

Abstract
Despite being mutated in cancer and RASopathies, the role of the activation segment (AS) has not been addressed for B‐Raf signaling in vivo. Here, we generated a conditional knock‐in mouse allowing the expression of the B‐RafAVKA mutant in which the AS phosphoacceptor sites T599 and S602 are replaced by alanine residues. Surprisingly, despite producing a kinase‐impaired protein, the Braf AVKA allele does not phenocopy the lethality of Braf‐knockout or paradoxically acting knock‐in alleles. However, Braf AVKA mice display abnormalities in the hematopoietic system, a distinct facial morphology, reduced ERK pathway activity in the brain, and an abnormal gait. This phenotype suggests that maximum B‐Raf activity is required for the proper development, function, and maintenance of certain cell populations. By establishing conditional murine embryonic fibroblast cultures, we further show that MEK/ERK phosphorylation and the immediate early gene response toward growth factors are impaired in the presence of B‐RafAVKA. Importantly, alanine substitution of T599/S602 impairs the transformation potential of oncogenic non‐V600E B‐Raf mutants and a fusion protein, suggesting that blocking their phosphorylation could represent an alternative strategy to ATP‐competitive inhibitors.
Keywords: BRAF fusion, Cre/loxP system, gene targeting, MAPK pathway, Ras
Subject Categories: Cancer, Signal Transduction
Introduction
The Ras/Raf/MEK/ERK pathway plays a pivotal role in controlling proliferation, survival, and differentiation. As this pathway is often deregulated in various diseases, in particular cancer and RASopathies, its components are pursued as targets for pharmacological intervention (Holderfield et al, 2014; Samatar & Poulikakos, 2014). Raf kinases represent a particularly important node as they are subject to a complex and tight regulation (Cseh et al, 2014). The Raf family comprises A‐Raf, B‐Raf, and Raf‐1 in vertebrates and single raf genes in invertebrates such as D‐Raf and LIN‐45 in Drosophila and Caenorhabditis, respectively. Distant relatives are the KSR proteins, which promote MEK activation as scaffolds, allosteric Raf activators, and potentially by their own kinase activity (Brennan et al, 2011). As described below, the presence of three Raf and two KSR isoforms allows fine‐tuning of Raf activity by forming homo‐ and heterodimers with distinct signaling potential (Cseh et al, 2014; Mooz et al, 2014). For example, B‐Raf/Raf‐1 heterodimers represent the most potent MEK activator (Rushworth et al, 2006; Freeman et al, 2013).
Genetic analyses demonstrated that Raf isoforms possess unique and overlapping functions. B‐Raf is required for maximum ERK activation and for placental development as it is illustrated by the embryonic lethality of B‐Raf‐deficient mice (Wojnowski et al, 2000; Brummer et al, 2002; Galabova‐Kovacs et al, 2006, 2008; Zhong et al, 2007). B‐Raf displays the most potent transforming activity among the three isoforms and is often activated by somatic alterations in cancer (Pritchard et al, 1995; Holderfield et al, 2014). Consequently, B‐Raf appears as an attractive therapeutic target and inhibitors such as vemurafenib yield unprecedented response rates in melanoma (Samatar & Poulikakos, 2014). However, the use of current B‐Raf selective inhibitors is restricted to BRAF mutant tumors, because binding of these ATP‐competitive compounds to wild‐type B‐Raf (B‐RafWT) provokes the so‐called paradoxical activation of the ERK pathway. This phenomenon involves active Ras and the allosteric activation of a drug free by an inhibitor‐bound or kinase‐dead Raf protomer via the dimer interface (DIF) (Hatzivassiliou et al, 2010; Heidorn et al, 2010; Poulikakos et al, 2010; Röring et al, 2012). This paradoxical action of B‐Raf inhibitors contributes to drug resistance and promotes secondary neoplasms ((Yaktapour et al, 2014) and references therein). Therefore, current B‐Raf inhibitors must not be used in tumors with aberrant Ras activity. Thus, alternative strategies are urgently needed for the inhibition of Raf kinases and are likely to emerge from a better understanding of B‐Raf regulation.
All Rafs share three highly conserved regions (CRs): the N‐terminal CR1 mediates the interaction with Ras‐GTP via R188 (amino acid positions refer to human B‐Raf). The CR2 recruits 14‐3‐3 proteins and is critical for autoinhibition. The CR3 harbors the kinase domain and phosphorylation sites within two areas, the N‐region and the activation segment (AS) consisting of the activation and P + 1 loops (Zhang & Guan, 2000; Nolen et al, 2004; Röring & Brummer, 2012). The CR3 also includes key residues controlling dimerization such as the DIF, including R509 in vicinity to the αC helix, and the C‐terminal 14‐3‐3 binding motif (Rajakulendran et al, 2009; Röring et al, 2012). The DIF also mediates the allosteric transactivation between Raf protomers (Röring et al, 2012; Hu et al, 2013).
Upon membrane recruitment by Ras‐GTP, B‐Raf undergoes conformational changes associated with increased dimerization, phosphorylation, and activity (Lavoie & Therrien, 2015). The Ras‐GTP‐ and dimerization‐triggered autophosphorylation of the AS at T599 and S602 in human B‐Raf is supposed to represent a key event in Raf activation (Zhang & Guan, 2000). Based on crystal structures, V600 of the AS engages in a hydrophobic interaction with the P‐loop that stabilizes the closed inactive conformation of the kinase domain (Wan et al, 2004). Phosphorylation of the T599VKS602 motif is supposed to disrupt this interaction and initiates the subsequent phosphotransferase reaction by ATP uptake (Thevakumaran et al, 2015). Alternatively, AS phosphorylation might contribute to conformational changes in the kinase domain, leading to catalysis and allosteric activation (Hu et al, 2015; Thevakumaran et al, 2015). The precise mechanism leading to AS phosphorylation remains elusive, although recent data on Raf‐1 suggest a dimerization‐induced autophosphorylation in cis (Hu et al, 2013).
The high frequency of point mutations and in‐frame insertion/deletions in the AS further underscore its critical role in B‐Raf regulation. Most alterations affect the N‐terminal portion of the AS, the activation segment helix 1 (AS‐H1). The dominating AS‐H1 mutation, the V600E substitution, occurs in about 7% of tumors and generates a constitutively active oncoprotein that is uncoupled from many regulatory processes controlling wild‐type B‐Raf activity (Garnett & Marais, 2004; Röring et al, 2012). Albeit at lower frequency, mutations of T599 and S602 are also found in a variety of cancers ((Eisenhardt et al, 2011; Thevakumaran et al, 2015) and the COSMIC database). Moreover, various in vitro studies using ectopically expressed proteins with T599 and S602 (or their equivalents) replaced by phosphomimetic residues further underscore the essential role of the TVKS motif for the activation of B‐Raf, Raf‐1, LIN‐45, and D‐Raf (Zhang & Guan, 2000; Chong et al, 2001; Rajakulendran et al, 2009; Röring et al, 2012). On the other hand, conversion of the TVKS into an AVKA motif by alanine substitution prevents the Ras‐induced increase in B‐Raf in vitro kinase activity (Zhang & Guan, 2000). However, the phosphoacceptor sites of the TVKS motif have not been assessed for their importance in oncogenic B‐Raf mutants and most importantly, as it was also pointed out recently (Lavoie & Therrien, 2015), not for the activation of B‐Raf in vivo. Furthermore, although crystallography provides essential insights into the regulation of the kinase domain, these studies all rely on truncated, dephosphorylated, and drug‐bound proteins in which the precise orientation of the complete AS remains unresolved (Hu et al, 2015; Thevakumaran et al, 2015). Thus, structural studies are best complemented by biochemical and genetic approaches interrogating full‐length B‐Raf in its cytoplasmic environment.
Here, we evaluated the relevance of TVKS motif phosphorylation for normal and oncogenic B‐Raf signaling. By using a knock‐in approach, we show that a B‐Raf mutant lacking the two phosphoacceptor residues of the TVKS motif permits long‐term postnatal development. Nevertheless, TVKS motif phosphorylation is essential for optimal signaling output of wild‐type B‐Raf and several non‐V600E oncoproteins. Thus, blocking TVKS motif phosphorylation could represent an alternative strategy to the ATP‐competitive compounds that are currently in use for B‐Raf inhibition.
Results
The TVKS motif plays an important role for normal and aberrant B‐Raf signaling
As little functional characterization of the TVKS motif has been conducted since its first description (Zhang & Guan, 2000), we revisited the function of its phosphoacceptor sites in more detail and in the context of the advances of the field. First, we asked how much this sequence motif has been conserved during evolution. Indeed, the AS, which spans the region between the DFG and APE motifs in subdomains VII and VIII, respectively, has been highly conserved in Raf orthologues, in particular if the AS‐H1 is considered. Importantly, the phosphoacceptor sites of the TVKS motif are not only found in eumetazoans, but also in the sponge Amphimedon suggesting a conserved function from the emergence of multicellular animals onwards (Fig 1A). Interestingly, while the V600 equivalent is substituted by other aliphatic residues such as alanine or isoleucine in some species, the threonine and lysine residues were maintained throughout evolution and the S602 equivalent has been subject to a conservative exchange to threonine in Raf proteins of protostomic invertebrates and in A‐Raf.
Figure 1. Loss of the phosphoacceptor sites of the TVKS motif impairs the signaling potential of wild‐type B‐Raf and some of its gain‐of‐function mutants.
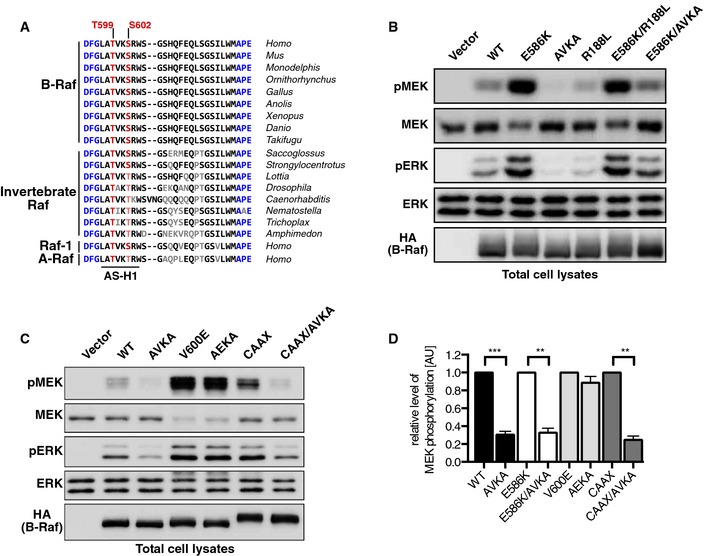
- The TVKS/T motif has been conserved throughout metazoan evolution. The phosphoacceptor sites are indicated in red, and the DFG and APE motif flanking the activation loop (AL) are indicated in blue. Residues differing from human B‐Raf are shown in gray or in red/blue with reduced intensity.
- B‐RafE586K signals independently from Ras‐GTP, but requires an intact AL. The MEK‐ERK activation potential of the indicated HA‐tagged B‐Raf mutants was assessed by Western blotting using TCLs from transiently transfected Plat‐E cells.
- B‐RafCAAX but not the B‐RafV600E oncoprotein signals independently from Ras‐GTP, but requires an intact AL. Same experimental setup as in (B).
- Quantification of experiments shown in (B) and (C). The signal elicited by the individual reference proteins (B‐Rafwt, B‐RafE586K, B‐RafV600E, and B‐RafCAAX) was set in each analysis to 100%. n = 3, mean + SEM, t‐test, **P < 0.01, ***P < 0.001.
