

Abstract
In the past decade our understanding of idiosyncratic drug induced liver injury (IDILI) and the contribution of genetic susceptibility and the adaptive immune system to the pathogenesis of this disease process has grown tremendously. One of the characteristics of IDILI is that it occurs rarely and only in a subset of individuals with a presumed susceptibility to the drug. Despite a clear association between single nucleotide polymorphisms in human leukocyte antigen (HLA) genes and certain drugs that cause IDILI, not all individuals with susceptible HLA genotypes develop clinically significant liver injury when exposed to drugs. The adaptation hypothesis has been put forth as an explanation for why only a small percentage of susceptible individuals develop overt IDILI and severe injury, while the majority with susceptible genotypes develop only mild abnormalities that resolve spontaneously upon continuation of the drug. This spontaneous resolution is referred to as clinical adaptation. Failure to adapt or defective adaptation leads to clinically significant liver injury. In this review we explore the immuno-tolerant microenvironment of the liver and the mechanisms of clinical adaptation in IDILI with a focus on the role of immune-tolerance and cellular adaptive responses.
Keywords: Hepatotoxicity, HLA, Immune-tolerance, T cells, DILI
Introduction
Understanding the pathogenesis of drug-induced liver injury (DILI) has greatly advanced in the recent years largely due to the contribution of genetic studies and improved animal models. DILI can occur as a result of dose related direct drug toxicity, as seen with acetaminophen, mitochondrial poisons, or certain chemotherapy drugs. On the other hand, the majority of DILI due to many drugs occurs in only a small proportion of patients and is therefore considered idiosyncratic due to individual susceptibility. Although not dose related in a strict sense, these reactions largely occur in drugs given at daily doses of greater than 50–100 mg suggesting a threshold exposure is required for either cumulative damage or eliciting an immune response. Much attention has been directed at understanding idiosyncratic DILI (IDILI) and the role of an immune pathogenesis. One of the characteristic features of IDILI is that mild injury (e.g. ALT< 3 upper limit of normal) occurs far more frequently than severe and that in most instances mild injury subsides despite continuation of the drug. We refer to this phenomenon as clinical adaptation, not to be confused with various biochemical adaptive responses (e.g., ER and mitochondrial unfolded protein responses, cellular stress responses such as activation of the mitogen activated protein kinase, MAPK, pathway). Consequently, the hypothesis has emerged that severe IDILI resulting in jaundice and liver failure, may be due to defective clinical adaptation or failure to dampen the initiating mechanisms of injury due to diminished adaptive responses.
The liver is an immune-privileged organ
The liver has evolved as an immune-privileged organ with striking capacity for immune-tolerance (1). Ingested antigens are constantly introduced to the liver where they are metabolized and either enter the systemic circulation or lymphatics or get excreted via the biliary system (2). Given the perpetual need for clearance of unwanted and harmful compounds, immune-tolerance is a necessary adaptation to protect hepatocytes from damage resulting from a constant inflammatory state in the liver. Immune-tolerance induced outside of primary lymph organs such as the Thymus is referred to as peripheral immune-tolerance. The liver manages to control the level of inflammatory activity by the induction of peripheral immune-tolerance towards incoming antigens (1). The role of the liver in inducing peripheral immune-tolerance to antigens has been well studied. For example, portovenous application of an immunogenic antigen, such as 1-chloro 2,4-dinitrobenzene, to adult dogs before subcutaneous injection suppresses the expected allergic skin reaction and the formation of specific circulating antibodies(3). Furthermore, diversion of portal flow from the liver abolishes this protective effect (3–5). The tolerogenic capacity of the liver has been a focus of scientific interest since early experimental transplantation studies in the 1960s demonstrated allogeneic liver grafts can be established and maintained in animal models without immunosuppression (6). Human Leukocyte Antigens (HLA) matching is not necessary for successful liver allograft transplantation and liver transplantation is the only solid organ transplant in which complete weaning of immunosuppression can be achieved in up to 20% of patients, a phenomenon known as spontaneous operational tolerance (7). It is important to point out that despite this high tolerogenic capacity to foreign antigens the liver also fights incoming pathogens via induction of an effective immune response when necessary.
The mechanisms of immune-tolerance can be broken down to the following key events: control of antigen presentation, clonal deletion (apoptosis of antigen-specific T cells) and immune deviation (switching from Th2 to Th1 predominance) (1). Liver Sinusoidal Endothelial Cells (LSECs) are scavenger cells and take up antigens in the sinusoids for processing and antigen-presentation and they can induce proliferation and cytokine expression in CD4+ T cells resulting in their activation (8, 9). Kupffer cells (KC) are resident liver macrophages that are strategically located in the periportal sinusoids of the liver to phagocytose and eliminate unwanted antigens entering the liver. KC also play a key role in the regulation of the immune response, or lack there of, to maintain homeostasis in response to the liver’s constant exposure to gut-derived antigens. The tolerogenic liver microenvironment is largely dependent on the autocrine and paracrine effects of cytokines secreted by the KC as well as the constant effect of low levels of LPS stimulation on immune cells and antigen presenting cells, in particular the KCs and LSECs (10–12). Cytokines such as IL-10, TGF-β, TNF-α and prostaglandins expressed by either KC and LSECs (constitutively or in response to LPS) result in down regulation of leukocyte adhesion to LSECs, expansion of regulatory T cells (T-regs), and abrogation of T cell activation that contributes to the immune-tolerant environment in the liver (10–16). T-regs are key regulatory subpopulation of CD4+ T cells that can curb both innate and adaptive immune responses, contributing to immune-tolerance in the liver. The impairment or deficiency of T-regs has been implicated in autoimmune hepatitis (17, 18). Furthermore, KCs have been shown to stimulate the expansion of IL-10 producing T-regs to induce immune-tolerance in the liver (18, 19). Hepatic stellate cells have a capacity to serve as antigen presenting cells, expand T-regs, and promote T cell apoptosis (via B7-H1, PDL-1) or inhibit cytotoxic CD8+ T cells, and therefore may also play a role in the tolerogenic liver microenvironment (20–22).
