

Abstract
Vascular endothelial growth factor (VEGF) increases endothelial barrier permeability, an effect that may contribute to macular edema in diabetic retinopathy. Since vitamin C, or ascorbic acid, can tighten the endothelial permeability barrier, we examined whether it could prevent the increase in permeability due to VEGF in human umbilical vein endothelial cells (HUVECs). As previously observed, VEGF increased HUVEC permeability to radiolabeled inulin within 60 min in a concentration-dependent manner. Loading the cells with increasing concentrations of ascorbate progressively prevented the leakage caused by 100 ng/ml VEGF, with a significant inhibition at 13 μM and complete inhibition at 50 μM. Loading cells with 100 μM ascorbate also decreased basal generation of reactive oxygen species and prevented the increase caused by both 100 ng/ml VEGF. VEGF treatment decreased intracellular ascorbate by 25%, thus linking ascorbate oxidation to its prevention of VEGF-induced barrier leakage. The latter was blocked by treating the cells with 60 μM L-NAME (but not D-NAME) as well as by 30 μM sepiapterin, a precursor of tetrahydrobiopterin that is required for proper function of endothelial nitric oxide synthase (eNOS). These findings suggest that VEGF-induced barrier leakage uncouples eNOS. Ascorbate inhibition of the VEGF effect could thus be due either to scavenging superoxide or to peroxynitrite generated by the uncoupled eNOS, or more likely to its ability to recycle tetrahydrobiopterin, thus avoiding enzyme uncoupling in the first place. Ascorbate prevention of VEGF-induced increases in endothelial permeability opens the possibility that its repletion could benefit diabetic macular edema.
Keywords: Ascorbic acid, VEGF, endothelial cells, oxidative stress, endothelial nitric oxide synthase
Introduction
One of the earliest known yet still enigmatic functions of vascular endothelial growth factor (VEGF) is to increase leakage of plasma components through the endothelial permeability barrier [1]. Barrier leakage due to VEGF has been demonstrated as early as 30 min after VEGF addition to endothelial cells cultured on semi-porous membranes [2, 3]. Other studies suggest a delayed effect requiring several hours [4, 5]. The latter is supported by the results of earlier studies suggest that prolonged treatment with VEGF sustains opening of fenestrae through which plasma components can diffuse [2, 6].
The mechanism of the acute VEGF-induced increase in endothelial permeability is controversial. Some authors suggest that it is due to VEGF induction of caveoli to form vesicular vacuolar organelles that move plasma components across the cells [6-8]. Others provide data showing that VEGF activation of the VEGF receptor-2 rapidly activates a series of steps leading to internalization of VE-cadherin and breakdown of the adherens junctions between endothelial cells [9, 10]. It seems likely that VEGF-induced increases in both transcytosis and paracellular transfer contribute to barrier leakiness, depending on the cell type, vascular bed (capillary or venule) and on treatment conditions, such as duration of stimulation.
The short-term increases endothelial barrier permeability induced by VEGF may be due, at least in part, to generation of reactive oxygen or nitrogen species (ROS/RNS). For example, it has been shown that scavenging of endogenous superoxide (but not H2O2) blunts VEGF-induced increases in endothelial barrier permeability [11]. The source of this superoxide is uncertain, with NADPH oxidase and uncoupled endothelial nitric oxide synthase activation as possible candidates. Although blockade of eNOS in brain [12] and retinal [7] microvascular endothelial cells, as well as in human umbilical vein endothelial cells (HUVECs) [13], prevented barrier permeability increases due to VEGF, the opposite effect was observed in brain retinal endothelial cells [3]. Whatever the source of oxidative stress, it is clear that increasing cellular antioxidant capacity might be beneficial in decreasing VEGF-induced hyperpermeability. In this regard, we have shown that vitamin C, or ascorbic acid, prevents endothelial barrier leakage due to oxidized low density lipoprotein [14] and more recently due to thrombin [15]. Although ascorbate is considered an important cellular antioxidant, in both basal [16] and thrombin-activated cells [15], the vitamin enhanced the function of eNOS to generate nitric oxide (NO). It is known from a previous study that ascorbate promotes NO generation by recycling the crucial eNOS co-factor tetrahydrobiopterin [17]. Subsequent activation of guanylate cyclase by NO to produce cyclic GMP resulted in increased cyclic AMP, which is known to tighten the endothelial barrier [18-20].
