

Abstract
Epistasis is a key factor in evolution, since it determines which combinations of mutations provide adaptive solutions and which mutational pathways towards these solutions are accessible by natural selection. There is growing evidence for the pervasiveness of sign epistasis – a complete reversion of mutational effects, particularly in protein evolution, yet its molecular basis remains poorly understood. We describe the structural basis of sign epistasis between G238S and R164S, two adaptive mutations in the antibiotic-resistance enzyme TEM-1 β-lactamase. Separated by 10Å, these mutations initiate two separate trajectories towards increased hydrolysis rates and resistance towards second and third-cephalosporins antibiotics. Both mutations allow the enzyme’s active-site to adopt alternative conformations and accommodate the new antibiotics. By solving the corresponding set of crystal structures we found that whereas G238S induces discrete conformations, R164S causes local disorder. When combined, the mutations in 238 and 164 induce local disorder whereby nonproductive conformations that perturb the enzyme’s catalytic pre-organization dominate. Specifically, Asn170 that coordinates the deacylating water molecule is misaligned, in both the free and the inhibitor-bound double mutant. This local disorder is not restored by stabilizing, global suppressor mutations and thus leads to an evolutionary cul-de-sac. Conformational dynamism therefore underlines the reshaping potential of proteins structures and functions, but also limits protein evolvability because of the fragility of the interactions networks that maintain protein structures.
Keywords: protein evolution, enzyme evolution, protein disorder, protein folds, Conformational diversity, Interactions network
Epistasis, i.e., non-additive effects of mutations, determines the topography of fitness landscapes1;2. Hence, epistasis describes which combinations of mutations provide adaptive solutions, and which stepwise mutational pathways leading to these solutions are selectively accessible3,4,5; 6. In doing so, epistasis determines the capacity of organisms, and of their proteins, to evolve and adapt, i.e., their evolvability. Interestingly, by increasing the dependence of mutational pathways on early mutations, epistasis causes mutational pathways to be historically contingent upon initial mutations7; 8. This influence is particularly strong in case of sign epistasis, where the sign of the fitness effect of a mutation (beneficial or deleterious) depends on its genetic background3. For example, mutations with individual beneficial effects, but combined deleterious effect, show reciprocal sign epistasis. They create a rugged adaptive landscape, where different trajectories may lead to adaptive peaks of different heights1; 3; 4; 9; 10; 11, and whereby some of the trajectories can turn into evolutionary dead ends, or cul-de-sacs12; 13; 14.
Epistasis, including sign epistasis, is pervasive2; 3. The phenomenon itself is measured and relates to organismal fitness. It is clear, however, that its origins lie in antagonistic interactions between mutations, either in separate genes (or proteins) or within the same gene/protein. However, the molecular basis of epistasis remains poorly understood10. We have better understanding of positive epistasis at the single gene level, such as in deer-mouse hemoglobin adapted to high altitude15. Typically, stabilizing mutations, which have no effect on protein fitness on their own, are revealed as beneficial when they compensate for new-function mutations that are typically destabilizing16. Such stabilizing mutations can have local, specific compensatory effects17; 18, or global effects, when they can compensate for a whole range of destabilizing mutations16 in non-contacting residues19; 20; 21. One case study of positive epistasis in the vertebrate glucocorticoid receptor revealed a mutation that was initially neutral, yet by reorienting an alpha helix, enabled the acceptance of an adaptive mutation that would be deleterious on its own22. Overall, it appears that stabilization of the protein’s configuration promotes its adaptive potential and underlines positive epistasis21; 22. However, a detailed, structural understanding of negative sign epistasis is lacking: Why do certain combinations of mutations, each of which is beneficial individually, become deleterious when combined23; 24?
Here, we explored a case of negative reciprocal sign epistasis between two adaptive mutations in TEM-1 β-lactamase, an enzyme present in numerous antibiotics resistant bacteria. These studied mutations, G238S and R164S, are amongst the most common mutations in clinically isolated variants, and therefore represent a highly relevant case study25; 26. Wild-type TEM-1 confers resistance to natural penicillin antibiotics. However, in the clinic, strains with new resistance mutations are frequently isolated. This increased resistance is the outcome of adaptive evolution of TEM-1 for new, so-called 2nd and 3rd cephalosporins antibiotics. Adaptive evolution of TEM-1 has occurred in numerous parallel events and can be readily reproduced in the laboratory25. The increased catalytic activity of TEM-1 to 2nd and 3rd cephalosporins antibiotics therefore comprises a broadly accepted model for enzyme evolution3; 25; 26. Owing to the large sampling of TEM-1 sequences by natural and directed evolution, the absence of certain combinations of mutations likely indicates their negative epistatic interaction. This is the case with G238S and R164S, which were individually found in 91 and 51 different variants, respectively, both in laboratory and clinical contexts25. However, their combination has only been observed in one clinical sample27. Indeed, the combination of G238S and R164S results in in vivo resistance levels to cefotaxime that are lower than each of the single mutants (4-fold lower than R164S, and 8-fold lower than G238S) and are similar to wild-type TEM-1 (ref. 7; 27). Laboratory experiments attempting to evolve variants carrying the R164S mutation toward high cefotaxime activity indicated that R164S blocks the path to maximal cefotaxime degradation activity7. As detailed in the Results section, one mutation, E104K, improves R164S’s activity, but the activity of the R164S/E104K double mutant was still inferior to G238S/E104K24. The only traceable trajectory to higher cefotaxime resistance in these laboratory evolution experiments was through reversion of R164S and take-over by G238S (ref. 7). Thus, R164S seems to comprise an evolutionary cul-de-sac7. However, the molecular basis underlining the limited adaptive potential of R164S and of the reciprocal sign epistasis with G238S remain unclear.
The strong, non-additive interaction between R164S and G238S is surprising for two reasons: firstly, the mutation sites are not in direct contact (>10Å apart), and secondly, combining these mutations does not further compromise TEM-1’s global configurational stability7; 16. To determine the structural basis for this classical case of reciprocal sign epistasis, and to understand why R164S might lead to an evolutionary cul-de-sac, we solved the crystal structures of the R164S, G238S and double mutants R164S/G238S, in their free forms and bound to an inhibitor. Although these mutations are key first-step mutations in the adaptation of TEM-1 to 2nd and 3rd cephalosporins antibiotics, the structure of enzymes harboring these mutations with no additional active-site mutations have not been available thus far. The combination of free vs. inhibitor bound and of variable temperature X-ray data allowed for increased sampling of the conformational ensemble of TEM-1, thus unraveling the structural basis of negative epistasis and evolvability in TEM-1.
Results
Mutations that confer TEM-1 with the ability to hydrolyze new antibiotics are located mostly on the loops surrounding the active site: the ‘Ω-loop’ (residues 164–179 (ref. 28)), the ‘238-loop’ (residues 238–242 (ref. 29)) and the ‘101-loop’ (residues 101–111 (ref. 30)). The mutations studied here, G238S and R164S, enhance cefotaxime hydrolysis (3rd cephalosporins antibiotic) and reside on the 238-loop and the Ω-loop, respectively7; 29. Mutation G238S increases cefotaxime resistance levels by ~16-fold: the MIC, the minimal inhibitory concentration, increases from 0.06μg/ml to 1μg/ml (Table 1). Mutation R164S increases cefotaxime resistance by only ~8-fold7; 31 (MIC = 0.5 μg/ml), but exhibits increased resistance levels with other, primarily 2nd-generation antibiotics31; 32, as well as a weaker tradeoff than G238S with ampicillin resistance (Table 1 (ref. 7)). In the absence of epistasis, the combined effect of the R164S and G238S mutations is expected to be multiplicative at the level of MIC (or additive at the level of log MIC33), thus resulting in up to ~120-fold higher cefotaxime resistance levels relative to wild-type. However, the double mutant’s MIC is only increased ~2-fold relative to wild type (Table 1). Accordingly, the catalytic efficiency (kcat/KM) of the R164S/G238S mutant with cefotaxime is essentially identical to that of the single mutant R164S, and about 20-fold lower than that of G238S (Table 1 (ref. 7)). These losses in catalytic efficiency, combined with a more minor effect on TEM-1’s conformational stability7, result in the in vivo MIC levels of the R164S/G238S mutant being lower than each of the single mutants (Table 1 (see also Ref. 7)). In addition, the R164S/G238S mutant exhibits a very low kcat value with ampicillin, the original substrate, and accordingly, much reduced MIC levels. The KM of the double mutant with ampicillin was improved. However, given the covalent intermediate mechanism of TEM-1, this change may in fact relate to a drastic slowdown in the rate of deacylation, and not improved substrate affinity34. Further, in both the wild-type and the mutants, the KM is much lower than the applied antibiotic concentrations, and therefore the improvement in KM has little effect on resistance levels. Thus, R164S and G238S exhibit negative reciprocal epistasis: the expected positive effect of both individual mutations is inverted to negative when they are combined.
