

Abstract
In addition to the appearance of senile plaques and neurofibrillary tangles, Alzheimer's disease (AD) is characterized by aberrant lipid metabolism and early mitochondrial dysfunction. We recently showed that there was increased functionality of mitochondria‐associated endoplasmic reticulum (ER) membranes (MAM), a subdomain of the ER involved in lipid and cholesterol homeostasis, in presenilin‐deficient cells and in fibroblasts from familial and sporadic AD patients. Individuals carrying the ε4 allele of apolipoprotein E (ApoE4) are at increased risk for developing AD compared to those carrying ApoE3. While the reason for this increased risk is unknown, we hypothesized that it might be associated with elevated MAM function. Using an astrocyte‐conditioned media (ACM) model, we now show that ER–mitochondrial communication and MAM function—as measured by the synthesis of phospholipids and of cholesteryl esters, respectively—are increased significantly in cells treated with ApoE4‐containing ACM as compared to those treated with ApoE3‐containing ACM. Notably, this effect was seen with lipoprotein‐enriched preparations, but not with lipid‐free ApoE protein. These data are consistent with a role of upregulated MAM function in the pathogenesis of AD and may help explain, in part, the contribution of ApoE4 as a risk factor in the disease.
Keywords: ApoE, cholesterol, cholesteryl esters, endoplasmic reticulum, lipoproteins, MAM, mitochondria, phospholipids
Subject Categories: Membrane & Intracellular Transport, Molecular Biology of Disease
Introduction
Alzheimer's disease (AD), the most common form of dementia in the elderly, is characterized by progressive neuronal loss accompanied by the formation of extracellular plaques containing β‐amyloid (Aβ) and of intracellular tangles composed of hyperphosphorylated tau 1. Aβ results from the processing of the C‐terminus of the amyloid precursor protein (APP), first by β‐secretase (BACE1) and then by γ‐secretase. The active components of γ‐secretase are presenilin‐1 (PS1) and/or presenilin‐2 (PS2), which are aspartyl proteases 1.
In addition to plaques and tangles, AD is associated with an array of biochemical alterations, including perturbations in cholesterol homeostasis 2, 3, phospholipid metabolism 4, calcium trafficking 5, and mitochondrial function 6. Notably, these functions share a common feature in that they are associated with a subcompartment of the endoplasmic reticulum (ER) known as mitochondria‐associated ER membranes (MAM). MAM is a region of the ER that communicates with mitochondria, both physically and biochemically, but it has different biophysical properties than bulk ER (e.g., it is a lipid raft‐like domain 7, 8, 9). We recently showed that PS1 and PS2, and γ‐secretase activity itself, are localized predominantly in MAM 10 and that pathogenic mutations in, or genetic ablation of, PS1 and PS2 increase MAM activity significantly 7. The convergence of these upregulated MAM functions in AD, and the finding that MAM contains active γ‐secretase complexes, has led us to propose that MAM dysfunction is a key event in the pathogenesis AD 7, 11, 12.
Mutations in APP, PS1, or PS2 result in the early‐onset or familial form of AD (FAD), although these mutations account for only 1% of total AD incidence; the vast majority of AD cases are late‐onset or sporadic (SAD), and are not inherited in an autosomal dominant manner, as is the case with FAD 13. Nevertheless, SAD is characterized by the inheritance of genetically determined risk factors, polymorphisms that do not positively determine AD but rather confer an increased likelihood of the disease.
Different isoforms of APOE are the most common and validated of these risk factors 14. APOE encodes apolipoprotein E (ApoE), a component of the lipoproteins that transport cholesterol and lipids throughout the body. In the brain, cholesterol is synthesized mainly by astrocytes but not neurons, with astrocyte‐derived cholesterol delivered to neurons via high‐density lipoproteins (HDL) 15, 16. The ε4 variant of APOE, denoted here as ApoE4 (arginines at positions 112 and 158; allele frequency in Europeans of ~10–30% 17), confers an increased gene dose‐dependent risk for developing AD of ~fourfold with one copy and ~12‐fold with 2 copies compared to individuals carrying ApoE3, the most common isotype (Cys112/Arg158; allele frequency of ~70–90%), while the rarest variant, ApoE2 (Cys112/Cys158; allele frequency of ~3–12%), is protective against AD 18. While it is unclear exactly how the two coding mutations that determine the APOE genotype modulate AD risk, a proposed mechanism implicates differential ApoE‐mediated aggregation and clearance of Aβ 14, but it is still unknown whether the normal physiological function of ApoE in cholesterol and lipid homeostasis relates to increased AD risk 14.
Given that ApoE4 is the main genetic risk factor for AD and that MAM alterations in cholesterol and lipid homeostasis are features of AD, we investigated the possibility that relative to ApoE3, ApoE4 exerts effects that lead to MAM dysfunction. Specifically, we compared MAM activity of cells treated with astrocyte‐conditioned media (ACM) generated from gene replacement mouse astrocytes expressing either ApoE3 or ApoE4. We found that, compared to ApoE3 ACM, ApoE4 ACM increased the MAM activity of target cells significantly. These findings imply that ApoE can modulate ER–mitochondrial communication, which may help explain, in part, the contribution of ApoE4 to the risk of developing AD.
Results and Discussion
We developed a protocol to treat fibroblasts with ACM derived from knock‐in mice expressing either human ApoE3 or ApoE4 under the control of the endogenous mouse ApoE promoter 19. We cultured the astrocytes for 3 days 20, and then incubated human fibroblasts, which, like neurons, normally do not express ApoE 21 (Fig EV1), in ACM for 1 day 22 (see scheme in Fig 1A). Prior to all experiments, we measured the amount of ApoE present in the ACM, which typically contained more ApoE3 than ApoE4, as expected 23, 24 (Fig EV1), and then applied the ACM's on an “equal ApoE” basis. Following treatment with the ACM's, we measured various aspects of MAM function.
Figure EV1. Quantification of ApoE.
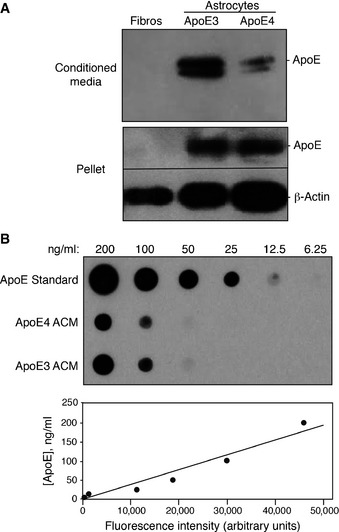
- The ApoE content of whole‐cell lysates (30 μg total protein loaded in each lane) and conditioned media (15 μl loaded in each lane) from cultured human fibroblasts and astrocytes from ApoE3‐ and ApoE4‐targeted gene replacement mice were analyzed by Western blotting.
- ApoE content of ACM and fibroblast‐conditioned media was analyzed by dot blot (upper panel). The amount of ApoE was calculated by comparison with a serial dilution of recombinant ApoE standard (lower panel). From this standard curve, the ApoE content of ApoE3 and ApoE4 ACM was determined to be ˜125 and 100 ng/ml, respectively.
Figure 1. Phospholipid synthesis in ApoE ACM‐treated cells.