If ascorbate can prevent increased endothelial barrier permeability due to thrombin, it might also do so with VEGF, either by scavenging ROS/RNS directly or by helping to prevent uncoupling of eNOS with subsequent superoxide generation. To examine whether intracellular ascorbate might modulate permeability-inducing effects of VEGF, we studied the effect of ascorbate on the permeability of HUVECs in response to VEGF, assessing possible antioxidant functions, as well as functions dependent on preservation of NO.
Materials and Methods
Materials
Sigma/Aldrich Chemical Co. (St. Louis, MO) supplied the reagent chemicals, including ascorbate, 2′,7′-dichlorofluorescein diacetate, Nω-nitro-D-arginine methyl ester hydrochloride (D-NAME), Nω-nitro- L-arginine methyl ester hydrochloride (L-NAME), N-2-hydroxyethylpiperazine N′-2-ethanesulfonic acid (Hepes), sepiapterin (S-(−)-2-amino-7,8-dihydro-6-(2-hydroxy-1-oxopropyl)-4(1 H)-pteridinone), and recombinant human VEGF (Catalog # V7259). Perkin-Elmer Life and Analytical Sciences, Inc. (Boston, MA) supplied the [carboxyl-14C]inulin (molecular weight range 5000-5500, 2 mCi/g).
Cell Culture
HUVECs were supplied by ScienCell Research Laboratories (Carlsbad, CA) and cultured in their Endothelial Cell Medium (Cat. #1001), but without antioxidant supplements. As noted by the manufacturer, this medium contains 5% (v/v) fetal bovine serum (Cat. #0025), 1% (v/v) endothelial cell growth supplement (Cat. #1052), and penicillin/streptomycin (Cat. #0503). Cells were used between passages 3-10. Cells were cultured at 37 °C in humidified air containing 5% CO2.
Assay of ascorbate
Ascorbate was measured in cells cultured in 6-well plates following 3 rinses with Krebs-Ringer Hepes buffer (KRH) that contained 20 mM Hepes, 128 mM NaCl, 5.2 mM KCl, 1 mM NaH2PO4, 1.4 mM MgSO4, and 1.4 mM CaCl2 and at pH 7.4. The cell monolayer was then treated with 0.1 ml of 25 % (w/v) metaphosphoric acid, scraped from the plate with a rubber spatula, and the acidic lysate was partially neutralized with 0.35 ml of 0.1M Na2HPO4 containing 0.05 mM EDTA, pH 8.0. After centrifuging the cell lysate at 4 °C for 1 minutes at 13,000 × g, the supernatant was taken for assay of ascorbate in duplicate by high performance liquid chromatography as previously described [21]. Intracellular ascorbate concentrations were calculated based on calculation of the intracellular distribution space of 3-O-methylglucose relative to the cell protein content [22], which was taken as 3.6 ± 1.2 μl/mg protein [23].
Assay of cellular oxidative stress
To measure intracellular ROS/RNS, confluent HUVECs in 96-well plates were rinsed free of culture medium and incubated for 30 minutes in KRH that contained 5 mM glucose and 20 μM 2′,7′-dichlorofluorescein diacetate. The cells were then rinsed twice with the KRH/glucose medium and the indicated reagents were added to wells in a total volume of 200 μL. Fluorescence of intracellular 2′,7′-dichlorofluorescen was measured in a fluorescence microtiter plate reader (Synergy HT multimode microtiter reader, BioTek, Winooski, VT) at the start of incubation and after 120 minutes of incubation using excitation and emission wavelengths of 480 and 530 nm, respectively. The cells were maintained at 37°C throughout the incubation period. Preliminary studies revealed that there was a linear increase in cell fluorescence with time (results not shown), so the background cell fluorescence at time zero was subtracted from the reading at the 120 minute time point for presentation.
Assay of endothelial permeability
HUVECS were cultured to confluence on polyethylene terephthalate cell culture inserts (6-well format, 0.4 micron pores at a density of 2 ± 0.2 × 106 pores per cm2, Falcon BD Biosciences, Franklin Lakes, NJ). Cells were cultured to confluence with 1.7 ml of medium in the upper well and 2.8 ml of medium in the lower well. After several days at confluence, agents were added as indicated, followed by duplicate measurements of permeability of the endothelial cell layer to [carboxyl-14C]inulin over 60 minutes at 37°C as previously described [24], as derived from [25]. The permeability coefficient of [carboxyl-14C]inulin was calculated with correction for the rate of [carboxyl-14C]inulin transfer across filters after removal of cells by treatment with ammonium hydroxide [26].