Table 1.
Kinetic and stability parameters, and E. coli resistance levels of the TEM-1 variants.
| Variant | Antibiotic | KM (μM) | kcat (sec−1) | kcat\KM (M−1sec−1) | MIC (μg/ml) |
|---|---|---|---|---|---|
| Wild-type | Amp | 71±9 | 1857±27 | 26±1.3 x106 | 4096 |
| Ctx | n.d. | n.d. | 0.1x104±6 | 0.06 | |
|
| |||||
| Stabilized v13a | Amp | 63±13 | 1040±21 | 16.5±1.3x106 | n.m. |
| Ctx | n.d. | n.d. | 0.07 x104±10 | ||
|
| |||||
| Wild-type + G238S | Amp | 26±4 | 42±0.4 | 1.4±0.08x106 | 1024 |
| Ctx | 403±146 | 50±3 | 13±4x104 | 1 | |
|
| |||||
| Stabilized v13+ G238S | Amp | 18±5 | 72±0.5 | 4±0.36 x106 | n.m. |
| Ctx | 502±65 | 75±1.4 | 14.5±1 x104 | ||
|
| |||||
| Wild-type+ R164S | Amp | 37±7 | 75±25 | 2.0±0.8x106 | 4096 |
| Ctx | 536±51 | 2.5±0.1 | 4.7±0.4x103 | 0.5 | |
|
| |||||
| Stabilized v13+ R164S | Amp | 140±36 | 164±24 | 1.1±0.7x106 | n.m. |
| Ctx | 887±173 | 5±0.5 | 5.3±2.5x103 | ||
|
| |||||
| Wild-type + G238S+R164S | Amp | 11±11 | 7±3 | 0.9±0.5x106 | 512 |
| Ctx | 578±26 | 2.6±0.1 | 4.5±0.5x103 | 0.125 | |
|
| |||||
| Stabilized v13 +G238S+R164S | Amp | 6±0.85 | 11±0.2 | 1.8±0.4x106 | n.m. |
| Ctx | 1300±137 | 12.5±0.85 | 9.6±2x103 | ||
v13 is a stabilized variant of TEM-1 described in Ref. 16 that exhibits essentially the same enzymatic parameters as the wild-type TEM-1.
n.d. – not detectable. A linear dependency of initial rates on substrate concentrations was observed (while applying the highest substrate concentrations possible within maximal absorbance of OD264nm ≤ 1.5), and thus only kcat/KM could be determined.
n.m. – not measured. The increased stability may affect the export rates and yields of TEM-1 to the periplasm, and thus the MIC values of stabilized variants were not measured.
Amp - ampicilline; Ctx - cefotaxime.
Crystal structures
Due to their compromised configurational stability, the single mutants, let alone the double mutant, were poorly expressed in E. coli 31. Their tendency to crystallize was also poor, especially of the R164S/G238S double mutant. Indeed, although a whole range of TEM-1 mutant structures is available, a structure of this double mutant has not been thus far attainable, not even at the background of a stabilizing mutation, M182T, that is routinely observed in clinical variants. We therefore made use of a stabilized variant of TEM-1 dubbed v13, that carries 7 stabilizing mutations located on the surface and away from the active-site, including mutations that are commonly observed in clinical variants such as M182T (for a list of mutations, and their structural locations, see supplementary Fig. 1). This variant is essentially identical in structure to wild-type TEM-1 (ref. 16), and the kinetic parameters of the studied 164 and 238 mutants in the background of v13 are essentially identical to the same mutations in the wild-type background (Table 1). Further, structures of v13 show the same active-site loop configurations, and the same levels of loop mobility as wild-type TEM-1 (ref. 16). The individual effects of G238S and R164S, and the non-additive effects of combining them, on the kcat/KM values for both cefotaxime and ampicillin are essentially identical to the effects of these mutations at the wild-type’s background (Table 1). The in vivo effects could not be measured because the hyper-stability of v13 has major effects on periplasmic export. However, the inconsistencies in the MIC level are the outcome of differences in expression levels and export rates, and are therefore irrelevant to issues under study here. The comparison of the in vitro measured kinetic parameters and structures of the wild-type background (Table 1; Ref. 16) indicate that v13 comprises a valid model for studying the structural and mechanistic origins of the loss of TEM-1’s catalytic function when G238S and R164S are combined as manifested in kcat/KM values, and especially in kcat (discussed below).
Overall, we solved 10 different structures of stabilized variant carrying the studied active-site mutations, individually and in combination (R164S/G238S) at both cryogenic and room temperatures. We obtained structures of the free enzyme form, and structures bound to EC25 (ref. 35), a covalent borate-based inhibitor that mimics the deacylation tetrahedral intermediate II36 (supplementary Fig. 2). EC25’s structure resembles ampicillin and not cefotaxime. However, a suitable boronate mimic of cefotaxime is not available (the one available, LP08, only mimics the thiazolyl part but not the lactam ring and its bulky substituents35). However, in the double mutant, cefotaxime and ampicillin show the very same trend of loss of activity relative to the single G238S mutant, both in relation to MIC values in vivo and to the in vitro kinetic parameters (Table 1). Additionally, as shown below, the origins of negative epistasis are in the disturbance of TEM-1’s catalytic machinery, and the mechanism of catalysis is essentially identical for both cefotaxime and ampicillin37.
Structures were solved by molecular replacement and refined to 1.05–2.4Å resolutions. The anisotropic B-factors were refined in the high resolution data as detailed in supplementary Table 1. All of the newly solved structures align well to one another and to the published wild-type TEM-1 structure (PDB code: 1ZG4 (ref. 38)). The only significant conformational differences between the structures are in the loops surrounding the active-site, foremost in the Ω-loop where R164S resides and in the 238-loop that includes G238S. Analyzing the main loop conformations in each of these structures reveals that the G238S mutation samples between two well-defined discrete 238-loop conformations, but that R164S results in a diverse Ω-loop conformational ensemble (Fig. 1). It should be noted that the crystal packing of the TEM-1 structures obtained here involves the omega-loop where R164S resides. Nonetheless, all structures presented in this study exhibit the same space group C2, although the published wild-type TEM-1 structure relates to a different one (P212121 (ref. 38). The comparison of the single to the double mutant is therefore based on all structures belonging to the same space group. Additionally, packing interactions tend to be stabilizing and thus to favor a single conformation. Hence, if anything, our data underestimates the conformational ensembles of the mutants, and specifically of the double mutant.
Fig. 1. Superposition of active-site loops in all structures.

(A) Superposition of the main Ω-loop conformation in all wild-type structures (gray) and in the different v13-R164S mutant structures (blue). Room-temperature structures are in green; note that in the double-mutant (R164S/G238S) room temperature structure, the Ω-loop could not be modeled and is therefore missing. Shown in sticks are the side-chains of N170 that aligns the de-acylating water (W1; gray for wild-type, red for mutants) and the catalytic S70 for reference. (B) Superposition of the main 238-loops conformation in all v13 structures containing the G238S mutation (blue), in wild-type (gray). The mutants structures determined at room temperature are in green. Shown is A237 whose backbone amide positions the oxyanion-hole water (W2). The loop conformations shown relate to the main clearly observed in the electron density of individual structures. Structures included are: A 4OPY, 4OPQ, 4OP5, 4OQI, 4OQH, 4OQ0, 4OQG, 1ZG4; B 4OPQ, 4OP8, 4OP4, 4OQI, 4OPZ, 4OQ0, 4OQG, 1ZG4 (ref. 38) (supplementary Table 1).