Since diversion of portal flow results in the loss of immune-tolerance (3–5) and intestinal LPS accompanies ingested antigens in the portal vein, LPS may play a role in modulating immune-tolerance. Supporting this hypothesis are observations that mice unresponsive to LPS (C3H/HeJ), lack tolerance to dietary antigens (23). Another proposed mechanism of peripheral immune-tolerance induction by the liver is the phenomenon of clonal deletion or induction of antigen-specific T cell apoptosis in the liver (24). While the elimination of T cell populations seems an attractive explanation, it does not seem to be the whole story since tolerance can be transferred from one animal to another. This can be achieved by adoptive transfer of γδ T cells, which suggests that the mechanism of this phenomenon is more complex and that tolerance is mediated, at least in part, by immune deviation as opposed to mass T cell elimination (25, 26).
Immune mediated IDILI
Unlike classic direct-acting hepatotoxins (e.g. acetaminophen), IDILI is largely dose independent above a threshold dose and is unpredictable with a variable and somewhat long latency. The exact pathophysiology of IDILI is likely multi-factorial involving drug/pharmacological factors (lipophilicity, dose, etc), environmental factors, and host factors (genetics, immunity, possibly microbiome) (27, 28). IDILI occurs only in a small subset of individuals exposed to a drug reflecting the fact that individual host factors contribute to the injurious outcome. In the past decade polymorphisms in Human Leukocyte Antigens (HLA) molecules have been linked to many drugs that cause IDILI raising the interesting possibility that the immune system is involved in the pathophysiologic process leading to liver injury (29). Interestingly, DILI due to certain drugs such as halothane, dihydralazine and anticonvulsants can present with classic allergic features such as rash and eosinophilia and in many cases the liver injury recurs with re-challenge (30). Other notable drugs that can exhibit increased eosinophil counts and an immuno-allergic phenotype are: trimethoprim/sulfamethoxazole, cefazolin and ciprofloxacin (31). Severe allergic skin reactions such as toxic epidermal necrosis (TEN) and Stevens Johnson syndrome (SJS) have also been described with IDILI and carry a poor prognosis (31). Furthermore, drugs or drug-protein adducts from IDILI compounds can activate peripheral blood lymphocytes (lymphocyte stimulation test), supporting the notion that host immune factors contribute to IDILI (32, 33). DILI can mimic, induce or unmask autoimmune hepatitis (AIH). Classical examples include nitrofurantoin and minocycline, two drugs that are frequently associated with the induction of antinuclear antibodies and the histological appearance of AIH (31). Despite these examples of an obvious role for drug allergy, most examples of IDILI occur in the absence of systemic immuno-allergic features; nevertheless, many are strongly HLA-linked suggesting a liver specific immune response. Additionally, since not all drug sensitivities are restricted to one or a few HLA haplotypes, more promiscuous immune reactions are very likely to occur and one cannot discount an immune mechanism (because no single HLA allele is associated with risk).
Phenomenon of Adaptation
Drugs that cause IDILI often induce mild abnormalities in liver tests among the general population which resolve with continued exposure, a phenomenon known as adaptation. A classic example of adaptation was described by Mitchell et al. in a seminal paper in 1975 where patients in a psychiatric hospital with a tuberculosis outbreak were treated with isoniazid (INH) and followed prospectively with liver enzymes every four weeks (34). Blood was collected prior to initiation of treatment with INH up to one week after termination of the study but results were not analyzed until many months after they were collected. INH resulted in elevated liver enzymes in 38% of patients, and interestingly this subsided in the majority of patients despite continued treatment with INH (Fig.1A), including several who developed concomitant hyperbilirubinemia (Hy’s Law, Fig 1B) (34, 35). Although INH toxicity can result in severe liver injury and even liver failure (36), no cases of liver failure were reported in this study. In light of current knowledge about potential immunologic mechanisms of INH hepatotoxicity, adaptation becomes a likely explanation for these findings (35, 36). Clinical adaptation, in this context may be related to the development and induction of a state of immune-tolerance against the drug or its metabolite as a hapten/immunogen. Since most IDILI is likely immune mediated, the presentation of overt liver injury in certain individuals with predisposition might be thought of as “defective adaptation”(37).
Figure 1.