So far, a role of the phosphoacceptor sites in the TVKS motif has been demonstrated for the activation of A‐Raf, B‐Raf, and Raf‐1 in the context of oncogenic Ras, phorbol esters, EGF, and the GPCR agonist carbachol (Barnard et al, 1998; Zhang & Guan, 2000; Chong et al, 2001; Tsukamoto et al, 2004; Baljuls et al, 2008). These studies, which differ in design and experimental approaches, identified distinct requirements of the individual isoforms for an intact TVKS motif, for example, in response to EGF (Baljuls et al, 2008). However, the question whether oncogenic B‐Raf mutants require the phosphoacceptor residues of the AS has not been addressed so far. Therefore, we introduced alanine substitutions of T599 and S602 of the TVKS motif, termed as AVKA mutation in the following, into wild‐type B‐Raf (B‐RafWT) and three of its gain‐of‐function mutants. We first turned to B‐RafE586K, which occurs in human tumors and displays oncogenic activity due to its enhanced dimerization potential and ability to activate other Raf protomers in trans (Emuss et al, 2005; Rajakulendran et al, 2009; Röring et al, 2012). Interestingly, while blocking Ras/B‐Raf interaction with the R188L substitution had no effect on B‐RafE586K, the AVKA mutation reduced the MEK/ERK phosphorylation potential of this B‐Raf oncoprotein by 70% (Fig 1B and D). In light of its enhanced dimerization behavior (Rajakulendran et al, 2009; Freeman et al, 2013) and the notion that B‐Raf dimerization is strongly promoted by Ras (Heidorn et al, 2010), the impaired signaling potential of B‐RafE586K/AVKA further supports the recent model in which Ras promotes full Raf activity by dimerization and subsequent AS phosphorylation (Hu et al, 2013). The AVKA mutation also reduced the signaling potential of B‐RafWT and B‐RafCAAX (Fig 1B–D). The latter displays transforming activity due to its constitutive membrane localization conferred by the polybasic region and CAAX‐box of K‐Ras (Papin et al, 1998; Röring et al, 2012). In sharp contrast, a B‐Raf mutant introducing the V600E mutation into B‐RafAVKA (B‐RafAEKA) displayed a comparable MEK/ERK phosphorylation potential as B‐RafV600E (Fig 1C and D). This demonstrates that the AVKA mutation does not inactivate the kinase, but prevents its activation following membrane recruitment and dimerization.
As truly kinase‐dead B‐Raf mutants like B‐RafD594A or B‐RafK483M act as allosteric activators for wild‐type Raf proteins and thereby provoke paradoxical activation of the MEK/ERK pathway in the context of oncogenic Ras (Heidorn et al, 2010; Röring et al, 2012), we next compared them with B‐RafAVKA in this setting. To this end, we expressed these B‐Raf mutants in a Braf‐deficient murine embryonic fibroblast (MEF) reconstitution system (Braf −/−;ERTmHRasG12V MEFs) in which oncogenic H‐Ras signaling can be initiated by 4‐hydroxy‐tamoxifen (4‐HT)‐mediated release of an ERTmH‐RasG12V fusion protein from its inhibitory chaperone complex (Röring et al, 2012). As shown in Fig 2A and B, B‐RafWT and its kinase‐dead mutants augment MEK and ERK phosphorylation in B‐Raf‐deficient MEFs, which is further increased by 4‐HT‐induced Ras signaling. These data are in full agreement with the paradoxical MEK/ERK activation triggered by kinase‐dead B‐Raf (Heidorn et al, 2010; Röring et al, 2012). In contrast, complementation of Braf −/− MEFs with B‐RafAVKA leads only to a slight improvement of steady‐state MEK/ERK phosphorylation compared to empty vector infected cells, indicating again that it lacks the full signaling potential of B‐RafWT. Most importantly, B‐RafAVKA, despite its impaired signaling potential, does not provoke paradoxical MEK/ERK phosphorylation as the B‐RafD594A mutant (Fig 2A and B).
Figure 2. Release of oncogenic H‐Ras leads to hyperphosphorylation of MEK by kinase‐dead B‐Raf mutants, but not by B‐RafAVKA .
- Braf −/−;ERTmHRasG12V MEFs were reconstituted with HA‐tagged hBRAF constructs or empty vector (EV). Infected cells were either treated with 4‐HT (1 μM) to induce oncogenic Ras activation or with ethanol (OH) as a control.
- Quantification of pMEK (left) and pERK (right) normed to WT level. n = 3, mean + SEM, t‐test, **P < 0.01.
- The AVKA mutation has no discernible influence on Ras‐induced paradoxical ERK activation triggered by the D594A mutation. Experimental setup as in (A), shown are lysates from 4‐HT‐treated MEFs.
- Quantification of pMEK and pERK levels normed to HA expression. n = 3, mean + SEM, t‐test, **P < 0.01.
- Top: Braf −/−;ERTmHRasG12V MEFs were reconstituted with the indicated B‐Raf constructs. Following induction of Ras signaling, HA‐B‐Raf proteins were purified using anti‐HA antibodies and subjected to Western blotting to analyze immunoprecipitation of Raf‐1. Bottom: Quantification of co‐immunoprecipitated Raf‐1 normed to HA‐B‐Raf levels. n = 3, mean + SEM, t‐test, *P < 0.05.
Next, we asked whether the AVKA mutation was able to reduce the paradoxical MEK/ERK phosphorylation of B‐RafD594A. As shown in Fig 2C and D, the AVKA mutation had only a marginal effect on the paradoxical effect of B‐RafD594A. This result is in sharp contrast to the effect of the R509H mutation on B‐RafD594A (Röring et al, 2012) and indicates that this kinase‐dead mutant does not require AS phosphorylation but an intact DIF for allosteric transactivation of Raf‐1. Accordingly, the B‐RafD594A/AVKA double mutant still recruited more Raf‐1 than B‐RafWT, although there was a significant reduction compared to B‐RafD594A proper (Fig 2E). Interestingly, the AVKA mutation also reduced the prominent phosphorylation‐dependent electrophoretic mobility shift of B‐RafD594A (Figs 2C and E, and EV1A), suggesting that TVKS motif phosphorylation either directly or indirectly contributes to this phenomenon.
Figure EV1. Analysis of the electromobility shift (EMS) of different B‐Raf mutants, related to Figure 2 .

- Braf −/−; ERTmHRasG12V MEFs were reconstituted with the indicated HAhBRAF constructs or empty vector (EV). After induction of Ras with 4‐HT, HAB‐Raf proteins were immunoprecipitated. Immunoprecipitates (IP) were first washed with normal lysis buffer and then with lambda phosphatase buffer, and IPs were then divided and incubated in the absence or presence of 400 U lambda phosphatase for 1 h. Afterward, the electrophoretic mobility of the B‐Raf proteins was assessed by Western blotting analysis.
Generation of a conditional Braf AVKA‐knock‐in mouse model
So far, the role of the phosphoacceptor residues of the AS has been only investigated in tissue culture experiments involving the ectopic expression of mutant Raf kinases with alanine (AVKA) or phosphomimetic (EVKD) substitutions (Zhang & Guan, 2000; Chong et al, 2001; Baljuls et al, 2008; Rajakulendran et al, 2009; Röring et al, 2012), but not by in vivo approaches analyzing endogenous B‐Raf. Therefore, we addressed the role of the phosphoacceptor sites of the TVKS motif by a knock‐in approach producing a B‐Raf protein in which T599 and S602 were replaced by alanine residues. As B‐Raf expression and activity are subject to a tight spatiotemporal control and are regulated by alternative splicing and presumably two alternative promoters (Barnier et al, 1995), a conditional mutagenesis allowing the expression of B‐RafAVKA from its endogenous locus appeared necessary (Fig 3A). This strategy maintains B‐RafWT expression until Cre excises the loxP‐site‐flanked (floxed) minigene cassette. This places the mutated exon 15AVKA downstream of exon 14, thereby allowing the production of a transcript encoding B‐RafAVKA.
Figure 3. Generation of the conditional Braf AVKA‐knock‐in mouse.

- Targeting strategy. Top: Targeting vector pSC‐floxAVKA and a part of the murine Braf locus containing exons E14–E16 (not drawn to scale). Middle: Braf locus after homologous recombination with the targeting vector replacing E15 with a loxP‐flanked (floxed) minigene (MG; E15–20) segment. pA, polyadenylation signals; SA, splice acceptor. LoxP and Frt sites are indicated by red triangles and blue hexagons, respectively. Bottom: The modified Braf locus after Flp‐e and Cre‐mediated recombination.
- Genomic PCR with the primers indicated as red arrows in (A) showing 3 out of 9 clones being positive for homologous recombination.
- Southern blot analysis of genomic DNA of parental W4 ES cells, wild‐type MEFs, and the ES cell clones #286 and #273 (with recombined Braf floxAVKA allele). Genomic DNA was digested by AseI and analyzed using the external probe indicated in (A).
- Genomic PCR showing Cre‐mediated recombination of the Braf floxAVKA allele in vivo using the primer pair E14fwd and E15rev (indicated as blue arrows in A).
- Electropherograms of sequenced RT–PCR amplicons generated using splenic RNA of a Braf +/floxAVKA (left) and a Braf +/AVKA;Cre mouse (right). RT–PCR amplicons were generated using the primers E14fwd and E16revNEW. Note the double peaks at the first position corresponding to the threonine/serine and alanine codons (indicated by red arrows).
Using the targeting vector shown in Fig 3A, we obtained several correctly targeted ES cell clones (Fig 3B and C) of which two (#215, #273) successfully contributed to germline chimera (see Appendix for further details). Their progeny was then mated with Flp‐e transgenic mice to remove the neoR cassette and to generate the Braf floxAVKA allele (Fig 3A).
For initial characterization of the targeted locus, we crossed homozygous Braf floxAVKA/floxAVKA mice of a mixed 129SVxC57/Bl6 background with CMV::Cre deleter mice. Depending on inheritance of the Cre transgene, this generates either Braf +/floxAVKA or Braf +/AVKA mice, as confirmed by genotyping PCR (Fig 3D). Using RNA from a Braf +/AVKA; Cre− mouse and Braf +/floxAVKA; Cre+ littermate, we confirmed the co‐expression of Braf AVKA and Braf WT transcripts in the latter (Fig 3E). We also crossed homozygous Braf floxAVKA/floxAVKA animals with Sox2::Cre transgenic mice (Hayashi et al, 2002). Mating Braf floxAVKA/floxAVKA mice with both Cre deleter strains yielded normal sized litters with the expected allele frequencies. Thus, in contrast to Braf V600E/+‐, Braf Q257R/+‐, Braf D594A/+‐, and Braf L597V/+‐knock‐in mice (Mercer et al, 2005; Kamata et al, 2010; Andreadi et al, 2012; Inoue et al, 2014), which all display a severe phenotype with pre‐ or postnatal lethality, heterozygous Braf +/AVKA mice appear normal, are fertile, and were kept without any obvious abnormalities.