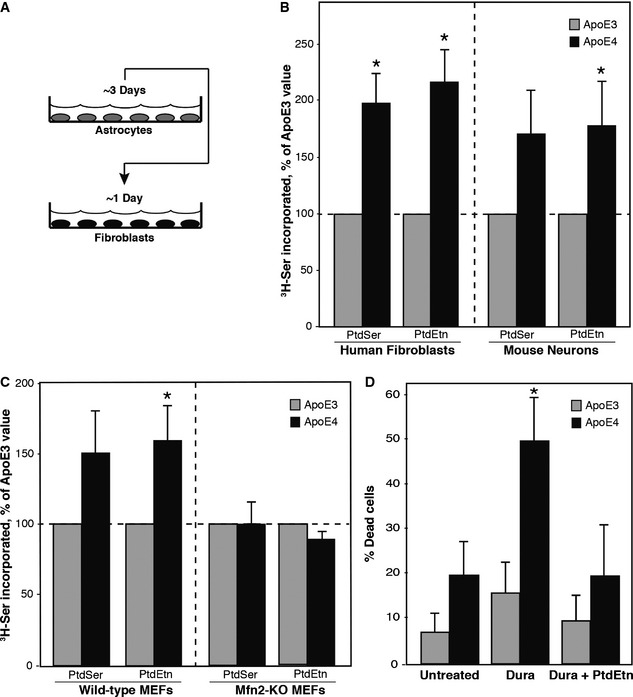
- Experimental scheme. See text for details.
- Phospholipid transport/synthesis, as measured by 3H‐serine incorporation, in human fibroblasts (mean ± SE; n = 9, with 5 replicates/experiment) and in primary mouse hippocampal neurons (mean ± SE; n = 3, with 4 or 5 replicates/experiment). *P < 0.05 vs. E3.
- Comparison of 3H‐serine incorporation in ApoE ACM‐treated WT and Mfn2‐KO MEFs (mean ± SE; n = 3, with 3 replicates/experiment). *P < 0.05 vs. E3.
- Duramycin sensitivity in ApoE ACM‐treated fibroblasts. Note increased sensitivity after 20 min to 5 μM duramycin in ApoE4‐treated cells, which was blocked upon the addition of 15 μM exogenous PtdEtn (mean ± SD; n = 3, with 3 replicates/experiment). *P < 0.05 vs. E3.
Phospholipid synthesis is a major function of MAM 25. In particular, phosphatidylserine (PtdSer) is synthesized in MAM 26. PtdSer is then transported from the ER to mitochondria, where it is decarboxylated to produce phosphatidylethanolamine (PtdEtn); PtdEtn can then be transported back to the ER, where it is either methylated to produce phosphatidylcholine (PtdCho) or distributed to other membranes of the cell 27. Although some PtdEtn is produced on the cytosolic face of the ER by CDP‐ethanolamine exchange via the Kennedy pathway 28, the majority of PtdEtn is produced by the conversion of PtdSer to PtdEtn in mitochondria 25; this conversion is an established marker for ER–mitochondrial communication 25.
We had shown previously that presenilin‐mutant cells, including AD fibroblasts, synthesized significantly more PtdSer and PtdEtn via the MAM pathway than did wild‐type controls 7. We therefore incubated normal human fibroblasts in ACM medium containing 3H‐Ser and measured the incorporation of the label into newly synthesized 3H‐PtdSer and 3H‐PtdEtn 29.
Relative to the values obtained with ApoE3 ACM, we found a significant increase in the synthesis and transport of both 3H‐PtdSer (fold increase of ~2.0 ± 0.3) and 3H‐PtdEtn (~2.2 ± 0.3) in human fibroblasts treated with ApoE4 ACM (Fig 1B). We also performed the same experiment on explanted hippocampal neurons derived from 1‐ to 3‐day‐old mice and found a significant increase in ApoE4‐mediated PtdEtn synthesis (~1.8 ± 0.5), while the increase in PtdSer trended to significance (~1.7 ± 0.7) (Fig 1B). Notably, this increase in PtdEtn synthesis did not appear to be the result of increased expression of the PtdEtn biosynthetic machinery, as the expression of phosphatidylserine decarboxylase (PISD), a key enzyme of PtdEtn synthesis that converts PtdSer to PtdEtn within the mitochondrial matrix 30, was similar in ApoE3 and ApoE4 ACM‐treated fibroblasts (Fig EV2).
Figure EV2. Western blot analysis of relevant MAM‐related proteins.
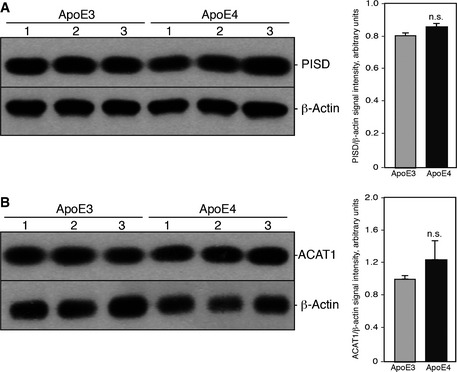
-
A, BWestern blots of 20 μg of whole‐cell lysates of human fibroblats treated with ApoE3 or ApoE4 ACM for 24 h analyzed for the detection of mitochondrial phosphatidylserine decarboxylase (PISD) (A) or ACAT1 (B) in triplicate. Quantitation is shown on the right; n.s., not significant.
To establish that the increase in phospholipid production was indeed mediated by MAM, we performed the same assay using mouse embryonic fibroblasts (MEFs) in which the mitofusin 2 gene (Mfn2) had been knocked out 30. Mfn2, in addition to its role in mitochondrial fusion, tethers ER to mitochondria 31; importantly, genetic ablation of Mfn2 reduces this connectivity 31. Consistent with this loss in interorganellar communication, we had previously established that PtdSer and PtdEtn production is reduced significantly in Mfn2‐KO MEFs 7. We therefore asked whether the ApoE4‐mediated increase in phospholipid synthesis could be abrogated in Mfn2‐KO cells. Similar to what we observed in the human fibroblasts 31, treatment of wild‐type MEFs with ApoE4 ACM resulted in an increase in PtdSer (~1.5 ± 0.3) and PtdEtn (~1.6 ± 0.2) production compared to ApoE3 ACM (Fig 1C). However, we found no such increase in either PtdSer (~1.0 ± 0.2) or PtdEtn (~0.9 ± 0.1) production in the Mfn2‐KO MEFs (Fig 1C), strongly suggesting that the ApoE4‐mediated effect on phospholipid metabolism was indeed the result of increased ER–mitochondrial communication.
