from (A, C, E, G, I) is shown in (B, D, F, H, J), respectively.
Figure EV4. Inhibiting the exocyst does not induce ectopic autophagy in fat bodies.
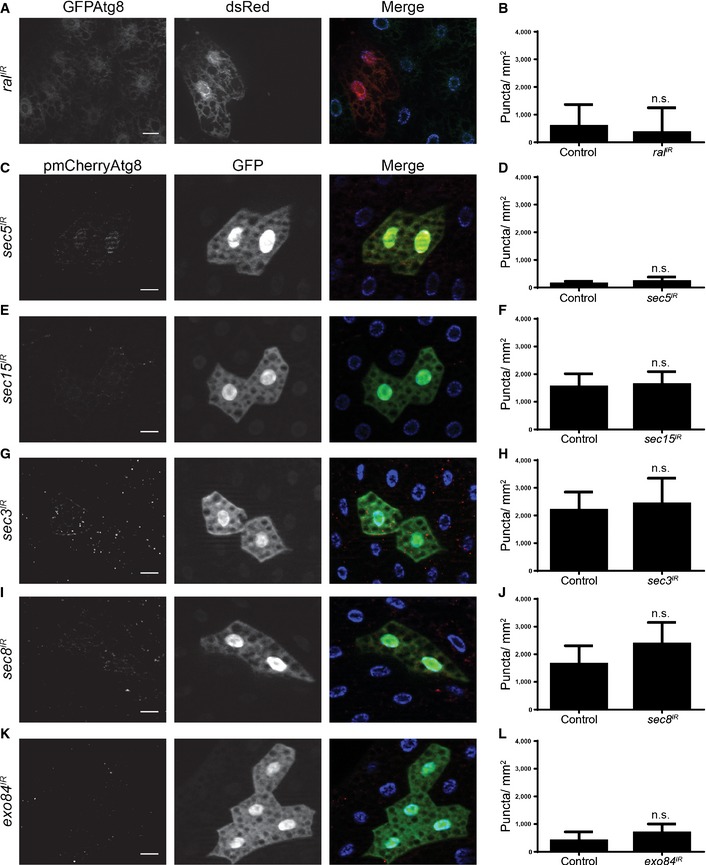
-
A, BFat body dissected from feeding larvae expressing eGFP‐Atg8a in all cells and ral IR specifically in dsRed‐marked clone cells (hsflp/w; peGFP‐Atg8a/+; act < FRT, cd2, FRT > GAL4, UAS‐dsRed/+) analyzed for eGFP‐Atg8a puncta. Quantification of the data is shown in (B).
-
C–LFat bodies dissected from feeding larvae expressing mCherry‐Atg8a in all cells, and (C) sec5 IR, (E) sec15 IR, (G) sec3 IR, (I) sec8 IR, (K) exo84 IR specifically in GFP‐marked clone cells (hsflp/w; pmCherry‐Atg8a/+; act < FRT, cd2, FRT > GAL4, UAS‐GFP/+) analyzed for mCherry‐Atg8a puncta. Quantification of the data from (C, E, G, I, K) is shown in (D, F, H, J, L), respectively.
Since the exocyst, like Ral, is not required for starvation‐induced autophagy, we next asked whether it is required for cell death‐associated autophagy. We tested this by knocking down several exocyst subunits in salivary glands and assaying for mCherry‐Atg8a puncta formation during salivary gland degradation. We found that in dying salivary glands from animals with fkh‐GAL4‐driven expression of either sec5 IR, sec15 IR, sec3 IR, sec8 IR, or exo84 IR, there were significantly fewer mCherry‐Atg8a punctae when compared to salivary glands from control animals lacking RNAi to any of the tested exocyst components (Fig 5). These data indicate that the exocyst is required for autophagy in dying salivary gland cells. In addition, we tested whether inhibition of the exocyst in salivary glands would cause a salivary gland degradation defect. Expression of either sec5 IR (Fig 6A and B), sec15 IR (Fig 6C and D), sec3 IR (Fig 6E and F), sec8 IR (Fig 6G and H), or exo84 IR (Fig 6I and J) resulted in a significantly higher percentage of animals with a salivary gland degradation defect when compared to their control animals. These results suggest that the exocyst is necessary for proper salivary gland degradation by autophagy during Drosophila development.