Next, we assessed the phenotype of homozygous Braf AVKA mice. To this end, we exploited the cytoplasmic Cre expression in Sox2::Cre oocytes to generate Braf +/AVKA animals without a Cre transgene in the F1 generation. Subsequently, Cre‐negative Braf +/AVKA mice were mated with each other to generate Braf +/+, Braf +/AVKA, and Braf AVKA/AVKA mice. As shown in Table 1, these matings produced litters with genotype frequencies at the expected Mendelian ratios in progenies derived from ES cell clones #215 and #273. At weaning, there was no significant difference in weights (Fig 4A), although we observed a tendency for Braf AVKA/AVKA mice to be lighter. In sharp contrast, when Braf +/− animals (also generated by the Sox2::Cre strategy), were mated with each other, we observed among the 73 weaned pups 42% Braf +/+ and 58% Braf +/−, but no Braf −/− animals. This is in full agreement with the embryonic lethal phenotype reported for three independently generated Braf‐knockout lines (Wojnowski et al, 1997; Galabova‐Kovacs et al, 2006; Zhong et al, 2007). Thus, despite producing a B‐Raf protein with impaired MEK phosphorylation potential (Fig 1B and C), the Braf AVKA‐knock‐in allele does not phenocopy the effects of the knockout allele. Homozygous Braf AVKA mice did not display any shortened life span, and both males and females were fertile and able to raise pups.
Table 1.
Litter genotype distribution from Braf +/AVKA intercrossed derived the two different founder knock‐in strains # 215 and #273
| Observed | Observed (%) | Expected | Expected (%) | |
|---|---|---|---|---|
| #215 | ||||
| +/+ | 49 | 26.63 | 46 | 25 |
| +/AVKA | 91 | 49.46 | 92 | 50 |
| AVKA/AVKA | 44 | 23.91 | 46 | 25 |
| Total | 184 | 100 | 184 | 100 |
| #273 | ||||
| +/+ | 111 | 23.57 | 117.8 | 25 |
| +/AVKA | 232 | 49.26 | 235.5 | 50 |
| AVKA/AVKA | 128 | 27.18 | 117.8 | 25 |
| Total | 471 | 100 | 471 | 100 |
Figure 4. Basic comparison and immune phenotyping between homozygous Braf WT and AVKA mice.

-
AWeight at weaning of Braf AVKA mice from two different founders showed no significant differences. 215: n = 24 (WT). 32 (Heterozygous), 19 (AVKA); 273: n = 114 (WT), 117 (Heterozygous), 52, t‐test.
-
BHomozygous Braf AVKA mice show reduced splenic cellularity. n = 7, t‐test.
-
C–ERelative number of CD8+ T cells. FACS analysis shows an increase in naïve cells (C), whereas differentiated TEM (D) and Tregs (E) reveal a reduction in Braf AVKA mice compared to WT. n = 8, t‐test.
-
FPrimary B cells of Braf AVKA mice show reduced CD69 surface expression upon LPS and anti‐IgM stimulation. n = 3, t‐test.
-
GPrimary B cells also show reduced proliferation upon stimulation compared with WT B cells. n = 3, t‐test.
Phenotypic characterization of Braf AVKA mice
As loss of B‐Raf expression impairs the development, activation, and effector functions of cells of the immune and hematopoietic systems (Brummer et al, 2002; Kamata et al, 2005; Tsukamoto et al, 2008; Dillon et al, 2013), we next analyzed the cellular composition of lymphatic organs. First, we noticed that spleens of homozygous Braf AVKA mice contained significantly lower amounts of cells (Fig 4B). Although all major hematopoietic subsets were present (Fig EV2A–E), certain subpopulations differed in size between wild‐type and knock‐in mice. The number of naive CD8+ T cells was significantly increased in B‐RafAVKA mice compared to wild‐type littermates. This observation was in line with a significant reduction in CD8+ T effector memory cells (TEM) and CD8+ T regulatory cells (Tregs) and suggests that differentiation into effector populations requires B‐Raf activity (Fig 4C–E). To evaluate whether this maturation defect is directly linked to B‐Raf signaling output, we isolated splenic T cells, stimulated them with a combination of anti‐CD3 and anti‐CD28 antibodies, and analyzed pERK levels by flow cytometry. We observed a slight but significant reduction in pERK levels in CD8+ cells 15 min after addition of the stimulus (Fig EV2F). Consistent with their normal population size in Braf AVKA mice, pERK levels were not significantly affected CD4+ T cells (Fig EV2G). Furthermore, Braf AVKA B cells were impaired in their activation ex vivo upon B‐cell antigen receptor (BCR) and Toll‐like receptor 4 (TLR4) stimulation as measured by surface expression of the activation marker and ERK target gene product CD69 (Minguet et al, 2005) (Fig 4F). Also, impaired LPS responses were associated with the failure to induce proliferation upon stimulation (Fig 4G).
Figure EV2. Blood and leukocytes analysis and pERK signaling in T cells related to immune phenotyping in Figure 4 .

- White blood, red blood count, and hemoglobin content of mouse blood. n = 3.
- FACS analysis of CD4‐positive T cells revealed no significant changes in the naïve or further differentiated subpopulations. n = 8.
- Relative amount of B cells in the spleen. n = 8.
- Relative amount of neutrophils in the spleen. n = 8.
- Relative amount of monocytes in the spleen. n = 8.
- pERK levels of CD8+ T cells stimulated with anti‐CD3/anti‐CD28 for the indicated times, assessed by flow cytometry. B‐RafAVKA‐expressing cells showed significant reduction in pERK 15 min after stimulation. WT: n = 4, AVKA: n = 5, *P < 0.05.
- pERK levels of CD4+ T cells stimulated with anti‐CD3/anti‐CD28 for indicated times, assessed by flow cytometry. WT: n = 4, AVKA: n = 5.
Data information: Mean + SEM, t‐test.
With increasing age, we noticed two further abnormalities. Firstly, homozygous Braf AVKA mice presented with shorter snouts and a more upward orientation of the eyes (Fig 5A). Secondly, homozygous Braf AVKA animals could be readily detected in cages by their distinct gait accompanied by a hunchback posture that is often displayed by mice with ataxia (Video EV1).
Figure 5. MEK and ERK activity is drastically impaired in the brain of AVKA‐knock‐in mice.

- Facial appearance of Braf AVKA mice.
- Top: In vitro kinase (IVK) assay of B‐Raf complexes purified from either Braf WT or Braf AVKA‐knock‐in brains from both founder strains revealed a reduced MEK phosphorylation potential regardless of their purification from NLB or RIPA lysates. Bottom: Quantification of IVK differentials: #215 samples indicated in black, #273 samples in gray. n = 5, mean + SEM, t‐test, **P < 0.01.
- Western blot analysis showing reduced pMEK and pERK levels in Braf AVKA brains.
- Immunohistochemistry showing reduced pERK levels in Purkinje cells of homozygous Braf AVKA mice. Brain sections imaged at 10× magnification.
The later observation, together with the long‐known fact that B‐Raf is highly expressed in the brain and fulfills multiple functions in neurons and macroglia (Chen et al, 2006; Zhong et al, 2007; Galabova‐Kovacs et al, 2008; Tien et al, 2012; Pfeiffer et al, 2013), prompted us to compare the B‐Raf signaling output in total brain extracts of Braf WT and Braf AVKA littermates. Firstly, we performed in vitro kinase (IVK) assays and could confirm for the first time that endogenously expressed B‐RafAVKA possesses < 50% of the MEK phosphorylation potential of B‐RafWT, irrespective of its purification under mild (NLB; 0.5% NP‐40) or harsh (RIPA) buffer conditions (Fig 5B). This decrease in IVK activity was also reflected in vivo as MEK and ERK phosphorylation was consistently reduced in Braf AVKA brain lysates (Figs 5C and EV3A and B). This reduction was not affected by sex or age. To obtain further insights into the brains of Braf AVKA mice, we performed immunohistochemistry. Macroscopic and histological examination did not reveal gross differences between brains from Braf WT and Braf AVKA littermates. However, Purkinje cells of Braf AVKA mice presented with reduced phospho‐ERK staining (Fig 5D), a phenotype that was also reported for Braf −/− mice (Galabova‐Kovacs et al, 2008). As Braf −/− mice display myelination defects, we analyzed the expression of myelin basic protein (MBP), whose expression was strongly reduced in Braf −/− animals (Galabova‐Kovacs et al, 2008). Interestingly, however, we could not observe significant differences in MBP levels in brain extracts and the staining intensity of myelinated fibers in cerebellar sections from Braf AVKA and Braf +/+ mice (Fig EV3C–E). This lack of an obvious myelination defect could explain why Braf AVKA‐knock‐in mice show a less severe and delayed neurological phenotype than Braf −/− animals.
Figure EV3. Further phenotypic analysis of Braf AVKA mice, related to Figure 5 .

- Quantification of total brain lysates of the 273 strain. pMEK and pERK levels were drastically reduced in B‐Raf AVKA lysates. pMEK: n = 8, pERK: n = 18. **P < 0.01, ***P < 0.001.
- Quantification of total brain lysates of the 215 strain. As in 273, pMEK and pERK levels were drastically reduced in B‐Raf AVKA lysates. pMEK: n = 6, pERK: n = 6, **P < 0.01.
- Western blot analysis of myelin basic protein (MBP) levels in total brain lysates of WT and mutant mice.
- Quantification of MBP levels in total brain lysates of the two founder strains (215 and 273). 215: n = 6, 273: n = 7.
- Representative images of Luxol fast blue (LFB) staining of the cerebellum. LFB (blue) is specifically bound to myelin sheath in the central nervous system. Note the equally blue staining in the white matter regions in the cerebella of a 4‐month‐old wild‐type and Braf AVKA mouse. Brain sections imaged at 10× magnification.
Data information: Mean + SEM, t‐test.
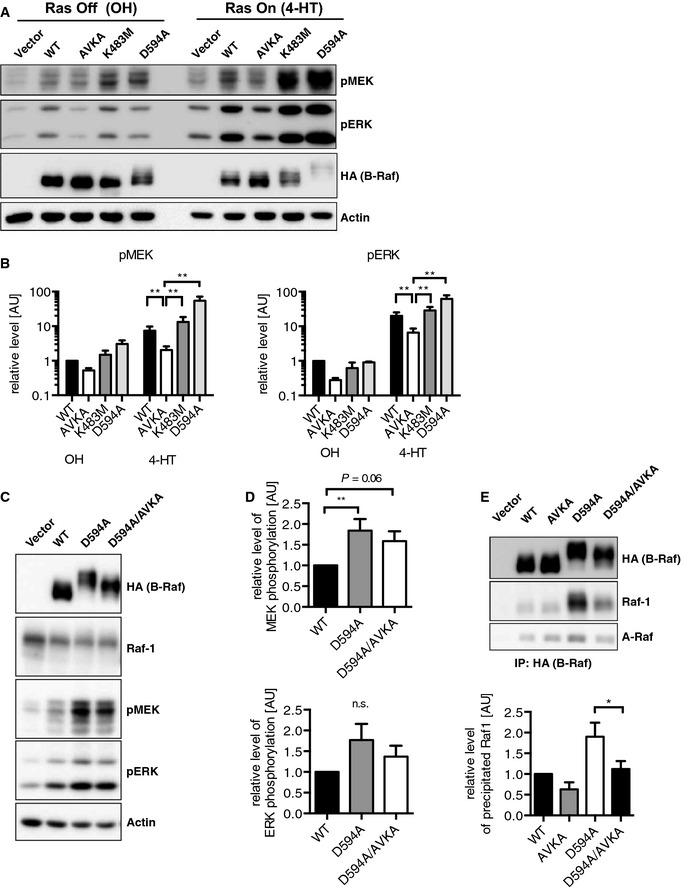
As B‐RafAVKA is potentially expressed in homozygous knock‐in animals from the zygotic stage onwards, it was conceivable that other Raf isoforms could be up‐regulated to compensate for the impaired signaling output by B‐RafAVKA. Nevertheless, B‐Raf, Raf‐1, A‐Raf, or KSR1 expression levels were not different between Braf WT and Braf AVKA mice (Figs 5C and EV4A and B). Interestingly, however, we observed a significant reduction in KSR1 by 40% in B‐RafAVKA compared to B‐RafWT immunoprecipitates, while the amount of co‐immunoprecipitated 14‐3‐3 proteins did not differ between B‐RafWT and B‐RafAVKA (Fig 6A). This suggests that the interaction of B‐Raf with KSR1 is promoted by AS phosphorylation. In contrast, the heterodimerization between B‐Raf and Raf‐1 was not affected by the AVKA mutation in mouse brain lysates (Fig 6A) or in a co‐immunoprecipitation assay (Fig 6B). By co‐expressing HA‐tagged and Myc‐tagged B‐RafAVKA proteins, we could also show that an intact TVKS motif is not essential for the formation of B‐Raf homodimers (Fig 6C). Interestingly, however, the phosphomimetic substitutions of T599 and S602 (EVKD) enhanced the homodimerization potential of B‐Raf to a similar degree as the E586K mutation, which promotes homodimerization (Rajakulendran et al, 2009; Freeman et al, 2013), although this effect was not as pronounced as observed for B‐RafV600E (Fig 6C).