Phospholipids produced at the MAM, including PtdEtn, are subsequently transported to the other membranous compartments of the cell, including the plasma membrane. Cell surface PtdEtn can be detected using cinnamycin and duramycin, two highly related lantibiotics that bind specifically to PtdEtn on the cell surface and induce cell death in a PtdEtn concentration‐dependent manner 32, 33. We had previously shown that the elevated levels of PtdEtn produced by presenilin‐mutant cells, including AD fibroblasts, were significantly more sensitive to both cinnamycin and duramycin than were wild‐type controls 7. To test whether there was also increased lantibiotic sensitivity in our experimental paradigm, we applied duramycin to ACM‐treated fibroblasts and measured cell death. We found that ApoE4 ACM‐treated fibroblasts were significantly more sensitive to duramycin treatment (~50 ± 20% cell death) than were ApoE3 ACM‐treated cells (~15 ± 7%; Fig 1D). The ability of duramycin to induce cell death was abrogated by the addition of exogenous PtdEtn to the medium (Fig 1D), supporting the specificity of the assay. Thus, these data are consistent with the 3H‐serine incorporation data and support the conclusion that ApoE4 plays a role in MAM‐mediated phospholipid synthesis and transport.
ApoE is present in the brain as a component of HDL‐like particles that are secreted by astrocytes and are taken up by neurons by various lipoprotein receptors 15, 16. Following receptor‐mediated endocytosis, the ApoE‐containing HDL‐like particles are hydrolyzed in the lysosome, and the remaining ApoE is either degraded intracellularly or re‐lipidated in the secretory pathway or at the cell surface 34. The activity of ApoE largely depends upon its lipidation state, as the lipid‐free form of ApoE (“free ApoE”) affects cellular activities differently than does ApoE as a component of assembled lipoproteins 34, 35, 36.
We therefore examined whether the increase in MAM‐mediated phospholipid synthesis seen in ApoE4 ACM‐treated cells was the result of the effect of ApoE4 as a component of HDLs as opposed to an effect due to ApoE as the free protein. Specifically, we prepared lipoprotein‐rich and lipoprotein‐free fractions from ApoE3 ACM and ApoE4 ACM by density ultracentrifugation (purification scheme in Fig 2A) 20, isolated successive fractions from the gradient, and then added selected fractions either containing (e.g., fraction 5, Fig 2B) or not containing (e.g., fraction 3, Fig 2B) ApoE (Fig EV1) to apolipoprotein‐free medium, in order to generate a “surrogate” ACM that was then applied to the cells, followed by assays for phospholipid synthesis as before. Compared to a lipoprotein‐rich fraction derived from ApoE3 ACM (i.e., fraction 5), lipoproteins enriched from ApoE4 ACM increased PtdEtn synthesis significantly (~2.4 ± 0.5) (Fig 2C). Importantly, this effect was not seen with a lipoprotein‐negative fraction from the same preparations (i.e., fraction 3), nor when equimolar amounts of free (i.e., unlipidated) recombinant ApoE3 and ApoE4 were applied to the cells (Fig 2C). Thus, we ascribe ApoE4's role in increasing MAM activity (as measured by phospholipid synthesis) to its function as a component of lipoproteins.
Figure 2. Effect of lipoprotein‐rich and lipoprotein‐poor fractions on phospholipid synthesis.
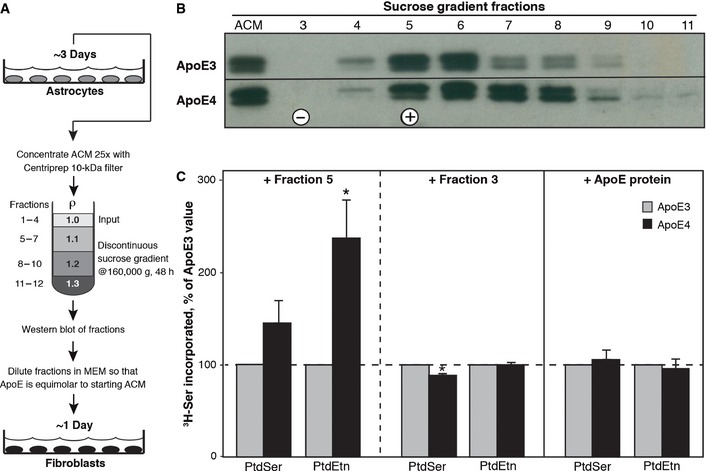
- Experimental scheme. See text for details.
- Fractions from the discontinuous gradient were blotted with anti‐ApoE (WUE‐4) antibody; the doublet is a characteristic feature on ApoE Western blots due to ApoE sialylation 16.
- An ApoE‐containing fraction (fraction 5 [+]), an ApoE‐negative fraction (fraction 3 [−]), and recombinant lipid‐free ApoE protein were applied to human fibroblasts. Note increase in phospholipid synthesis using an ApoE4‐containing lipoprotein fraction, but not with either an ApoE4‐negative fraction or with lipoprotein‐free recombinant ApoE3 or ApoE4 protein (mean ± SE; n = 3, with 3 replicates/experiment). *P < 0.05 vs. E3.
Mitochondria‐associated ER membranes have the properties of an intracellular lipid raft that is rich in cholesterol and sphingolipids 7, 8, 9. Membrane cholesterol is under tight control, and excess cholesterol may either be oxidized by cholesterol hydroxylases (e.g., CYP46A1) or converted into cholesteryl esters (CE) by acyl‐coenzyme A:cholesterol acyltransferase 1 (ACAT1; gene SOAT1), a resident MAM protein 37. ACAT1 controls the equilibrium between membrane‐bound free cholesterol and CE stored in cytoplasmic lipid droplets 38. Remarkably, that equilibrium plays a key role in regulating the production of Aβ, via a currently unknown mechanism 39. We also note that skin fibroblasts 2, 7 and neurons 40 from AD patients contain more lipid droplets than controls.
We had shown previously that presenilin‐mutant cells, including AD fibroblasts, synthesized significantly more CE and lipid droplets than did wild‐type controls 7. To test the contribution of ApoE to CE synthesis, we applied ACM to wild‐type fibroblasts as above and determined CE production (via incorporation of labeled oleate into CE) and lipid droplet formation (via staining with LipidTox™). We added 3H‐oleate to ACM‐treated fibroblasts for 24 h, extracted lipids from these samples, and separated cholesteryl esters from triglycerides by thin layer chromatography (TLC). Compared to ApoE3 ACM‐treated fibroblasts, there was a significant increase in the levels of 3H‐cholesteryl oleate in the ApoE4 ACM‐treated cells (~4.1 ± 1.2) (Fig 3A). Notably, this increase in CE synthesis did not appear to be the result of increased ACAT1 expression, as the levels of this key cholesterol metabolism enzyme were similar in ApoE3 and ApoE4 ACM‐treated fibroblasts (Fig EV2). As with the phospholipid assays, we had established previously that cholesteryl ester synthesis is reduced significantly in Mfn2‐KO MEFs 7. Consistent with this observation, the ApoE4‐mediated increase in cholesterol ester synthesis was abrogated in Mfn2‐KO cells (Fig 3B), supporting the view that the ApoE4‐mediated effect on cholesterol metabolism was indeed the result of increased MAM functionality. In addition, the ApoE4 ACM‐treated cells contained significantly more lipid droplets per cell (~39 ± 4) than did the ApoE3 ACM‐treated samples (~25 ± 3) (Fig 3C). LipidTox also stains triglycerides, another component of lipid droplets. Importantly, we detected essentially no difference in the incorporation of 3H‐oleate into 3H‐triglycerides (Fig 3A), indicating that the ApoE4‐specific increase in lipid droplets was primarily the result of elevated CE production (Fig 3C).
Figure 3. Cholesteryl ester production in ApoE ACM‐treated cells.