The effects of G238S
In v13-G238S mutant, the dominant conformation of the 238-loop is perturbed, while the Ω-loop conformation more closely resembles the wild-type conformation (Fig. 2). Inspection of the 238-loop Fourier difference density maps of both the free and EC25-bound structures revealed alternative loop conformations that resemble a wild-type-like conformation (Fig. 2 and Fig. 3 G238S – middle top and bottom; density for the loop is fit by the dominant “major” conformation shown in thick sticks and a secondary “minor” conformation shown as thin sticks). The alternative, open conformation, presumably allows access to the much larger cefotaxime substrate, and thereby increases the kcat/KM of G238S by ≥130-fold relative to wild-type, in which, the closed conformation prevails. However, the perturbed, open conformation also results in the loss of a key interaction between the Ω-loop and 238-loop (E240’s backbone amide with N170’s backbone carbonyl oxygen– Fig. 2 and Fig. 3 shown as red dashed-circle). Apart from connecting the two loops, the interaction with E240 seems to be critical for stabilizing N170, a key catalytic residue that coordinates the de-acylating water molecule39. Concordantly, there is electron density consistent with a second N170 side chain conformation in a non-catalytic orientation. However, unlike v13-R164S, in the v13-G238S structure, the main chain conformation of N170 closely resembles the catalytically competent wild-type conformation (Fig. 2). Upon ligand binding, the minor conformation of the 238-loop shifts to restore the interaction between the loops (Fig. 3 G238S - middle bottom: the red-dashed circle encompasses the wild-type and minor 238-loop conformations). Supplementary Movie 2 illustrates the relatively small changes in the Ω-loop’s configuration upon inhibitor binding in comparison to the R164 mutant.
Fig. 2. The Ω-loop and 238-loop interactions network.

A snapshot of the Ω-loop, the 238-loop, and the key catalytic residues in the free structures (left column) and in structures with the inhibitor EC25 bound (right column). Shown are wild-type TEM-1 (gray), the single v13 mutants R164S (yellow) and G238S (blue), and the double v13 mutant R164S/G238S (green). Catalytic residues are in smoked pink and EC25 is in light pink ball-sticks. Minor conformations of the loops are shown in light blue. Dashed lines represent putative interactions involving R164 and G238, and interactions lost in the R164S/G238S double mutant are marked in blue in the wild-type. The non-native interaction in the double mutant between E171 and S238 is in red. A putative stacking interaction between R164 and R178 (ref. 71) is also marked.
Fig. 3. Electron density maps of the 238 and Ω-loops.
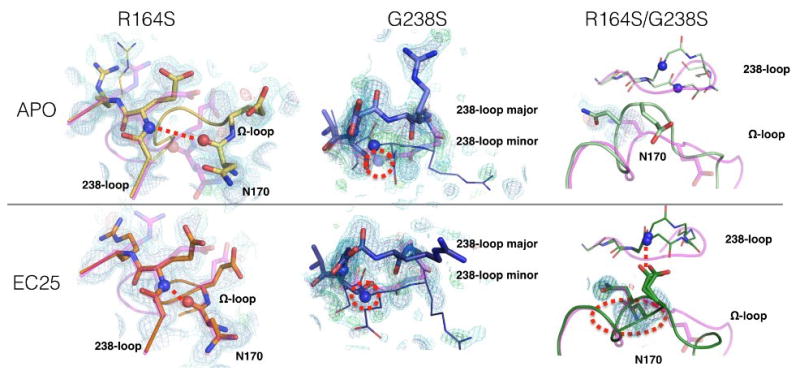
The wild-type structure is shown for comparison in partially transparent magenta. The 2mFo-DFc electron density maps for the two v13 single mutants, and the v13 double mutant, are contoured at 0.5 (cyan) and 1 (blue) sigma, and mFo-DFc electron density maps are contoured at +3.0 (green) and −3.0 (red) sigma. (The m and D terms indicate that the maps were calculated while correcting for down-weight poorly phased reflections, and including a scaling factor to account for overall scattering differences due to missing components such as partially ordered waters).
The incomplete shift of the minor conformation to the catalytically competent conformation suggests that formation of the transition state(s) leading to and/or from the acyl-enzyme intermediate involves a higher entropic penalty for G238S than for wild-type. As shown below, this cost seems far more pronounced in the R164S mutant that samples between many perturbed Ω-loop conformations in the free enzyme.
The effects of R164S
As previously reported28; 40; 41, the R164S mutant increased the conformational freedom of the Ω-loop, as indicated by high B-factors and a weak electron density (Table 2, supplementary Fig. 4 and 5) relative to other parts of the protein. Indeed, the increased conformational freedom of the Ω-loop was suggested to increase TEM-1’s active-site accessibility for larger substrates such as cefotaxime25; 31. The other active-site loops, including the 238-loop, are well ordered in the R164S free and inhibitor-bound v13 structures (Fig. 2). The dominant conformation of the Ω-loop is distinct from the wild-type conformation (C-alpha RMSD is ~2Å; Fig. 2). Accordingly, the interaction between E240-NH and N170-O is lost (Fig. 3 shown as the red dashes). Indeed, in the inhibitor-free structure, N170 exhibits an outward rotation relative to the wild-type conformation, away from the deacylating water (Fig. 2). The deacylating water is accordingly shifted, although its other ligating residue, E166, exhibits wild-type conformation. Upon EC25 binding, the mobility of the Ω-loop is reduced, the critical interaction with the 238-loop is restored, and N170, as well as E166, adopt wild-type conformation (Table 2 and Fig. 2). Supplementary Movie 1 illustrates the changes in the Ω-loop’s configuration upon inhibitor binding, the concomitant gain of interaction between the Ω-loop and 238-loop and the relocation of N170.
Table 2.
The Ω- and 238-loops B-factorsa
| Relative loops B-factors (Å2)b | ||
|---|---|---|
| Structurec | Free enzyme | EC25 complex |
| Wild-type 238-loop | 0.98 | - |
| Wild-type omega-loop | 1.24 | - |
| G238S 238-loop | 1.18 | 1.06 |
| G238S omega-loop | 2.56 | 1.75 |
| R164S 238-loop | 1.02 | 1.12 |
| R164S omega-loop | 2.54 | 2.22 |
| R164S/G238S 238-loop | 1.46 | 1.24 |
| R164S/G238S omega-loop | 4.75 | 3.34 |
B-factors indicate the level of thermally-induced mobility of the loops’ backbone atoms.
Calculated for the set of the structures obtained at cryo temperature (supplementary Table 1). Noted are the average B-factors for backbone atoms of all loop residues, normalized to the average B-factor for all backbone atoms in a given structure.
All structures here are on the background of stabilized v13 and belong to the same space group, thus minimizing biases due to crystal packing, particularly in the Ω-loop.
The binding of EC25 to TEM-1 is covalent (supplementary Fig. 2), thereby promoting its ability to shift the equilibrium towards the well-organized conformation of the Ω-loop and N170. The cost of N170’s alternative, non-catalytic conformation in the case of the actual substrate is therefore likely to be much higher. These results suggest that substrate binding to the R164S mutant involves an increased entropic penalty due to the poor preorganization of the Ω-loop, as indicated by N170’s perturbed conformation, and the elevated B-factors for the entire Ω-loop (a detailed analysis is provided below). However, the perturbed Ω-loop conformational dynamics of the free enzyme are likely required to accommodate larger 2nd and 3rd cephalosporins antibiotics.
