

Abstract
Recent developments in sequencing techniques have enabled rapid and high-throughput generation of sequence data, democratizing the ability to compile information on large amounts of genetic variations in individual laboratories. However, there is a growing gap between the generation of raw sequencing data and the extraction of meaningful biological information. Here, we describe a protocol to use the ANNOVAR (ANNOtate VARiation) software to facilitate fast and easy variant annotations, including gene-based, region-based and filter-based annotations on a variant call format (VCF) file generated from human genomes. We further describe a protocol for gene-based annotation of a newly sequenced nonhuman species. Finally, we describe how to use a user-friendly and easily accessible web server called wANNOVAR to prioritize candidate genes for a Mendelian disease. The variant annotation protocols take 5–30 min of computer time, depending on the size of the variant file, and 5–10 min of hands-on time. In summary, through the command-line tool and the web server, these protocols provide a convenient means to analyze genetic variants generated in humans and other species.
INTRODUCTION
With the development and deployment of high-throughput sequencing platforms, DNA sequencing data can now be generated at unprecedented rates by individual laboratories, thus allowing for the collection of large amounts of genetic data. However, the massive amounts of data pose substantial challenges for downstream studies, as highly specialized software tools and expertise are necessary to analyze and interpret the sequence data. A variety of bioinformatics tools have been developed to handle DNA sequencing data, including those to align the raw sequencing reads to a reference genome1 (such as BWA2 and Bowtie3), to assemble new genomes4 (such as FERMI5, Abyss6 and SoapDenovo7), to perform data quality control8 and to call single-nucleotide variants (SNVs)9 (such as Genome Analysis Toolkit (GATK)10) or structural variants (SVs)11 (such as CNVnator12 and ERDS13). However, after variant calls are generated, researchers need to understand the functional content within the data and therefore perform prioritization analysis on all variants for functional follow-up on selected variants. To address these challenges, we previously developed a tool called ANNOVAR14 to rapidly annotate genetic variants and predict their functionalities. Besides ANNOVAR, several other similar annotation tools have also been developed, such as VEP15, snpEff16, VAAST17, AnnTools18 and others.
Over the past few years, ANNOVAR has been widely adopted in a variety of research studies on human genomes ranging from studies on population samples19,20 to studies on a single pedigree21,22. In addition, ANNOVAR is widely used in clinical genome-sequencing studies. On the basis of the 2014 CLARITY report (an international effort toward developing standards for best practices in analysis, interpretation and reporting of clinical genome sequencing results), 52% of the clinical genome sequencing laboratories and 63% of the finalists used ANNOVAR to find disease-related mutations23. Furthermore, ANNOVAR is used in the genome annotation of several nonhuman species as well, including mice24 and chimpanzees25. To facilitate easy access to some of the most widely used functionalities in ANNOVAR, we developed a web server called wANNOVAR26. The web server takes VCF files as input, with optional configuration settings, to allow biologists without informatics skills to run variant annotation easily. More recently, we also developed a phenotype-based variant annotation and prioritization tool called Phenolyzer27, and we incorporated it within wANNOVAR to allow phenotype-based prioritization of disease variants. In this article, we provide several step-by-step protocols for using ANNOVAR and wANNOVAR in genome annotation and analysis.
Overview of ANNOVAR
ANNOVAR can be accessed from http://annovar.openbioinformatics.org/. It is a command-line tool written in the Perl programming language, which can be executed on a variety of operating systems with a Perl interpreter installed. It can take a variety of input formats, including the most commonly used VCF format, and it outputs an annotated variant file in several different formats (such as annotated VCF file, tab-delimited text file or comma-delimited text file), which contains annotations for each variant in the input file. For example, the input file contains one line as ‘1 881627 881627 G A’, which means that there is a variant at chromosome 1, position 881627, with a nucleotide change of G to A. The tab-delimited output file contains a line combining the original input information with additional annotation information, such as ‘exonic’ (genomic function), ‘NOC2L’ (the gene being affected), ‘NM_015658’ (the transcript being affected), ‘synonymous SNV’ (functional role of the coding variant), ‘c.2334G>C’ (the nucleotide change in the transcript) and ‘p.A328G’ (the amino acid change in the protein).
In general, ANNOVAR offers three types of annotations: gene-based annotation, region-based annotation and filter-based annotation (Fig. 1). Gene-based annotation refers to the manner in which a variant affects known genes, by inferring several types of information: (i) whether the variant is exonic, intronic, splicing, 3′-untranslated region (UTR), 5′-UTR, intergenic, etc.; (ii) when the variant is exonic, what functional role it has on protein coding—synonymous, nonsynonymous, frameshift insertion/deletion, etc.; and (iii) what transcripts are affected and what changes occur in the corresponding amino acids. Region-based annotation can be useful when we want to know whether the variants overlap with certain regions of interest, such as conserved genomic elements28, cytogenetic bands, microRNA target sites29 and Encyclopedia of DNA Elements (ENCODE)-annotated regions30. Furthermore, we can use filter-based annotation to annotate and filter a list of variants, such as finding the alternative allele frequency for variants in the 1000 Genomes Project31 and the US National Institutes of Health–National Heart, Lung, and Blood Institute (NIH-NHLBI) ESP6500 exome-sequencing project32; finding the SIFT33 and PolyPhen34 scores for nonsynonymous variants; and identifying the presence or absence of a variant in the dbSNP database35.
