

Abstract
It is well established that TDP‐43 accumulates in degenerating neurons in patients with ALS/FTLD, which might affect normal TDP‐43 function. In this issue of The EMBO Journal Xia et al (2016) show a novel connection between TDP‐43 loss of function and autophagy failure. Using knockdown models of TDP‐43, they observed enhanced autophagosome and lysosome biogenesis through mTORC1 activity inhibition and TFEB activation. Impaired autophagosome–lysosome fusion was also observed, however in an mTORC1‐independent manner. The data identify dysfunctions at multiple stages of the autophagic pathway following TDP‐43 depletion that might represent possible targets of future therapeutic interventions.
Subject Categories: Autophagy & Cell Death, Neuroscience, RNA Biology
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are neurodegenerative diseases characterized by the deposition of pathological protein aggregates composed of TAR DNA‐binding protein 43 (TDP‐43) (Neumann et al, 2006). A chicken‐and‐egg question for neurodegeneration is whether TDP‐43 aggregation per se causes neurotoxicity or is a by‐product of the disease process. The fact that the loss of TDP‐43 results in motor dysfunction and neuropathological alterations—common to ALS—in different animal models such as Drosophila (Feiguin et al, 2009) and mouse (Iguchi et al, 2013) indicates that TDP‐43 loss of function appears to play a central role in ALS/FTLD pathogenesis. After all, it is known that eggs came before chickens (the reptilians, cold‐blooded bird's ancestors, laid eggs), so there should be a plausible order of the events in TDP‐43‐associated neurodegeneration. However, as one tries to go beyond this analogy, answers begin to become not so straightforward. Studies of the cellular targets of TDP‐43 have yielded more than 6,000 brain RNAs targets and TDP‐43 depletion causes the change of the pattern of splicing of about 1,500 RNAs in the mammalian brain (Polymenidou et al, 2011; Tollervey et al, 2011) consequently changing the proteome. Among the targets are those of ALS‐linked genes such as Ang1, Atxn2 and FUS/TLS as well as Atg7 and p62/SQSTM1, which are components of the autophagy system, the protein clearance pathway essential for normal neuronal functioning.
The ensuing TDP43‐induced disturbance in RNA and protein homeostasis therefore also seems highly probable to play a central feature in ALS/FTLD pathogenesis (Ling et al, 2013). In regard to autophagy, the accumulation of TDP‐43 in patients with ALS/FTLD suggests that dysfunction in this process may be involved in the disease pathogenesis. In line with this, it was observed in a FTLD mouse model based on TDP‐43 overexpression that autophagy activators successfully inhibit formation of TDP‐43‐positive inclusions and enhance neuronal survival (Wang et al, 2012). But surprisingly, in fact, autophagy appears to be already upregulated in ALS. Indeed, autophagosomes have been observed to accumulate in motor neurons of patients with ALS (Sasaki, 2011), in agreement with similar findings in the knockout TDP‐43 mouse (Iguchi et al, 2013).
Xia et al (2016) now demonstrate how TDP‐43 knockdown enhances global autophagy–lysosome pathway (ALP) gene expression leading to an increase in autophagy and lysosomal biogenesis in an mTORC1‐dependent manner. Their results reconcile the previous observations, proving that TDP‐43 absence from the cell can induce upregulation of the autophagosome pathway but at the same time inhibits autophagosome–lysosome fusion in an mTORC1‐independent way.
The study shows, both in vitro and in vivo, that TDP‐43 knockdown results in downregulation of mTORC1 adaptor protein raptor, resulting in lower mTOR lysosomal localization and mTORC1 activity, which in turn leads to an increased Transcription factor EB (TFEB) nuclear translocation and activity. TFEB, the master regulator of ALP (Settembre et al, 2011), in turn drives the expression of autophagy and lysosomal genes (see Fig 1).
Figure 1. RNA and protein homeostasis derangements underlie TDP‐43‐associated neuro‐degenerative pathology.
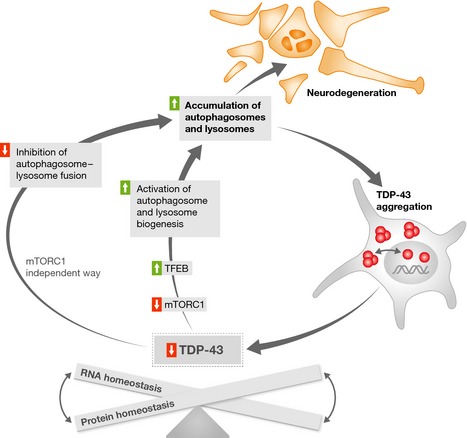
Aggregation of TDP‐43 causes its loss of function, which in turn is linked to autophagosome and lysosome biogenesis activation through mTORC1 inhibition and TFEB activation. At the same time, it leads to autophagosome–lysosome fusion impairment in an mTORC1‐independent manner. The resulting accumulation of autophagic vacuoles contributes to TDP‐43 aggregation and neurode‐generation.