The observed differences in loop configurations, and particularly of the Ω-loop, may relate to crystal packing, and may thus, fail to report the enzyme’s actual configuration(s) in solution. Note, however, that all v13 structures presented here belong to the same space group (Supplementary Table 2). Additionally, the Ω-loop’s configuration does not seem to vary differently between structures belonging to different space groups, suggesting that crystal packing does not significantly alter this loop’s configuration (Supplementary Figure 3).
The combined effect of R164S and G238S
A systematic analysis of the v13 double mutant’s structure did not reveal potential effects of the mutations on substrate binding. Indeed, neither R164S nor G238S are in direct contact with the substrate (as far as can be judged from the EC25 complex) and neither takes part in catalysis. Therefore, we examined the structures for longer-range effects. Both single mutants exhibit conformational changes within the active-site loops that are necessary for accommodating the larger cefotaxime substrate29; 41. However, these two mutations have different side-effects on the active-site conformational ensemble, suggesting that the mechanism underlining TEM-1’s adaptation towards cefotaxime is more complex than previously proposed42; 43. In the v13 R164S/G238S double mutant, a wider ensemble of conformations is seen that incudes nonproductive conformations in both the free and inhibitor-bound states (Fig. 2). Specifically, like in the v13-R164S mutant, the Ω-loop of the double mutant is conformationally heterogeneous. Additionally, like in the v13-G238S mutant, the perturbed conformation of the 238-loop dominates, and the near-wild-type conformation is scarcely populated (Fig. 2 and Fig. 3 R164S/G238 – right top: the major and minor conformations of the 238-loop are shown as thin sticks with the position of E240-N indicated by spheres in both minor and major conformations). However, in the double mutant the 238-loop and Ω-loop fail to establish interactions that stabilize the catalytically competent conformation of N170. Further, N170’s side-chain carbonyl oxygen takes the place of the deacylating water that is not present in the double mutant’s free structure, although E166 exhibits a wild-type-like conformation. Even covalent inhibitor binding is insufficient to drive the rearrangement. Supplementary Movies 3 and 4 address the minor and major 238-loop conformations, respectively. The morph movies illustrate the profound changes in the Ω-loop’s configuration upon inhibitor binding, the loss of interaction between the Ω-loop and 238-loop in both the free and inhibitor bound structures, and the displacement of N170.
The loss of the catalytically competent N170 conformation relates to a new, nonproductive interaction seen exclusively in the double mutant: in the bound structure E171’s side chain interacts with the backbone amide of E240 within the perturbed conformation of the 238-loop (Fig. 2 and Fig. 3 R164S/G238 – right top: The interaction between E171 and E240 is indicated by red dashes). The non-productive 171–240 interaction originates to the loss of E171’s side-chain interaction with R164 upon mutating this residue to Ser. However, the 171–240 interaction does not occur in the v13-R164S single mutant, simply because the perturbed 238-loop conformation is only populated when G238 is mutated to Ser. Therefore, negative epistasis between these two mutations has two primary structural causes: 1) Excessive conformational flexibility when the two mutations are combined decreases the pre-organization of the enzyme active site in the free state; 2) a non-native 171–240 interaction between the omega and 238 loops seems to shift the equilibrium against the productive active-site conformation. As discussed in detail below, the shift towards nonproductive conformations is also apparent in the double mutant’s free structure, where the deacylating water molecule is missing and, further, N170’s side-chain and the catalytic S70’s hydroxyl are within contacting distance. This interaction is likely to severely hamper the active-site (Fig. 2).
The disturbance of active site pre-organization
Enzyme active-sites, including TEM-1’s, are pre-organized in a catalytically optimal conformation44; 45. However, the high mobility of the Ω-loop, in both the v13 single R164S mutant and especially in the v13-R164S/G238S double mutant, induced alternative, suboptimal configurations (Fig. 4A). To determine the magnitude of conformational change the apo-enzyme exhibit in order to get to the catalytically optimal conformation, we examined the differences between the free and the inhibitor-bound enzyme structures. Specifically, we observed the displacement of N170, a residue that coordinates the de-acylating water molecule39. In wild-type and G238S, the overall deviations in the active-site’s structure upon binding are minimal (RMSD≤0.39Å Table 3) and thus N170 is largely pre-aligned for catalysis. In v13-R164S and R164S/G238S, however, N170 is misaligned in the free structures, and the Ω-loop exhibits a large conformational change upon inhibitor binding (RMSD≈1.85Å; Table 3 Fig. 4). This displacement dominates the conformational ensembles of the v13-R164S mutant and the v13 double mutant R164S/G238S, but is only a minor conformation in the v13-G238S mutant (Figure 4A and Table 4). Consistent with this structural analysis, the R164S mutation was shown to impair de-acylation in class-A β-lactamases40; 41. Thus, the prevailing conformation(s) in the free enzyme states of v13-R164S and v13-R164S/G238S probably exhibit no, or poor catalytic activity. The entropic cost of selecting from multiple conformations, combined with the impaired configuration of N170 even in the inhibitor bound structure, are manifested in the very low kcat value of the double mutant. As expected, inhibitor binding shifts the equilibrium toward the catalytic conformation in all mutants (Fig. 4B, Table 4). However, even in the inhibitor-bound v13-R164S/G238S structure, a wild-type-like configuration of N170 is only observed at 0.5 occupancy alongside an alternative, highly perturbed conformation that is unlikely to be catalytically active (Fig. 4B, in green; Table 4).
Fig. 4. The mutations impair TEM-1’s catalytic pre-organization.

Superposition of the key active-sites residues in wild-type (gray), the v13 single G238S (blue) and R164S (yellow) mutants, and the v13 double mutant R164S/G238S (green). (A) Ligand-free structures. (B) The EC25 inhibitor complexes (EC25 is shown in ball-sticks). In the free structures, W1 represents the de-acylating water and W2 in the oxyanion-hole water44. In the EC25-bound structures, the borate’s oxygen atoms, marked as O1 and O2, sit in the locations of W1 and W2, respectively. Note the dual conformation of N170 in the EC25 complex of R164S/G238S.
Table 3. Structural deviations between the free and inhibitor-bound structures.
Noted are average RMSD values for main-chain atoms (in Å)
| Structure | All residues (H25-W286) | Ω-loop (E164–R179) | 238-loop (S238–S242) |
|---|---|---|---|
| wild-type | 0.37 | 0.18 | 0.22 |
| G238S | 0.34 | 0.39 | 0.20 |
| R164S | 0.39 | 1.83 | 0.07 |
| R164S/ G238S | 0.33 | 1.86 | 0.12 |
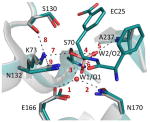
Table 4. The key interactions underlining TEM-1’s catalysis.
The figure below depicts a superposition of the key active site residues in free (gray, PDB:1ZG4 (ref. 38)) and bound (green, PDB: 4OQG) wild-type structures. The distances for the indicated bonds are provided in the table below.
| Interaction # | Interaction distances (Å) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| Free enzyme | |||||||||
|
| |||||||||
| Wild-type | 2.5 | 2.8 | 2.8 | 2.4 | 3.2 | 2.9 | 2.8 | 3.0 | 2.7 |
|
| |||||||||
| R164S | 2.7 | 3.5 | 3.2 | 2.9 | 2.9 | 3.1 | 2.7 | 2.9 | 2.7 |
|
| |||||||||
| G238S | 2.5 |
2.3a 6.1 |
2.9 | 2.8 | 2.4 | 3.0 | 2.7 | 2.9 | 2.8 |
|
| |||||||||
| R164S/G238S | n.d. | n.d. | n.d. | 2.9 | 2.9 | 3.1 | 2.7 | 2.8 | 2.7 |
|
| |||||||||
| EC25-bound | |||||||||
|
| |||||||||
| Wild-type | 2.8 | 2.9 | 2.2 | 2.6 | 3.8 | 3.1 | 2.9 | 2.5 | 3.1 |
|
| |||||||||
| R164S | 2.5 | 2.6 | 2.6 | 2.5 | 2.8 | 2.8 | 2.8 | 2.8 | 2.6 |
|
| |||||||||
| G238S | 2.5 | 2.7 | 2.5 | 2.5 | 2.8 | 2.8 | 2.7 | 2.8 | 2.8 |
|
| |||||||||
| R164S/G238S | 2.5 |
2.7a 6.1 |
2.4 | 2.4 | 3.0 | 2.8 | 2.7 | 2.8 | 2.7 |
|
| |||||||||
| Acceptor | E166 Oε |
N170 Oδ |
n.a. | n.a. | W2/O2b | W2/O2b | K73 NζH3 |
S130 Oγ |
N13 2 Oδ |
|
| |||||||||
| Donor | W1/O1c | W1/O1c | n.a. | n.a. | A237 Nα |
S70 Nα |
S70 Oγ |
K73 NζH3 |
K73 NζH3 |
|
| |||||||||
| |||||||||
n.d. – Not detected: the interaction is not observed because at least one of the interacting TEM-1 side-chain atoms is disordered.
n.a. – not applicable. These contacts are not hydrogen bonds.