Figure 1.
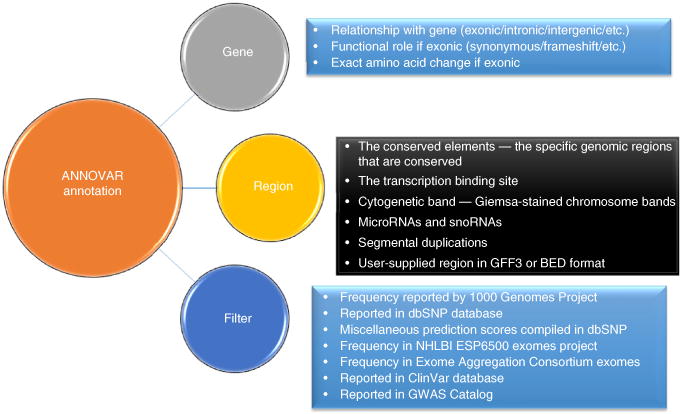
The three different types of annotations supported by ANNOVAR are gene-based, region-based and filter-based annotations. Different annotations focus on different aspects of each variant: gene-based annotation tells its relationship and functional impact on known genes; region-based annotation tells its relationship with different specific genomic regions, such as whether it falls within a known conserved genomic region; and filter-based annotation gives a variety of information on this variant, such as population frequency in different populations and various types of variant-deleteriousness prediction scores, which can be used to filter the common and probably nondeleterious variants.
Overview of wANNOVAR
To facilitate convenient and fast access to ANNOVAR for researchers who prefer a graphical user interface, we have developed a web server called wANNOVAR26 (http://wannovar.usc.edu), implementing the most commonly used functionalities of ANNOVAR for human genomes in a web interface. By visiting the wANNOVAR website, users can directly upload their variant files and obtain their results back via web interface. Currently, a few commonly used functional annotations are included in the results, such as different types of gene annotations, alternative allele frequency in the 1000 Genomes Project, conserved element annotation, dbSNP annotation, deleteriousness prediction scores for nonsynonymous variants, ClinVar variant annotation and genome-wide association study (GWAS) variant annotation. Moreover, wANNOVAR implements a variant-reduction pipeline based on commonly used filters and disease models, such as selecting only the non-synonymous variants and splicing variants, selecting only the rare or novel variants in the 1000 Genomes Project database, and selecting predicted deleterious variants. In addition, wANNOVAR implements phenotype-based variant prioritization, which is helpful in scenarios in which a sample’s specific phenotype or disease information is available and may help identify causal variants.
Comparison with other methods
Besides ANNOVAR, a number of other similar tools are available to annotate and prioritize genetic variants, some of which have been reviewed previously36. In general, these tools can be accessed either through command line (for example, VEP15, VAAST17, pVAAST37, AnnTools18, SnpEff38 and GEMINI39) or on the web (for example, VAT40, SeattleSeq41, AVIA42 and VARIANT43). Compared with other command-line tools, ANNOVAR is easy to install; it only requires unpacking the downloaded file, and it provides rich documentation and extensive flexibility for users to retrieve a variety of annotation types for their own needs. We note that a recent report compared ANNOVAR with VEP and claimed that ANNOVAR mis-annotated variants (such as annotating SNVs as indels)44; we obtained this list of variants from the original authors and found that none of these variants was annotated incorrectly by ANNOVAR as reported (Supplementary Data). Therefore, the previously reported results may be generated on an outdated transcript FASTA file that does not correspond to the gene definition file used in the annotation. This example demonstrated the need to keep gene definition files and transcript FASTA files synchronized with each other when using ANNOVAR—a caveat that we stress here. In addition, unlike some other tools, ANNOVAR is designed to be flexible to annotate newly sequenced species as long as the users have access to the genome assembly (such as a FASTA file) and a gene definition file (such as a GTF file) to generate the transcript FASTA file. A more detailed comparison with several other software tools is given in Supplementary Table 1.
Compared with several other web servers for functional annotation of genetic variants, wANNOVAR is user-friendly and simple to use, but it provides similar sets of functionalities. With the default settings, users simply supply a VCF file from the human genome, and then all the commonly used functional annotations will be generated in the results, which the users can directly download and examine. In addition, unlike many other web servers, wANNOVAR also implements a variant reduction pipeline, as well as a phenotype-based variant prioritization scheme, which help users quickly identify disease-relevant variants from an input VCF file on the human genome. When a custom filter is selected, wANNOVAR also displays additional configuration options to help fine-tune the variant reduction procedure, to improve the performance on finding causal variants. A more detailed comparison with several other web servers is given in Supplementary Table 2.