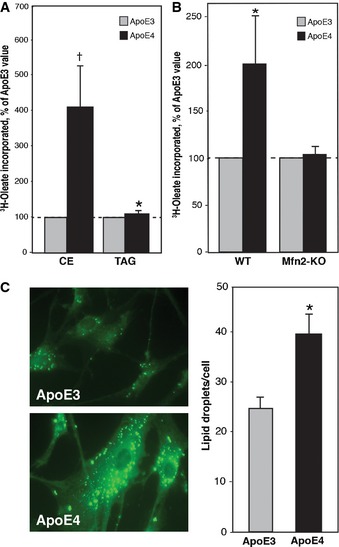
- Cholesteryl ester synthesis, as measured by 3H‐oleate incorporation. Note increased CE in ApoE4‐treated cells, whereas there was essentially no difference in triglyceride (TAG) production (mean ± SE; n = 3, with 3 replicates/experiment). *P < 0.05, † P = 0.06 vs. E3.
- Comparison of 3H‐oleate incorporation in ApoE ACM‐treated WT and Mfn2‐KO MEFs (mean ± SE; n = 3, with 5 replicates/experiment). *P < 0.05 vs. E3.
- ApoE ACM‐treated fibroblasts were stained with LipidTox green neutral lipid stain, and lipid droplets were visualized (example at left) and counted (right). Note significantly more lipid droplets in ApoE4‐treated cells (mean ± SD, n = 3). *P < 0.05 vs. E3.
The stability and capacity of mitochondrial oxidative phosphorylation (OxPhos) complexes (which is reduced in AD cells 6) can be affected by the phospholipid composition of the mitochondrial inner membrane, especially by PtdEtn and the related molecule cardiolipin 41. Nevertheless, we found that respiration in ApoE4 ACM‐treated cells was similar to that in ApoE3 ACM‐treated cells (Fig EV3).
Figure EV3. Bioenergetics of ApoE ACM‐treated fibroblasts.
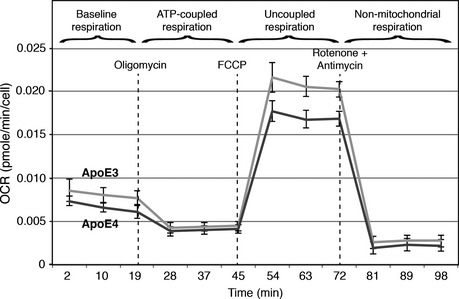
ApoE ACM‐treated human fibroblasts were analyzed using a Seahorse XF‐24 Flux Analyzer. Oxygen consumption rate (OCR) was determined (in pmol O2/min/cell) after the addition of oligomycin (to inhibit ATP synthesis), FCCP (to uncouple ATP synthesis from respiration), and rotenone and antimycin A (to inhibit complexes I and III, respectively). The overall pattern of respiration was essentially similar between ApoE3 and ApoE4 ACM‐treated cells, although maximal respiration of ApoE4 ACM‐treated cells (the plateau between FCCP and rotenone/antimycin addition) was somewhat lower than that of ApoE3 ACM‐treated cells (mean ± SD, n = 3).
We had shown previously that ER–mitochondrial apposition is increased in AD fibroblasts 42. We therefore measured ER–mitochondrial co‐localization in cells after 24 h of treatment with ApoE ACM, using confocal microscopy to visualize ER in green (transfection with GFP‐Sec61‐β) and mitochondria in red (transfection with DsRed‐Mito). While there was no significant difference in the overall degree of co‐localization between ApoE3 ACM vs. ApoE4 ACM treatment after 24 h, there was a clear trend indicating that the median degree of co‐localization was higher in ApoE4‐treated cells as compared to ApoE3‐treated cells (Fig 4).
Figure 4. ER–mitochondrial co‐localization in ApoE ACM‐treated cells.
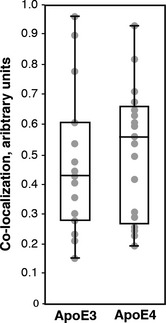
HeLa cells were transfected with plasmids to visualize ER (in green) and mitochondria (in red) and then were treated with ApoE3 ACM and ApoE4 ACM for 24 h. Shown is a box‐and‐whisker plot (showing the median and upper and lower quartiles) comparing co‐localization in ApoE3 ACM (n = 15 images analyzed; gray circles, average of co‐localization in ~5 cells in each image field) compared to that in ApoE4 ACM (n = 18 images).
The role of ApoE4 as a major risk factor in AD has been enigmatic since the initial discovery of its linkage to the disease in 1993 42. Broadly speaking, studies of ApoE's role in AD pathogenesis have focused on amyloid‐dependent or amyloid‐independent pathways. Amyloid‐dependent theories make use of the fact that ApoE, either by direct binding to Aβ 42 or by competition with Aβ receptors, such as the lipoprotein receptor‐related protein‐1 43, is able to modify Aβ production 44 or clearance 45. Amyloid‐independent theories implicate the cytotoxicity of ApoE4 fragments 46, its role in cholesterol homeostasis 35, neuroprotection/apoptosis 20, neurite outgrowth 19, inflammation 47, and vascular integrity 48. AD is clearly a multifaceted disease, with changes in amyloid metabolism occurring alongside these amyloid‐unrelated phenomena.
We reported previously that PS1, PS2, and γ‐secretase activities are highly enriched in the MAM 10 and that ER–mitochondrial communication and MAM function, including phospholipid and cholesteryl ester synthesis, are increased dramatically in presenilin‐mutant and presenilin‐deficient cells and in cells from AD patients 7. Both lines of evidence support the view that PS1 and PS2, in addition to generating Aβ, are negative regulators of MAM function 7 and that these alterations play a critical role in the pathogenesis of the disease (the “MAM hypothesis”) 11, 12. If the hypothesis has any validity, it should accommodate ApoE4's role as a risk factor in the disease, that is, ApoE4 should play a role in mediating MAM behavior, either directly or indirectly. We therefore treated cells with ApoE4 vs. ApoE3 ACM and measured two key aspects of MAM behavior, namely ER–mitochondrial communication and MAM function, as measured by the synthesis of phospholipids and of cholesteryl esters, respectively. We found that in both cases, ApoE4 ACM, like the presenilin mutants, was indeed able to increase MAM functionality. We note that in general, the degrees of increase in phospholipid and cholesterol ester synthesis, while significant, were not as pronounced as those reported previously in presenilin‐mutant cells 7, consistent with ApoE4's role as a risk factor, not a determinative factor, in AD. This latter point may also help explain why the degree of ER–mitochondrial apposition, as measured by confocal microscopy, showed only a trend toward greater median apposition in ApoE4‐treated cells as compared to ApoE3‐treated cells.
The mechanism of this ApoE4‐mediated increase in MAM activity is unclear. However, given that ApoE4 exerted its effects on MAM when incorporated into lipoprotein particles, but not when present as the “free” protein, we suspect that the lipoprotein metabolism pathway plays a role in ApoE4‐mediated pathogenesis. Support for this idea comes from the numerous points of intersection between lipoprotein metabolism and alterations in APP metabolism by γ‐secretase, which is a known regulator of MAM function 7 and which is central to AD pathology 42.