Figure 5. The exocyst is required for autophagy in dying salivary gland cells.
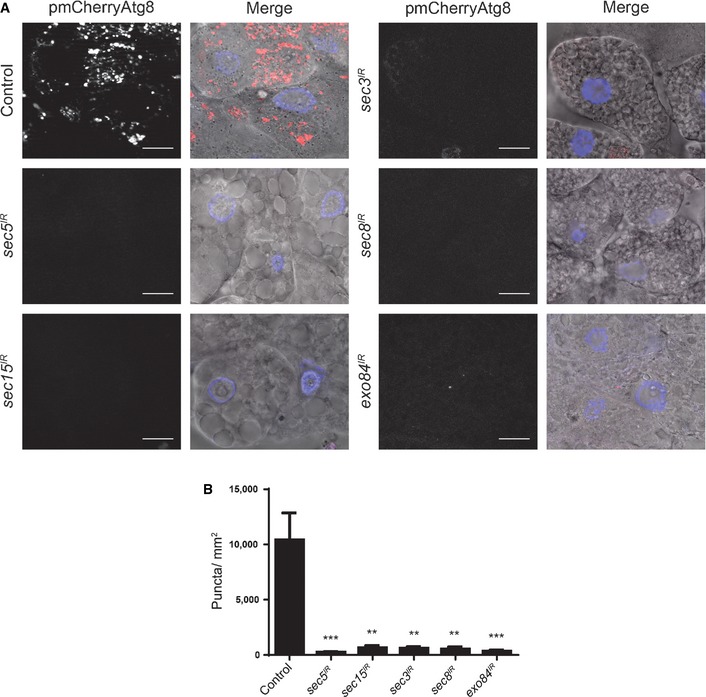
-
A, BSalivary glands dissected from 14 h after puparium formation (top left) control (fkh‐GAL4/+; pmCherry‐Atg8a/+), and those with salivary gland‐specific knockdown of (middle left) sec5 (fkh‐GAL4/w; pmCherry‐Atg8a/+; UAS‐sec5 IR/+), (bottom left) sec15 (fkh‐GAL4/w; pmCherry‐Atg8a/+; UAS‐sec15 IR/+), (top right) sec3 (fkh‐GAL4/w; pmCherry‐Atg8a/UAS‐sec3 IR), (middle right) sec8 (fkh‐GAL4/w; pmCherry‐Atg8a/UAS‐sec8 IR), (bottom right) exo84 (fkh‐GAL4/w; pmCherry‐Atg8a/UAS‐exo84 IR) analyzed for mCherry‐Atg8a puncta. Scale bars represent 20 μm. Quantification of the data is shown in (B). Data are represented as means ± SEM; n ≥ 10. Statistical significance was determined using a Student's t‐test (**P < 0.001, ***P < 0.0001).
Figure 6. The exocyst is required for salivary gland degradation.
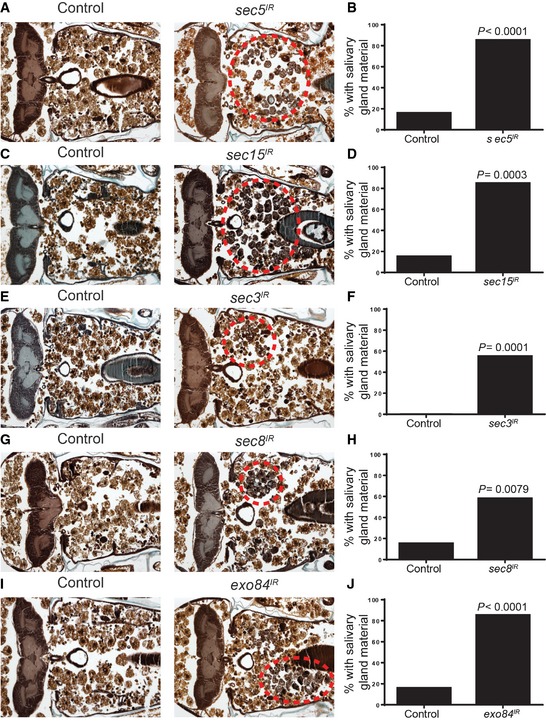
-
A, BControl animals (+/w; UAS‐sec5 IR/+, n = 19) and those with salivary gland‐specific knockdown of sec5 (fkh‐GAL4/w; UAS‐sec5 IR/+, n = 20) were analyzed by histology for the presence of salivary gland material (red dotted circle) 24 h after puparium formation. Quantification of the data is shown in (B).