Limitations of ANNOVAR and wANNOVAR
Although ANNOVAR and wANNOVAR are widely used to identify and prioritize disease-causing genetic variants, variants need to be prioritized and filtered carefully, and both software programs may require tuning for different scenarios to obtain optimal results. For example, the wANNOVAR server implements several default ‘variant-reduction’ schemes (disease models) that help users retrieve a small subset of functionally important variants from the input files. However, these criteria may not be optimal for the specific use case, and they may eliminate true causal variants in some scenarios during the filtering procedure. Compared with rule-based ‘hard’ filtering, a probabilistic prioritization approach may work better to re-rank variants, especially for complex diseases in which disease causal variants may not fit the hard filtering criteria.
Second, ANNOVAR can generate a variety of different annotations for each variant (such as several different types of deleteriousness-prediction scores), and it leaves the choice of selecting annotations to users. Sometimes this presents overwhelming information to users, and they may be confused with regard to which piece of information to choose from. For example, for nonsynonymous variants, ANNOVAR and wANNOVAR can generate more than ten different types of deleteriousness-prediction scores, occasionally with discordant predictions. Recently, we developed an ensemble score called MetaSVM for nonsynonymous SNVs, using an SVM (support vector machine) model that combines multiple scoring systems45. We demonstrated the improved performance of this scoring system over individual scores. In general, we recommend that users use one popular prediction score (such as SIFT) and one meta-score (such as metaSVM) to judge whether a variant is likely to be deleterious. For noncoding variants, a similar ensemble score called CADD (combined annotation–dependent depletion) score46 was reported to have the best performance. Similarly, we recommend that users use one popular prediction score (such as PhyloP47) and one meta-score (such as CADD) for predicting the deleteriousness of noncoding variants.
Third, owing to the lack of a common standard for denoting functional annotations, ANNOVAR uses its own nomenclature on functional annotations, and it has its own output file formats (such as tab-delimited files or annotated VCF files). Sometimes, this may make it difficult to communicate results with other users, or to compare results generated by different annotation software tools or analyze results by downstream association tools. We now provide a translation table between ANNOVAR terms and Sequence Ontology48 terms (Supplementary Table 3). A unified annotation format is being developed by several authors of annotation software tools (see an initial version at http://snpeff.sourceforge.net/VCFannotationformat_v1.0.pdf). If these annotation standards are adopted by the broad community, we plan to modify the ANNOVAR software to conform to these standards.
Fourth, ANNOVAR and wANNOVAR were originally developed as annotation tools for genetic variants from a single genome, with limited functionality on analyzing multiple genomes. Therefore, these tools do not support case-control association analysis, or family-based association analysis, to identify variants that are associated with specific phenotypic traits or diseases. However, to facilitate the analysis of genomes of individual patients, we implemented the variant-reduction pipeline, as well as a phenotype-based gene-prioritization scheme, to help users better identify variants associated with diseases.
Fifth, ANNOVAR’s support for large-scale SVs needs improvements. Currently, ANNOVAR is limited to identifying genes contained within deletions or duplications, but it cannot infer complex SVs and translocations. For small SVs, such as indels of <50 bp, ANNOVAR can perform functional annotations, as long as the variants conform to the VCF format.
Experimental design
In this section, we describe how to prepare input file and database files for annotation by ANNOVAR or wANNOVAR. ANNOVAR needs to use an input file with genetic variants to conduct the functional annotation. The recommended file format is the VCF format version 4.0 and above. The VCF format is now supported by most variant-calling software tools, such as GATK10, SAMtools49 and many others. The ANNOVAR package contains an ‘example’ directory, which contains example VCF files for testing.
For gene-based annotation in ANNOVAR, you need to use a gene definition file in genePred format, as well as one transcript sequence file in FASTA format. For the human genome and many other genomes, the gene definition file can be directly downloaded using a command in ANNOVAR from the University of California Santa Cruz (UCSC) annotation database (see PROCEDURE for details). For genomes that are not available from the UCSC annotation database, a GFF3 or GTF file downloaded from Ensembl or compiled by the user is needed to be converted to the genePred format. The conversion can be performed by the ‘gff3ToGenePred’ or ‘gtfToGenePred‘ tools, which are available at http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/. In our experience, occasionally some GFF3 files from Ensembl cannot be converted correctly. Thus, for nonhuman species that are not available from the UCSC annotation database, we recommend using the GTF file to generate the gene definition file. For an example on how to annotate variants for Arabidopsis thaliana, please refer to Box 1.
Box 1. Preparing annotation files for Arabidopsis thaliana.
- Go to http://plants.ensembl.org/info/website/ftp/index.html to download the GTF file and the genome FASTA file for Arabidopsis into a folder called ‘atdb’.
mkdir atdb cd atdb wget ftp://ftp.ensemblgenomes.org/pub/release-27/plants/fasta/arabidopsis_thaliana/dna/Arabidopsis_thaliana.TAIR10.27.dna.genome.fa.gz wget ftp://ftp.ensemblgenomes.org/pub/release-27/plants/gtf/arabidopsis_thaliana/Arabidopsis_thaliana.TAIR10.27.gtf.gz
- Decompress both files:
gunzip Arabidopsis_thaliana.TAIR10.27.dna.genome.fa.gz gunzip Arabidopsis_thaliana.TAIR10.27.gtf.gz
- Use the gtfToGenePred tool to convert the GTF file to a GenePred file:
gtfToGenePred -genePredExt Arabidopsis_thaliana.TAIR10.27.gtf AT_refGene.txt
- Generate a transcript FASTA file with our provided script:
perl ../retrieve_seq_from_fasta.pl --format refGene --seqfile Arabidopsis_thaliana.TAIR10.27.dna.genome.fa AT_refGene.txt --out AT_refGeneMrna.fa
After this step, the annotation database files needed for gene-based annotation are ready. Now you can annotate a given VCF file using the procedure starting from Step 2B(iii). Please note that the ‘--buildver’ argument should be set to ‘AT’.