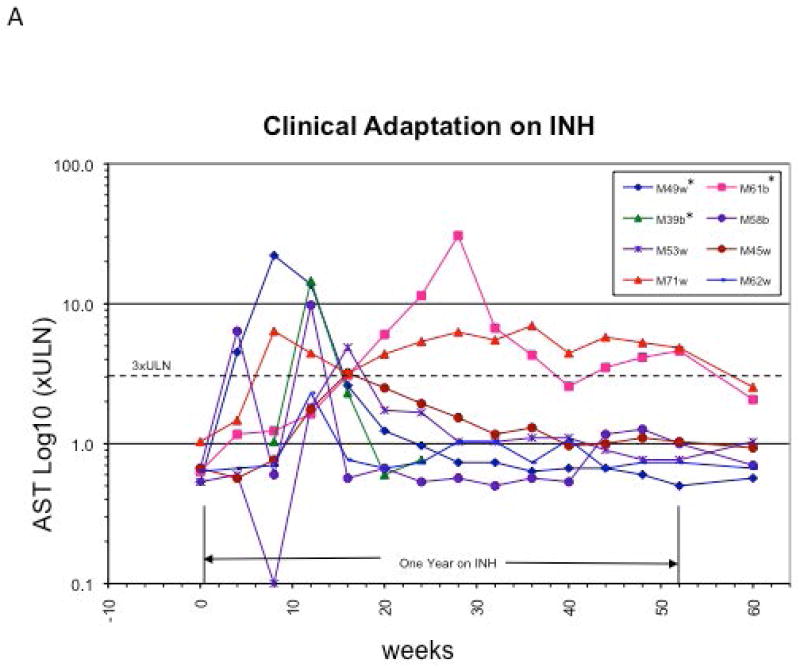
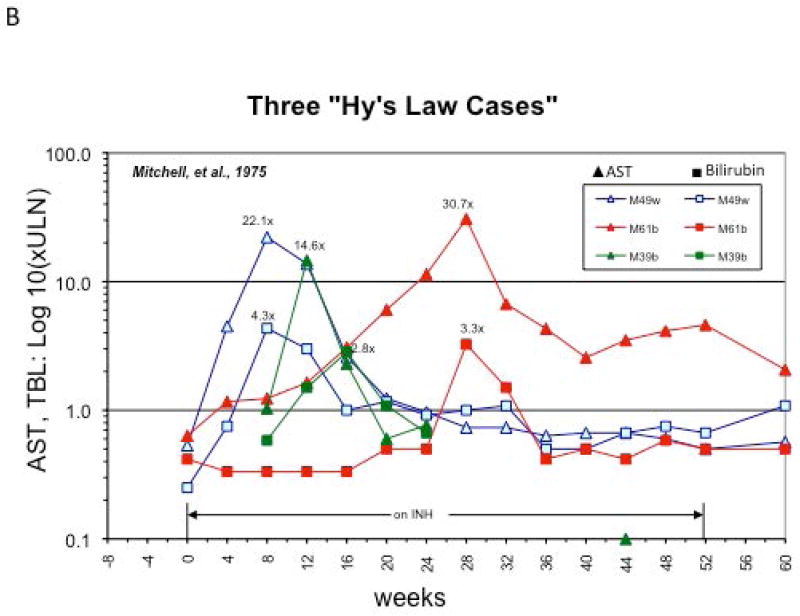
Clinical Adaptation to INH. Aspartate Aminotransferase (AST) levels of eight patients described in the original paper by Mitchell et al (Ref. 34) as plotted by Dr. John Senior (FDA). A) Data from cases with AST > 60 and bilirubin >1.2 mg/dl are included. Three patients with elevated bilirubin >2 times upper limit of normal (Hy’s law cases) are noted with an *. Notice the early rise in AST in the first few weeks on the drug followed by normalization of serum AST despite continued INH therapy. B) AST and Bilirubin trends of the three Hy’s Law cases. AST: Aspartate amino transferase.
HLA associations clearly point to the involvement of the adaptive immune system in IDILI. However only a small percentage of patients with known genetic polymorphisms or known drug risk alleles develop toxicity, underscoring the complexity and multi-factorial nature of IDILI (38). For example, lumiracoxib, a cyclooxygenase-2 inhibitor and analgesic, was withdrawn from the market due to safety concerns and reports of IDILI (39). A pharmacogenetic retrospective case control study was conducted to look for HLA associations between 41 patients with lumiracoxib toxicity (ALT or AST >5 times the upper of limit of normal) and 176 matched lumiracoxib treated patients without liver injury. Several single nucleotide polymorphisms (SNP) in the MHC II region of chromosome 6 (6p21.32) were associated with IDILI from the drug. Four alleles were highly associated with toxicity, and interestingly these alleles lie on a common haplotype (HLA-DRB1*1501- HLA-DQB1*0602-HLA-DRB5*0101-HLA-DQA1*0102) that has been associated with multiple sclerosis (39, 40). It has been hypothesized that a lumiracoxib metabolite acts as a “hapten” causing a T cell immunogenic response by reacting with certain HLA complexes (39). Interestingly the positive predictive value of the SNP alleles for lumiracoxib DILI was low due to the high frequency of the allele. These results demonstrate that being positive for a known associated SNP or risk allele is not sufficient to induce hepatotoxicity and in addition to genetic susceptibility other factor(s), such as defective adaptation, must be present for ensuing toxicity. However, in the absence of HLA risk alleles, IDILI usually does not occur underscoring that HLA restricted susceptibility is critical.
The unpredictable and experimentally irreproducible nature of idiosyncratic hepatotoxicity has made it particularly difficult to study in animal models. Recently two independent groups have used the concept of defective adaptation to develop mouse models for IDILI (41, 42). Metushi et al. examined toxicity from amodiaquine in female mice defective in two genes implicated in immune-tolerance: Casitas B-lineage Lymphoma (Cbl-b1), an E3 ubiquitin ligase; and programmed cell death 1 (PD-1), a negative regulator of T cell activation (41). Mice knockout for either Cbl-b1 or PD-1 exhibited liver injury with amodiaquine greater than that of wild type mice. However, the injury resolved upon continued treatment with amodiaquine. In other words the mice “adapted”. The authors speculated that since there are multiple pathways involved in immune-tolerance, interference with additional pathways is necessary to circumvent adaptation and induce persistent and more severe toxicity. They then blocked cytotoxic T-lymphocyte-associate protein 4 (CTLA4), a key inducer of immune-tolerance, by using an anti-CTLA4 antibody. Indeed, treatment of PD-1−/− mice with CTLA4 antibody prior to amodiaquine resulted in more severe injury both histologically and with greater ALT elevation, and this did not resolve with continuous exposure to the drug (41). The authors observed an increase in CD8+ T cells and macrophages and an increased response in regulatory T cells presumably in an attempt to dampen and control the injury. Subsequently they depleted CD8+ T cells in this model and showed prevention of injury. Thus, the immune injury was mediated by cytotoxic CD8+ T lymphocytes (43). This work demonstrates the many redundancies built into ensuring an immune-tolerant state in the liver, as the combination of PD-1 knockout along with inhibition of CTLA4 was necessary to overcome or break the immune-tolerance. This may also explain the relative rarity of clinically overt IDILI, as multiple drug, host, and immune factors need to line up for the toxicity event to occur.