-
C, DControl animals (+/w; UAS‐sec15 IR/+, n = 20) and those with salivary gland‐specific expression sec15 (fkh‐GAL4/w; UAS‐sec15 IR/+, n = 20) were analyzed by histology for the presence of salivary gland material (red dotted circle) 24 h after puparium formation. Quantification of the data is shown in (D).
-
E, FControl animals (+/w; UAS‐sec3 IR/+, n = 20) and those with salivary gland‐specific expression sec3 (fkh‐GAL4/w; UAS‐sec3 IR/+, n = 20) were analyzed by histology for the presence of salivary gland material (red dotted circle) 24 h after puparium formation. Quantification of the data is shown in (F).
-
G, HControl animals (+/w; UAS‐sec8 IR/+, n = 20) and those with salivary gland‐specific knockdown of sec8 (fkh‐GAL4/w; UAS‐sec8 IR/+, n = 19) were analyzed by histology for the presence of salivary gland material (red dotted circle) 24 h after puparium formation. Quantification of the data is shown in (H).
-
I, JControl animals (+/w; UAS‐exo84 IR/+, n = 19) and those with salivary gland‐specific knockdown of exo84 (fkh‐GAL4/w; UAS‐exo84 IR/+, n = 20) were analyzed by histology for the presence of salivary gland material (red dotted circle) 24 h after puparium formation. Quantification of the data is shown in (J).
Notch is regulated by Ral and required for autophagy in salivary glands
Previous work in Drosophila identified both the Janus Kinase/STAT and Notch pathways as regulatory targets of Ral 28, 29. Therefore, we tested whether reporters of STAT signaling 30 and Notch signaling 31 are induced in dying salivary glands. Although reporters of both of these pathways are active in salivary glands, the reporter of Notch exhibited a significant induction prior to salivary gland destruction (Fig 7A and B). Moreover, decreased function of Ral by expression of an RNAi specifically in salivary glands significantly reduced Notch reporter activity (Fig 7A and B). These data prompted us to investigate whether Notch influences autophagy in salivary glands. Significantly, clonal knockdown of notch function by expression of notch IR in salivary gland cells 14 h after puparium formation resulted in a decrease in autophagy reporter mCherry‐Atg8a puncta formation compared to neighboring control cells (Fig 7C and D). These data indicate that Ral may regulate autophagy via Notch.
Figure 7. Notch activity is regulated by ral and is required for autophagy in dying salivary gland cells.
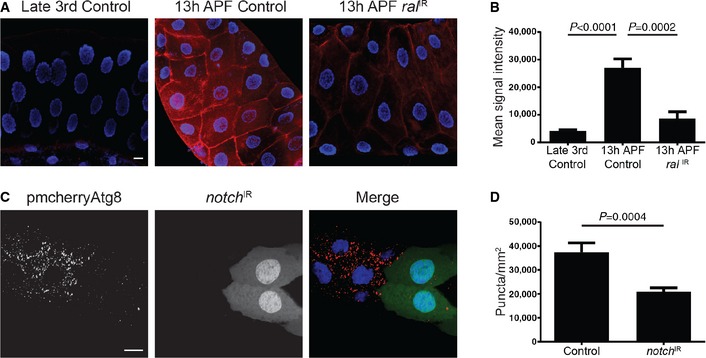
-
A, BSalivary glands dissected from late third‐instar larvae (Gbe Su(H)‐lac‐Z/w −;+;Fkh‐GAL4/+) show less notch activity (left) compared to glands from 14 h after puparium formation (APF) animals (middle). Downregulation of ral in salivary glands from 14 h APF animals (Gbe Su(H)‐lac‐Z/w −; UAS‐ral IR/+; Fkh‐GAL4/+) reduces the levels of notch activity (right). Scale bar represents 20 μm. Quantification of the data is shown in (B).