For region-based and filter-based annotation in ANNOVAR, the annotation database files need to be downloaded by the user using the command line. The protocols below include some examples, but more extensive instructions are available from the ANNOVAR documentation site at http://annovar.openbioinformatics.org/, when describing each annotation database.
For the wANNOVAR server, the user only needs to prepare a variant file for the human genome in VCF format in the desired genome build. Please note that the phenotype-based variant- discovery and disease-inheritance models only work on single samples. For example, the ‘hemolytic anemia’ instance shown in the PROCEDURE is for one individual. In the future, wANNOVAR may support family-based analysis such that a VCF file containing multiple samples within the same family, together with phenotype information for all family members, can be analyzed for finding disease-associated genes.
MATERIALS
EQUIPMENT
Computer with internet connection (see Equipment Setup)
EQUIPMENT SETUP
Software requirements
To use the command-line version of ANNOVAR, a computer with Perl interpreter is required (most Linux distributions and Mac OS already have it built in; for Windows visit https://www.perl.org/get.html). To use the wANNOVAR web server, a computer with an Internet connection and a modern browser (e.g., the latest versions of Chrome, Safari or Firefox) is required. Microsoft Excel can be used to view the results of annotation in comma-delimited text files.
PROCEDURE
Preparation of input variant files ● TIMING 5 min
-
1| Please prepare your variants in the commonly used VCF format. The VCF file requires at least eight tab-delimited columns, which represent chromosome, position, identifier, reference allele, alternative allele, variant quality, filter and other information. A more detailed description of the VCF format is given at http://vcftools.sourceforge.net/specs.html. One example line in a VCF file is shown below (each column should be tab-delimited in the actual file):
CHROM POS ID REF ALT QUAL FILTER INFO
20 17330 . T A 3 q10 NS=3;DP=11;AF=0.017
▲ CRITICAL STEP You can also prepare your file in ANNOVAR’s input format, which requires a minimum of five space- or tab-delimited columns, including chromosome, start, end, reference allele and alternative allele, as well as any number of extra columns.
Perform variant annotation with ANNOVAR or wANNOVAR
- 2| The protocols described below are separated into three sections. The first two sections (Step 2, options A and B) demonstrate how to use the ANNOVAR command-line tool to annotate variants in the genomes of humans and other species, respectively. The third section (option C) describes the procedure of using wANNOVAR to annotate and filter human genomic variants and to identify disease causal genes on the basis of specific disease models.
- Annotation of human genomic variants by ANNOVAR ● TIMING ~10 min
-
Fill in the registration form, download the ANNOVAR software and save it into a directory. After downloading the ‘annovar.latest.tar.gz’ file, extract the file by the command:
tar xvfz annovar.latest.tar.gz
▲ CRITICAL STEP You can also add the ANNOVAR directory into the PATH environmental variable in your operating system, so that all ANNOVAR scripts can be executed directly by typing the command name. -
Download all the necessary annotation databases. The downloaded ANNOVAR package already comes with the refGene databases for gene annotation. For other annotations, download the databases to the ‘humandb/’ directory with the commands below:
perl annotate_variation.pl --downdb --buildver hg19 cytoBand humandb/ perl annotate_variation.pl --downdb --webfrom annovar --buildver hg19 1000g2014oct humandb/ perl annotate_variation.pl --downdb --webfrom annovar --buildver hg19 exac03 humandb/ perl annotate_variation.pl --downdb --webfrom annovar --buildver hg19 ljb26_all humandb/ perl annotate_variation.pl --downdb --webfrom annovar --buildver hg19 clinvar_20140929 humandb/ perl annotate_variation.pl --downdb --webfrom annovar --buildver hg19 snp138 humandb/
Here we download several commonly used databases: ‘cytoBand’ for the chromosome coordinate of each cytogenetic band, ‘1000g2014oct’ for alternative allele frequency in the 1000 Genomes Project (version October 2014), ‘exac03’ for the variants reported in the Exome Aggregation Consortium (version 0.3)50, ‘ljb26_all’ for various functional deleteriousness prediction scores from the dbNSFP database (version 2.6)51, ‘clinvar_20140929’ for the variants reported in the ClinVar database (version 20140929)52 and ‘snp138’ for the dbSNP database (version 138)53. Note that the first command does not have the ‘--webfrom annovar’ argument, so it downloads the file from the UCSC Genome Browser annotation database54. The ‘--buildver hg19’ argument specifies that the genome build is hg19.You will find that several additional files with the ‘hg19’ prefix are saved in the ‘humandb/’ directory after running the commands above.? TROUBLESHOOTING -
Annotate variants with the ‘table_annovar.pl’ script, which allows custom selection of multiple annotation types in the same command with specified order of the output.Please enter the command below to annotate variants in VCF format with the annotation databases downloaded previously:
perl table_annovar.pl <variant.vcf> humandb/--outfile final –buildver hg19 --protocol refGene,cytoBand,1000g2014oct_eur,1000g2014oct_afr,exac03, ljb26_all,clinvar_20140929,snp138 --operation g,r,f,f,f,f,f,f --vcfinput
<variant.vcf> refers to the name of the input VCF file. Please follow the ‘--protocol’ argument by the exact names of the annotation sources, and follow the ‘--operation’ argument by the annotation type: ‘g’ for gene-based annotation, ‘r’ for region-based annotation and ‘f’ for filter-based annotation. The ‘--outfile’ argument specifies the prefix of the name of your output file.▲ CRITICAL STEP Make sure that the annotation database names are entered correctly and in the order in which you want them to be shown in the output. Make sure that the annotation types following ‘--operation’ are in the same order as the annotation database names following ‘--protocol’. Make sure that there is only one comma and no space between individual protocol names or annotation types.? TROUBLESHOOTING - Check your result in the file ‘final.hg19_multianno.vcf’. This will be a VCF file in which the INFO column has extra fields in the form ‘key=value’ separated by ‘;’. For example, ‘Func.refGene=intronic;Gene.refGene=SAMD11’. Each key-value pair represents one piece of ANNOVAR annotation. The output file can be further processed by genetic analysis software tools that are designed for the VCF file format.