Chakraborty el al, examined halothane induced allergic hepatitis in mice with compromised immune-tolerance due to depletion of myeloid derived suppressor cells (MDSCs). Halothane re-exposure in humans often causes an allergic drug induced liver injury with rash and eosinophilia. In this model, female BALB/cJ mice received two doses of halothane fourteen days apart to sensitize them to the drug. In normal mice, an acute direct toxicity occurred after each dose, which rapidly subsided. The authors had noted that intra-peritoneal halothane induced infiltration of leukocytes in the liver, a significant proportion of which were CD11b and Gr1high positive (42). CD11b+Gr1high MDSCs isolated from halothane treated mice had a significant immune-suppressive effect on CD4+ and CD8+ T cells in a dose dependent manner. To examine the functional relevance of these cells, mice were given antibody to Gr1 which resulted in depletion of the CD11b+Gr1high immune-suppressor cells and accompanied by increased liver injury 9 days post halothane re-challenge (after the second dose). The hapten-drug metabolite, which is the immune target, persisted after the initial bouts of hepatitis resolved, allowing a rebound in injury when the tolerance was abrogated. Anti-Gr1 treated mice showed delayed severe inflammation, necrosis and increased eosinophil infiltration and T cell response to halothane and its metabolite (42). In order to demonstrate which T cell population contributes to the injury phenotype, mice were treated with anti-CD4 and anti-CD8 antibodies prior to halothane rechallenge, interestingly only anti-CD4 antibody abrogated the response to halothane. In contrast to the amodiaquine model, in this instance injury was mediated by antibody dependent cytotoxicity.
These recent studies demonstrate the importance of immune-tolerance in protecting against IDILI. While different tolerance pathways were targeted in each work, both animal models achieved an IDILI phenotype by manipulating the liver’s capacity for immune-tolerance. This also demonstrates that despite the manipulation of a common concept to replicate an IDILI model, each drug had a unique signature of injury. For example eosinophilia is a feature of halothane that is not seen in amodiaquine DILI. Interestingly CD4+ T cell depletion abrogated the toxic phenotype in halothane DILI while the same was true of depletion of CD8+ T cells in the amodiaquine model (44). These differences may point to the different underlying mechanisms of injury and signaling pathways involved in each model. In addition, these two distinct models demonstrate that the regulation of immune-tolerance is multi-factorial and involves multiple pathways. Furthermore, tolerance is dynamic in both modulating the severity of the initial injury in the long latency amodiaquine model and the duration of the injury in both models.
The exact underlying mechanism of defective clinical adaptation in IDILI remains unknown; however, many interesting associations have been made. For example, 9 out of the top 10 agents causing IDILI in the DILIN database are antimicrobials and as previously discussed LPS plays an important role in inducing immune-tolerance (31). Given the strong evidence for the tolerogenic properties of LPS on antigen presentation and cytokine secretion, it is intriguing to hypothesize that the antibiotic effects on gut microflora and changes in LPS exposure may contribute to defective adaptation. The changes in gut microbiota may also have effects on the phenotypes and functions of regulatory immune cells in the liver immune system e.g. KC polarization (pro- or anti-inflammatory), T-reg expansion/frequency, hepatic stellate cells, etc. Furthermore, it is important to point out that the microbiome can influence drug toxicity and the liver not only by the tolerogenic properties of LPS, but by affecting drug metabolism (45).
IL-10 is a key immunomodulatory cytokine which functions to inhibit antigen specific T cell activation. IL-10 polymorphisms have been associated with susceptibility to diclofenac DILI (46). In this small cohort of patients treated with diclofenac, all patients with DILI had positive serum adducts for diclofenac and a strong association was noted in polymorphisms resulting in low IL-10 (Odds Ratio of 2.8) suggesting an immune mediated role for toxicity and supporting the hypothesis that loss of immune-tolerance may be an contributing factor to the disease process (46). Another intriguing association with regards to clinical adaptation is the observation by the Spanish DILI network that patients with worse outcomes carried IL-10 polymorphisms resulting in lower IL-10 levels (47). Since IL-10 is a potent anti-inflammatory cytokine, it functions to induce immune-tolerance by acting directly on macrophages reducing IL-8, IFN-γ, IL-12 levels as well as inhibiting T cell activation (48, 49). IL-10 is also a major effector cytokine by which T-regs exert their inhibitory effects in both the innate and adaptive immune responses. Thus lower, ineffective IL-10 may be a surrogate for a state of defective clinical adaptation in certain patients. It is important to point out that these are only associations and intriguing as hypotheses but no evidence of causality exists.
Clinical Implications
The clinical implications of IDILI in the context of defective immune-tolerance are becoming increasingly important. One intriguing clinical context is cancer immunotherapy targets and their potential adverse effects. Recently much attention has been directed towards “immune check point therapy” as an important tool to combat cancer, especially those neoplasms well versed in immune evasion such as melanoma (50). There are currently three FDA approved antibodies that aim to inhibit T cell immune-tolerant states in order to increase effective anti-tumor T cell responses. These drugs include ipilimumab (anti-CTLA4), as well as, pembrolizumab and nivolumab (anti-PD-1). Ipilimumab, an anti-CTLA4 antibody has been FDA approved since 2011 for metastatic melanoma (51). Hepatitis with features of autoimmunity and responsiveness to steroids has been reported at a rate of 3–9% with ipilimumab and one case of fatal fulminant liver failure (steroid refractory) has been described (51–53). Histologic features of AIH such as hepatocyte rosettes, interface hepatitis, confluent necrosis, increased eosinophils, granulomas, lymphoplasmacytic infiltrate have been reported in cases of ipilimumab DILI (54, 55). Similar side effects have been reported with the PD-1 inhibitors (56). Though difficult to ascertain, we can hypothesize that ipilimumab induced autoimmune hepatitis may in fact be an IDILI manifestation. Ipilimumab can block the liver’s capacity for immune regulation and adaptation, thus rendering it susceptible to a whole host of antigens and haptens that can elicit an immune response. In this scenario it would be extremely difficult to tease out the inciting antigen, especially in cases of poly-pharmacy or auto-immunity.