-
C, DSalivary glands dissected from 14 h after puparium formation animals expressing mCherry‐Atg8a in all cells, and notch IR specifically in GFP‐marked clone cells (yw, hsflp/w,UAS‐notch IR; pmCherry‐Atg8a/+; act < FRT, cd2, FRT > GAL4, UAS‐GFP/+) analyzed for mCherry‐Atg8a puncta. Scale bar represents 20 μm. Quantification of the data is shown in (D).
Discussion
In this study, we investigated the role of Ral GTPase in the regulation of autophagy in vivo and demonstrated that Ral regulates autophagy in the context of developmentally programmed cell death. Our data indicate that the exocyst, a downstream Ral effector, is also required for developmentally programmed cell death.
Previous studies have implicated the Ral/exocyst effector complex in the regulation of autophagy 18, 27. In mammalian cells, RalB is proposed to regulate starvation‐induced autophagy through binding of an Exo84 subcomplex of the exocyst. In contrast, we found that neither Ral nor the exocyst has an observable role in autophagy triggered by nutrient deprivation in vivo. This could be due to the apparent cell type specificity in which Ral regulates autophagy. Additionally, our data contradicts the idea of Ral regulating autophagy through distinct exocyst subcomplexes. When we knocked down the different exocyst subunits in salivary glands, we observed a similar inhibition of pmCherry‐Atg8a puncta formation across all subunits, suggesting that the exocyst may function as a whole complex during autophagic cell death. In addition, knockdown of Sec5, a key component of the so‐called autophagy suppression complex 18, failed to cause premature autophagy in the fly fat body (Fig EV4). These discrepancies could be due to either organism differences or differences in the regulation of autophagy depending on context; exocyst subcomplexes could regulate starvation‐induced autophagy, while the octameric exocyst complex regulates autophagic cell death.
It remains unclear why Ral regulates autophagy in a context‐dependent manner. Ral can be activated by Ras or Rap via its Ral‐GEF or by Ca2+/calmodulin binding 21, 32, 33, 34, 35, 36. Our data indicate that the Ral‐GEF, Rgl, is necessary for salivary gland cell death; however, this does not preclude Ca2+/calmodulin binding from being involved. Several studies have linked calcium signaling to autophagy and recent results indicate that calmodulin functions downstream of IP3 signaling during salivary gland cell death, but not during starvation‐induced autophagy in the fat body 24, 37. It could be possible that in the context of cell death, Ral is activated by both Ral‐GEF and Ca2+/calmodulin binding and potentiates these signals to promote autophagy.
Signaling downstream of Ral could also affect how Ral regulates autophagy in different cell and tissue contexts. In mammalian cells, it has been proposed that Ral regulates autophagy through mTOR 18, 27. However, mTOR is not the only factor that has been implicated in both Ral signaling and autophagy. Notch signaling has been shown to both regulate autophagy 38, 39, and to be regulated by autophagy 40, 41. Interestingly, there is evidence that Ral regulates Notch in Drosophila 29. Here, we have shown that Notch is required for autophagy in degrading salivary glands with Notch activity increasing prior to salivary gland degradation (Fig 7). Additionally, knockdown of ral reduces Notch reporter activity in salivary glands (Fig 7). These results raise the possibility that Ral regulates autophagy through the modulation of Notch levels in degrading salivary glands. Further studies are required to determine the mechanistic link between Ral, Notch, and autophagy during salivary gland cell death.
The Ral/exocyst effector complex is an important spatiotemporal regulator of several membrane trafficking processes. Our findings indicate a previously unknown role for the Ral/exocyst effector complex in the regulation of autophagy that is involved in cell death. Importantly, we have shown that Ral and the exocyst regulate autophagy in a context‐dependent manner, and question a global role for these factors in their regulation of autophagy in animals. Given recent interest in the targeting of Ral 42 and autophagy 43 for cancer therapy, it will be important to understand how Ral regulates autophagy in a variety of cell contexts.
