

Abstract
Phosphorylation of the activation loop in RAF kinases has been suggested to be critical for changes in activity. The extent to which the activation segment is phosphorylated, the specific structural consequences, and the in vivo relevance have however remained elusive. In this issue of the The EMBO Journal, Köhler et al (2015) addressed these questions by generating a knock‐in mouse expressing a B‐Raf mutant with a non‐phosphorylatable activation loop. The mutant causes a range of developmental phenotypes; intriguingly, it also impairs the tumorigenic potential of a subset of BRAF mutants, suggesting potential new strategies for RAF inhibition.
Subject Categories: Cancer, Signal Transduction
The RAS/RAF/MEK/ERK pathway is one of the oldest paradigms of a cytosolic signal transduction cascade. Many studies have implicated the pathway in the control of cell proliferation, and two of its components, RAS and BRAF, are mutated with different frequencies in human tumors. As a consequence, major efforts are being made to target the “druggable” nodes, that is, the kinases RAF, MEK, and ERK, using both ATP‐competitive and allosteric inhibitors to block pathway activation. A better understanding of kinase regulation will be instrumental in reaching this goal.
Protein kinases can be considered molecular switches oscillating between inactive and active conformations. For many enzymes, including RAF, these changes in activity correlate with the phosphorylation status of crucial residues and with the formation of protein–protein complexes that enable productive signaling. A growing number of structural studies show that protein kinase activation is governed essentially by the status of two hydrophobic “spines” in the kinase domain, the “C” or catalytic spine, and the “R” or regulatory spine. These arrays of hydrophobic residues are “broken”, or disordered, in the inactive state, and aligned in the active state. When the spines are aligned, the N and C lobes of the kinase are in close proximity, and the kinase is in a stable, active conformation. The C spine is completed by ATP, while the alignment of the “R” spine can be brought about by different mechanisms (Shaw et al, 2014; Lavoie & Therrien, 2015). In a recent structure of monomeric BRAF, the activation loop was found organized in a helical conformation (AS‐H1), which blocked both the catalytic site and the essential inward motion of the αC helix. Side‐to‐side RAF dimerization is predicted to stabilize the “in” conformation of the αC helix structure; similarly, phosphorylation of the activation segment (or loop as in Köhler et al, 2016) would promote an extended conformation, stabilizing an active, dimeric kinase structure (Thevakumaran et al, 2015). Based on structure–function studies, phosphorylation of the activation loop is proposed to occur in cis, upon allosteric activation of one RAF molecule (receiver) by another (activator), which may or may not be catalytically active (Hu et al, 2013). Indeed, allosteric activation of a receiver RAF molecule by an activator bound to a chemical inhibitor (therefore catalytically inactive) is the basis for the paradox activation of the ERK pathway by drugs favoring RAF dimerization (reviewed in Gibney et al, 2013).
The model is very attractive and it is supported by elegant structural data and mutational analysis. It is also worth noting that the AS‐H1 is the element of BRAF most frequently mutated in disease and that the activation segment, in particular the phosphoacceptor sites, is evolutionary conserved in RAF orthologues from human to the sponge Amphimedon (Köhler et al, 2016). Both facts strongly suggest an important function of these residues in RAF signaling. Nevertheless, activation loop phosphorylation has remained elusive, possibly because of technical limitations, and evidence of its relevance in vivo was so far missing.
Köhler et al (2016) now close the latter gap by showing the consequences of mutating both phosphorylation sites in the activation loop of BRAF to alanine. Both mouse development and tumorigenic potential of BRAF mutants in cells in culture are affected in the presence of this mutant (Fig 1).
Figure 1. Preventing BRAF activation loop phosphorylation reduces MEK phosphorylation in mouse brain and impairs the transforming potential of a subset of BRAF mutants in vitro .
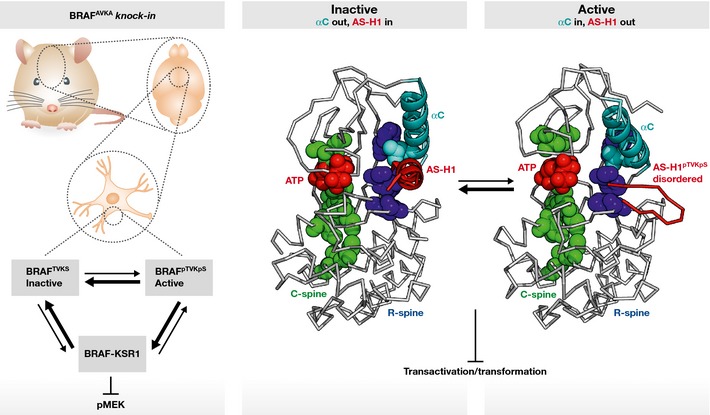
Brain lysates of BRAFAVKA knock‐in mice show reduced BRAF–KSR1 interaction, which might be responsible for the reduced MEK phosphorylation in the tissue. Activation loop phosphorylation can potentially influence the conformation of the activation segment helix 1 (AS‐H1) and contribute to the alignment of the R‐spine residues important for the active conformation of BRAF, which is necessary for the transactivation of the receiver kinase within the RAF dimer.