Figure EV4. Further phenotypic analysis of mice, related to Fig 5 .

- Quantification of the expression of Raf isoforms (A‐Raf, B‐Raf, and Raf‐1) and KSR1 in littermates of the 273 strain. There was no significant difference in total protein levels. B‐Raf: n = 8, A‐Raf/Raf‐1/KSR1: n = 5.
- Quantification of total brain lysates from littermates of the 215 strain. As for the 273 strain, Braf AVKA mice derived from ES cell clone 215 show no significant differences in Raf isoform or KSR1 expression levels. n = 6.
Data information: t‐test. Shown is the mean + SEM.
Figure 6. Characterization of the homo‐ and heterodimerization potential of B‐RafAVKA .

- Total brain lysates of WT and Braf AVKA mice were subject to immunoprecipitation (IP) using either anti‐B‐Raf antibodies or non‐specific IgG as a negative control. The co‐purified proteins were visualized using the indicated antibodies and quantified. Ksr1 is significantly decreased in B‐RafAVKA complexes, while the other proteins are equally bound to B‐RafWT or B‐RafAVKA. n = 3, mean + SEM, t‐test, *P < 0.05.
- Anti‐Myc IP of Plat‐E cells transfected with Myc‐tagged Raf‐1 and different HA‐B‐Raf constructs. Western blot analysis showed that B‐RafAVKA is not impaired in its ability to interact with Raf‐1.
- Anti‐Myc IP of Plat‐E cells expressing different Myc‐ and HA‐B‐Raf proteins. Note that B‐RafAVKA is not impaired in its ability to interact with other B‐Raf proteins, while homodimerization of B‐RafEVKD is enhanced.
- Top: Exemplary Western blot analysis of EGF‐stimulated primary astrocytes. Mutant astrocytes show drastic reduction in basic and stimulated levels of pMEK and pERK. Bottom: Quantification of all Western blot samples. WT: n = 7, AVKA: n = 6, mean + SEM, t‐test, *P < 0.05, **P < 0.01.
As astrocytes represent the most abundant cell lineage in the brain and because B‐Raf signaling is of growing importance for our understanding of normal and neoplastic macroglia (Eisenhardt et al, 2011; Tien et al, 2012), we compared MEK and ERK phosphorylation in short‐term cultured primary astrocytes isolated from newborn Braf WT and Braf AVKA animals (Fig 6D). This revealed that both pMEK and pERK levels were significantly reduced under normal growth conditions and in the presence of EGF.
TVKS motif phosphorylation is required for efficient ERK signaling
To obtain more detailed biochemical insights into the signaling properties of B‐RafAVKA‐expressing cells, we required an experimental isogenic system allowing the generation and analysis of large quantities of homogenous cells. To this end, we generated fibroblasts from homozygous Braf floxAVKA (#273) embryos (murine embryonic fibroblast = MEF) and immortalized them by retroviral transduction with an expression vector for the SV40 large T antigen (TAg). Subsequently, two pools of these MEFs (#5 and #6) were transduced with the expression vector pMIBerry/CreERT2 encoding the 4‐HT‐regulated Cre recombinase CreERT2 and the fluorescent dsRed2 protein (Röring et al, 2012). Infected cells were sorted to homogeneity by flow cytometry, expanded, and then either treated with ethanol (OH) or 4‐HT to induce recombination of the Braf floxAVKA locus. As shown in Fig EV5A, a pulse of 30–100 nM 4‐HT for 48 h was sufficient to induce recombination at the DNA level and an exchange of the wild‐type against the mutant transcript. Based on data from an analogous MEF system generated from Braf floxE12 mice in which we observed the loss of B‐Raf protein within 3–5 days (Fig EV5B), we estimated that 5 days of culture after 4‐HT addition was sufficient to exchange B‐RafWT against B‐RafAVKA.
Figure EV5. Further analysis of signaling properties and proliferation of B‐RafAVKA and B‐Raf fusion protein FAM131‐B‐Raf, related to Figs 7 and 8 .
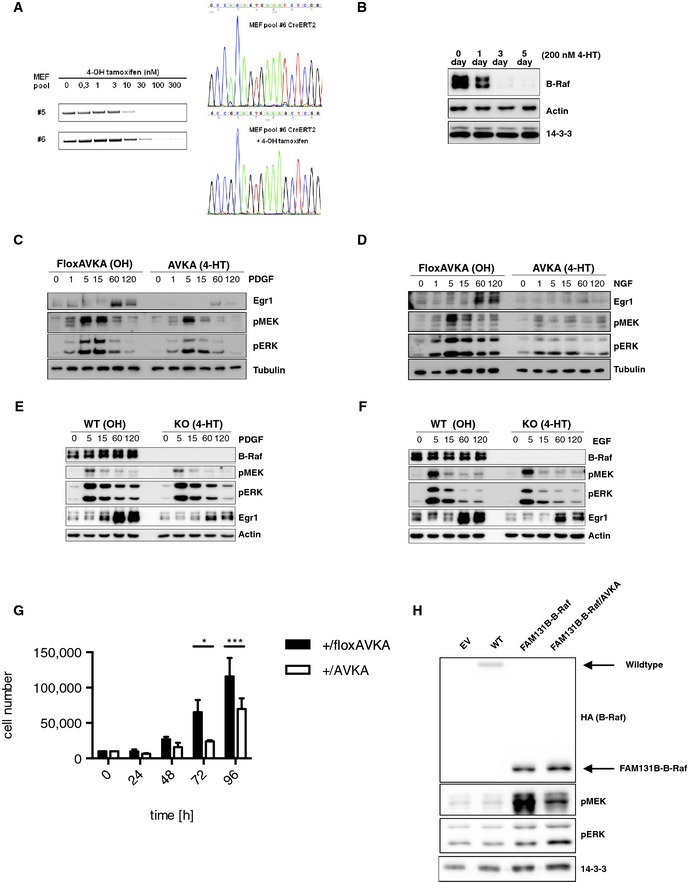
-
ASequencing of mRNA of MEF with floxed Braf locus. Upon 4‐HT treatment, the CreERT2 was activated and recombined the floxed Braf locus. Sequencing reveals that 300 nM of 4‐HT are sufficient for recombination.
-
BTotal cell lysates of MEF with floxed knockout Braf locus. Western blotting reveals that B‐Raf protein is lost between three and five days.
-
C, DWestern blot analysis of MEFs expressing either B‐RafWT or B‐RafAVKA upon stimulation with PDGF or NGF. Similar to EGF stimulation, pMEK and pERK levels were strongly reduced. Also, expression of Egr1 was significantly impaired.
-
E, FEGF and PDGF stimulation experiments of Braf‐knockout MEFs lead to comparable results as stimulation of B‐RafAVKA MEFs.
-
GProliferation of MEFs heterozygous for the Braf locus. Cells have only one AVKA‐knock‐in allele. Similar to homozygous knock‐in MEFs, these cells show reduced proliferation compared to WT. n = 3, mean + SEM, t‐test, *P < 0.05, **P < 0.01.
-
HWestern blot analysis of the FAM131B‐B‐Braf and FAM131B‐B‐RafAVKA signaling. Please note that FAM131B‐B‐Raf as an N‐terminally truncated fusion protein consisting predominantly of the B‐Raf CR3 runs faster in the SDS–PAGE (indicated by arrows) than full‐length B‐RafWT. In agreement with their focus‐forming potentials (Fig 8A and B), pMEK levels, which represent the direct readout of B‐Raf activity, were drastically reduced in MEFs expressing FAM131B‐B‐RafAVKA compared to those producing FAM131B‐B‐Raf. The lack of an effect on pERK in FAM131B‐B‐RafAVKA‐expressing cells compared to those expressing FAM131B‐B‐Raf is best explained by the action of compensatory feedback loops.
Firstly, we stimulated MEFs retaining B‐RafWT or expressing B‐RafAVKA with EGF for various time points (Fig 7A). In both populations, EGF‐induced MEK/ERK phosphorylation, although pERK returned faster to baseline levels in B‐RafAVKA‐expressing MEFs than in their B‐RafWT‐expressing counterparts. Importantly, expression of the immediate early gene (IEG) product c‐Fos was completely blocked in B‐RafAVKA‐expressing MEFs. Similarly, expression of Egr1, another IEG product, was reduced in B‐RafAVKA‐expressing MEFs. Similar findings for pMEK/pERK were obtained for MEFs stimulated with PDGF or NGF (Fig EV5C and D). Interestingly, the reduction in Egr1 induction of the B‐RafAVKA‐expressing MEFs is reminiscent of our observations with TAg‐immortalized Braf‐deficient MEFs (Fig EV5E and F). This indicates that AS phosphorylation and hence full B‐Raf activity is required for optimal downstream signaling and an IEG response toward activated growth factor receptors.
Figure 7. Phenotypic characterization of conditional Braf AVKA MEFs.
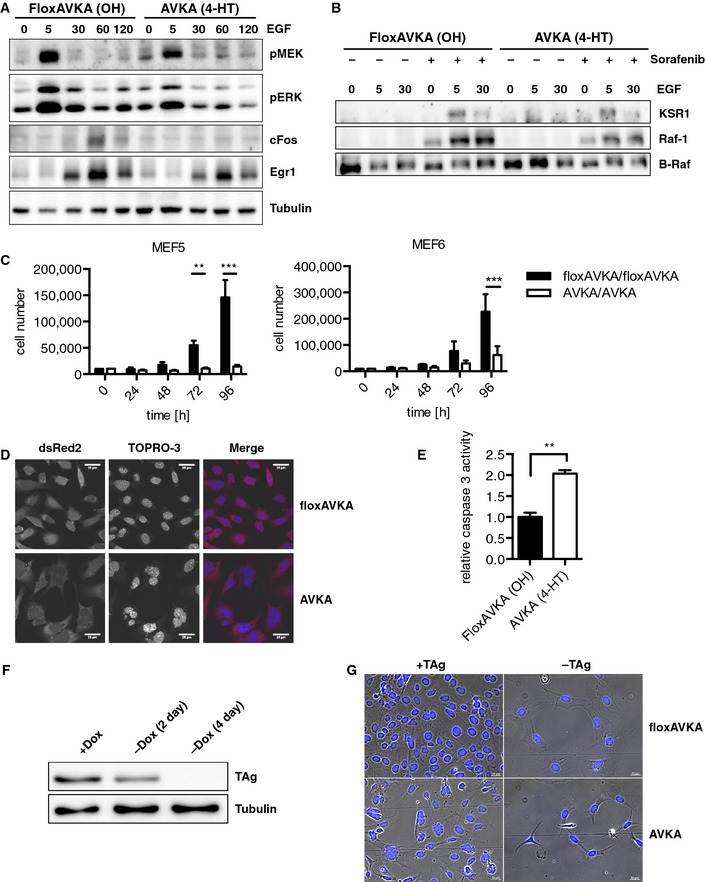
- MEFs expressing B‐RafWT or B‐RafAVKA were stimulated with EGF for the indicated times (min) and analyzed by Western blotting.