Isoform‐specific differences in lipoprotein metabolism, and, by association, cholesterol trafficking, may provide some insight as to how ApoE4 leads to MAM dysfunction. One of these differences is the relative affinity of each isoform for the lipoprotein receptors at the cell surface, thereby affecting the total amount of cholesterol imported 14. Upon receptor‐mediated endocytosis, the ApoE‐containing HDL‐like particles are transported to lysosomes, where they are hydrolyzed. Some ApoE is degraded and some is recycled back to the cell surface 34, 35, where it is re‐lipidated by ABCA1 or ABCG1 and secreted. Notably, ApoE4 is recycled much less efficiently than is ApoE3 34, 35, resulting in an accumulation of cholesterol in endosomes and lysosomes, reminiscent of Niemann–Pick disease type C, a neurodegenerative disorder characterized by the accumulation of cholesterol in lysosomes. Thus, it may be that the increase in cholesteryl esters seen in ApoE4 ACM‐treated cells reflects a concomitant increase in available substrate (i.e., cholesterol) at the MAM.
Cellular cholesterol handling affects APP processing at numerous points in cell metabolism 49 and vice versa 50. First, the internalization of cholesterol‐containing lipoproteins from the extracellular space may affect the rate of APP internalization, which is a requirement for its amyloidogenic processing 51. Indeed, the bulk uptake of cholesterol is important in AD, as diet‐induced hypercholesterolemia in rabbits resulted in amyloid pathology 52. Second, the cholesterol content of intracellular membranes likely affects APP processing, since the C‐terminal fragment of APP binds cholesterol 53, 54. Third, each of the three proteases that process APP is also influenced by cholesterol. A fraction of BACE1 localizes to, and is active in, lipid rafts, and an increase in cholesterol enhances the β‐cleavage of APP 55. γ‐Secretase is also localized to rafts 56 and is affected by cholesterol levels, particularly by the ratio of free cholesterol:cholesteryl esters, as inhibition of ACAT1, either pharmacologically or by genetic ablation, reduces Aβ production 39. Conversely, the non‐amyloidogenic arm of APP processing by α‐secretase is inhibited by the addition of exogenous cholesterol 57. These observations point to a complex biology in which the subcellular amount and distribution of cholesterol are linked to APP metabolism and MAM function.
It is unclear at present why ApoE4 should exert differential effects on MAM function compared to ApoE3. We note that the two main risk factors for developing AD are aging and ApoE4. Perhaps, the two are related, with potentially increased ApoE4‐mediated cholesterol levels affecting cellular processes that are known to be altered in aging (and in AD) 58.
We also note that from an evolutionary point of view, the ApoE4 genotype is apparently present only in the primate line, including ancestors to humans 59, implying that the maintenance of the ε4 allele in this genus may actually confer a selective advantage. Besides the aforementioned role of ApoE4 in apoptosis 20 (a MAM‐mediated function 60), another possible advantage might be in resistance to infection 61, 62. Since it is known that MAM mediates the innate immune response 63 and that pathogens gain entry into the host cell via lipid rafts 64, perhaps ApoE4 mitigates infectivity 65. More research on the relationship of MAM to infection is needed to elucidate the differential contribution of ApoE isotypes to this process.
In summary, we have demonstrated that ApoE4‐containing ACM increases MAM function relative to ApoE3. This report extends our previous findings demonstrating upregulation of MAM function in AD and may help explain why ApoE4 is a risk factor in the disease.
Materials and Methods
Cells and reagents
Telomerase (hTERT)‐transformed human fibroblasts were a kind gift of Drs. Orhan Akman and Sonja Brosel (Columbia University). Immortalized APOE3‐ and APOE4‐targeted gene replacement astrocytes 66 were prepared as described 67. Cells were cultured in minimal essential media (MEM [lacking serine]) (Gibco 11095) or Dulbecco's MEM (DMEM [containing serine]) (Gibco 11995‐065). Immortalized cells were cultured in DMEM containing 10% fetal bovine serum (FBS) (Sigma F2442) and 1% antibiotic–antimycotic (Gibco 15240).
Mouse hippocampal neuronal culture techniques were adapted from our previous approaches 68. The hippocampus was dissected from 1‐ to 3‐day‐old wild‐type C57BL6/J pups, the tissue digested in a papain solution (Worthington Biochemical, Lakewood, NJ) at 20 units/ml, the cells triturated, spun down, and resuspended in NbActiv4 media (Brain Bits Nb4‐500) supplemented with 1% heat‐inactivated FBS (Life Technologies 16140‐063). Cells were plated at a density of ~320,000 cells per 35‐mm diameter petri dishes pre‐coated with laminin (Millipore, 10 μg/ml) for 1 h. The medium was changed 3 days after plating the cells. Cultures were maintained at 37°C and 5% CO2. For all experiments with ACM, the cultured neurons were incubated for 10 days prior to assay.
We used antibodies against ACAT1 (polyclonal aa6‐23; Abcam 39327), ApoE (monoclonal aa140–160; Novus WUE‐4 NB110‐60531), clusterin (polyclonal C‐terminus; Novus NBP1‐19637), β‐actin (monoclonal; Sigma AC‐15 A5541), and PISD (Sigma HPA031091), which were used according to manufacturers' instructions. Thin layer chromatography (TLC) silica plates were from EMD Biosciences (5748‐7). Phosphatidylserine (PtdSer) (Sigma P7769), phosphatidylethanolamine (PtdEtn) (Sigma 60660), cholesteryl palmitate (Sigma C6072), and soybean phospholipid mixtures (Sigma P3817) were used as standards for TLC in experiments with radiolabeled 3H‐serine (Perkin Elmer NET248005MC) and 3H‐oleate (Perkin Elmer, NET289001MC). Bovine serum albumin (Sigma A3803) was used to solubilize 3H‐oleate 69.
Astrocyte‐conditioned media generation and lipoprotein enrichment
APOE3 and APOE4 astrocytes were grown to confluency, washed once with MEM, and incubated in MEM for 3 days. Astrocyte‐conditioned medium (ACM) was spun at 3,000 g for 3 min to remove debris and analyzed first by Western blot against ApoE to confirm the presence of lipoproteins. Prior to all experiments, we measured the amount of ApoE present in the ACM by dot blot (Bio‐Rad Bio‐Dot™ Apparatus) (it typically contained more ApoE3 than ApoE4, consistent with the finding that the concentration of ApoE3 in the brain homogenates 23 and in the interstitial fluid of human ApoE knock‐in mice 24 is ~50% greater than that of ApoE4) and then added the ACM on an “equal ApoE” basis, by adding MEM to the ApoE3 ACM as appropriate.
Density ultracentrifugation was used to prepare enriched fractions of lipoproteins 70. Briefly, 100 ml ACM was concentrated to ~4 ml using Centriprep 10K filters (EMD Millipore). Four milliliters of ACM concentrate was layered on top of 3 ml 1.1 g/ml, 3 ml 1.2 g/ml, 2 ml 1.3 g/ml discontinuous sucrose gradient. Samples were centrifuged at 160,000 g in a Sw44‐Ti rotor (Beckman) for 48 h at 4°C. One‐millilitre fractions were removed from the top of the gradient, and ApoE content was analyzed by Western blot.