N170 appears in two alternative conformations in this structure.
The oxyanion hole water or the borate oxygen atom mimicking it.
The de-acylating water or the borate oxygen atom mimicking it.
Table 4’s figure: TEM-1’s active-site configuration
B-factors analysis
To further test the idea that increased conformational flexibility underlies the sign negative epistasis between R164S and G238S, we examined the refined crystallographic temperature factors (B-factors) along TEM-1’s polypeptide chain. B-factors model how much an atom deviates from its average position in the crystal46; 47. However, this model includes both dynamic and static disorder and is sensitive to model errors. B-factors are especially useful reporters of molecular flexibility when discrete conformations cannot be distinguished in the electron density. Since alternative conformations for the 238-loop are apparent at low electron density levels (supplementary Fig. 4 and 5), we carefully modeled these fragments and then normalized the B-factors within each structure to indicate the relative mobility of atoms47. The normalized B-factors indicate that in the free structures of the mutants, both the omega and G238 loops exhibit higher flexibility in the mutants than in wild-type TEM-1. Interestingly, while R164S mutation induces only local changes in the flexibility of the Ω-loop, the G238S mutation induces alternative conformations of the 238-loop and increased B-factors in both the 238 and the Ω-loop. In the double mutant, we observed that both of the loops exhibit increased B-factors. However, the Ω-loop exhibits normalized B-factors that are almost 2-fold higher than in each of the single mutants (Table 2). To determine how flexibility changes upon inhibitor binding, we analyzed the patterns of B-factors in the inhibitor-bound structures. In both of the single mutants, inhibitor binding reduced the B-factor values. However, in the double mutant, the reduction in the loop’s B-factors upon binding is much lower than in any of the single mutants (Table 2). The B-factor analysis therefore supports the hypothesis that the double mutant exhibits a higher degree of conformational freedom compared to the single mutants. Hence, we suggest that both the negative epistasis between R164S and G238S mutant, and the decreased evolvability of alleles carrying R164S, result from the mutations widening the conformational ensembles to a level that penetrates into the active-site and hampers catalysis.
Discussion
Sign epistasis modulates the fitness landscape such that traversing from one peak (i.e., a given sequence and function) to another may occur via only few trajectories3,48; 49. Understanding the underlying causes of this particular form of epistasis may make evolutionary processes, and enzyme design and engineering, easier to understand and predict2. Here, we revealed the basis of reciprocal sign epistasis between two mutations in TEM-1 β-lactamase that initiate alternative resistance pathways to second and third-cephalosporins antibiotics including cephalosporins. As nearly all other new-function mutations in enzymes, R164S and G238S occur not in the catalytic residues, and not even in residues that directly contact the substrate50. Rather, they are located in loops that surround the active-site, and in residues that are >10Å apart. Nonetheless, these mutations strongly interact phenotypically at the level of enzyme function. Individually, each of the studied adaptive mutations introduces conformational changes in the active site loops that enable TEM-1’s active-site to better accommodate a new larger substrate. However, the level of conformational diversity introduced by these mutations, and the side effects of widening the enzyme’s conformational ensemble, differ between these two mutations. While one mutation, G238S, introduces new well-defined conformations, the other, R164S, introduces disorder along large segments of the Ω-loop (Fig. 1). Further, in the G238S mutant, the 238-loop conformation dominates in both the free and inhibitor-bound enzyme forms – hence, this mutation simply reshapes the loop (Table 3). In contrast, R164S induces an ensemble of different conformations, most of which are of nearly equal stability and are hence rarely represented (manifested in the weak electron density of the Ω-loop in the crystal). Binding of the inhibitor, and accordingly, substrate binding and formation of the transition state, shift the equilibrium towards the catalytically active conformation (Table 3).
The existence of multiple non-active conformations is manifested in the kcat value of the R164S/G238S double mutant with ampicillin (TEM-1’s preferred substrate) being ~10-fold lower than that of R164S, and 260-fold lower than that of wild-type (Table 1). Assuming ‘conformational-selection’, i.e., multiple pre-existing conformations and substrate binding shifting the equilibrium towards the functional conformation, the measured kcat relates to the kcat of individual conformers multiplied by their relative representation. Thus, a loss of ~100-fold in kcat upon combining R164S and G238S (relative to the expected additive effect) suggests that the fraction of the catalytically optimal conformer(s) dropped to ~1%. The role of multiple pre-existing conformers in mediating the multi-functionality of proteins51; 52 and their evolutionary potential53 is widely recognized. But the evolutionary cost of excessive conformational freedom, as observed here, is generally overlooked. One case highlighting the cost of increased conformational freedom relates to a laboratory engineered trypsin/chymotrypsin loop swap54. This swap resulted in an enzyme that was not only catalytically impaired but also lacked the expected chymotrypsin-like specificity. The low turnover rate and specificity related to the high mobility of active-site loops, and their stabilization via a single point mutation led to the recovery of the enzyme’s rate and specificity. In triosephosphate isomerase55, mutations destabilizing the loops surrounding the active-site, severely hamper catalytic activity, and especially the kcat., as observed here. Similarly, in RnaseH56, insertion/deletion of a glycine altered the ensemble of loop conformers and modulated the catalytic rate. Here we addressed the effects of mutations that occurred along adaptive evolutionary trajectories. We found that both G238S and R164S increase the representation of a new active-site conformation that better accommodates cefotaxime. However, combining these two mutations results in non-catalytic conformers dominating the conformational ensemble, and in non-native interactions between the loops such as the hydrogen bond between E171 and E240 (Fig. 2; R164S/G238S red dashes).
Thus, in TEM-1, sign epistasis between mutations that increase enzyme function seems to be the outcome of ‘conformational anarchy’ whereby the catalytically optimal conformation becomes diluted to a degree that severely hampers catalytic efficiency. Previous work described the role of compensatory mutations with global stabilizing effects in the evolution of new enzyme functions, and specifically in TEM-119; 31; 50. However, as far as the catalytic parameters are concerned, the negative epistasis effect between R164S and G238S in TEM-1 is not alleviated by global, stabilizing, compensatory mutations – not by M182T (ref.7; 24; 31), TEM-1’s most frequently observed stabilizing mutation, and not even in v13 that carries 7 stabilizing mutations including another known global suppressor, L201A (ref. 57). The inability to evolve a more stable and rigid core and scaffold to compensate for increased mobility of the active-site loops relates to the polarized architecture of TEM-1 (ref. 16).
Our results therefore reveal a delicate balance between the adaptive benefit associated with increased structural freedom and its cost. An example of this tradeoff is antibody affinity maturation. Germ-line antibodies exhibit high conformational plasticity yet low affinity, which is compensated by the higher avidity of IgM antibodies. Maturation involves the funneling of conformations towards the one that binds the antigen, and hence increase affinity 58,59.