- Check your result in the file ‘final.hg19_multianno.txt’. Each line in the file represents one variant from the input file. It is a tab-delimited file with added annotations represented as extra columns, by the same order as the annotation types following the ‘--protocol’ argument.
-
-
Gene-based annotation of nonhuman species by ANNOVAR ● TIMING ~10 min▲ CRITICAL STEP In this protocol, we will demonstrate how to use ANNOVAR to annotate variants in the chimpanzee genome, with the genome build identifier as panTro2. The instructions for installing ANNOVAR are the same as in Step 2A(i).
-
Download the chimpanzee genome gene definition file and FASTA file for genome sequence in a directory called ‘chimpdb/’, by entering the commands below:
perl annotate_variation.pl --downdb --buildver panTro2 gene chimpdb/ perl annotate_variation.pl --downdb --buildver panTro2 seq chimpdb/panTro2_seq
? TROUBLESHOOTING -
Note that as ANNOVAR database repository contains only prebuilt transcript FASTA files for the human genome, you need to build a transcript FASTA file for the chimpanzee genome using the following command:
perl retrieve_seq_from_fasta.pl chimpdb/panTro2_refGene.txt –seqdir chimpdb/panTro2_seq --format refGene --outfile chimpdb/panTro2_refGeneMrna.fa
The ‘--seqdir’ specifies the directory in which the downloaded sequence files reside. The ‘--format’ specifies the format of the gene definition file. The ‘--outfile’ specifies the name of the output mRNA sequence file.▲ CRITICAL STEP The output file name following ‘--outfile’ argument should be in the ‘<buildver>_refGeneMrna.fa’ format; otherwise, the next step will not be able to find the correct transcript FASTA sequence file.? TROUBLESHOOTING -
Annotate variants with the chimpanzee gene annotation:
perl table_annovar.pl <variant.vcf> chimpdb/--vcfinput --outfile final –buildver panTro2 --protocol refGene --operation g
Here <variant.vcf> is the input VCF file, ‘chimpdb/’ is the directory of the downloaded databases and other arguments are the same as in the human genome annotation. -
Check your result in the ‘final.panTro2_multianno.txt’ file. The gene annotation for chimpanzee is added after the input variants.▲ CRITICAL STEP When the gene definition file is not directly available, the GFF3 or GTF file generated by gene prediction tools can be used and converted to the gene definition file. Please refer to the Experimental design section in the INTRODUCTION and Box 1 for examples, and refer to http://annovar.openbioinformatics.org/en/latest/user-guide/gene/ for more details.
-
-
Annotation and prioritization of human variants by wANNOVAR ● TIMING 10–30 min▲ CRITICAL STEP To facilitate the annotation and analysis on human genomic variants, we established a web server called wANNOVAR26. In the protocol described below, we illustrate how to discover disease-related candidate genes from a patient affected with idiopathic hemolytic anemia with a recessive pattern of inheritance.
-
To use wANNOVAR, visit the website at http://wannovar.usc.edu/. Enter the basic submission information, including your e-mail address and a sample identifier, so that you can receive an e-mail after the job is completed and identify different submissions easily. See Figure 2 for an overview of the wANNOVAR interface.▲ CRITICAL STEP If you do not provide an e-mail address, please record the results URL after uploading the VCF file, as results will be available at this URL once annotation is completed.
-
Download the example VCF file from http://wannovar.usc.edu/download/anemia.vcf. Upload this variant file by clicking the ‘Input File’ button.▲ CRITICAL STEP If the input file size is small, you may directly paste the variant into the ‘Paste Variant Calls’ text area, which is helpful when you are interested in examining only a few specific variants.! CAUTION If you upload a variant file and past variants in the text area at the same time, only the uploaded variant file will be processed.