Data Analysis
Results are shown as mean + standard error. Statistical comparisons were made using GraphPad Prism version 6.05 for Windows (GraphPad Software, San Diego, CA). Differences between treatments were assessed by one-way ANOVA with post-hoc testing using Tukey's or Dunnett's test, as appropriate.
Results
VEGF-induced endothelial permeability barrier leakiness is prevented by ascorbate
Endothelial cells cultured 5-6 days on semi-porous filter supports formed a barrier to the passive diffusion of radioactive inulin across the matrix and cell layers, measured over 60 minutes at 37 °C (Fig. 1). Although not shown, this basal transfer rate was stable for several hours. Treatment of the cells in the upper chamber with 100 ng/ml VEGF for 0-120 min prior to the inulin transfer assay increased barrier leakiness at all time points (Fig. 1A), indicating that the effect of VEGF to increase permeability becomes maximal at a time point less than 60 min, which was the limiting duration of the transfer assay. When cells were exposed to increasing concentrations of VEGF for 30 min before the inulin transfer assay, there was a progressive increase in barrier leakage, which became significant at 13 ng/ml VEGF and increased slightly further at 100 ng/ml (Fig. 1B). To assess the effect of ascorbate on VEGF-induced increases in barrier permeability, cells were treated without (Fig. 1C, square) or with 100 ng/ml VEGF (Fig. 1C, circles) for 30 min followed by addition of increasing concentrations of ascorbate for 30 min before the inulin transfer assay. As expected, VEGF increased permeability as much as two-fold in the absence of ascorbate (Fig. 1C, compare square and circle at zero ascorbate). However, increasing amounts of ascorbate progressively tightened the endothelial barrier, with statistical significance reached at 13 μM ascorbate (Fig. 1C, circles). This shows that ascorbate addition to VEGF-treated cells potently inhibited barrier leakiness due to VEGF, although not to levels below the initial basal level. In other studies not shown, preloading the cells with ascorbate before VEGF treatment also blocked the increase in barrier leakiness due to VEGF.
Figure 1. VEGF-induced increases in endothelial permeability are prevented by intracellular ascorbate.
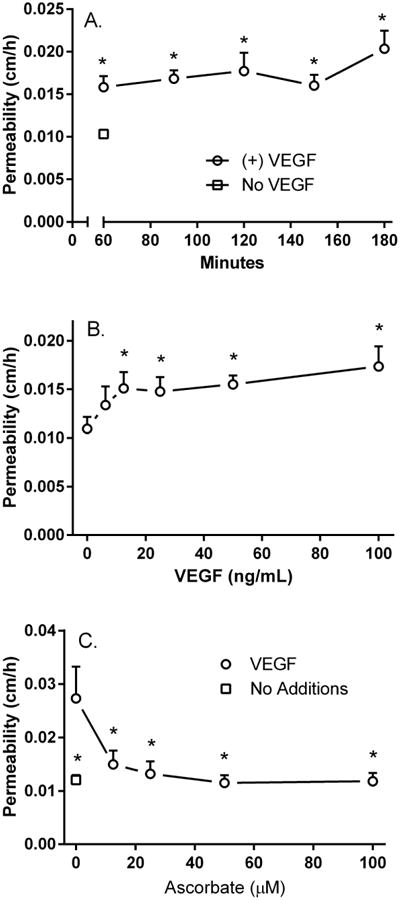
Panel A. Time-dependence of VEGF-induced barrier opening. Cells were treated without (square) or with 100 ng/ml VEGF (circles) in the upper chamber for the times indicated at 37°C before assay of radiolabeled inulin permeability as described in Methods. Results are shown from 6 experiments, with “*” indicating p < 0.05 compared to no VEGF. The error bar for control lies within the symbol. Panel B. Concentration dependence of the VEGF effect. Confluent HUVECs on filter inserts were treated for 30 min at 37°C with the indicated concentration of VEGF, followed by addition of radiolabeled inulin and the 60 minute inulin transfer assay. Results are shown from 4 experiments, with “*” indicating p < 0.05 compared to cells without added VEGF. Panel C. Ascorbate inhibits of VEGF-induced endothelial permeability. Confluent HUVECs were treated for 30 minutes without (square) or with 100 ng/ml VEGF (circles), followed by addition of the indicated concentration of ascorbate in the upper well for 30 minutes, then by the radiolabeled inulin transfer assay, all at 37°C. Results are shown from 6 experiments, with “*” indicating p < 0.05 compared to VEGF-treated cells that received no ascorbate.