- IP of EGF‐stimulated MEFs expressing B‐RafWT or B‐RafAVKA. Sorafenib was used to increase complex build up.
- Proliferation of two different MEF pools expressing either B‐RafWT or B‐RafAVKA. MEFs expressing B‐RafAVKA shows a strong reduction in growth rate. n = 3, mean + SEM, t‐test, **P < 0.01, ***P < 0.001.
- Cell morphology of B‐RafAVKA‐expressing MEFs is enlarged and present with abnormal nuclei.
- MEFs expressing B‐RafAVKA show a significant increase in caspase‐3 activity. n = 3, mean + SEM, t‐test, **P < 0.01.
- Decay of TAg expression in MEFs following dox withdrawal.
- Morphology of MEFs expressing B‐Rafwt or B‐RafAVKA in the presence or absence of TAg.
The signaling defect of B‐RafAVKA‐expressing MEFs prompted us to investigate the heterodimerization potential of the activation loop mutant in EGF‐stimulated MEFs (Fig 7B), either in the absence or presence of sorafenib, which is a potent inducer of B‐Raf heterodimers compared to other Raf inhibitors (Röring et al, 2012). Although we failed to detect heterodimers between endogenous B‐Raf and Raf‐1 or KSR1, probably due to their low abundance under these conditions, we could detect them for both B‐RafWT and B‐RafAVKA in the presence of sorafenib, indicating again that the latter is able to interact with Raf‐1 or KSR1.
The Braf AVKA allele impairs the fitness of immortalized MEFs
Next, we measured the effect of the Braf AVKA allele on the proliferation in the aforementioned TAg‐immortalized MEF pools originating from Braf floxAVKA/floxAVKA embryos (Fig 7C). As another control, we used MEFs from a heterozygous Braf floxAVKA/+ animal (Fig EV5G). Although we noticed a significant twofold reduction in Braf AVKA/+ MEFs numbers compared to their isogenic counterpart, this difference was by far more pronounced in homozygous Braf AVKA/AVKA MEFs (4‐ to 10‐fold reduction) (Fig 7C). In the course of these experiments, we noticed changes in the morphology of Braf AVKA/AVKA MEFs, which we further evaluated by microscopy using the cytoplasmic expression of dsRed2 introduced with the CreERT2 construct and the DNA stain TOPRO‐3 as markers for overall cell shape and nuclear morphology, respectively. This analysis revealed that B‐RafAVKA‐expressing MEFs were often enlarged and poly‐nucleated (Fig 7D). This aberrant morphology was accompanied by an increase in caspase‐3 activity (Fig 7E). These observations suggest that the inducible expression of B‐RafAVKA induces endomitotic events in MEFs accompanied with the induction of cell death. Given the relatively good fitness of Braf AVKA mice, we reasoned that B‐RafAVKA‐expressing MEFs only display this phenotype in the presence of TAg, which enforces cell cycle progression by the inactivation of E2F and p53 (Ahuja et al, 2005). Therefore, we took an early passage aliquot of primary Braf floxAVKA/floxAVKA MEF pool #5 and infected them with a doxycycline (dox) inducible TAg expression system. This experimental setup allows the expansion of MEFs in the presence of dox and TAg and the subsequent withdrawal of the immortalizing oncoprotein by dox withdrawal (Fig 7F). Indeed, B‐RafAVKA‐expressing MEFs presented with aberrant nuclei only in the presence of TAg, while B‐RafAVKA MEFs in which dox was withdrawn prior to CreERT2‐mediated recombination continued to display smooth regularly shaped nuclei as they were observed in B‐RafWT‐expressing MEFs, regardless whether they have been exposed to dox or not (Fig 7G). Taken together, these data suggest that the coordination of events permitting cellular proliferation enforced by TAg requires maximum B‐Raf signaling.
The AVKA mutation impairs transformation by non‐V600E B‐Raf mutants
As we observed that the AVKA mutation reduces cellular proliferation and is able to disrupt the signaling potential of transforming non‐V600E B‐Raf mutants (Fig 1B–D), we tested whether the lack of T599 and S602 also affected their transformation potential. To this end, we transduced the mutants described in Fig 1 into TAg‐immortalized MEFs and conducted focus‐forming assays. We also included the FAM131B‐BRAF fusion oncogene that has been recurrently found in low‐grade astrocytomas (Fig EV5A) (Cin et al, 2011; Roth et al, 2015). This oncoprotein was chosen as a representative of the growing spectrum of oncogenic B‐Raf fusion proteins, which have been discovered in various tumor entities and all share the possession of an intact kinase domain and the loss of the autoinhibitory N‐terminal moiety.
As shown in Fig 8A and B, MEFs expressing B‐RafV600E and B‐RafE586K, but not those transduced with an empty control vector or expressing B‐RafWT, generated multiple foci, indicative of impaired contact inhibition. Transformation efficiency was further enhanced in MEFs expressing B‐RafEVKD, B‐RafCAAX, or FAM131B‐B‐Raf. This finding can be explained by our previous observations that these mutants trigger lower MEK/ERK phosphorylation levels than B‐RafV600E (Fig 1; Cin et al, 2011; Röring et al, 2012), which might be more compatible with proliferation. Strikingly, the AVKA mutation drastically reduced the transformation potential of all mutants except B‐RafV600E. These findings further underscore the relevance of the AS for the signaling output of non‐V600E B‐Raf mutants and identify it as a promising target for the therapy of these oncoproteins.
Figure 8. The AVKA mutation is able to block transformation potential of B‐Raf mutants and model of B‐Raf activation incorporating the roles of the activation segment and the DIF.

- MEFs were then infected with retroviral vectors encoding the indicated B‐Raf proteins. Giemsa staining revealed transformed and hyper‐proliferated cells by dark purple color.
- Quantification of focus formation potential of the B‐Raf mutants used in (A). n = 3, mean + SEM, t‐test, ***P < 0.001.
- Following recruitment by Ras‐GTP, B‐Raf forms homo‐ or heterodimers with the assistance of 14‐3‐3 proteins. This loosely dimerized and membrane‐bound state is imitated by the B‐RafE586K and B‐RafCAAX mutants, which still require an intact DIF and AL phosphorylation. Within the dimer, one protomer becomes phosphorylated at its TVKS motif in cis (depicted by black bent arrow) leading to reorientation of the DIF and induction of a conformational change in the other protomer (blue bent arrow) followed by phosphorylation of the AS in the other protomer in cis. B‐RafV600E is locked in the highly active state and thus signals independently of an intact TVKS motif or DIF. See text for further details.
Discussion
Since its first description 15 years ago, the role of the TVKS motif phosphorylation sites in the activation of Raf isoforms has been predominantly deduced from in vitro kinase assays (Zhang & Guan, 2000; Baljuls et al, 2008; Holderfield et al, 2013) and from the large number of mutations affecting the AS in cancer, most notably V600E, and in RASopathies (Garnett & Marais, 2004; Andreadi et al, 2012) and references therein or in the COSMIC database. However, very little is known about its role for B‐Raf signaling in intact cells and no evaluation of the TVKS motif has been conducted in vivo. Here, we report for the first time the phenotype of mice expressing a B‐Raf protein lacking the highly conserved AS phosphorylation sites T599 and S602. Surprisingly and in sharp contrast to mice with Braf‐knock‐out (Wojnowski et al, 1997; Galabova‐Kovacs et al, 2006; Zhong et al, 2007) and Braf‐knock‐in alleles causing increased or paradoxical B‐Raf activity (Mercer et al, 2005; Kamata et al, 2010; Andreadi et al, 2012; Inoue et al, 2014), B‐RafAVKA‐expressing mice are viable, despite a significant reduction in MEK/ERK signaling output in and ex vivo. As we could neither observe a paradoxical behavior of B‐RafAVKA in our Braf −/− MEF complementation assay nor in tissues from Braf AVKA mice, we conclude that mutation of the TVKS motif and the associated reduction in kinase activity does not provoke a paradoxical action like it is observed for the kinase‐dead B‐RafD594A mutant in tissue culture (Fig 2 and Röring et al, 2012) and in vivo (Heidorn et al, 2010; Kamata et al, 2010). Moreover, our observation that Braf AVKA mice do not represent a phenocopy of Braf‐deficient animals supports the growing notion that B‐Raf fulfills critical biological functions that are not directly proportional to its enzymatic activity, for example, a scaffolding role independent of AS phosphorylation.
However, the signaling deficits and phenotypic alterations observed in B‐RafAVKA expressing MEFs, primary astrocytes, and mice clearly demonstrate the relevance of the TVKS motif. Indeed, by comparing the growth factor‐induced Egr1 expression in conditional Braf AVKA/AVKA MEFs with Braf −/− MEFs (Figs 7A and EV5C–F), it became evident that the AVKA knock‐in might confer similar effects like B‐Raf deficiency on growth factor‐induced IEG expression. Furthermore, the differential in pERK phosphorylation observed in brain lysates of Braf AVKA animals (Figs 5C and EV3A and B) was similar to that reported for 18‐d‐old Braf −/− mice (Galabova‐Kovacs et al, 2008). In further analogy to the study by Galabova‐Kovacs et al (2008) in mice with Nestin::Cre‐mediated Braf deletion, we observed a reduction in pERK staining intensity in the Purkinje cells in cerebella of Braf AVKA mice. This defect or other potential functional impairments caused by reduced ERK activity in macroglial cell types could also account for the ataxia observed in older Braf AVKA mice. However, it should be noted that the neurological deficits observed in Braf AVKA mice were never as severe as reported for Braf −/− animals, which, depending on the Cre transgene, did not live beyond P21‐P35 (Zhong et al, 2007; Galabova‐Kovacs et al, 2008; Pfeiffer et al, 2013). This could be explained by the observation that Braf AVKA animals show no obvious effects in myelination levels compared to wild‐type mice. This is in strong contrast to Braf‐deficient mice, suggesting that the loss of the B‐Raf protein shapes the knockout phenotype by MEK/ERK‐dependent and MEK/ERK‐independent mechanisms. Thus, Braf AVKA mice will be useful to study the role of B‐Raf in the development, function, and maintenance of the nervous system at stages that are not reached by Braf −/− animals.