Immune-independent Factors
Despite immune mechanisms driving DILI, it is likely that other intrinsic or environmental factors modulate adaptation. As listed in Table 1, a number of biochemical and organelle stressors can elicit adaptive responses that can dampen or turn off injury, perpetuating stress. Alternatively, if these responses are maladaptive injury may worsen and ultimately even lead to liver failure. Independent of the immune system most drugs are converted to reactive metabolites and if the reactive metabolite binds to its CypP450, it can inactivate it (suicide substrate) suppressing further production of a toxic metabolite. Oxidative stress due to effects of drugs on mitochondria can elicit an adaptive anti-oxidant defense, which enhances GSH synthesis and expression of phase 2 detoxification enzymes; thus, the ability to mitigate oxidative stress contributes to clinical adaptation in immune-independent toxicity.
Table 1.
| Cellular adaptive responses |
|---|
| Dampening reactive metabolite production or exposure |
| Enhanced detoxification/antioxidant defense (e.g. Nrf2) |
| Mitochondrial adaptation: mitophagy, biogenesis, fission/fusion |
| Upregulation of chaperones: ER-UPR and mitochondrial UPR |
| Anti-inflammatory balance (innate immunity) |
| Regeneration |
Alternatively, intracellular adaptive responses when not successful in restoring homeostasis might sensitize “stressed hepatocytes” to greater immune-mediated toxicity and T cell or cytokine induced apoptosis. Similar hypothesis can be generated for adaptation to ER stress, commonly seen as response to covalent binding, as well as adaptive responses to mitochondrial stress. In both instances the cell responds to organelle specific stress through unfolded protein responses (ER or mitochondria), which up regulate the transcription of chaperones and improve protein folding. In addition damaged mitochondria elicit other adaptive responses such as mitophagy to remove them or biogenesis to replace them. Finally, a key factor in severity and clinical adaptation might be the ability to regenerate or an effect of the culprit drug/metabolite on the liver’s regenerative responses.
In summary, idiosyncratic drug induced liver injury is the result of a complex interplay between potentially immunogenic drugs or metabolites and the host’s immune response. In most instances, the liver’s natural propensity to maintain a state of immune-tolerance results in adaptation to foreign antigens, even in individuals with HLA associations favoring an immunogenic response (Figure 2). Recently, animal models have elegantly demonstrated that blocking adaptive compensatory mechanisms through any number of pathways can result in IDILI when exposed to certain drugs or immunogens. Defective adaptation in humans has not been extensively studied but some clinical studies have implicated associations between IDILI and levels of immune tolerogenic cytokines such as IL-10 (46, 47). Interestingly, the association of LPS with immune-tolerance in the liver raises the possibility of the contribution of the microbiome to protection against immunogenicity and the fact that many top IDILI drugs are antibiotics makes this hypothesis even more plausible. Whatever the trigger for immune deregulation, it is clear that T cell response is key to the development of toxicity and prevention or suppression of T cell activation may be the pivotal event. Drugs that induce immune-tolerance such as the CTLA-4 agonists, abatacept, are indicated for rheumatoid arthritis and are currently in clinical trials for transplant immunosupression. Interestingly, abatacept itself has been implicated in case reports causing DILI, underscoring the delicate balance between pro and anti-immune pathways (57, 58). The recent spotlight on the role of immune-tolerance, as a key pathogenic contributor to the development of IDILI brings up intriguing implications regarding diagnosing and predicting IDILI and future areas of investigation. One could speculate that approaches comparing gene expression profiles in study subjects that adapt vs those who develop overt injury (non-adaptors) may help uncover new and relevant pathways. Closely examining for mutations in genes relevant to immune-tolerance and monitoring the study subjects’ gene expression profiles before, during or after recovery may aid in determining which pathways are actively participating in the adaptors vs non-adaptors. Aside from potential acquired and genetic variation of regulatory factors in immune-tolerance, there are many potential physiological responses, which may dampen or worsen the severity of liver injury at the outset, or lead to persistence of injury or subsequent adaptation. Among these are responses to oxidative stress, ER, mitochondrial stress as well as the liver’s regenerative capacity. All of these adaptive stress responses are potentially influenced by: genetics, epigenetics, concomitant diseases and medications, infections, inflammation as well as diet and the microbiome. Clearly, a very complex picture emerges with a rich landscape of factors, which can influence the various outcomes in IDILI, be it no injury, mild injury, very severe injury, or clinical adaptation and its failure. This complex interplay explains the rarity of clinically significant and severe IDILI.
Figure 2.

Role of Adaptation in IDILI. Certain drugs produce reactive metabolites, which can act as haptens or antigenic peptides. When subjects with susceptible HLA haplotypes are exposed to these antigenic peptides some immediately become tolerant and others develop mild injury, which can resolve with continued exposure to the drug, a phenomenon known as adaptation. In rare instances due to defective adaptation, mild liver injury can progress to severe IDILI and acute liver failure. SNP: Single Nucleotide Polymorphism, HLA: Human Leukocyte Antigen, IDILI: Idiosyncratic Drug Induced Liver Injury, ALF: Acute Liver Failure.
Key Points.
IDILI can result in mild abnormalities in liver tests in individuals with HLA associations favoring an immunogenic response.
In most people, liver test abnormalities are transient and resolve with continued exposure, a phenomenon known as clinical adaptation.
In a minority of patients, “defective adaptation” leads to persistent liver injury and ultimately results in clinically significant hepatotoxicity.
Various host genetic and immune factors can contribute to effective or ineffective adaptation.
Acknowledgments
Support: This work was supported by NIH grant RO1DK067215 (NK), P30DK48522 (NK), and 5U01AA021857 (ZXL)
The authors thank Dr. John Senior for his guidance and contribution of Figure 1.