Why do these TEM-1 mutations behave so differently – R164S leading to a local adaptive peak versus G238S leading towards higher fitness? Both R164 and G238 are part of an elaborate network that shapes TEM-1’s active-site (Fig. 2). However, whereas R164 has 7 contacts with neighboring residues, G238 has only two. Contacts density correlates with how buried a residue is60, yet R164 and G238 exhibit the same relative solvent accessibility. Thus, R164 exhibits higher than average contact density for its degree of burial (mean = 4.8 contacts per residue) whereas G238 is far below average (supplementary Fig. 6). This difference goes beyond the expected for the different sizes of these amino acids. The high connectivity of R164 results in the collapse of this interaction network upon mutating to serine (Fig. 2) and underlines the appearance of non-catalytic conformations (Fig. 4). Negative epistasis in TEM-1 may therefore relate to the network properties of protein folds. These so-called ‘small-world’ networks61; 62 are characterized by most residues (nodes) being connected via few hubs. In such networks, highly connected residues as R164 are relatively rare, and their perturbation severely undermines the network62. These networks, however, are highly robust to changes in weakly connected nodes as G238. Accordingly, position 238 shows ~10-fold faster evolutionary rate than 164 (238 evolves 3 time slower than the protein’s average, and R164 30-fold slower as calculated by rate4site63). Owing to its limited connectivity within the active-site’s interactions network, position 238 exhibits high robustness as well as innovability – the potential to promote new enzymatic functions64.
Overall, our analysis suggests a tight link between protein structural networks, conformational ensembles and evolvability. At least for the case of TEM-1 beta-lactamase, we observed a delicate balance between the potential of mutations to introduce new conformations that may initiate new functions, and their cost in over-broadening the protein’s conformational ensemble. The latter appears to result in negative epistasis and what seems to be an evolutionary cul-de-sacs in TEM-1’s adaptation via the R164S mutation. Better understanding of how a protein’s architecture, structural networks and conformational dynamics at different time scales affect this balance will promote our understanding of protein evolution and also advance the design and engineering of new proteins65; 66.
Materials and Methods
Crystallization
Wild-type TEM-1 and mutants were expressed and purified as previously described16. For crystallization, concentrated solutions of TEM-1 variants (60 mg/ml) in 25 mM Tris pH 8.4 and 100 mM NaCl were submitted to crystallization trials. Crystallization was performed using the hanging-drop vapor diffusion method. Equal volumes (0.5 μl) of protein and reservoir solutions were mixed, and the resulting drops were equilibrated at 293 K against a 400μl reservoir solution made of 9% (wt/vol) polyethylene glycol (PEG) 8000, 100 mM MES pH 6.2, and 200mM Ca(OAc)2 for all mutants; and 6% PEG 8000, 100 mM MES buffer pH 6.2, 200mM Ca(OAc)2 and 50μM NaF for the wild-type. Microseeding was performed to obtain larger crystals. For the structures bound to EC25 (ref. 35), the crystals were soaked for 1 hour at 293 K in a solution containing 50mM EC25, 20% (wt/vol) PEG 8000, 25% PEG 600, 100 mM MES pH 6.5, and 200mM Ca(OAc)2 for all mutants. The wild-type TEM1 crystals were soaked using the same protocol in a solution containing 50mM EC25, 25% PEG 600, 100 mM MES pH 6.5, and 200mM Ca(OAc)2.
Data collection at 100 K
Crystals of ligand-free proteins were mounted on microloops (MiTeGen, USA) and transferred into a cryo-protectant solution containing 20% PEG 8000, 25% PEG 600, 100 mM MES pH 6.5, and 200mM Ca(OAc)2 for 1 min, and then flash-cooled in liquid nitrogen. For the bound wild-type and v13 structures, the soaking solution was used as cryo-protectant. X-ray diffraction data of mutant G238S free and in complex with EC25, R164S and R164S/G238S in complex with EC25 were collected on a Rigaku R-AXIS IV++ imaging plate area detector mounted on a Rigaku RU-H3R generator with CuKα radiation focused by Osmic confocal mirrors (in-house source). The X-ray diffraction data of the wild-type crystals in complex with EC25, of v13 mutants R164S and R164S/G238S (free forms) were collected at ID29 beam-line (European Synchrotron Facility (ESRF), Grenoble, France) using a PILATUS 6M detector. The diffraction data of R164S crystals were collected on ID14-1 beam-line (ESRF) using an ADSC Q210 detector.
Data collection at 293 K
Crystals were mounted on microloops (MiTeGen) and transferred into capillaries (Hampton, UK). Diffraction data were collected on a Rigaku R-AXIS IV++ imaging plate area detector mounted on our in-house source.
Data processing and refinement
Diffraction data were integrated and scaled using XDS package 67. Molecular replacement was performed using MOLREP68 using the structure of wild-type TEM-1 (PDB: 1ZG4 (ref. 38)) as the starting model. Manual model improvement was performed using Coot69 and the refinement was performed using REFMAC5 (ref. 68) and PHENIX70. Data collection and refinement statistics are provided in supplementary Table 1. Figures depicting structures were prepared with PyMOL.
Relative loops B-factors
The occupancies of all residues in all analyzed structures were set at a value of 1 (for residues with alternative conformations, the sums of occupancies were set at 1). The structures were re-refined using REFMAC5 (ref. 68). The relative loops B-factor values were obtained by normalizing the B-factor values of the loops main chain atoms by the average B-factor of main chain atoms of the whole structure as described in 16.
RMSD between free and bound structures
RMSD calculations were done using the Swiss-PDB viewer, using main chain atoms of the loops or the entire structures.
Evolutionary rates
Evolutionary rates were calculated using rate4site63 as described in 18. This method computes the relative evolutionary rate for individual protein positions across the entire phylogeny, via Bayesian Estimation. The rates are normalized with respect to the phylogeny such that the expected rate over all sites is 1. Hence, the rate of each site indicates how rapidly the site evolves relative to the mean. The sequence alignment was taken from ConSurf, and contained 150 sequences with 95%-35% identity to wild-type TEM-1.
Number of contacts and ASA values were calculated as in Ref. 16.
Supplementary Material
Acknowledgments
Financial support by the Israel Science foundation, and EMBO visiting fellowship and NIH (OD009180) to JSF, are gratefully acknowledged. DST is the Nella and Leon Benoziyo Professor. We thank Linda Shimon for invaluable assistance with the home source and room temperature X-ray data collections.
Footnotes
Accession codes
Protein Data Bank: Atomic coordinates and structure factors have been deposited with the accession codes PDB ID: 4OPY, 4OPQ, 4OP5, 4OQI, 4OQH, 4OQ0, 4OQG, 4OP8, 4OPR, 4OPZ.