Analysis of phospholipid synthesis
Fibroblasts were grown to confluency (5 replica plates per condition) and washed once with MEM. ACM, recombinant ApoE, or enriched lipoprotein fractions containing 2.5 μCi/ml of 3H‐serine (30.5 Ci/mmol) were applied to the cells grown in 60‐mm dishes for 24 h. The cells were washed twice with 1 ml ice‐cold phosphate‐buffered saline (PBS) and collected on ice in 1 ml PBS, pelleted at 3,000 g for 3 m at 4°C, and resuspended in 0.5 ml water. Twenty microliters of suspension was stored at −20°C for protein quantification by Bradford analysis. Total lipids were extracted as follows: Three volumes of 2:1 chloroform: methanol v/v were added to the samples which were then vortexed and centrifuged at 8,000 g for 5 min, and the aqueous phase was discarded. The organic phase was washed with two volumes of 1:1 methanol/water v/v, centrifuged at 8,000 g for 5 min, and then washed again in the same manner. The organic phase was then transferred to a fresh tube and incubated at 50°C until dry. After resuspension in 40 μl of chloroform, total lipids were spotted onto a TLC plate. Plates were run through a series of two solvents, first 84:15:1 petroleum ether/diethyl ether/acetic acid v/v/v, and then 60:50:1:4 chloroform/methanol/acetic acid/water v/v/v/v. Separated phospholipids and the corresponding standards were visualized after overnight exposure to iodine vapor. Phospholipid species were cut from the plate, suspended in scintillation liquid (Ultima Gold XR Perkin Elmer), and counted by scintillation to detect 3H (Packard Tri‐Carb 2900TR).
Duramycin assay
Fibroblasts were treated with ACM for 24 h, washed once with MEM, and treated with the specified concentration of duramycin (Sigma D3168) or duramycin and PtdEtn diluted in MEM (1:3 duramycin:PtdEtn mol/mol) for 20 min at 37°C. Cells were washed twice with PBS and stained with the LIVE/DEAD® Viability/Cytotoxicity Kit (Life Technologies L3224) according to manufacturer's instructions. Cells were visualized by fluorescent microscopy and hand‐scored. Percent death was calculated as a ratio of ethidium‐stained red cells to total cells.
Analysis of cholesteryl ester synthesis
Fibroblasts were grown to confluency and washed once with MEM. ACM containing 2.5 μCi/ml of 3H‐oleate (54.6 Ci/mmol) solubilized in MEM‐2% BSA were applied to the cells for 24 h. Sample processing and total lipid extraction were performed as described above. TLC plates were run using 190:9:1 chloroform/methanol/acetic acid v/v/v as a solvent. After exposure to iodine vapors, spots corresponding to the cholesteryl ester standards were scraped and counted by scintillation as described above.
Lipid droplet staining
Fibroblasts were grown to ~80% confluency on glass coverslips, washed once with MEM, and treated with either ApoE3 or ApoE4 ACM for 24 h. Cells were washed once in PBS, and lipid droplets were stained with HCS LipidTox™ Deep Green neutral lipid stain (Invitrogen H34475) according to manufacturer's instructions. Lipid droplets were visualized by fluorescent microscopy, and the number of lipid droplets per cell was quantified by hand.
Mitochondrial respiration measurements
Mitochondrial respiration was determined using the Seahorse XF‐24 analyzer (Seahorse Bioscience). WT fibroblasts were seeded at 8 × 104 cells per well in a XF24 cell culture microplate and treated with ACM for 24 h. Cells were washed with XF Assay Media (Seahorse Bioscience), pre‐incubated for 1 h at 37°C and 0% CO2 in XF Assay Media, and supplemented with 2 mM pyruvate and 25 mM glucose. For the assay, cells were treated sequentially with 1 μM oligomycin, 0.75 μM carbonyl cyanide‐4‐trifluoromethoxy phenylhydrazone, and 1 μM rotenone plus 1 μM antimycin A.
Analysis of ER–mitochondrial apposition
ER–mitochondrial apposition was assayed as described previously 7. In brief, following ACM treatment for 24 h, HeLa cells were co‐transfected with GFP‐Sec61‐β (Addgene plasmid #15108) and DsRed‐Mito (Clontech, #632421) at a 1:1 ratio in serum‐free DMEM, and 12 h post‐transfection were analyzed in a single‐plane with a Zeiss LSM510 microscope. Interactions between ER (green) and mitochondria (red) were calculated using ImageJ software (http://rsbweb.nih.gov/ij/), determining the area occupied by one organelle and using its signal as a mask for the other one. The co‐localization data sets were compared using Mander's coefficient.
Author contributions
MDT, EA‐G, and EAS designed the research; MDT, MP, HY, and CG‐L performed the experiments; MDT, MP, and EAS analyzed the data; EK, DS, DH, and HY provided key reagents and critical advice on their use and preparation; MDT and EAS wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Drs. Liza Pon, Tae‐Wan Kim, Gilbert Di Paolo, and James Goldman for their helpful comments. WT and Mfn2‐KO MEFs were the kind gift of David Chan (California Institute of Technology). This work was supported by the U.S. National Institutes of Health (K01 AG045335 to EA‐G and NS38370 to DS, and R01 NS090934 and R01AG047644 to DMH); the JPB Foundation (to DMH and DS); the Parkinson's Disease Foundation (to DS); and The Alzheimer's Drug Discovery Foundation, The Ellison Medical Foundation, the U.S. Department of Defense (W911NF‐12‐1‐9159 and W911F‐15‐1‐0169), and the J. Willard and Alice S. Marriott Foundation (to EAS).