- (Optional) Enter disease or phenotype terms in the ‘Enter Disease or Phenotype Terms’ text area, with short and simplified phrases (such as ‘cancer’ or ‘lung cancer’) rather than long phrases (such as ‘somatic cancer late onset’ or ‘lung cancer caused by smoking’). Please separate different phrases by a semicolon or the return character. For this specific example, you can enter ‘hemolytic anemia’. The genes and variants from the input file will be further prioritized on the basis of their association with the input disease or phenotype term, where the association is calculated by integrating multiple disease-gene databases, disease- phenotype databases, disease ontology databases and gene-gene interaction databases through a machine-learning framework27.
-
Please choose ‘hg19’ as the ‘Reference Genome’ (currently only three builds of human genomes are supported: hg18, hg19 and hg38), the correct input format (VCF file) and the gene definition (RefGene). These options are already selected by default.Please note that the gene definition refers to different gene build annotation systems, including RefGene from the National Center for Biotechnology Information (NCBI)55, UCSC Known Gene from UCSC56, ENSEMBL gene from the ENSEMBL project57 and GENCODE gene from the GENCODE consortium58.▲ CRITICAL STEP Make sure that your input file is consistent with your parameter settings, as the wrong input format will cause an error message. The use of a wrong genome build may not generate an error message immediately, but it will make your results completely wrong.
-
Please choose ‘rare recessive Mendelian’ as the disease model. The commonly used models are ‘rare recessive Mendelian’ and ‘rare dominant Mendelian’ disease models.These two models first select the nonsynonymous and splicing variants, and then they filter out the variants with minor allele frequency (MAF) > 0.01 in the 1000 Genomes Project, the NHLBI ESP6500 exome data set and the ExAC 65,000 exomes data set. Finally, the candidate disease-causing genes and variants are selected on the basis of the recessive or dominant mode of inheritance: with the recessive model, only genes with two or more predicted deleterious alleles are selected; with the dominant model, all genes with a predicted deleterious allele are selected. (We acknowledge that two predicted deleterious alleles may be located in the same haplotype, in the absence of family information.) The filtration pipeline can also be customized by choosing ‘custom filtering’ options (Table 1).? TROUBLESHOOTING
-
Click the ‘Submit’ button. You will receive an e-mail to retrieve your result. If you did not enter a valid e-mail address, make sure to record the URL from the web page, as the results will be shown in this web page when they are ready.? TROUBLESHOOTING
-
Figure 2.

Screenshot of wANNOVAR, including the general steps to upload and prioritize variants. Please follow the steps (1–9) in the picture. If you want to quickly start the job with default parameters, please directly click submit (9) after the variant file is uploaded.
TABLE 1.
Description of custom filtering options in the wANNOVAR server.
| Filter | Description |
|---|---|
| MAF in variants filtering | Specify the MAF threshold for filtering variants. This threshold will be used for all the population frequency databases, such as 1000 Genomes Project, ESP6500 and ExAC03 databases. The variants with a MAF larger than this threshold will be filtered out |
| dbSNP version | Specify the dbSNP version for filtering. This version is separate from and will not affect the annotation step |
| Disease type | Choose a disease type for the final variant reduction step; ‘recessive’ and ‘dominant’ have been described previously, and ‘unknown’ indicates no selection |
| Variant filters | Choose different filer combinations for the variant reduction pipeline |
| Not in control | Upload the variant file on control subjects, or paste the variant calls. Control file should be in the same format as the input file, and all the variants in the control file will be filtered out from the input file |
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
TABLE 2.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 2A(ii) | You see ‘Failed’ for all the files to be downloaded | Internet connection or firewall settings issue | Please check your internet connection and firewall settings |
| 2A(iii) | You see ‘ERROR’ information displayed | The wrong command was entered, or some required databases are not downloaded properly | Please check the log printed in the terminal. First, make sure that you typed in or pasted exactly the same command as written. If the command is correct, it is possible that one or more databases are not downloaded properly. You should find the missing database from the log and go back to Step 2A(ii) to download the missing database |
| 2B(i), 2B(ii) | You see ‘ERROR’ about missing FASTA file | ANNOVAR cannot read the sequence files in the entered directory correctly | In this case, the ‘–seqfile <genome fasta file>’ argument should be used instead of the ‘—seqdir’ argument, to explicitly specify the genome FASTA file to be used |
| 2C(v) | There are 0 variants in the final result in the ‘ANNOVAR filtering result’ section | The filter and model selected may be too stringent in this step | Please choose ‘Dominant model’ rather than ‘Recessive model’; please increase the MAF value to include more variants. Please note that filters and models do not affect the output in the ‘Basic Information’ section |
| 2C(vi) | There is an error message right after you click ‘Submit’ | The uploaded variant file is different from the ‘Input Format’ selection | For VCF, please make sure that the VCF file conforms to a valid format. VCF files can be validated with VCF tools60 (see http://vcftools.sourceforge.net/perl_module.html#vcf-validator) |
● TIMING
For options A and B, with a modern Linux computer with 8 GB memory and a 2.3 GHz Intel processor, it takes ~10 min to process 100,000 variants. For option C, it takes 10–30 min to upload and process 100,000 variants, depending on the current server load.