The first set of data shows that mutating the phosphorylation sites in the activation loop reduces ERK signaling in the brain and in astrocytes and that this correlates with neurological defects. These data are reminiscent of those obtained in B‐Raf knockout mice (Zhong et al, 2007; Galabova‐Kovacs et al, 2008). Interestingly, the biochemical defects were associated with a reduction in the ability of the mutant to dimerize with kinase suppressor of RAS (KSR) rather than with RAF1. This particularity is noteworthy because Galabova et al had already shown that MEK can bind to RAF1, but not to KSR, in BRAF‐deficient glial cells. Together, the data point to a pivotal role of the BRAF:KSR complex in the implementation of strong, sustained ERK signaling in the CNS, in agreement with the function of this complex demonstrated by the Morrison laboratory in MEFs (McKay et al, 2009). In addition, the structure of KSR2 in complex with MEK shows tetramers comprising two KSR2 molecules involved in side‐to‐side dimers, with both subunits bound to MEK in a face‐to‐face manner (Brennan et al, 2011). The authors propose that KSR2, via the interaction with a regulatory RAF molecule, may promote the repositioning of the activation segment of MEK in a conformation accessible to catalytically active RAF. While this model needs further testing, it would explain how a reduction in BRAF:KSR binding can impact ERK signaling despite the continuing presence of BRAF:RAF1 dimers. Why the phosphorylation of the activation loop would have a higher impact on the formation of BRAF:KSR dimers rather than BRAF:RAF1 dimers remains to be determined.
A second significant outcome of this study is the mechanistic aspect. The authors show that mimicking activation loop phosphorylation in BRAF promotes ERK activation (Röring et al, 2012) and induces cell proliferation to the same levels as the highly oncogenic V600E BRAF mutant. V600E BRAF, in turn, is refractory to the ablation of phosphorylation in the activation loop. Thus, activation loop phosphorylation and the V600E mutation have the same purpose, namely repositioning the activation loop in the “out” conformation. The authors further show that a BRAF mutant tethered to the membrane by a CAAX motif still requires phosphorylation of the activation loop. However, dimerization interface BRAF mutants (i.e., those which owe their oncogenic potential to the increased interaction with other RAF or KSR molecules) are differentially sensitive to the ablation of activation loop phosphorylation: the kinase‐dead D594A mutant is refractory and the hyperactive E586K mutant is sensitive to phosphoablation. This is consistent with the idea that phosphorylation of the activation loop is still necessary after membrane recruitment and that it occurs mostly in cis.
Therrien and colleagues recently showed that the destabilization of the activation loop favors a dimer‐competent conformation (Thevakumaran et al, 2015). This suggests that a non‐phosphorylatable protomer may have a dominant negative effect on the dimer. However, Köhler et al (2016) show that the ablation of the activation loop phosphorylation sites does not impact ERK activation by the kinase‐dead mutant—although it reduces its dimerization with RAF1. Similarly, heterozygous mice do not show a phenotype, which would be expected from a dominant negative allele. Be that as it may, the hypothesis of a dominant negative effect is further testable. We will certainly learn more about RAF regulation via phosphorylation of the RAF activation loop in the future. A very important milestone in this context would be the availability of methods allowing the detection of activation loop sites phosphorylation in tissues, which would clarify their relevance as well as their potential use as biomarkers or druggable targets.
See also: M Köhler et al (January 2016)
References
- Brennan DF, Dar AC, Hertz NT, Chao WC, Burlingame AL, Shokat KM, Barford D (2011) A Raf‐induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature 472: 366–369 [DOI] [PubMed] [Google Scholar]
- Galabova‐Kovacs G, Catalanotti F, Matzen D, Reyes GX, Zezula J, Herbst R, Silva A, Walter I, Baccarini M (2008) Essential role of B‐Raf in oligodendrocyte maturation and myelination during postnatal central nervous system development. J Cell Biol 180: 947–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney GT, Messina JL, Fedorenko IV, Sondak VK, Smalley KSM (2013) Paradoxical oncogenesis[mdash]the long‐term effects of BRAF inhibition in melanoma. Nat Rev Clin Oncol 10: 390–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJ, Kornev AP, Taylor SS, Shaw AS (2013) Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154: 1036–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler M, Röring M, Schorch B, Heilmann K, Stickel N, Fiala GJ, Schmitt LC, Braun S, Ehrenfeld S, Uhl FM, Kaltenbacher T, Weinberg F, Herzog S, Zeiser R, Schamel WW, Jumaa H, Brummer T (2016) Activation loop phosphorylation regulates B‐Raf in vivo and transformation by B‐Raf mutants. EMBO J 35: 143–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H, Therrien M (2015) Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 16: 281–298 [DOI] [PubMed] [Google Scholar]
- McKay MM, Ritt DA, Morrison DK (2009) Signaling dynamics of the KSR1 scaffold complex. Proc Natl Acad Sci USA 106: 11022–11027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röring M, Herr R, Fiala GJ, Heilmann K, Braun S, Eisenhardt AE, Halbach S, Capper D, von Deimling A, Schamel WW, Saunders DN, Brummer T (2012) Distinct requirement for an intact dimer interface in wild‐type, V600E and kinase‐dead B‐Raf signalling. EMBO J 31: 2629–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw AS, Kornev AP, Hu J, Ahuja LG, Taylor SS (2014) Kinases and pseudokinases: lessons from RAF. Mol Cell Biol 34: 1538–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevakumaran N, Lavoie H, Critton DA, Tebben A, Marinier A, Sicheri F, Therrien M (2015) Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat Struct Mol Biol 22: 37–43 [DOI] [PubMed] [Google Scholar]
- Zhong J, Li X, McNamee C, Chen AP, Baccarini M, Snider WD (2007) Raf kinase signaling functions in sensory neuron differentiation and axon growth in vivo . Nat Neurosci 10: 598–607 [DOI] [PubMed] [Google Scholar]