List of Abbreviations
- DILI
Drug Induced Liver Injury
- HLA
Human Leukocyte Antigen
- LSECs
Liver Sinusoidal Endothelial Cells
- KC
Kupffer Cells
- LPS
lipopolysaccharide
- T-regs
Regulatory T cells
- TEN
Toxic epidermal necrosis
- SJS
Stevens Johnson syndrome
- AIH
autoimmune hepatitis
- INH
isoniazid
- SNP
single nucleotide polymorphisms
- Cbl-b1
Casitas B-lineage Lymphoma
- PD-1
programmed cell death 1
- CTLA4
cytotoxic T-lymphocyte-associate protein 4
- MDSCs
myeloid derived suppressor cells
- CypP450
Cytochrome P450
Footnotes
Conflict of Interest: No direct conflicts with this review. However, NK consults for the following pharmaceutical companies: Takeda, GSK, Pfizer, Daiichi-Sankyo, Johnson& Johnson, Geron and Ono.
References
- 1.KNOLLE PA, GERKEN G. Local control of the immune response in the liver. Immunol Rev. 2000;174:21–34. doi: 10.1034/j.1600-0528.2002.017408.x. [DOI] [PubMed] [Google Scholar]
- 2.WILLIAMS GM, IATROPOULOS MJ. Alteration of liver cell function and proliferation: differentiation between adaptation and toxicity. Toxicol Pathol. 2002;30(1):41–53. doi: 10.1080/01926230252824699. [DOI] [PubMed] [Google Scholar]
- 3.CANTOR HM, DUMONT AE. Hepatic suppression of sensitization to antigen absorbed into the portal system. Nature. 1967;215(5102):744–5. doi: 10.1038/215744a0. [DOI] [PubMed] [Google Scholar]
- 4.CALLERY MP, KAMEI T, FLYE MW. The effect of portacaval shunt on delayed-hypersensitivity responses following antigen feeding. J Surg Res. 1989;46(4):391–4. doi: 10.1016/0022-4804(89)90208-4. [DOI] [PubMed] [Google Scholar]
- 5.YANG R, LIU Q, GROSFELD JL, PESCOVITZ MD. Intestinal venous drainage through the liver is a prerequisite for oral tolerance induction. Journal of pediatric surgery. 1994;29(8):1145–8. doi: 10.1016/0022-3468(94)90297-6. [DOI] [PubMed] [Google Scholar]
- 6.CALNE RY, SELLS RA, PENA JR, DAVIS DR, MILLARD PR, HERBERTSON BM, et al. Induction of immunological tolerance by porcine liver allografts. Nature. 1969;223(5205):472–6. doi: 10.1038/223472a0. [DOI] [PubMed] [Google Scholar]
- 7.ADAMS DH, SANCHEZ-FUEYO A, SAMUEL D. From immunosuppression to tolerance. J Hepatol. 2015;62(1 Suppl):S170–85. doi: 10.1016/j.jhep.2015.02.042. [DOI] [PubMed] [Google Scholar]
- 8.LOHSE AW, KNOLLE PA, BILO K, UHRIG A, WALDMANN C, IBE M, et al. Antigen-presenting function and B7 expression of murine sinusoidal endothelial cells and Kupffer cells. Gastroenterology. 1996;110(4):1175–81. doi: 10.1053/gast.1996.v110.pm8613007. [DOI] [PubMed] [Google Scholar]
- 9.SORENSEN KK, MCCOURT P, BERG T, CROSSLEY C, LE COUTEUR D, WAKE K, et al. The scavenger endothelial cell: a new player in homeostasis and immunity. Am J Physiol Regul Integr Comp Physiol. 2012;303(12):R1217–30. doi: 10.1152/ajpregu.00686.2011. [DOI] [PubMed] [Google Scholar]
- 10.KNOLLE PA, UHRIG A, PROTZER U, TRIPPLER M, DUCHMANN R, MEYER ZUM BUSCHENFELDE KH, et al. Interleukin-10 expression is autoregulated at the transcriptional level in human and murine Kupffer cells. Hepatology. 1998;27(1):93–9. doi: 10.1002/hep.510270116. [DOI] [PubMed] [Google Scholar]
- 11.KNOLLE PA, SCHMITT E, JIN S, GERMANN T, DUCHMANN R, HEGENBARTH S, et al. Induction of cytokine production in naive CD4(+) T cells by antigen-presenting murine liver sinusoidal endothelial cells but failure to induce differentiation toward Th1 cells. Gastroenterology. 1999;116(6):1428–40. doi: 10.1016/s0016-5085(99)70508-1. [DOI] [PubMed] [Google Scholar]
- 12.BISSELL DM, WANG SS, JARNAGIN WR, ROLL FJ. Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest. 1995;96(1):447–55. doi: 10.1172/JCI118055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.KNOLLE P, SCHLAAK J, UHRIG A, KEMPF P, MEYER ZUM BUSCHENFELDE KH, GERKEN G. Human Kupffer cells secrete IL-10 in response to lipopolysaccharide (LPS) challenge. J Hepatol. 1995;22(2):226–9. doi: 10.1016/0168-8278(95)80433-1. [DOI] [PubMed] [Google Scholar]
- 14.DIETER P, SCHULZE-SPECKING A, KARCK U, DECKER K. Prostaglandin release but not superoxide production by rat Kupffer cells stimulated in vitro depends on Na+/H+ exchange. European journal of biochemistry / FEBS. 1987;170(1–2):201–6. doi: 10.1111/j.1432-1033.1987.tb13687.x. [DOI] [PubMed] [Google Scholar]
- 15.RIEDER H, RAMADORI G, ALLMANN KH, MEYER ZUM BUSCHENFELDE KH. Prostanoid release of cultured liver sinusoidal endothelial cells in response to endotoxin and tumor necrosis factor. Comparison with umbilical vein endothelial cells. J Hepatol. 1990;11(3):359–66. doi: 10.1016/0168-8278(90)90222-d. [DOI] [PubMed] [Google Scholar]
- 16.KNOLLE PA, GERMANN T, TREICHEL U, UHRIG A, SCHMITT E, HEGENBARTH S, et al. Endotoxin down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells. J Immunol. 1999;162(3):1401–7. [PubMed] [Google Scholar]