Materials and Methods
Drosophila strains
We used the following fly stocks: Canton‐S as wild‐type control, fkh‐GAL4, w; SgsΔ3‐GFP (Bloomington Drosophila Stock Center), yw,hs‐Flp; pmCherry‐Atg8a; act > CD2 > GAL4, UAS‐GFP/TM6B, fkh‐GAL4; pmCherry‐Atg8a, yw, hs‐Flp, FRT19A, Ubi‐RFP and yw, hs‐Flp, FRT19A, Ubi‐RFP; pGFP‐Atg8a. For loss‐of‐function studies, we used ral 35d provided by J. Camonis 21 and ral PG89,FRT19A provided by J. Fischer 28. For RNAi studies, we used the following stocks from the Vienna Drosophila RNAi Center (VDRC): UAS‐ral IR VDRC Transformant ID (TID) 43622, UAS‐rgl IR TID 23639, UAS‐sec5 IR TID 28873, UAS‐sec15 IR TID 35162, UAS‐sec3 IR TID 35806, UAS‐sec8 IR TID 45032, UAS‐exo84 IR TID 108650. For ectopic expression studies, we used UAS‐ral S25N and UAS‐p35 (Bloomington Drosophila Stock Center). For the notch analyses, we used the Gbe Su(H)‐lac‐Z line 31 provided by N. Tapon and the UAS‐notch IR 44 (Bloomington Drosophila Stock Center).
Histology
Histology was performed as described previously 45.
Immunostaining and microscopy
For immunostaining, salivary glands were dissected in PBS and fixed overnight in 4% formaldehyde in PBS at 4°C. They were blocked in PBS, 1% BSA, and 0.1% Tween (PBSBT) and incubated with primary antibody (rabbit anti‐cleaved caspase‐3 1:400, Cell Signaling Technology; rabbit anti‐Ref(2)p 1:2,000, gift from G. Juhasz; rabbit anti‐beta galactosidase 1:3,000, Cappel/MP Biomedicals) overnight at 4°C. Then, salivary glands were washed in PBSBT for 2 h at room temperature, incubated with the appropriate secondary antibody for 2 h at room temperature, and washed for 1 h in PBSBT. Salivary glands were mounted in Vectashield (Vector Laboratories). For mCherry‐Atg8a and GFP‐Atg8a analysis, salivary glands and fat bodies were dissected from animals staged at 25°C in PBS and mounted in 50% glycerol in PBS containing 2 μM Hoescht stain. mCherry‐Atg8a, GFP‐Atg8a, and Ref(2)p puncta were quantified using ImageJ software. Imaging was performed on either a Zeiss Axiophot II microscope or on a Zeiss LSM700 confocal microscope.
Induction of cell clones
The induction of RNAi‐expressing cell clones was performed as described previously 22. For ral loss‐of‐function clones, females ral PG89/Fm7‐GFP flies were crossed to yw, hs‐Flp, FRT19A, Ubi‐RFP or yw, hs‐Flp, FRT19A, Ubi‐RFP; pGFP‐Atg8a male flies and were left to lay eggs for 8 h. The egg lay was heat shocked at 37°C for 1 h to induce cell clones.
Protein secretion assay
Salivary glands were dissected at 6 h after pupariation, fixed briefly with 4% formaldehyde, and stained with Hoescht.
Starvation
Feeding third‐instar larvae either remained in the food (fed), or were removed from the food and placed in a moist Petri dish for 4 h (starved) at 25°C.
Author contributions
KT, PDV, and EHB designed the experiments and wrote the manuscript. Experiments were performed by KT and PDV.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank J. Camonis, N. Tapon, J. Fischer, the Bloomington Stock Center, the Drosophila Genetic Resource Center, and the VDRC for flies, and T. Fortier for technical support. This work was supported by NIH grant GM079431 to E.H.B. E.H.B. is an Ellison Medical Foundation Scholar.