References
- 1.Breen MS, Kemena C, Vlasov PK, Notredame C, Kondrashov FA. Epistasis as the primary factor in molecular evolution. Nature. 490:535–8. doi: 10.1038/nature11510. [DOI] [PubMed] [Google Scholar]
- 2.de Visser JA, Krug J. Empirical fitness landscapes and the predictability of evolution. Nat Rev Genet. 2014;15:480–90. doi: 10.1038/nrg3744. [DOI] [PubMed] [Google Scholar]
- 3.Weinreich DM, Delaney NF, Depristo MA, Hartl DL. Darwinian evolution can follow only very few mutational paths to fitter proteins. Science. 2006;312:111–4. doi: 10.1126/science.1123539. [DOI] [PubMed] [Google Scholar]
- 4.Bridgham JT, Ortlund EA, Thornton JW. An epistatic ratchet constrains the direction of glucocorticoid receptor evolution. Nature. 2009;461:515–9. doi: 10.1038/nature08249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borman AM, Paulous S, Clavel F. Resistance of human immunodeficiency virus type 1 to protease inhibitors: selection of resistance mutations in the presence and absence of the drug. J Gen Virol. 1996;77(Pt 3):419–26. doi: 10.1099/0022-1317-77-3-419. [DOI] [PubMed] [Google Scholar]
- 6.Maisnier-Patin S, Berg OG, Liljas L, Andersson DI. Compensatory adaptation to the deleterious effect of antibiotic resistance in Salmonella typhimurium. Mol Microbiol. 2002;46:355–66. doi: 10.1046/j.1365-2958.2002.03173.x. [DOI] [PubMed] [Google Scholar]
- 7.Salverda ML, Dellus E, Gorter FA, Debets AJ, van der Oost J, Hoekstra RF, Tawfik DS, de Visser JA. Initial mutations direct alternative pathways of protein evolution. PLoS Genet. 2011;7:e1001321. doi: 10.1371/journal.pgen.1001321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blount ZD, Borland CZ, Lenski RE. Historical contingency and the evolution of a key innovation in an experimental population of Escherichia coli. Proc Natl Acad Sci U S A. 2008;105:7899–906. doi: 10.1073/pnas.0803151105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poelwijk FJ, Kiviet DJ, Weinreich DM, Tans SJ. Empirical fitness landscapes reveal accessible evolutionary paths. Nature. 2007;445:383–6. doi: 10.1038/nature05451. [DOI] [PubMed] [Google Scholar]
- 10.de Visser JA, Cooper TF, Elena SF. The causes of epistasis. Proc Biol Sci. 2011;278:3617–24. doi: 10.1098/rspb.2011.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poelwijk FJ, Tanase-Nicola S, Kiviet DJ, Tans SJ. Reciprocal sign epistasis is a necessary condition for multi-peaked fitness landscapes. J Theor Biol. 2011;272:141–4. doi: 10.1016/j.jtbi.2010.12.015. [DOI] [PubMed] [Google Scholar]
- 12.Burch CL, Chao L. Evolvability of an RNA virus is determined by its mutational neighbourhood. Nature. 2000;406:625–8. doi: 10.1038/35020564. [DOI] [PubMed] [Google Scholar]
- 13.Rozen DE, Habets MG, Handel A, de Visser JA. Heterogeneous adaptive trajectories of small populations on complex fitness landscapes. PLoS One. 2008;3:e1715. doi: 10.1371/journal.pone.0001715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woods RJ, Barrick JE, Cooper TF, Shrestha U, Kauth MR, Lenski RE. Second-order selection for evolvability in a large Escherichia coli population. Science. 2011;331:1433–6. doi: 10.1126/science.1198914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Natarajan C, Inoguchi N, Weber RE, Fago A, Moriyama H, Storz JF. Epistasis among adaptive mutations in deer mouse hemoglobin. Science. 2013;340:1324–7. doi: 10.1126/science.1236862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dellus-Gur E, Toth-Petroczy A, Elias M, Tawfik DS. What makes a protein fold amenable to functional innovation? Fold polarity and stability trade-offs. J Mol Biol. 2013;425:2609–21. doi: 10.1016/j.jmb.2013.03.033. [DOI] [PubMed] [Google Scholar]
- 17.Toth-Petroczy A, Tawfik DS. Protein insertions and deletions enabled by neutral roaming in sequence space. Mol Biol Evol. 2013;30:761–71. doi: 10.1093/molbev/mst003. [DOI] [PubMed] [Google Scholar]
- 18.Toth-Petroczy A, Tawfik DS. Slow protein evolutionary rates are dictated by surface-core association. Proc Natl Acad Sci U S A. 2011;108:11151–6. doi: 10.1073/pnas.1015994108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lunzer M, Golding GB, Dean AM. Pervasive cryptic epistasis in molecular evolution. PLoS Genet. 2010;6:e1001162. doi: 10.1371/journal.pgen.1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bloom JD, Silberg JJ, Wilke CO, Drummond DA, Adami C, Arnold FH. Thermodynamic prediction of protein neutrality. Proc Natl Acad Sci U S A. 2005;102:606–11. doi: 10.1073/pnas.0406744102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. Protein stability promotes evolvability. Proc Natl Acad Sci U S A. 2006;103:5869–74. doi: 10.1073/pnas.0510098103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ortlund EA, Bridgham JT, Redinbo MR, Thornton JW. Crystal structure of an ancient protein: evolution by conformational epistasis. Science. 2007;317:1544–8. doi: 10.1126/science.1142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin G, Elena SF, Lenormand T. Distributions of epistasis in microbes fit predictions from a fitness landscape model. Nat Genet. 2007;39:555–60. doi: 10.1038/ng1998. [DOI] [PubMed] [Google Scholar]
- 24.Schenk MF, Szendro IG, Salverda ML, Krug J, de Visser JA. Patterns of Epistasis between beneficial mutations in an antibiotic resistance gene. Mol Biol Evol. 2013;30:1779–87. doi: 10.1093/molbev/mst096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salverda ML, De Visser JA, Barlow M. Natural evolution of TEM-1 beta-lactamase: experimental reconstruction and clinical relevance. FEMS Microbiol Rev. 2010;34:1015–36. doi: 10.1111/j.1574-6976.2010.00222.x. [DOI] [PubMed] [Google Scholar]
- 26.Guthrie VB, Allen J, Camps M, Karchin R. Network models of TEM beta-lactamase mutations coevolving under antibiotic selection show modular structure and anticipate evolutionary trajectories. PLoS Comput Biol. 2011;7:e1002184. doi: 10.1371/journal.pcbi.1002184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giakkoupi P, Tzelepi E, Tassios PT, Legakis NJ, Tzouvelekis LS. Detrimental effect of the combination of R164S with G238S in TEM-1 beta-lactamase on the extended-spectrum activity conferred by each single mutation. J Antimicrob Chemother. 2000;45:101–4. doi: 10.1093/jac/45.1.101. [DOI] [PubMed] [Google Scholar]
- 28.Petrosino JF, Palzkill T. Systematic mutagenesis of the active site omega loop of TEM-1 beta-lactamase. J Bacteriol. 1996;178:1821–8. doi: 10.1128/jb.178.7.1821-1828.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cantu C, 3rd, Palzkill T. The role of residue 238 of TEM-1 beta-lactamase in the hydrolysis of extended-spectrum antibiotics. J Biol Chem. 1998;273:26603–9. doi: 10.1074/jbc.273.41.26603. [DOI] [PubMed] [Google Scholar]
- 30.Petit A, Maveyraud L, Lenfant F, Samama JP, Labia R, Masson JM. Multiple substitutions at position 104 of beta-lactamase TEM-1: assessing the role of this residue in substrate specificity. Biochem J. 1995;305(Pt 1):33–40. doi: 10.1042/bj3050033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Minasov G, Shoichet BK. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J Mol Biol. 2002;320:85–95. doi: 10.1016/S0022-2836(02)00400-X. [DOI] [PubMed] [Google Scholar]
- 32.Stapleton PD, Shannon KP, French GL. Construction and characterization of mutants of the TEM-1 beta-lactamase containing amino acid substitutions associated with both extended-spectrum resistance and resistance to beta-lactamase inhibitors. Antimicrob Agents Chemother. 1999;43:1881–7. doi: 10.1128/aac.43.8.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mani R, St Onge RP, Hartman JLt, Giaever G, Roth FP. Defining genetic interaction. Proc Natl Acad Sci U S A. 2008;105:3461–6. doi: 10.1073/pnas.0712255105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fersht A. Enzyme Structure and Mechanism. W. H. Freeman; 1977. [Google Scholar]