Given the well‐documented role of B‐Raf in normal and malignant hematopoiesis and immune cell function (Brummer et al, 2002; Tsukamoto et al, 2004; Kamata et al, 2005; Röring & Brummer, 2012; Yaktapour et al, 2014), we initially characterized the cell populations in peripheral blood and lymphatic organs. These analyses revealed a significant reduction in CD8+ T effector memory cells (TEM) and T regulatory cells (Tregs) in Braf AVKA mice suggesting that optimal B‐Raf signaling is required to differentiate progenitors into these CD8+ T‐cell subpopulations. These findings could be explained by the critical role of ERK signaling in the differentiation of T‐cell effector lineages and the emerging role of B‐Raf in T‐cell antigen receptor (TCR) signaling (Tsukamoto et al, 2004, 2008; Deswal et al, 2013; Dillon et al, 2013). However, our flow cytometry analyses revealed only a very subtle reduction in ERK phosphorylation in CD8+ T cells raising the question for its biological significance. Among several possibilities, this could be caused by feedback loops in the mutant cells conferring robustness to pathway perturbation (Fritsche‐Guenther et al, 2011). In any case, our in vivo data further extend the experiments by Tsukamoto et al (2004), who showed that overexpression of B‐RafAVKA in the Jurkat T‐cell line impaired the sustained phase of TCR‐induced MEK/ERK phosphorylation and IL‐2 production. Likewise, B lymphocytes from Braf AVKA mice responded with reduced proliferation and impaired CD69 induction toward BCR or TLR4 stimulation. In summary, Braf AVKA mice show no obvious signs of immune deficiency. Thus, it remains to be investigated whether the observed changes in immune cell population size and signaling impact on the functionality of the immune system.
Our data demonstrate that endogenously expressed B‐RafAVKA displays significantly reduced kinase activity and impairs the ERK signaling pathway in vivo and in various ex vivo systems. This raises the question whether these signaling defects are caused by impaired B‐Raf activity, thereby mimicking the functional loss of an important MEK/ERK activator, like it is observed in B‐Raf deficiency (Brummer et al, 2002; Galabova‐Kovacs et al, 2006, 2008), or whether B‐RafAVKA exerts a dominant‐negative effect, for example, by being incorporated into Raf heterodimers. Although both possibilities are not mutually exclusive, the latter scenario appears quite realistic. Firstly, B‐RafAVKA entertains interactions with itself, Raf‐1 and KSR1, albeit the number of stable B‐Raf/KSR1 complexes was significantly reduced. Based on these findings and our previous observation that B‐RafR509H is still able to interact with but fails to transactivate Raf‐1 (Röring et al, 2012), a dominant‐negative action of B‐RafAVKA appears feasible, in particular given the recent structural insights into the role of the AS‐H1 (Thevakumaran et al, 2015). Here, Thevakumaran et al (2015) show in B‐RafV600E crystals that the salt bridge between E600 in the mutant AS‐H1 and K507 strengthens the nearby “αC helix‐in” conformation and thereby promotes phosphotransferase activity and dimerization. They also emphasize that, due to the close positioning of T599 and S602 position to V600, a similar conformational change is induced by TVKS motif phosphorylation. This concept is supported by our observation that the B‐RafE586K and B‐RafCAAX gain‐of‐function mutants, but not the B‐RafV600E oncoprotein, which can form its mutation‐specific E600‐K507 salt bridge and is thus exempted from AS‐H1 phosphorylation, require an intact TVKS motif (Figs 1 and 8A and B). Moreover, B‐RafEVKD, which carries phosphomimetic substitutions of T599 and S602, displayed enhanced homodimerization compared to B‐RafWT (Fig 6C). Importantly, the model by Thevakumaran et al further suggests that dimerization and thereby allosteric transactivation via R509 is promoted by phosphorylation‐induced reorientation of the AS‐H1. Thus, it appears very unlikely that the aforementioned interaction between the AS‐H1 and the αC helix can be formed in B‐RafAVKA as it lacks the negative charges provided either by pT599/pS602, their phosphomimetic substitutions in the B‐RafEVKD mutant, or the glutamic acid in B‐RafV600E. Consequently, enzymatic activity of B‐RafAVKA cannot be increased and the αC helix stays in the outward orientation leaving R509 in a position in which it cannot mediate efficient dimerization with KSR1 and/or transactivation of Raf‐1. According to this model, Raf‐1, which we observed in B‐RafAVKA complexes in MEFs and in brain lysates, cannot be transactivated and remains trapped in a functionally compromised complex, ultimately resulting in impaired downstream signaling. Thus, residual ERK signaling in Braf AVKA mice is probably driven by Raf‐1 homo‐ and Raf‐1/A‐Raf heterodimers (Mooz et al, 2014), while Ras‐mediated recruitment of B‐RafAVKA containing homo‐ or heterodimers represents a futile approach. This would lower the capacity of the cell to respond to extracellular signals with maximum MEK/ERK activation and IEG induction, but would still allow enough pathway activity to promote a relatively normal development as displayed by Braf AVKA mice. Certain cell types, however, in particular those with a high expression or dependence of the B‐Raf isoform, could still be impaired in their development, function, or maintenance as it is suggested by the phenotype of Braf AVKA mice.
Our approach to introduce the AVKA substitution into various B‐Raf mutants with increased kinase activity provides new insights into the B‐Raf activation cycle (Fig 8C). For example, we investigated the tumor‐associated B‐RafE586K mutant that displays elevated MEK phosphorylation and transforming potential (Emuss et al, 2005), most likely due to its increased dimerization potential (Rajakulendran et al, 2009; Freeman et al, 2013). In agreement with the model that Raf dimerization follows recruitment by Ras (Lavoie & Therrien, 2015), the R188L substitution had little to no effect on the MEK phosphorylation potential of B‐RafE586K (Fig 1B). However, the activity of this oncoprotein is sensitive to N‐region neutralization (Emuss et al, 2005), the R509H (Röring et al, 2012), and AVKA mutations (Figs 1B and 8A and B). Based on the aforementioned concept that AS‐H1 phosphorylation leads to DIF reorientation and the model by Hu et al (2013) incorporating the charged N‐region into the extended DIF, these observations suggest that dimer initiation by Ras and the phosphorylation events in the N region and AS cooperate in reorientating the DIF in the activator to induce optimal transactivation and trans‐phosphorylation of the receiver protomer. The concept that TVKS motif phosphorylation occurs after membrane recruitment by Ras is also supported by our observation that the MEK phosphorylation and focus formation provoked by B‐RafCAAX is reduced by 80% and 90% by the AVKA mutation, respectively.
Lastly, our observation that the AVKA mutation reduces the signaling output and transforming potential of B‐Raf and some of its gain‐of‐function mutants identifies AS phosphorylation as a potential therapeutic target, for example, for tumors expressing B‐Raf oncoproteins, which are activated by mutations outside of the AS or by N‐terminal truncation. Since B‐RafAVKA does not behave paradoxically, a therapeutic approach blocking AS phosphorylation could represent an alternative strategy to the current ATP‐competitive inhibitors that must not be used in the context of dysregulated Ras signaling and that also disappoint in the context of oncogenic B‐Raf fusion proteins (Hutchinson et al, 2013; Sievert et al, 2013). Indeed, we demonstrate here that the AVKA mutation strongly impairs transformation by the FAM131B‐B‐Raf fusion, which, like all other oncogenic B‐Raf fusions reported so far, possesses a non‐mutated kinase domain and exhibits oncogenic properties due to the loss of the CR1/CR2 and its inability for autoinhibition (Cin et al, 2011). The drastic effect of the AVKA mutation on FAM131B‐B‐Raf demonstrates the relevance of the AS for the signaling output of N‐terminally truncated B‐Raf oncoproteins. As FAM131B‐B‐Raf lacks an RBD, our data indicate that Ras‐mediated displacement of the autoinhibitory N‐terminal moiety is not sufficient for full B‐Raf activation, but requires the aforementioned conformational changes within the kinase domain that are induced by AL phosphorylation (Thevakumaran et al, 2015). Thus, even the kinase domain of oncogenic B‐Raf fusion proteins appears to toggle between an inactive and active conformation, depending on the degree of AL phosphorylation. This concept might contribute to our understanding how existing B‐Raf inhibitors bind to oncogenic B‐Raf fusions.
In summary, our study shows that T599 and S602 are dispensable for the development and normal life span and cycle of laboratory mice, while being essential for oncogenic signaling by non‐V600E B‐Raf mutants. These findings appear as ideal prerequisites to design therapies exploiting oncogene addiction while minimizing the risk for side effects. The conditional Braf AVKA‐knock‐in model will be invaluable to further address this concept in future studies.
Materials and Methods
Mouse strains
Mice were housed in the specific pathogen‐free barrier facility of the University Medical Center Freiburg in accordance with the institutional guidelines and approved by the local animal ethics committee (G‐10/51). The generation and genotyping of Braf floxAVKA mice is described in detail in the Appendix. Braf floxE12 mice (Chen et al, 2006) were kindly provided by Dr. Manuela Baccarini (Vienna) and backcrossed onto a C57/Bl6 background. Sox2::Cre deleters were kindly provided by Dr. Sebastian Arnold (Freiburg) and backcrossed on a C57/Bl6 background. Mice were kept under standard conditions (12‐h light/dark cycle) with food and water ad libitum. Littermates were used throughout the experiments, and investigators were blinded for genotypes previous to data analysis except for Western blot and video analyses.
Tissue culture
The cultivation and transfection of Plat‐E cells as well as the generation and cultivation of TAg‐immortalized Braf floxE12/floxE12 MEFs was described previously (Röring et al, 2012). Braf floxAVKA/floxAVKA MEFs were generated using the same procedures. Primary astrocytes were isolated from brains of 4‐d‐old mice and dissociated mechanically in DMEM. Cells were washed and plated in Plat‐E culture medium on poly‐L‐ornithine‐coated dishes. When cells reached confluency, EGF was added for 5 min and cells were lysed.
Plasmids
The pMIG/HA‐B‐Raf, pMIG/FAM131B‐B‐Raf, and pMIBerry/B‐raf‐Myc/His expression vectors and the pMIBerry/CreERT2 plasmid have been described previously (Cin et al, 2011; Röring et al, 2012). Mutations were introduced using standard site‐directed mutagenesis (oligonucleotides sequences can be provided upon request). For construction of the dox‐inducible TAg expression vector, the TAg cDNA from pQCXIH/TAg (Röring et al, 2012) was subcloned into the retroviral vector SH570, which combines the elements of a tightly regulated dox‐inducible cDNA expression system (Herr et al, 2011) and which was similarly constructed as the retroviral two‐component system for dox‐inducible cDNA expression (Haug et al, 2015).
Antibodies
Antibodies used in this study were as follows: anti‐Egr1 (C‐19), anti‐Raf‐A (C‐20), anti‐Raf‐B (F‐7 or H‐145), anti‐Raf‐1 (C‐12 or E‐10), anti‐pan 14‐3‐3 (H‐8), anti‐β‐actin (C‐4), anti‐tubulin (B‐5‐1‐2; all from Santa Cruz Biotechnology, USA), anti‐HA 3F10 (Roche, Germany), anti‐c‐FOS (9F6), anti‐phospho‐MEK1/2 (pS217/221), anti‐phospho‐MEK1/2 (pS221), anti‐MEK1/2, anti‐p42/p44 MAPK, anti‐phospho‐MAPK (pT202/pY204; ERK1/2; all from Cell Signaling Technologies, USA), and anti‐KSR‐1 (BD, USA). Detailed information on antibodies used for immune phenotyping is provided in the Appendix.