- 17.LONGHI MS, MA Y, BOGDANOS DP, CHEESEMAN P, MIELI-VERGANI G, VERGANI D. Impairment of CD4(+)CD25(+) regulatory T-cells in autoimmune liver disease. J Hepatol. 2004;41(1):31–7. doi: 10.1016/j.jhep.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 18.ERHARDT A, BIBURGER M, PAPADOPOULOS T, TIEGS G. IL-10, regulatory T cells, and Kupffer cells mediate tolerance in concanavalin A-induced liver injury in mice. Hepatology. 2007;45(2):475–85. doi: 10.1002/hep.21498. [DOI] [PubMed] [Google Scholar]
- 19.BREOUS E, SOMANATHAN S, VANDENBERGHE LH, WILSON JM. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology. 2009;50(2):612–21. doi: 10.1002/hep.23043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DUNHAM RM, THAPA M, VELAZQUEZ VM, ELROD EJ, DENNING TL, PULENDRAN B, et al. Hepatic stellate cells preferentially induce Foxp3+ regulatory T cells by production of retinoic acid. J Immunol. 2013;190(5):2009–16. doi: 10.4049/jimmunol.1201937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.YANG HR, CHOU HS, GU X, WANG L, BROWN KE, FUNG JJ, et al. Mechanistic insights into immunomodulation by hepatic stellate cells in mice: a critical role of interferon-gamma signaling. Hepatology. 2009;50(6):1981–91. doi: 10.1002/hep.23202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.SCHILDBERG FA, WOJTALLA A, SIEGMUND SV, ENDL E, DIEHL L, ABDULLAH Z, et al. Murine hepatic stellate cells veto CD8 T cell activation by a CD54-dependent mechanism. Hepatology. 2011;54(1):262–72. doi: 10.1002/hep.24352. [DOI] [PubMed] [Google Scholar]
- 23.KIYONO H, MCGHEE JR, WANNEMUEHLER MJ, MICHALEK SM. Lack of oral tolerance in C3H/HeJ mice. J Exp Med. 1982;155(2):605–10. doi: 10.1084/jem.155.2.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.HUANG L, SOLDEVILA G, LEEKER M, FLAVELL R, CRISPE IN. The liver eliminates T cells undergoing antigen-triggered apoptosis in vivo. Immunity. 1994;1(9):741–9. doi: 10.1016/s1074-7613(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 25.GORCZYNSKI RM. Adoptive transfer of unresponsiveness to allogeneic skin grafts with hepatic gamma delta + T cells. Immunology. 1994;81(1):27–35. [PMC free article] [PubMed] [Google Scholar]
- 26.GORCZYNSKI RM, CHEN Z, HOANG Y, ROSSI-BERGMAN B. A subset of gamma delta T-cell receptor-positive cells produce T-helper type-2 cytokines and regulate mouse skin graft rejection following portal venous pretransplant preimmunization. Immunology. 1996;87(3):381–9. doi: 10.1046/j.1365-2567.1996.481554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.STEPHENS C, ANDRADE RJ, LUCENA MI. Mechanisms of drug-induced liver injury. Current opinion in allergy and clinical immunology. 2014;14(4):286–92. doi: 10.1097/ACI.0000000000000070. [DOI] [PubMed] [Google Scholar]
- 28.CHEN M, BORLAK J, TONG W. High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology. 2013;58(1):388–96. doi: 10.1002/hep.26208. [DOI] [PubMed] [Google Scholar]
- 29.KAPLOWITZ N. Avoiding idiosyncratic DILI: two is better than one. Hepatology. 2013;58(1):15–7. doi: 10.1002/hep.26295. [DOI] [PubMed] [Google Scholar]
- 30.LIU ZX, KAPLOWITZ N. Immune-mediated drug-induced liver disease. Clin Liver Dis. 2002;6(3):755–74. doi: 10.1016/s1089-3261(02)00025-9. [DOI] [PubMed] [Google Scholar]
- 31.CHALASANI N, BONKOVSKY HL, FONTANA R, LEE W, STOLZ A, TALWALKAR J, et al. Features and Outcomes of 899 Patients With Drug-Induced Liver Injury: The DILIN Prospective Study. Gastroenterology. 2015;148(7):1340–52. e7. doi: 10.1053/j.gastro.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.TSUTSUI H, TERANO Y, SAKAGAMI C, HASEGAWA I, MIZOGUCHI Y, MORISAWA S. Drug-specific T cells derived from patients with drug-induced allergic hepatitis. J Immunol. 1992;149(2):706–16. [PubMed] [Google Scholar]
- 33.MONSHI MM, FAULKNER L, GIBSON A, JENKINS RE, FARRELL J, EARNSHAW CJ, et al. Human leukocyte antigen (HLA)-B*57:01-restricted activation of drug-specific T cells provides the immunological basis for flucloxacillin-induced liver injury. Hepatology. 2013;57(2):727–39. doi: 10.1002/hep.26077. [DOI] [PubMed] [Google Scholar]
- 34.MITCHELL JR, LONG MW, THORGEIRSSON UP, JOLLOW DJ. Acetylation rates and monthly liver function tests during one year of isoniazid preventive therapy. Chest. 1975;68(2):181–90. doi: 10.1378/chest.68.2.181. [DOI] [PubMed] [Google Scholar]
- 35.SENIOR J. Antitargets and Dryg safetty. 1. Wiley-VCH Verlag GmbH & Co. KGaA; 2015. Hepatic Side Effects; pp. 81–106. [Google Scholar]
- 36.BLACK M, MITCHELL JR, ZIMMERMAN HJ, ISHAK KG, EPLER GR. Isoniazid-associated hepatitis in 114 patients. Gastroenterology. 1975;69(2):289–302. [PubMed] [Google Scholar]
- 37.WATKINS PB. Idiosyncratic liver injury: challenges and approaches. Toxicol Pathol. 2005;33(1):1–5. doi: 10.1080/01926230590888306. [DOI] [PubMed] [Google Scholar]
- 38.AITHAL GP. Pharmacogenetic testing in idiosyncratic drug-induced liver injury: current role in clinical practice. Liver international : official journal of the International Association for the Study of the Liver. 2015;35(7):1801–8. doi: 10.1111/liv.12836. [DOI] [PubMed] [Google Scholar]