Step 1, preparation of input variant files: 5 min
Step 2A, annotation of human genomic variants by ANNOVAR: ~10 min (plus time for manually checking the variants)
Step 2A(i), decompression takes <10 s
Step 2A(ii), downloading all databases takes <5 min
Step 2A(iii), running the annotation script takes 5–10 min
Step 2A(iv), manually checking the variants takes 5 min to 1 h
Step 2B, gene-based annotation of nonhuman species by ANNOVAR: ~10 min
Step 2B(i), downloading the chimpanzee database takes <2 min
Step 2B(ii), generating the mRNA sequence takes <5 min
Step 2B(iii), running the annotation script takes 5–10 min
Step 2C, annotation and prioritization of human variants by wANNOVAR: 10–30 min
Step 2C(i,ii), visiting the website and downloading the example VCF takes <1 min
Step 2C(iii), each added phenotype or disease term costs 20 s to 1 min additionally
Step 2C(iv,v), selecting the correct parameters takes <2 min
Step 2C(vi), after submission, it takes 5–25 min for wANNOVAR server to generate results
ANTICIPATED RESULTS
For the command-line tool of ANNOVAR, make sure that no ‘ERROR’ message is displayed after each command. After running the ‘table_annovar.pl’ script, there should be multiple ‘NOTICE’ messages showing the current progress of the program. One example is shown below:
|
|
| NOTICE: Processing operation=g protocol=refgene |
| NOTICE: Reading gene annotation from humandb/hg19_refGene.txt … Done with 46966 transcripts (including 9362 without coding sequence annotation) for 24933 unique genes |
| NOTICE: Reading FASTA sequences from humandb/hg19_refGeneMrna.fa … Done with 29 sequences |
|
|
After submitting VCF files to wANNOVAR, you will receive an e-mail with the URL of the results page when the job is completed (if an e-mail address is provided during submission). In addition, the URL that is displayed after submission can be recorded to check the status of the submission. In any case, there should be at least two sections in the result page: the submission information and the basic information. The submission information includes the submission ID, sample identifier, file name, file format, processed variant number and others. The basic information includes the link to view annotated variants on the web, and the links to download the comma- or tab-delimited file with the annotated variants (Fig. 3). If a disease model was selected, then an additional section called ‘ANNOVAR filtering results’ will be shown, including the remaining variants after each filtration step. If any disease or phenotype terms have been entered, an additional section called ‘Phenotype/disease prioritization result’ will be shown, including the input gene list, the output prioritized-gene list and the link to the disease-gene network (Fig. 3). In our wANNOVAR example, the PKLR gene should be prioritized as the top disease gene in the result list. In addition, if you click the ‘Show’ link to see the gene network, PKLR is the largest gene in the network (Supplementary Fig. 1).
Figure 3.

Screenshot of the wANNOVAR result page. (a) The basic information section includes the submission ID, submission information and the annotated variant list, which can be accessed by clicking ‘View’, or it can be downloaded by clicking ‘CSV file’ or ‘TXT file’. (b) The additional information section includes the filtered variant files, the phenotype-based gene prioritization and annotations. If you have selected a disease model, the download links of the results after each filtering step will be shown. For example, Step 1 identifies missense, nonsense and splicing variants from the input variant list, and it provides the VCF file through the ‘download’ link. If you have entered any disease or phenotype terms, the prioritized gene list can be retrieved from the ‘Result Gene List’ link and the network visualization can be retrieved by clicking ‘Show’.
Please note that the disease gene prioritization only prioritizes genes based on the input disease or phenotype term, without considering the variant information. Thus, to effectively use this score, please first select a disease model for variant reduction or combine this score with other variant prediction scores such as SIFT score59, MetaSVM score45 and CADD scores46.
There are dozens of columns in the results page with annotations, including gene annotations, different types of allele frequencies and miscellaneous variant prediction scores (Supplementary Fig. 2). For more details about these annotations, please visit http://annovar.openbioinformatics.org/. For announcements on new software releases and urgent bug fixes, please subscribe to the ANNOVAR mailing list at https://groups.google.com/forum/#!forum/annovar.
Supplementary Material
Acknowledgments
Development of the ANNOVAR/wANNOVAR tool is supported by US National Institutes of Health (NIH) grant R01 HG006465. We thank X. Chang for the initial development of the wANNOVAR server. We thank all ANNOVAR and wANNOVAR users for their helpful suggestions, comments and bug reports to improve the software tools and web servers.