- 35.Drawz SM, Taracila M, Caselli E, Prati F, Bonomo RA. Exploring sequence requirements for C(3)/C(4) carboxylate recognition in the Pseudomonas aeruginosa cephalosporinase: Insights into plasticity of the AmpC beta-lactamase. Protein Sci. 2011;20:941–58. doi: 10.1002/pro.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strynadka NC, Martin R, Jensen SE, Gold M, Jones JB. Structure-based design of a potent transition state analogue for TEM-1 beta-lactamase. Nat Struct Biol. 1996;3:688–95. doi: 10.1038/nsb0896-688. [DOI] [PubMed] [Google Scholar]
- 37.Fabre H, Eddine NH, Berge G. Degradation kinetics in aqueous solution of cefotaxime sodium, a third-generation cephalosporin. J Pharm Sci. 1984;73:611–8. doi: 10.1002/jps.2600730508. [DOI] [PubMed] [Google Scholar]
- 38.Stec B, Holtz KM, Wojciechowski CL, Kantrowitz ER. Structure of the wild-type TEM-1 beta-lactamase at 1.55 A and the mutant enzyme Ser70Ala at 2.1 A suggest the mode of noncovalent catalysis for the mutant enzyme. Acta Crystallogr D Biol Crystallogr. 2005;61:1072–9. doi: 10.1107/S0907444905014356. [DOI] [PubMed] [Google Scholar]
- 39.Brown NG, Shanker S, Prasad BV, Palzkill T. Structural and biochemical evidence that a TEM-1 beta-lactamase N170G active site mutant acts via substrate-assisted catalysis. J Biol Chem. 2009;284:33703–12. doi: 10.1074/jbc.M109.053819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levitt PS, Papp-Wallace KM, Taracila MA, Hujer AM, Winkler ML, Smith KM, Xu Y, Harris ME, Bonomo RA. Exploring the role of a conserved class A residue in the Omega-Loop of KPC-2 beta-lactamase: a mechanism for ceftazidime hydrolysis. J Biol Chem. 2012;287:31783–93. doi: 10.1074/jbc.M112.348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vakulenko SB, Taibi-Tronche P, Toth M, Massova I, Lerner SA, Mobashery S. Effects on substrate profile by mutational substitutions at positions 164 and 179 of the class A TEM(pUC19) beta-lactamase from Escherichia coli. J Biol Chem. 1999;274:23052–60. doi: 10.1074/jbc.274.33.23052. [DOI] [PubMed] [Google Scholar]
- 42.Orencia MC, Yoon JS, Ness JE, Stemmer WP, Stevens RC. Predicting the emergence of antibiotic resistance by directed evolution and structural analysis. Nat Struct Biol. 2001;8:238–42. doi: 10.1038/84981. [DOI] [PubMed] [Google Scholar]
- 43.Shimamura T, Ibuka A, Fushinobu S, Wakagi T, Ishiguro M, Ishii Y, Matsuzawa H. Acyl-intermediate structures of the extended-spectrum class A beta-lactamase, Toho-1, in complex with cefotaxime, cephalothin, and benzylpenicillin. J Biol Chem. 2002;277:46601–8. doi: 10.1074/jbc.M207884200. [DOI] [PubMed] [Google Scholar]
- 44.Strynadka NC, Adachi H, Jensen SE, Johns K, Sielecki A, Betzel C, Sutoh K, James MN. Molecular structure of the acyl-enzyme intermediate in beta-lactam hydrolysis at 1.7 A resolution. Nature. 1992;359:700–5. doi: 10.1038/359700a0. [DOI] [PubMed] [Google Scholar]
- 45.Warshel A. Electrostatic origin of the catalytic power of enzymes and the role of preorganized active sites. J Biol Chem. 1998;273:27035–8. doi: 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- 46.Parthasarathy S, Murthy MR. Analysis of temperature factor distribution in high-resolution protein structures. Protein Sci. 1997;6:2561–7. doi: 10.1002/pro.5560061208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yuan Z, Bailey TL, Teasdale RD. Prediction of protein B-factor profiles. Proteins. 2005;58:905–12. doi: 10.1002/prot.20375. [DOI] [PubMed] [Google Scholar]
- 48.Weinreich DM, Watson RA, Chao L. Perspective: Sign epistasis and genetic constraint on evolutionary trajectories. Evolution. 2005;59:1165–74. [PubMed] [Google Scholar]
- 49.Povolotskaya IS, Kondrashov FA. Sequence space and the ongoing expansion of the protein universe. Nature. 2010;465:922–6. doi: 10.1038/nature09105. [DOI] [PubMed] [Google Scholar]
- 50.Tokuriki N, Stricher F, Serrano L, Tawfik DS. How protein stability and new functions trade off. PLoS Comput Biol. 2008;4:e1000002. doi: 10.1371/journal.pcbi.1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phillips AH, Zhang Y, Cunningham CN, Zhou L, Forrest WF, Liu PS, Steffek M, Lee J, Tam C, Helgason E, Murray JM, Kirkpatrick DS, Fairbrother WJ, Corn JE. Conformational dynamics control ubiquitin-deubiquitinase interactions and influence in vivo signaling. Proc Natl Acad Sci U S A. 2013;110:11379–84. doi: 10.1073/pnas.1302407110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weng JK, Philippe RN, Noel JP. The rise of chemodiversity in plants. Science. 2012;336:1667–70. doi: 10.1126/science.1217411. [DOI] [PubMed] [Google Scholar]
- 53.Tokuriki N, Tawfik DS. Protein dynamism and evolvability. Science. 2009;324:203–7. doi: 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- 54.Hedstrom L, Perona JJ, Rutter WJ. Converting trypsin to chymotrypsin: residue 172 is a substrate specificity determinant. Biochemistry. 1994;33:8757–63. doi: 10.1021/bi00195a017. [DOI] [PubMed] [Google Scholar]
- 55.Sampson NS, Knowles JR. Segmental movement: definition of the structural requirements for loop closure in catalysis by triosephosphate isomerase. Biochemistry. 1992;31:8482–7. doi: 10.1021/bi00151a014. [DOI] [PubMed] [Google Scholar]
- 56.Butterwick JA, Palmer AG., 3rd An inserted Gly residue fine tunes dynamics between mesophilic and thermophilic ribonucleases H. Protein Sci. 2006;15:2697–707. doi: 10.1110/ps.062398606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marciano DC, Pennington JM, Wang X, Wang J, Chen Y, Thomas VL, Shoichet BK, Palzkill T. Genetic and structural characterization of an L201P global suppressor substitution in TEM-1 beta-lactamase. J Mol Biol. 2008;384:151–64. doi: 10.1016/j.jmb.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.James LC, Tawfik DS. Conformational diversity and protein evolution--a 60-year-old hypothesis revisited. Trends Biochem Sci. 2003;28:361–8. doi: 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- 59.Foote J, Milstein C. Conformational isomerism and the diversity of antibodies. Proc Natl Acad Sci U S A. 1994;91:10370–4. doi: 10.1073/pnas.91.22.10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Richards FM. The interpretation of protein structures: total volume, group volume distributions and packing density. J Mol Biol. 1974;82:1–14. doi: 10.1016/0022-2836(74)90570-1. [DOI] [PubMed] [Google Scholar]
- 61.Bode C, Kovacs IA, Szalay MS, Palotai R, Korcsmaros T, Csermely P. Network analysis of protein dynamics. FEBS Lett. 2007;581:2776–82. doi: 10.1016/j.febslet.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 62.Greene LH, Higman VA. Uncovering network systems within protein structures. J Mol Biol. 2003;334:781–91. doi: 10.1016/j.jmb.2003.08.061. [DOI] [PubMed] [Google Scholar]
- 63.Mayrose I, Graur D, Ben-Tal N, Pupko T. Comparison of site-specific rate-inference methods for protein sequences: empirical Bayesian methods are superior. Mol Biol Evol. 2004;21:1781–91. doi: 10.1093/molbev/msh194. [DOI] [PubMed] [Google Scholar]
- 64.Payne JL, Wagner A. The robustness and evolvability of transcription factor binding sites. Science. 2014;343:875–7. doi: 10.1126/science.1249046. [DOI] [PubMed] [Google Scholar]
- 65.Gobeil SM, Clouthier CM, Park J, Gagne D, Berghuis AM, Doucet N, Pelletier JN. Maintenance of Native-like Protein Dynamics May Not Be Required for Engineering Functional Proteins. Chem Biol. 2014;21:1330–40. doi: 10.1016/j.chembiol.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 66.Tokuriki N, Jackson CJ. Enzyme dynamics and engineering: one step at a time. Chem Biol. 2014;21:1259–60. doi: 10.1016/j.chembiol.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 67.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. Journal of Applied Crystallography. 1993;26:795–800. [Google Scholar]
- 68.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 69.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 70.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Neves MA, Yeager M, Abagyan R. Unusual arginine formations in protein function and assembly: rings, strings, and stacks. J Phys Chem B. 2012;116:7006–13. doi: 10.1021/jp3009699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.