Western blot analysis of tissue culture cells
Total cell lysates (TCLs) were generated and analyzed as described previously (Röring et al, 2012). In brief, cells were lysed in either normal lysis buffers (NLB: 50 mM Tris/HCl, pH 7.5; 1% Triton X‐100; 137 mM NaCl; 1% glycerin; 1 mM sodium orthovanadate; 0.5 mM EDTA; 0.01 μg/μl leupeptin, 0.1 μg/μl aprotinin, 1 mM AEBSF) or RIPA buffer (NLB supplemented with 0.5% sodium deoxycholate and 0.1% sodium dodecyl sulfate). Blotted proteins were visualized with a Fusion Solo chemiluminescence reader and quantified using FusionCapt software (Vilber Lourmat, Germany).
B‐cell stimulation and proliferation assay
Magnetic cell sorting–sorted (Miltenyi Biotec, Germany) CD19+ B cells were stimulated with indicated concentration of IgM or LPS (Southern Biotech, USA). CD69 expression was measured 12 h later by FACS analysis. For proliferation, B cells were labeled with 1 μM CFSE (Molecular Probes) and stimulated with LPS (10 μM). Cells were cultured for 96 h and cell proliferation was determined by the extent of CFSE dye dilution, as measured by flow cytometry (Stickel et al, 2014).
T‐cell stimulation and pERK phosphoflow
Magnetic cell sorting–sorted (Miltenyi Biotec, Germany) untouched T cells were stimulated with 5 μg/ml anti‐CD3/anti‐CD‐28 for indicated time points. Cell were fixed with 4% formaldehyde solution for 30 min and stained with anti‐CD4/CD8 antibodies. Afterward, cells were permeabilized, stained for pERK, and measured by flow cytometry.
Brain lysates and in vitro kinase assays
Individual brains were lyzed in 2 ml ice‐cold lysis buffer (NLB in which 1% Triton X‐100 was replaced by 0.5% NP‐40 or RIPA buffer as indicated) using a Dounce homogenizer. Following clearance by centrifugation, brain lysates were mixed with 2× sample buffer and boiled or incubated with either 2 μg anti‐B‐Raf H‐145 antibodies or rabbit control IgG for 30 min followed by addition of protein G‐sepharose beads (approx. 60 μl bead volume; GE Healthcare). After incubation on a rotator at 4° C for 3 h, the beads were washed 6 times with lysis buffer. In vitro kinase assays using recombinant GST‐MEK1 were performed with the anti‐B‐Raf H‐145 immunoprecipitates as described previously (Röring et al, 2012) with some minor modifications. In brief, after four washes with kinase assay buffer (KAB; 20 mM 4‐morpholinepropanesulfonic acid (MOPS), pH 7.2; 25 mM β‐glycerol phosphate; 5 mM EGTA, 1 mM sodium orthovanadate, 1 mM dithiothreitol), the beads were resuspended in 150 μl KAB. Then, 10 μl of this suspension were mixed with 2 μg recombinant GST‐MEK1 and 5 mM ATP and 18.75 mM MgCl2 in a total reaction volume of 40 μl. GST‐MEK1 was expressed in E. coli Rosetta cells using the pGEX‐MEK1 plasmid (Coles & Shaw, 2002) and purified using standard procedures (Brummer et al, 2004). The IVK reaction was incubated at 30°C for 60 min and immediately stopped by addition of sample buffer and boiling for 5 min. Subsequently, IVK reactions were separated by SDS–PAGE and blotted with phospho‐MEK antibodies as a readout for the MEK kinase activity of the purified B‐Raf proteins.
Histology
Brains were fixed in 10% formaldehyde solution (Rotifix, Roth, Germany) and embedded into paraffin blocks. Embedded brain tissue was sectioned and rabbit anti‐pERK antibody (Cell signaling technology; 1:50 dilution) was used for detecting the phosphorylation in the cerebellum as described previously (Galabova‐Kovacs et al, 2008). Specificity was confirmed using rabbit control IgG (Santa Cruz, Biotechnology). Myelination was assessed by Luxol fast blue staining (Abcam) following the instructions by the manufacturer.
cDNA synthesis and sequencing
Murine embryonic fibroblasts were trypsinated and washed once with ice‐cold PBS. The cell pellet was used for RNA isolation (RNeasy kit, Qiagen, Germany) according to the manufacturer's recommendation. cDNA was generated using oligo‐dT primer and the RevertAid First‐strand cDNA synthesis kit (Fermentas, USA) employing the provided standard protocol. Following a PCR with the indicated oligonucleotides, the obtained amplicons were sequenced at GATC biotech (Konstanz, Germany).
Stimulation with growth factors
Murine embryonic fibroblasts were treated with 200 nM 4‐hydroxytamoxifen (4‐HT) or ethanol (OH) for 72 h. Cells were starved for 24 h in medium containing 0.5% FCS. Indicated growth factors were purchased from R&D Systems and added to a final concentration in the starvation medium for the indicated time points.
MEF proliferation assay
Murine embryonic fibroblasts were induced with 4‐HT or OH as control for 5 days. At day zero, cells were trypsinized and plated in 24‐well plates at a density of 10,000 cells per well. At indicated time points, cells were trypsinized and counted.
Caspase‐3 activity assay
Total cell lysates of the desired cell population were lysed and adjusted to the same protein concentration by BCA assay (Thermo Scientific, USA). For the reaction, 80 μl of reaction buffer (100 mM HEPES, 1 mM DTT) was mixed with 20 μl cell lysate and supplemented to a final concentration of 6 μM fluorogenic caspase‐3 substrate (Ac‐DEVD‐AMC, Enzo Life Science, Switzerland). Fluorescence measurement (excitation: 380 nm, emission: 460 nm) was performed every minute over a one hour time course in a plate reader (Infinite m200, Tecan, Switzerland).
Transformation Assay
TAg‐immortalized MEFs were infected with diluted virus supernatant of expression vectors of above‐described B‐Raf wild‐type and mutant proteins. Cells were cultured for 20 days with medium exchange every two to three days and stained with Giemsa to visualize foci of transformed cells.
Statistical analysis
Quantitative data are presented as mean ± SE. Pairwise comparisons were performed by Student's t‐test (two‐tailed), respectively. If data did not conform to a normal distribution, the Mann–Whitney U‐test was utilized. A P value of ≤ 0.05 was considered statistically significant (***P < 0.001; **P < 0.01; and *P < 0.05). Unless noted otherwise, bar diagrams report the mean of three independent experiments.
Further Materials and Methods are detailed in the Appendix.
Author contributions
MK, MR, and TB designed and conceived experiments, performed experimental work and data interpretation, and wrote the manuscript. BS, KH, NS, LCS, SB, GJF, SE, FMU, TK, and FW performed experimental work and data interpretation. SH, RZ, WWS, and HJ contributed to experimental design and data interpretation. TB supervised research and project planning.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Video EV1
Review Process File
Acknowledgements
We would like to thank Michael Reth for support during the initial phase of the project, Thomas Brabletz for discussion and Benoit Kanzler, Caroline Johner, Cathrin Eschbach, Marianne Eckert, and Erika von Donner‐Gromoff for advice and technical support. This work was funded by the Deutsche Forschungsgemeinschaft (DFG) via the Emmy‐Noether‐Program (TB), the Excellence Initiative of the German Federal and State Governments (EXC 294 BIOSS), and the Spemann Graduate School for Biology and Medicine (GSC‐4; SGBM) via fellowships to MK, NS and GJF. RZ and TB are also supported by the DFG‐funded SFB 850.
The EMBO Journal (2016) 35: 143–161
See also: A Varga & M Baccarini (January 2016)
References
- Ahuja D, Saenz‐Robles MT, Pipas JM (2005) SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 24: 7729–7745 [DOI] [PubMed] [Google Scholar]
- Andreadi C, Cheung LK, Giblett S, Patel B, Jin H, Mercer K, Kamata T, Lee P, Williams A, McMahon M, Marais R, Pritchard C (2012) The intermediate‐activity (L597V)BRAF mutant acts as an epistatic modifier of oncogenic RAS by enhancing signaling through the RAF/MEK/ERK pathway. Genes Dev 26: 1945–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baljuls A, Schmitz W, Mueller T, Zahedi RP, Sickmann A, Hekman M, Rapp UR (2008) Positive regulation of A‐RAF by phosphorylation of isoform‐specific hinge segment and identification of novel phosphorylation sites. J Biol Chem 283: 27239–27254 [DOI] [PubMed] [Google Scholar]
- Barnard D, Diaz B, Clawson D, Marshall M (1998) Oncogenes, growth factors and phorbol esters regulate Raf‐1 through common mechanisms. Oncogene 17: 1539–1547 [DOI] [PubMed] [Google Scholar]
- Barnier JV, Papin C, Eychene A, Lecoq O, Calothy G (1995) The mouse B‐raf gene encodes multiple protein isoforms with tissue‐specific expression. J Biol Chem 270: 23381–23389 [DOI] [PubMed] [Google Scholar]
- Brennan DF, Dar AC, Hertz NT, Chao WC, Burlingame AL, Shokat KM, Barford D (2011) A Raf‐induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature 472: 366–369 [DOI] [PubMed] [Google Scholar]
- Brummer T, Elis W, Reth M, Huber M (2004) B‐cell signal transduction: tyrosine phosphorylation, kinase activity, and calcium mobilization. Methods Mol Biol 271: 189–212 [DOI] [PubMed] [Google Scholar]
- Brummer T, Shaw PE, Reth M, Misawa Y (2002) Inducible gene deletion reveals different roles for B‐Raf and Raf‐1 in B‐cell antigen receptor signalling. EMBO J 21: 5611–5622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AP, Ohno M, Giese KP, Kuhn R, Chen RL, Silva AJ (2006) Forebrain‐specific knockout of B‐raf kinase leads to deficits in hippocampal long‐term potentiation, learning, and memory. J Neurosci Res 83: 28–38 [DOI] [PubMed] [Google Scholar]
- Chong H, Lee J, Guan KL (2001) Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J 20: 3716–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cin H, Meyer C, Herr R, Janzarik WG, Lambert S, Jones DT, Jacob K, Benner A, Witt H, Remke M, Bender S, Falkenstein F, Van Anh TN, Olbrich H, von Deimling A, Pekrun A, Kulozik AE, Gnekow A, Scheurlen W, Witt O et al (2011) Oncogenic FAM131B‐BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol 121: 763–774 [DOI] [PubMed] [Google Scholar]
- Coles LC, Shaw PE (2002) PAK1 primes MEK1 for phosphorylation by Raf‐1 kinase during cross‐cascade activation of the ERK pathway. Oncogene 21: 2236–2244 [DOI] [PubMed] [Google Scholar]
- Cseh B, Doma E, Baccarini M (2014) “RAF” neighborhood: protein‐protein interaction in the Raf/Mek/Erk pathway. FEBS Lett 588: 2398–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deswal S, Meyer A, Fiala GJ, Eisenhardt AE, Schmitt LC, Salek M, Brummer T, Acuto O, Schamel WW (2013) Kidins220/ARMS associates with B‐Raf and the TCR, promoting sustained Erk signaling in T cells. J Immunol 190: 1927–1935 [DOI] [PubMed] [Google Scholar]
- Dillon TJ, Takahashi M, Li Y, Tavisala S, Murray SE, Moran AE, Parker DC, Stork PJ (2013) B‐Raf is required for positive selection and survival of DP cells, but not for negative selection of SP cells. Int Immunol 25: 259–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhardt AE, Olbrich H, Roring M, Janzarik W, Anh TN, Cin H, Remke M, Witt H, Korshunov A, Pfister SM, Omran H, Brummer T (2011) Functional characterization of a BRAF insertion mutant associated with pilocytic astrocytoma. Int J Cancer 129: 2297–2303 [DOI] [PubMed] [Google Scholar]