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS H.Y. and K.W. drafted and revised this manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Li H, Homer N. A survey of sequence alignment algorithms for next-generation sequencing. Brief Bioinform. 2010;11:473–483. doi: 10.1093/bib/bbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagarajan N, Pop M. Sequence assembly demystified. Nat Rev Genet. 2013;14:157–167. doi: 10.1038/nrg3367. [DOI] [PubMed] [Google Scholar]
- 5.Li H. Exploring single-sample SNP and INDEL calling with whole-genome de novo assembly. Bioinformatics. 2012;28:1838–1844. doi: 10.1093/bioinformatics/bts280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simpson JT, et al. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xie Y, et al. SOAPdenovo-Trans: de novo transcriptome assembly with short RNA-seq reads. Bioinformatics. 2014;30:1660–1666. doi: 10.1093/bioinformatics/btu077. [DOI] [PubMed] [Google Scholar]
- 8.Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010 http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 9.Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet. 2011;12:443–451. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao M, Wang Q, Wang Q, Jia P, Zhao Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics. 2013;14:S1. doi: 10.1186/1471-2105-14-S11-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–984. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu M, et al. Using ERDS to infer copy-number variants in high-coverage genomes. Am J Hum Genet. 2012;91:408–421. doi: 10.1016/j.ajhg.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLaren W, et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Baets G, et al. SNPeffect 4.0: on-line prediction of molecular and structural effects of protein-coding variants. Nucleic Acids Res. 2012;40(Database issue):D935–D939. doi: 10.1093/nar/gkr996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu H, et al. VAAST 2.0: improved variant classification and disease-gene identification using a conservation-controlled amino acid substitution matrix. Genet Epidemiol. 2013;37:622–634. doi: 10.1002/gepi.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Makarov V, et al. AnnTools: a comprehensive and versatile annotation toolkit for genomic variants. Bioinformatics. 2012;28:724–725. doi: 10.1093/bioinformatics/bts032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Michaelson JJ, et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 2012;151:1431–1442. doi: 10.1016/j.cell.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Girard SL, et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat Genet. 2011;43:860–863. doi: 10.1038/ng.886. [DOI] [PubMed] [Google Scholar]
- 21.Weedon MN, et al. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet. 2011;89:308–312. doi: 10.1016/j.ajhg.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai C-C, et al. Whole-exome sequencing to identify a novel LMNA gene mutation associated with inherited cardiac conduction disease. PLoS ONE. 2013;8:e83322. doi: 10.1371/journal.pone.0083322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brownstein CA, et al. An international effort towards developing standards for best practices in analysis, interpretation and reporting of clinical genome sequencing results in the CLARITY Challenge. Genome Biol. 2014;15:R53. doi: 10.1186/gb-2014-15-3-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, et al. Regenerative phenotype in mice with a point mutation in transforming growth factor β type I receptor (TGFBR1) Proc Natl Acad Sci USA. 2011;108:14560–14565. doi: 10.1073/pnas.1111056108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nam K, et al. Strong selective sweeps associated with ampliconic regions in great ape X chromosomes. arXiv:1402.5790. 2014 [Google Scholar]
- 26.Chang X, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet. 2012;49:433–436. doi: 10.1136/jmedgenet-2012-100918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang H, Robinson PN, Wang K. Phenolyzer: phenotype-based prioritization of candidate genes for human diseases. Nat Methods. 2015 Jul 20; doi: 10.1038/nmeth.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siepel A, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis BP, Shih I-h, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 30.Birney E, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Consortium GP. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu W, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sherry ST, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyon GJ, Wang K. Identifying disease mutations in genomic medicine settings: current challenges and how to accelerate progress. Genome Med. 2012;4:58. doi: 10.1186/gm359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu H, et al. A unified test of linkage analysis and rare-variant association for analysis of pedigree sequence data. Nat Biotechnol. 2014;32:663–669. doi: 10.1038/nbt.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paila U, Chapman BA, Kirchner R, Quinlan AR. GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput Biol. 2013;9:e1003153. doi: 10.1371/journal.pcbi.1003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Habegger L, et al. VAT: a computational framework to functionally annotate variants in personal genomes within a cloud-computing environment. Bioinformatics. 2012;28:2267–2269. doi: 10.1093/bioinformatics/bts368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ng SB, et al. Exome sequencing identifies the cause of a Mendelian disorder. Nature Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vuong H, et al. AVIA v2.0: annotation, visualization and impact analysis of genomic variants and genes. Bioinformatics. 2015;31:2748–2750. doi: 10.1093/bioinformatics/btv200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Medina I, et al. VARIANT: command line, web service and web interface for fast and accurate functional characterization of variants found by next-generation sequencing. Nucleic Acids Res. 2012;40:W54–W58. doi: 10.1093/nar/gks572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McCarthy DJ, et al. Choice of transcripts and software has a large effect on variant annotation. Genome Med. 2014;6:26. doi: 10.1186/gm543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dong C, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole-exome sequencing studies. Hum Mol Genet. 2015;24:2125–2137. doi: 10.1093/hmg/ddu733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kircher M, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eilbeck K, et al. The Sequence Ontology: a tool for the unification of genome annotations. Genome Biol. 2005;6:R44. doi: 10.1186/gb-2005-6-5-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Consortium GP. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat. 2013;34:E2393–E2402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Landrum MJ, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(Database issue):D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Day IN. dbSNP in the detail and copy number complexities. Hum Mutat. 2010;31:2–4. doi: 10.1002/humu.21149. [DOI] [PubMed] [Google Scholar]
- 54.Karolchik D, et al. The UCSC genome browser database: 2014 update. Nucleic Acids Res. 2014;42:D764–D770. doi: 10.1093/nar/gkt1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hsu F, et al. The UCSC known genes. Bioinformatics. 2006;22:1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- 57.Hubbard T, et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Derrien T, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ng PC. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.