Xia et al (2016) also observe an unexpected effect: The induction of autophagy flux through starvation does not normalize the autophagy behavior in siTDP‐43 cells. This observation strongly suggests that the effect of TDP‐43 knockdown during starvation is mTORC1 independent. Specifically, TDP‐43 depletion induces autophagosome–lysosome fusion impairment with no involvement of mTORC1. An explanation provided by the authors is that Dctn1 gene expression level and Dynactin 1 protein level are reduced in TDP‐43‐depleted cells, which in turn affects lysosomal positioning and autophagosome–lysosome fusion.
Taken together, Xia and co‐workers suggest that TDP‐43 knockdown has both mTORC1‐dependent and mTORC1‐independent effects on the autophagy–lysosome pathway and that abnormal accumulation of autophagic cargoes contributes to TDP‐43‐mediated neurodegeneration (see Fig 1).
In accordance with their findings, mTOR inhibitor rapamycin worsens the phenotype of TDP‐43−/− flies, whereas phosphatidic acid, an mTOR agonist, treatment improves it. These findings are in agreement with earlier observations that the activation of autophagy can become harmful in the context of impaired autophagic flux, as previously observed in the SOD mouse model of ALS (Zhang et al, 2011).
In conclusion, Xia et al (2016) shed light on some critical points with regard to TDP43‐mediated abnormal regulation of autophagy in neurodegeneration. In addition, their observations are consistent with the hypothesis that TDP‐43 loss of function, caused by its sequestering in the characteristic inclusions observed in ALS neurons, is at the basis of ALS/FTLD pathogenesis through a negative loop: TDP‐43 depletion by sequestering aggregates leads to defective clearance pathways, which in turn will increase TDP‐43 sequestration/depletion, and eventually lead to neuronal dysfunction (Buratti & Baralle, 2009).
Most importantly, understanding the complexity behind the autophagocytosis failure induced by functional TDP‐43 depletion in ALS/FTLD neurodegeneration will be essential for the development of future therapeutic approaches targeting autophagy. In fact, irrespective of the role played by the aggregates, their reduction represents an important therapeutic route. The reduction will either remove the toxicity of the aggregates or abolish the “sink” that sequesters soluble TDP‐43.
The results are also helpful to point our research toward a direction that was not an easily foreseeable one. For neurodegenerative disorders, the widespread view is that stimulation of autophagy reduces protein inclusions and alleviates motor neuron degeneration. However, from the results of Xia et al (2016), it seems that for TDP‐43 proteinopathies, a generalized activation of autophagy might not be the best strategy after all. Rather, a perhaps much better choice would be to design compounds that specifically target the maturation of autophagosomes, which in turn will promote aggregate breakdown in lysosomes.
See also: Q Xia et al (January 2016)
References
- Buratti E, Baralle FE (2009) The molecular links between TDP‐43 dysfunction and neurodegeneration. Adv Genet 66: 1–34 [DOI] [PubMed] [Google Scholar]
- Feiguin F, Godena VK, Romano G, D'Ambrogio A, Klima R, Baralle FE (2009) Depletion of TDP‐43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett 583: 1586–1592 [DOI] [PubMed] [Google Scholar]
- Iguchi Y, Katsuno M, Niwa J, Takagi S, Ishigaki S, Ikenaka K, Kawai K, Watanabe H, Yamanaka K, Takahashi R, Misawa H, Sasaki S, Tanaka F, Sobue G (2013) Loss of TDP‐43 causes age‐dependent progressive motor neuron degeneration. Brain 136: 1371–1382 [DOI] [PubMed] [Google Scholar]
- Ling S‐C, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79: 416–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM‐Y (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (New York, N.Y.) 314: 130–133. [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Lagier‐Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling S‐C, Sun E, Wancewicz E, Mazur C, Kordasiewicz H, Sedaghat Y, Donohue JP, Shiue L, Bennett CF, Yeo GW, Cleveland DW (2011) Long pre‐mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP‐43. Nat Neurosci 14: 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S (2011) Autophagy in spinal cord motor neurons in sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 70: 349–359 [DOI] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011) TFEB links autophagy to lysosomal biogenesis. Science (New York, N.Y.) 332: 1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, König J, Hortobágyi T, Nishimura AL, Zupunski V, Patani R, Chandran S, Rot G, Zupan B, Shaw CE, Ule J (2011) Characterizing the RNA targets and position‐dependent splicing regulation by TDP‐43. Nat Neurosci 14: 452–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang I‐F, Guo B‐S, Liu Y‐C, Wu C‐C, Yang C‐H, Tsai K‐J, Shen C‐KJ (2012) Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA‐binding protein 43. Proc Natl Acad Sci USA 109: 15024–15029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Q, Wang H, Hao Z, Fu C, Hu Q, Gao F, Ren H, Chen D, Han J, Ying Z, Wang G (2016) TDP‐43 loss of function increases TFEB activity and blocks autophagosome–lysosome fusion. EMBO J 35: 121–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li L, Chen S, Yang D, Wang Y, Zhang X, Wang Z, Le W (2011) Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 7: 412–425 [DOI] [PubMed] [Google Scholar]