EMBO Reports (2016) 17: 27–36
References
- 1. Goedert M, Spillantini MG (2006) A century of Alzheimer's disease. Science 314: 777–781 [DOI] [PubMed] [Google Scholar]
- 2. Pani A, Dessì S, Diaz G, La Colla P, Abete C, Mulas C, Angius F, Cannas MD, Orru CD, Cocco PL et al (2009) Altered cholesterol ester cycle in skin fibroblasts from patients with Alzheimer's disease. J Alzheimers Dis 18: 829–841 [DOI] [PubMed] [Google Scholar]
- 3. Stefani M, Liguri G (2009) Cholesterol in Alzheimer's disease: unresolved questions. Curr Alz Res 6: 15–29 [DOI] [PubMed] [Google Scholar]
- 4. Pettegrew JW, Panchalingam K, Hamilton RL, McClure RJ (2001) Brain membrane phospholipid alterations in Alzheimer's disease. Neurochem Res 26: 771–782 [DOI] [PubMed] [Google Scholar]
- 5. Bezprozvanny I, Mattson MP (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci 31: 454–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang X, Su B, Zheng L, Perry G, Smith MA, Zhu X (2009) The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J Neurochem 109: 153–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Area‐Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia‐Laguarta C, de Groof AJC, Madra M, Ikenouchi J, Umeda M, Bird TD, Sturley SL et al (2012) Upregulated function of mitochondria‐associated ER membranes in Alzheimer disease. EMBO J 31: 4106–4123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hayashi T, Fujimoto M (2010) Detergent‐resistant microdomains determine the localization of sigma‐1 receptors to the endoplasmic reticulum‐mitochondria junction. Mol Pharmacol 77: 517–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Williamson CD, Zhang A, Colberg‐Poley AM (2011) The human cytomegalovirus protein UL37 exon 1 associates with internal lipid rafts. J Virol 85: 2100–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Area‐Gomez E, de Groof AJ, Boldogh I, Bird TD, Gibson GE, Koehler CM, Yu WH, Duff KE, Yaffe MP, Pon LA et al (2009) Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol 175: 1810–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schon EA, Area‐Gomez E (2010) Is Alzheimer's disease a disorder of mitochondria‐associated membranes? J Alzheimers Dis 20(Suppl 2): S281–S292 [DOI] [PubMed] [Google Scholar]
- 12. Schon EA, Area‐Gomez E (2013) Mitochondria‐associated ER membranes in Alzheimer disease. Mol Cell Neurosci 55: 26–36 [DOI] [PubMed] [Google Scholar]
- 13. Tanzi RE (2012) The genetics of Alzheimer disease. Cold Spring Harb Perspect Med 2: a006296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Holtzman DM, Herz J, Bu G (2012) Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med 2: a006312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lund‐Katz S, Phillips MC (2010) High density lipoprotein structure‐function and role in reverse cholesterol transport. Subcell Biochem 51: 183–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW (1987) Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E‐containing lipoproteins. Biochim Biophys Acta 917: 148–161 [DOI] [PubMed] [Google Scholar]
- 17. Gerdes LU (2003) The common polymorphism of apolipoprotein E: geographical aspects and new pathophysiological relations. Clin Chem Lab Med 41: 628–631 [DOI] [PubMed] [Google Scholar]
- 18. Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak‐Vance MA, Risch N, van Duijn CM (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. JAMA 278: 1349–1356 [PubMed] [Google Scholar]
- 19. DeMattos RB, Rudel LL, Williams DL (2001) Biochemical analysis of cell‐derived apoE3 particles active in stimulating neurite outgrowth. J Lipid Res 42: 976–987 [PubMed] [Google Scholar]
- 20. Hayashi H, Campenot RB, Vance DE, Vance JE (2007) Apolipoprotein E‐containing lipoproteins protect neurons from apoptosis via a signaling pathway involving low‐density lipoprotein receptor‐related protein‐1. J Neurosci 27: 1933–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu Q, Bernardo A, Walker D, Kanegawa T, Mahley RW, Huang Y (2006) Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci 26: 4985–4994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harris FM, Tesseur I, Brecht WJ, Xu Q, Mullendorff K, Chang S, Wyss‐Coray T, Mahley RW, Huang Y (2004) Astroglial regulation of apolipoprotein E expression in neuronal cells. Implications for Alzheimer's disease. J Biol Chem 279: 3862–3868 [DOI] [PubMed] [Google Scholar]
- 23. Ramaswamy G, Xu Q, Huang Y, Weisgraber KH (2005) Effect of domain interaction on apolipoprotein E levels in mouse brain. J Neurosci 25: 10658–10663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ulrich JD, Burchett JM, Restivo JL, Schuler DR, Verghese PB, Mahan TE, Landreth GE, Castellano JM, Jiang H, Cirrito JR et al (2013) In vivo measurement of apolipoprotein E from the brain interstitial fluid using microdialysis. Mol Neurodegen 8: 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Voelker DR (2005) Bridging gaps in phospholipid transport. Trends Biochem Sci 30: 396–404 [DOI] [PubMed] [Google Scholar]
- 26. Hayashi T, Rizzuto R, Hajnoczky G, Su TP (2009) MAM: more than just a housekeeper. Trends Cell Biol 19: 81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vance JE (2008) Phosphatidylserine and phosphatidylethanolamine in mammalian cells: two metabolically related aminophospholipids. J Lipid Res 49: 1377–1387 [DOI] [PubMed] [Google Scholar]
- 28. Gibellini F, Smith TK (2010) The Kennedy pathway–De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life 62: 414–428 [DOI] [PubMed] [Google Scholar]
- 29. Schumacher MM, Choi JY, Voelker DR (2002) Phosphatidylserine transport to the mitochondria is regulated by ubiquitination. J Biol Chem 277: 51033–51042 [DOI] [PubMed] [Google Scholar]
- 30. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160: 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605–610 [DOI] [PubMed] [Google Scholar]
- 32. Makino A, Baba T, Fujimoto K, Iwamoto K, Yano Y, Terada N, Ohno S, Sato SB, Ohta A, Umeda M et al (2003) Cinnamycin (Ro 09‐0198) promotes cell binding and toxicity by inducing transbilayer lipid movement. J Biol Chem 278: 3204–3209 [DOI] [PubMed] [Google Scholar]
- 33. Marki F, Hanni E, Fredenhagen A, van Oostrum J (1991) Mode of action of the lanthionine‐containing peptide antibiotics duramycin, duramycin B and C, and cinnamycin as indirect inhibitors of phospholipase A2. Biochem Pharmacol 42: 2027–2035 [DOI] [PubMed] [Google Scholar]
- 34. Heeren J, Grewal T, Laatsch A, Becker N, Rinninger F, Rye K‐A, Beisiegel U (2004) Impaired recycling of apolipoprotein E4 is associated with intracellular cholesterol accumulation. J Biol Chem 279: 55483–55492 [DOI] [PubMed] [Google Scholar]
- 35. Heeren J, Grewal T, Laatsch A, Rottke D, Rinninger F, Enrich C, Beisiegel U (2003) Recycling of apoprotein E is associated with cholesterol efflux and high density lipoprotein internalization. J Biol Chem 278: 14370–14378 [DOI] [PubMed] [Google Scholar]
- 36. Tokuda T, Calero M, Matsubara E, Vidal R, Kumar A, Permanne B, Zlokovic B, Smith JD, Ladu MJ, Rostagno A et al (2000) Lipidation of apolipoprotein E influences its isoform‐specific interaction with Alzheimer's amyloid beta peptides. Biochem J 348(Pt 2): 359–365 [PMC free article] [PubMed] [Google Scholar]
- 37. Rusinol AE, Cui Z, Chen MH, Vance JE (1994) A unique mitochondria‐associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre‐Golgi secretory proteins including nascent lipoproteins. J Biol Chem 269: 27494–27502 [PubMed] [Google Scholar]