Antioxidant role of ascorbate in preventing VEGF-induced barrier leakiness
VEGF is known to generate ROS/RNS early in its time course of action and ROS/RNS can increase endothelial barrier permeability. To test this, HUVECs were treated for 120 min at 37 °C with 100 ng/ml VEGF and the cellular fluorescence of 2′,7′-dichlorofluorescin was used to measure ROS/RNS. As shown in Fig. 2A, this increased significantly by 20% in response to VEGF and this increase was decreased by 42% by co-treatment of the cells with 100 μM ascorbate. Indeed ascorbate decreased basal fluorescence in untreated cells by about 33%. Further evidence favoring an increase in oxidative stress was the finding that VEGF concentrations of 50 ng/ml and greater decreased intracellular ascorbate in cells that had been treated for 30 min with 100 μM ascorbate (Fig. 2B).
Figure 2. Ascorbate inhibits oxidative stress generated by VEGF and prevents barrier leakage by H2O2.
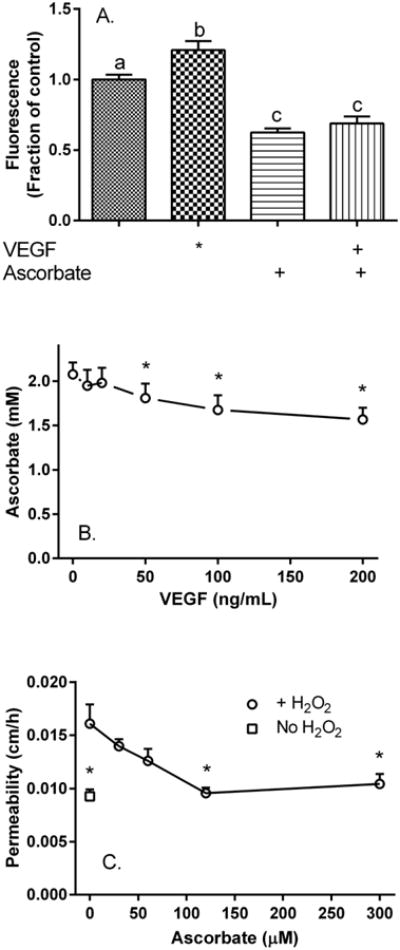
Panel A. HUVECs cultured to confluence in 96-well plates were treated for 30 min with 2′,7′-dichlorofluorescein diacetate, rinsed and incubated at 37°C where indicated with 100 ng/ml VEGF or 100 μM ascorbate with measurement of fluorescence as noted under Methods. Results from 16 replicates from several plates are expressed as a fraction of the fluorescence change seen in untreated cells, with bars not having the same small letters different at p < 0.05. Panel B. VEGF decreases intracellular ascorbate. HUVECs cultured to confluence in 6-well plates were treated with 100 μM ascorbate for 30 minutes, rinsed three times in 2 ml of KRH, and treated for 90 minutes with the indicated VEGF concentration at 37°C before removal for assay of ascorbate as described in Methods. Results are shown from 4 experiments, with an “*” indicating p < 0.05 compared to the sample not treated with VEGF. Panel C. Ascorbate prevents H2O2-induced endothelial barrier leakage. Cells cultured on filters were treated with the indicated concentrations of ascorbate for 30 minutes, rinsed twice in KRH, then treated in the absence (square) or presence (circles) of 250 μM H2O2 for 60 minutes at 37°C, followed by the radiolabeled inulin transfer assay. Results are shown from 6 experiments, with an “*” indicating p < 0.01 compared to the sample treated with H2O2 alone.
Finally, to test directly whether ascorbate can prevent endothelial barrier leak due to direct application of an oxidant, cells cultured on semi-porous filters were treated with increasing concentrations of ascorbate for 30 min, then treated for another 60 min with 250 μM H2O2, followed by the inulin transfer assay. As shown in Fig. 2C, H2O2 treatment increased endothelial barrier permeability by 73% and ascorbate prevented this leakiness, albeit at 2-3-fold higher concentrations than required for VEGF-induced leakiness. Together, these results suggest that at least part of the effect of VEGF could be due to increased oxidative stress and that ascorbate might inhibit the VEGF effect by functioning as an antioxidant.