- Emuss V, Garnett M, Mason C, Marais R (2005) Mutations of C‐RAF are rare in human cancer because C‐RAF has a low basal kinase activity compared with B‐RAF. Cancer Res 65: 9719–9726 [DOI] [PubMed] [Google Scholar]
- Freeman AK, Ritt DA, Morrison DK (2013) Effects of Raf dimerization and its inhibition on normal and disease‐associated Raf signaling. Mol Cell 49: 751–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche‐Guenther R, Witzel F, Sieber A, Herr R, Schmidt N, Braun S, Brummer T, Sers C, Bluthgen N (2011) Strong negative feedback from Erk to Raf confers robustness to MAPK signalling. Mol Syst Biol 7: 489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galabova‐Kovacs G, Catalanotti F, Matzen D, Reyes GX, Zezula J, Herbst R, Silva A, Walter I, Baccarini M (2008) Essential role of B‐Raf in oligodendrocyte maturation and myelination during postnatal central nervous system development. J Cell Biol 180: 947–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galabova‐Kovacs G, Matzen D, Piazzolla D, Meissl K, Plyushch T, Chen AP, Silva A, Baccarini M (2006) Essential role of B‐Raf in ERK activation during extraembryonic development. Proc Natl Acad Sci USA 103: 1325–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Marais R (2004) Guilty as charged: B‐RAF is a human oncogene. Cancer Cell 6: 313–319 [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, Morales T, Aliagas I, Liu B, Sideris S, Hoeflich KP, Jaiswal BS, Seshagiri S, Koeppen H, Belvin M, Friedman LS et al (2010) RAF inhibitors prime wild‐type RAF to activate the MAPK pathway and enhance growth. Nature 464: 431–435 [DOI] [PubMed] [Google Scholar]
- Haug S, Schnerch D, Halbach S, Mastroianni J, Dumit VI, Follo M, Hasenburg A, Kohler M, Dierbach H, Herzog S, Proske A, Werner M, Dengjel J, Brummer T, Lassmann S, Wasch R, Zeiser R (2015) Metadherin exon 11 skipping variant enhances metastatic spread of ovarian cancer. Int J Cancer 136: 2328–2340 [DOI] [PubMed] [Google Scholar]
- Hayashi S, Lewis P, Pevny L, McMahon AP (2002) Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech Dev 119(Suppl 1): S97–S101 [DOI] [PubMed] [Google Scholar]
- Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu‐Duvas I, Dhomen N, Hussain J, Reis‐Filho JS, Springer CJ, Pritchard C, Marais R (2010) Kinase‐dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140: 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr R, Wohrle FU, Danke C, Berens C, Brummer T (2011) A novel MCF‐10A line allowing conditional oncogene expression in 3D culture. Cell Commun Signal 9: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holderfield M, Deuker MM, McCormick F, McMahon M (2014) Targeting RAF kinases for cancer therapy: BRAF‐mutated melanoma and beyond. Nat Rev Cancer 14: 455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holderfield M, Merritt H, Chan J, Wallroth M, Tandeske L, Zhai H, Tellew J, Hardy S, Hekmat‐Nejad M, Stuart DD, McCormick F, Nagel TE (2013) RAF inhibitors activate the MAPK pathway by relieving inhibitory autophosphorylation. Cancer Cell 23: 594–602 [DOI] [PubMed] [Google Scholar]
- Hu J, Ahuja LG, Meharena HS, Kannan N, Kornev AP, Taylor SS, Shaw AS (2015) Kinase regulation by hydrophobic spine assembly in cancer. Mol Cell Biol 35: 264–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJ, Kornev AP, Taylor SS, Shaw AS (2013) Allosteric activation of functionally asymmetric RAF kinase dimers. Cell 154: 1036–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson KE, Lipson D, Stephens PJ, Otto G, Lehmann BD, Lyle PL, Vnencak‐Jones CL, Ross JS, Pietenpol JA, Sosman JA, Puzanov I, Miller VA, Pao W (2013) BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin Cancer Res 19: 6696–6702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue S, Moriya M, Watanabe Y, Miyagawa‐Tomita S, Niihori T, Oba D, Ono M, Kure S, Ogura T, Matsubara Y, Aoki Y (2014) New BRAF‐knock‐in mice provide a pathogenetic mechanism of developmental defects and a therapeutic approach in cardio‐facio‐cutaneous syndrome. Hum Mol Genet 23: 6553–6566 [DOI] [PubMed] [Google Scholar]
- Kamata T, Hussain J, Giblett S, Hayward R, Marais R, Pritchard C (2010) BRAF inactivation drives aneuploidy by deregulating CRAF. Cancer Res 70: 8475–8486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata T, Kang J, Lee TH, Wojnowski L, Pritchard CA, Leavitt AD (2005) A critical function for B‐Raf at multiple stages of myelopoiesis. Blood 106: 833–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H, Therrien M (2015) Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 16: 281–298 [DOI] [PubMed] [Google Scholar]
- Mercer K, Giblett S, Green S, Lloyd D, Darocha Dias S, Plumb M, Marais R, Pritchard C (2005) Expression of endogenous oncogenic V600EB‐raf induces proliferation and developmental defects in mice and transformation of primary fibroblasts. Cancer Res 65: 11493–11500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minguet S, Huber M, Rosenkranz L, Schamel WW, Reth M, Brummer T (2005) Adenosine and cAMP are potent inhibitors of the NF‐kappaB pathway downstream of immunoreceptors. Eur J Immunol 35: 31–41 [DOI] [PubMed] [Google Scholar]
- Mooz J, Oberoi‐Khanuja TK, Harms GS, Wang W, Jaiswal BS, Seshagiri S, Tikkanen R, Rajalingam K (2014) Dimerization of the kinase ARAF promotes MAPK pathway activation and cell migration. Sci Signal 7: ra73 [DOI] [PubMed] [Google Scholar]
- Nolen B, Taylor S, Ghosh G (2004) Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell 15: 661–675 [DOI] [PubMed] [Google Scholar]
- Papin C, Denouel‐Galy A, Laugier D, Calothy G, Eychene A (1998) Modulation of kinase activity and oncogenic properties by alternative splicing reveals a novel regulatory mechanism for B‐Raf. J Biol Chem 273: 24939–24947 [DOI] [PubMed] [Google Scholar]
- Pfeiffer V, Gotz R, Xiang C, Camarero G, Braun A, Zhang Y, Blum R, Heinsen H, Nieswandt B, Rapp UR (2013) Ablation of BRaf impairs neuronal differentiation in the postnatal hippocampus and cerebellum. PLoS ONE 8: e58259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N (2010) RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild‐type BRAF. Nature 464: 427–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard CA, Samuels ML, Bosch E, McMahon M (1995) Conditionally oncogenic forms of the A‐Raf and B‐Raf protein kinases display different biological and biochemical properties in NIH 3T3 cells. Mol Cell Biol 15: 6430–6442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakulendran T, Sahmi M, Lefrancois M, Sicheri F, Therrien M (2009) A dimerization‐dependent mechanism drives RAF catalytic activation. Nature 461: 542–545 [DOI] [PubMed] [Google Scholar]
- Röring M, Brummer T (2012) Aberrant B‐raf signaling in human cancer – 10 years from bench to bedside. Crit Rev Oncog 17: 97–121 [DOI] [PubMed] [Google Scholar]
- Röring M, Herr R, Fiala GJ, Heilmann K, Braun S, Eisenhardt AE, Halbach S, Capper D, von Deimling A, Schamel WW, Saunders DN, Brummer T (2012) Distinct requirement for an intact dimer interface in wild‐type, V600E and kinase‐dead B‐Raf signalling. EMBO J 31: 2629–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth JJ, Santi M, Pollock AN, Harding BN, Rorke‐Adams LB, Tooke LS, Biegel JA (2015) Chromosome band 7q34 deletions resulting in KIAA1549‐BRAF and FAM131B‐BRAF fusions in pediatric low‐grade Gliomas. Brain Pathol 25: 182–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushworth LK, Hindley AD, O'Neill E, Kolch W (2006) Regulation and role of Raf‐1/B‐Raf heterodimerization. Mol Cell Biol 26: 2262–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samatar AA, Poulikakos PI (2014) Targeting RAS‐ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 13: 928–942 [DOI] [PubMed] [Google Scholar]
- Sievert AJ, Lang SS, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N, Kellet M, Storm PB, Resnick AC (2013) Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci USA 110: 5957–5962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stickel N, Prinz G, Pfeifer D, Hasselblatt P, Schmitt‐Graeff A, Follo M, Thimme R, Finke J, Duyster J, Salzer U, Zeiser R (2014) MiR‐146a regulates the TRAF6/TNF‐axis in donor T cells during GVHD. Blood 124: 2586–2595 [DOI] [PubMed] [Google Scholar]
- Thevakumaran N, Lavoie H, Critton DA, Tebben A, Marinier A, Sicheri F, Therrien M (2015) Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat Struct Mol Biol 22: 37–43 [DOI] [PubMed] [Google Scholar]
- Tien AC, Tsai HH, Molofsky AV, McMahon M, Foo LC, Kaul A, Dougherty JD, Heintz N, Gutmann DH, Barres BA, Rowitch DH (2012) Regulated temporal‐spatial astrocyte precursor cell proliferation involves BRAF signalling in mammalian spinal cord. Development 139: 2477–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto H, Irie A, Nishimura Y (2004) B‐Raf contributes to sustained extracellular signal‐regulated kinase activation associated with interleukin‐2 production stimulated through the T cell receptor. J Biol Chem 279: 48457–48465 [DOI] [PubMed] [Google Scholar]
- Tsukamoto H, Irie A, Senju S, Hatzopoulos AK, Wojnowski L, Nishimura Y (2008) B‐Raf‐mediated signaling pathway regulates T cell development. Eur J Immunol 38: 518–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu‐Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R (2004) Mechanism of activation of the RAF‐ERK signaling pathway by oncogenic mutations of B‐RAF. Cell 116: 855–867 [DOI] [PubMed] [Google Scholar]
- Wojnowski L, Stancato LF, Larner AC, Rapp UR, Zimmer A (2000) Overlapping and specific functions of Braf and Craf‐1 proto‐oncogenes during mouse embryogenesis. Mech Dev 91: 97–104 [DOI] [PubMed] [Google Scholar]
- Wojnowski L, Zimmer AM, Beck TW, Hahn H, Bernal R, Rapp UR, Zimmer A (1997) Endothelial apoptosis in Braf‐deficient mice. Nat Genet 16: 293–297 [DOI] [PubMed] [Google Scholar]
- Yaktapour N, Meiss F, Mastroianni J, Zenz T, Andrlova H, Mathew NR, Claus R, Hutter B, Frohling S, Brors B, Pfeifer D, Pantic M, Bartsch I, Spehl TS, Meyer PT, Duyster J, Zirlik K, Brummer T, Zeiser R (2014) BRAF inhibitor‐associated ERK activation drives development of chronic lymphocytic leukemia. J Clin Investig 124: 5074–5084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang BH, Guan KL (2000) Activation of B‐Raf kinase requires phosphorylation of the conserved residues Thr598 and Ser601. EMBO J 19: 5429–5439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Li X, McNamee C, Chen AP, Baccarini M, Snider WD (2007) Raf kinase signaling functions in sensory neuron differentiation and axon growth in vivo. Nat Neurosci 10: 598–607 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Video EV1
Review Process File