- 38. Puglielli L, Konopka G, Pack‐Chung E, Ingano LA, Berezovska O, Hyman BT, Chang TY, Tanzi RE, Kovacs DM (2001) Acyl‐coenzyme A: cholesterol acyltransferase modulates the generation of the amyloid β‐peptide. Nat Cell Biol 3: 905–912 [DOI] [PubMed] [Google Scholar]
- 39. Puglielli L, Ellis BC, Ingano LA, Kovacs DM (2004) Role of acyl‐coenzyme A:cholesterol acyltransferase activity in the processing of the amyloid precursor protein. J Mol Neurosci 24: 93–96 [DOI] [PubMed] [Google Scholar]
- 40. Gómez‐Ramos P, Asunción Morán M (2007) Ultrastructural localization of intraneuronal Aβ‐peptide in Alzheimer disease brains. J Alzheimers Dis 11: 53–59 [DOI] [PubMed] [Google Scholar]
- 41. Bottinger L, Horvath SE, Kleinschroth T, Hunte C, Daum G, Pfanner N, Becker T (2012) Phosphatidylethanolamine and cardiolipin differentially affect the stability of mitochondrial respiratory chain supercomplexes. J Mol Biol 423: 677–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Strittmatter WJ, Saunders AM, Schmechel D, Pericak‐Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high‐avidity binding to β‐amyloid and increased frequency of type 4 allele in late‐onset familial Alzheimer disease. Proc Natl Acad Sci USA 90: 1977–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sagare AP, Bell RD, Srivastava A, Sengillo JD, Singh I, Nishida Y, Chow N, Zlokovic BV (2013) A lipoprotein receptor cluster IV mutant preferentially binds amyloid‐β and regulates its clearance from the mouse brain. J Biol Chem 288: 15154–15166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vincent B, Smith JD (2001) Astrocytes down‐regulate neuronal β‐amyloid precursor protein expression and modify its processing in an apolipoprotein E isoform‐specific manner. Eur J Neurosci 14: 256–266 [DOI] [PubMed] [Google Scholar]
- 45. Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, Bu G, Frieden C, Holtzman DM (2013) ApoE influences amyloid‐β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc Natl Acad Sci USA 110: E1807–E1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss‐Coray T, Fish JD, Masliah E, Hopkins PC, Scearce‐Levie K et al (2003) Carboxyl‐terminal‐truncated apolipoprotein E4 causes Alzheimer's disease‐like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci USA 100: 10966–10971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guo L, LaDu MJ, Van Eldik LJ (2004) A dual role for apolipoprotein E in neuroinflammation: anti‐ and pro‐inflammatory activity. J Mol Neurosci 23: 205–212 [DOI] [PubMed] [Google Scholar]
- 48. Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J et al (2012) Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485: 512–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Di Paolo G, Kim T‐W (2011) Linking lipids to Alzheimer's disease: cholesterol and beyond. Nat Rev Neurosci 12: 284–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pierrot N, Tyteca D, D'Auria L, Dewachter I, Gailly P, Hendrickx A, Tasiaux B, Haylani LE, Muls N, N'Kuli F et al (2013) Amyloid precursor protein controls cholesterol turnover needed for neuronal activity. EMBO Mol Med 5: 608–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schneider A, Rajendran L, Honsho M, Gralle M, Donnert G, Wouters F, Hell SW, Simons M (2008) Flotillin‐dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J Neurosci 28: 2874–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sparks DL, Scheff SW, Hunsaker JC 3rd, Liu H, Landers T, Gross DR (1994) Induction of Alzheimer‐like β‐amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp Neurol 126: 88–94 [DOI] [PubMed] [Google Scholar]
- 53. Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, Sanders CR (2012) The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336: 1168–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Beel AJ, Mobley CK, Kim HJ, Tian F, Hadziselimovic A, Jap B, Prestegard JH, Sanders CR (2008) Structural studies of the transmembrane C‐terminal domain of the amyloid precursor protein (APP): does APP function as a cholesterol sensor? Biochemistry 47: 9428–9446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Marquer C, Devauges V, Cossec J‐C, Liot G, Lecart S, Saudou F, Duyckaerts C, Leveque‐Fort S, Potier M‐C (2011) Local cholesterol increase triggers amyloid precursor protein‐Bace1 clustering in lipid rafts and rapid endocytosis. FASEB J 25: 1295–1305 [DOI] [PubMed] [Google Scholar]
- 56. Urano Y, Hayashi I, Isoo N, Reid PC, Shibasaki Y, Noguchi N, Tomita T, Iwatsubo T, Hamakubo T, Kodama T (2005) Association of γ‐secretase complex with lipid rafts. J Lipid Res 46: 904–912 [DOI] [PubMed] [Google Scholar]
- 57. Bodovitz S, Klein WL (1996) Cholesterol modulates α‐secretase cleavage of amyloid precursor protein. J Biol Chem 271: 4436–4440 [DOI] [PubMed] [Google Scholar]
- 58. Wirz KTS, Keitel S, Swaab DF, Verhaagen J, Bossers K (2014) Early molecular changes in Alzheimer disease: can we catch the disease in its presymptomatic phase? J Alzheimers Dis 38: 719–740 [DOI] [PubMed] [Google Scholar]
- 59. McIntosh AM, Bennett C, Dickson D, Anestis SF, Watts DP, Webster TH, Fontenot MB, Bradley BJ (2012) The apolipoprotein E (APOE) gene appears functionally monomorphic in chimpanzees (Pan troglodytes). PLoS ONE 7: e47760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R (2008) Calcium and apoptosis: ER‐mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27: 6407–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Roselaar SE, Daugherty A (1998) Apolipoprotein E‐deficient mice have impaired innate immune responses to Listeria monocytogenes in vivo. J Lipid Res 39: 1740–1743 [PubMed] [Google Scholar]
- 62. Martin GM (1999) APOE alleles and lipophylic pathogens. Neurobiol Aging 20: 441–443 [DOI] [PubMed] [Google Scholar]
- 63. Cloonan SM, Choi AMK (2013) Mitochondria: sensors and mediators of innate immune receptor signaling. Curr Opin Microbiol 16: 327–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vieira FS, Correa G, Einicker‐Lamas M, Coutinho‐Silva R (2010) Host‐cell lipid rafts: a safe door for micro‐organisms? Biol Cell 102: 391–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Price DA, Bassendine MF, Norris SM, Golding C, Toms GL, Schmid ML, Morris CM, Burt AD, Donaldson PT (2006) Apolipoprotein ε3 allele is associated with persistent hepatitis C virus infection. Gut 55: 715–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xu PT, Schmechel D, Rothrock‐Christian T, Burkhart DS, Qiu HL, Popko B, Sullivan P, Maeda N, Saunders AM, Roses AD et al (1996) Human apolipoprotein E2, E3, and E4 isoform‐specific transgenic mice: human‐like pattern of glial and neuronal immunoreactivity in central nervous system not observed in wild‐type mice. Neurobiol Dis 3: 229–245 [DOI] [PubMed] [Google Scholar]
- 67. Fagan AM, Holtzman DM, Munson G, Mathur T, Schneider D, Chang LK, Getz GS, Reardon CA, Lukens J, Shah JA et al (1999) Unique lipoproteins secreted by primary astrocytes from wild type, apoE (‐/‐), and human apoE transgenic mice. J Biol Chem 274: 30001–30007 [DOI] [PubMed] [Google Scholar]
- 68. Staal RGW, Rayport S, Sulzer D (2007) Amperometric detection of dopamine exocytosis from synaptic terminals In Electrochemical Methods for Neuroscience, Michael AC, Borland M. (eds), pp 340–349. Boca Raton, FL: CRC Press; [PubMed] [Google Scholar]
- 69. Saifer A, Goldman L (1961) The free fatty acids bound to human serum albumin. J Lipid Res 2: 268–270 [Google Scholar]
- 70. Vance JE, Hayashi H (2010) Formation and function of apolipoprotein E‐containing lipoproteins in the nervous system. Biochim Biophys Acta 1801: 806–818 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File