EMBO Reports (2016) 17: 110–121
References
- 1. Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147: 728–741 [DOI] [PubMed] [Google Scholar]
- 2. Mizushima N, Levine B, Cuervo AM, Klionsky DJ (2008) Autophagy fights disease through cellular self‐digestion. Nature 451: 1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. White E (2012) Deconvoluting the context‐dependent role for autophagy in cancer. Nat Rev Cancer 12: 401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB (2005) Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120: 237–248 [DOI] [PubMed] [Google Scholar]
- 5. Berry DL, Baehrecke EH (2007) Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 131: 1137–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Das G, Shravage BV, Baehrecke EH (2012) Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb Perspect Biol 4: 321–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y (2000) Tor‐mediated Induction of Autophagy Via an Apg1 Protein Kinase Complex. J Cell Biol 150: 1507–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Juhasz G, Hill JH, Yan Y, Sass M, Baehrecke EH, Backer JM, Neufeld TP (2008) The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J Cell Biol 181: 655–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schu PV, Takegawa K, Fry MJ, Stack JH, Waterfield MD, Emr SD (1993) Phosphatidylinositol 3‐kinase encoded by yeast VPS34 gene essential for protein sorting. Science 260: 88–91 [DOI] [PubMed] [Google Scholar]
- 10. Stack JH, Herman PK, Schu PV, Emr SD (1993) A membrane‐associated complex containing the Vps15 protein kinase and the Vps34 PI 3‐kinase is essential for protein sorting to the yeast lysosome‐like vacuole. EMBO J 12: 2195–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gentry LR, Martin TD, Reiner DJ, Der CJ (2014) Ral small GTPase signaling and oncogenesis: more than just 15 minutes of fame. Biochim Biophys Acta 1843: 2976–2988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moskalenko S, Henry DO, Rosse C, Mirey G, Camonis JH, White MA (2002) The exocyst is a Ral effector complex. Nat Cell Biol 4: 66–72 [DOI] [PubMed] [Google Scholar]
- 13. Moskalenko S, Tong C, Rosse C, Mirey G, Formstecher E, Daviet L, Camonis J, White MA (2003) Ral GTPases regulate exocyst assembly through dual subunit interactions. J Biol Chem 278: 51743–51748 [DOI] [PubMed] [Google Scholar]
- 14. Jin R, Junutula JR, Matern HT, Ervin KE, Scheller RH, Brunger AT (2005) Exo84 and Sec5 are competitive regulatory Sec6/8 effectors to the RalA GTPase. EMBO J 24: 2064–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Deretic V, Jiang S, Dupont N (2012) Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol 22: 397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shravage BV, Hill JH, Powers CM, Wu L, Baehrecke EH (2013) Atg6 is required for multiple vesicle trafficking pathways and hematopoiesis in Drosophila. Development 140: 1321–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Anding AL, Baehrecke EH (2015) Vps15 is required for stress induced and developmentally triggered autophagy and salivary gland protein secretion in Drosophila. Cell Death Differ 22: 457–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bodemann BO, Orvedahl A, Cheng T, Ram RR, Ou Y‐H, Formstecher E, Maiti M, Hazelett CC, Wauson EM, Balakireva M et al (2011) RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 144: 253–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee CY, Baehrecke EH (2001) Steroid regulation of autophagic programmed cell death during development. Development 128: 1443–1455 [DOI] [PubMed] [Google Scholar]
- 20. Wang L, Evans J, Andrews HK, Beckstead RB, Thummel CS, Bashirullah A (2008) A genetic screen identifies new regulators of steroid‐triggered programmed cell death in Drosophila. Genetics 180: 269–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mirey G, Balakireva M, L'Hoste S, Rossé C, Voegeling S, Camonis J (2003) A Ral guanine exchange factor‐Ral pathway is conserved in Drosophila melanogaster and sheds new light on the connectivity of the Ral, Ras, and Rap pathways. Mol Cell Biol 23: 1112–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McPhee CK, Logan MA, Freeman MR, Baehrecke EH (2010) Activation of autophagy during cell death requires the engulfment receptor Draper. Nature 465: 1093–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chang T‐K, Shravage BV, Hayes SD, Powers CM, Simin RT, Wade Harper J, Baehrecke EH (2013) Uba1 functions in Atg7‐ and Atg3‐independent autophagy. Nat Cell Biol 15: 1067–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nelson C, Ambros V, Baehrecke EH (2014) miR‐14 regulates autophagy during developmental cell death by targeting ip3‐kinase 2. Mol Cell 56: 376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Dam EM, Robinson PJ (2006) Ral: mediator of membrane trafficking. Int J Biochem Cell Biol 38: 1841–1847 [DOI] [PubMed] [Google Scholar]