- 39.SINGER JB, LEWITZKY S, LEROY E, YANG F, ZHAO X, KLICKSTEIN L, et al. A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nature genetics. 2010;42(8):711–4. doi: 10.1038/ng.632. [DOI] [PubMed] [Google Scholar]
- 40.OKSENBERG JR, BARANZINI SE, SAWCER S, HAUSER SL. The genetics of multiple sclerosis: SNPs to pathways to pathogenesis. Nature reviews Genetics. 2008;9(7):516–26. doi: 10.1038/nrg2395. [DOI] [PubMed] [Google Scholar]
- 41.METUSHI IG, HAYES MA, UETRECHT J. Treatment of PD-1(−/−) mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology. 2015;61(4):1332–42. doi: 10.1002/hep.27549. [DOI] [PubMed] [Google Scholar]
- 42.CHAKRABORTY M, FULLERTON AM, SEMPLE K, CHEA LS, PROCTOR WR, BOURDI M, et al. Drug-induced allergic hepatitis developed in mice when myeloid-derived suppressor cells were depleted prior to halothane treatment. Hepatology. 2015 doi: 10.1002/hep.27764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.MAK A, UETRECHT J. The Role of CD8 T Cells in Amodiaquine-Induced Liver Injury in PD1−/− Mice Cotreated with Anti-CTLA-4. Chemical research in toxicology. 2015 doi: 10.1021/acs.chemrestox.5b00137. [DOI] [PubMed] [Google Scholar]
- 44.UETRECHT J, KAPLOWITZ N. Inhibition of immune tolerance unmasks drug-induced allergic hepatitis. Hepatology. 2015;62(2):346–8. doi: 10.1002/hep.27824. [DOI] [PubMed] [Google Scholar]
- 45.CLAYTON TA, BAKER D, LINDON JC, EVERETT JR, NICHOLSON JK. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci U S A. 2009;106(34):14728–33. doi: 10.1073/pnas.0904489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.AITHAL GP, RAMSAY L, DALY AK, SONCHIT N, LEATHART JB, ALEXANDER G, et al. Hepatic adducts, circulating antibodies, and cytokine polymorphisms in patients with diclofenac hepatotoxicity. Hepatology. 2004;39(5):1430–40. doi: 10.1002/hep.20205. [DOI] [PubMed] [Google Scholar]
- 47.PACHKORIA K, LUCENA MI, CRESPO E, RUIZ-CABELLO F, LOPEZ-ORTEGA S, FERNANDEZ MA, et al. Analysis of IL-10, IL-4 and TNF-alpha polymorphisms in drug-induced liver injury (DILI) and its outcome. J Hepatol. 2008;49(1):107–14. doi: 10.1016/j.jhep.2008.03.017. [DOI] [PubMed] [Google Scholar]
- 48.DE WAAL MALEFYT R, HAANEN J, SPITS H, RONCAROLO MG, TE VELDE A, FIGDOR C, et al. Interleukin 10 (IL-10) and viral IL-10 strongly reduce antigen-specific human T cell proliferation by diminishing the antigen-presenting capacity of monocytes via downregulation of class II major histocompatibility complex expression. J Exp Med. 1991;174(4):915–24. doi: 10.1084/jem.174.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DE VRIES JE. Immunosuppressive and anti-inflammatory properties of interleukin 10. Annals of medicine. 1995;27(5):537–41. doi: 10.3109/07853899509002465. [DOI] [PubMed] [Google Scholar]
- 50.SHARMA P, ALLISON JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 51.VOSKENS CJ, GOLDINGER SM, LOQUAI C, ROBERT C, KAEHLER KC, BERKING C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 2013;8(1):e53745. doi: 10.1371/journal.pone.0053745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.CHENG R, COOPER A, KENCH J, WATSON G, BYE W, MCNEIL C, et al. Ipilimumab-induced toxicities and the gastroenterologist. J Gastroenterol Hepatol. 2015;30(4):657–66. doi: 10.1111/jgh.12888. [DOI] [PubMed] [Google Scholar]
- 53.AHMED T, PANDEY R, SHAH B, BLACK J. Resolution of ipilimumab induced severe hepatotoxicity with triple immunosuppressants therapy. BMJ case reports. 2015;2015 doi: 10.1136/bcr-2014-208102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.KIM KW, RAMAIYA NH, KRAJEWSKI KM, JAGANNATHAN JP, TIRUMANI SH, SRIVASTAVA A, et al. Ipilimumab associated hepatitis: imaging and clinicopathologic findings. Investigational new drugs. 2013;31(4):1071–7. doi: 10.1007/s10637-013-9939-6. [DOI] [PubMed] [Google Scholar]
- 55.KLEINER DE, BERMAN D. Pathologic changes in ipilimumab-related hepatitis in patients with metastatic melanoma. Digestive diseases and sciences. 2012;57(8):2233–40. doi: 10.1007/s10620-012-2140-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.POSTOW MA. Managing immune checkpoint-blocking antibody side effects. American Society of Clinical Oncology educational book / ASCO American Society of Clinical Oncology Meeting. 2015;35:76–83. doi: 10.14694/EdBook_AM.2015.35.76. [DOI] [PubMed] [Google Scholar]
- 57.IWANAGA N, ORIGUCHI T, TERADA K, UEKI Y, KAMO Y, KINOSHITA N, et al. Rheumatoid arthritis complicated with severe liver injury during treatment with abatacept. Modern rheumatology / the Japan Rheumatism Association. 2014;24(5):874–6. doi: 10.3109/14397595.2013.844399. [DOI] [PubMed] [Google Scholar]
- 58.HOOFNAGLE J. LiverTox. 2015 [cited 2015 8.20.2015]; Available from: http://livertox.nih.gov/Abatacept.htm.