- 26. Heider MR, Munson M (2012) Exorcising the exocyst complex. Traffic 13: 898–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martin TD, Chen X‐W, Kaplan REW, Saltiel AR, Walker CL, Reiner DJ, Der CJ (2014) Ral and Rheb GTPase activating proteins integrate mTOR and GTPase signaling in aging, autophagy, and tumor cell invasion. Mol Cell 53: 209–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ghiglione C, Devergne O, Cerezo D, Noselli S (2008) Drosophila RalA is essential for the maintenance of Jak/Stat signalling in ovarian follicles. EMBO Rep 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cho B, Fischer JA (2011) Ral GTPase promotes asymmetric Notch activation in the Drosophila eye in response to Frizzled/PCP signaling by repressing ligand‐independent receptor activation. Development 138: 1349–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bach EA, Ekas LA, Ayala‐Camargo A, Flaherty MS, Lee H, Perrimon N, Baeg G‐H (2007) GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo . Gene Expr Patterns 7: 323–331 [DOI] [PubMed] [Google Scholar]
- 31. Furriols M, Bray S (2001) A model Notch response element detects Suppressor of Hairless‐dependent molecular switch. Curr Biol 11: 60–64 [DOI] [PubMed] [Google Scholar]
- 32. Hofer F, Berdeaux R, Martin GS (1998) Ras‐independent activation of Ral by a Ca(2+)‐dependent pathway. Curr Biol 8: 839–842 [DOI] [PubMed] [Google Scholar]
- 33. Hofer F, Fields S, Schneider C, Martin GS (1994) Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator. PNAS 91: 11089–11093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kikuchi A, Demo SD, Ye ZH, Chen YW, Williams LT (1994) ralGDS family members interact with the effector loop of ras p21. Mol Cell Biol 14: 7483–7491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wolthuis RM, Zwartkruis F, Moen TC, Bos JL (1998) Ras‐dependent activation of the small GTPase Ral. Curr Biol 8: 471–474 [DOI] [PubMed] [Google Scholar]
- 36. Wang KL, Roufogalis BD (1999) Ca2+/calmodulin stimulates GTP binding to the ras‐related protein ral‐A. J Biol Chem 274: 14525–14528 [DOI] [PubMed] [Google Scholar]
- 37. Decuypere J‐P, Bultynck G, Parys JB (2011) A dual role for Ca(2+) in autophagy regulation. Cell Calcium 50: 242–250 [DOI] [PubMed] [Google Scholar]
- 38. Lapierre LR, Gelino S, Melendez A, Hansen M (2011) Autophagy and lipid metabolism coordinately modulate life span in germline‐less C. elegans. Curr Biol 21: 1507–1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL et al (2007) Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T‐cell leukemia. Nat Med 13: 1203–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kwon MH, Callaway H, Zhong J, Yedvobnick B (2013) A targeted genetic modifier screen links the SWI2/SNF2 protein domino to growth and autophagy genes in Drosophila melanogaster. G3 (Bethesda) 3: 815–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thumm M, Kadowaki T (2001) The loss of Drosophila APG4/AUT2 function modifies the phenotypes of cut and Notch signaling pathway mutants. Mol Genet Genomics 266: 657–663 [DOI] [PubMed] [Google Scholar]
- 42. Yan C, Liu D, Li L, Wempe MF, Guin S, Khanna M, Meier J, Hoffman B, Owens C, Wysoczynski CL et al (2014) Discovery and characterization of small molecules that target the GTPase Ral. Nature 515: 443–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cheng Y, Ren X, Hait WN, Yang J‐M (2013) Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol Rev 65: 1162–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Presente A, Shaw S, Nye JS, Andres AJ (2002) Transgene‐mediated RNA interference defines a novel role for notch in chemosensory startle behavior. Genesis 34: 165–169 [DOI] [PubMed] [Google Scholar]
- 45. Muro I, Berry DL, Huh JR, Chen CH, Huang H, Yoo SJ, Guo M, Baehrecke EH, Hay BA (2006) The Drosophila caspase Ice is important for many apoptotic cell deaths and for spermatid individualization, a nonapoptotic process. Development 133: 3305–3315 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File