Role of NO in VEGF-induced endothelial barrier leakiness
To assess whether eNOS is involved in acute VEGF-induced weakening the endothelial permeability barrier, we tested effects of L-NAME, using D-NAME as a negative control. In experiments not shown, concentrations of D-NAME of 100 μM and higher suppressed VEGF-induced barrier leakage. To obtain selectivity for eNOS, concentrations of 60 μM L- and D-NAME were used. As seen in Fig. 3A, neither L-NAME nor D-NAME alone had a significant effect on basal endothelial permeability (compare 1st, 3rd, and 5th bars). However, L-NAME prevented the VEGF-induced increase in permeability (compare 2nd and 4th bars), whereas D-NAME had no effect (compare 2nd and 6th bars). The results with L-NAME indicate that eNOS contributes to the barrier weakening properties of VEGF.
Figure 3. VEGF-induced increases in endothelial barrier permeability require eNOS.
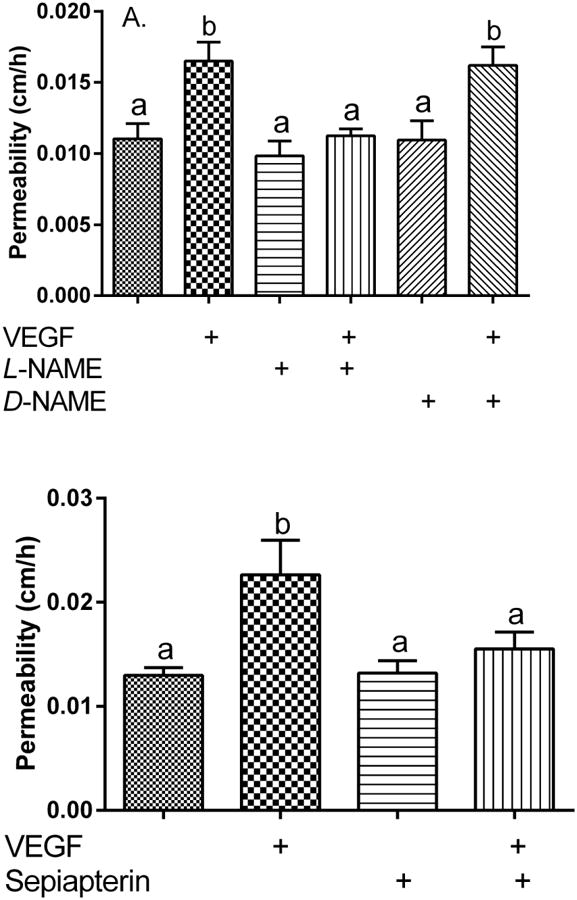
Panel A. L-NAME prevents VEGF-induced increases in endothelial barrier permeability. Cells in culture medium on filters were treated for 30 minute at 37°C with 60 μM L- or D-NAME as indicated, followed by addition of 100 ng/ml VEGF for another 30 minutes before assay of radiolabeled inulin permeability as described in Methods. Results are shown from 4 experiments, with bars not sharing the same small letters different at p < 0.05. Panel B. Sepiapterin inhibits VEGF-induced increases in endothelial permeability. HUVECs in culture medium on filters were treated at 37°C where indicated for 30 minutes with 30 μM sepiapterin, followed by 100 ng/ml VEGF. After 30 minutes the radiolabeled inulin transfer assay was carried out. Results are shown from 8 experiments, with not sharing the same small letters different at p < 0.05.
If ascorbate inhibits the VEGF by preserving NO through recycling of tetrahydrobiopterin, then increasing the cellular concentration of tetrahydrobiopterin by treating cells with its precursor sepiapterin should have a similar effect. As shown in Fig. 3B, sepiapterin had no effect on basal permeability (compare 1st and 3rd bars), but it prevented the increase in permeability due to VEGF (compare 2nd and 4th bars).
Discussion
In this work we found that treating HUVECs with VEGF for as short a time as one hour increased endothelial barrier permeability and that this increase was completely prevented by loading the cells with ascorbate. Loading ascorbate concentrations were well within the physiologic human plasma range of the vitamin (12-60 μM). Treating the cells with 100 μM ascorbate for 90 minutes resulted in an intracellular ascorbate concentration of 2 mM, which is similar to its concentration in circulating human leukocytes [27] and the maximal intracellular ascorbate concentration in vivo [28]. The presence of this or even lower concentrations in endothelial cells would therefore likely blunt or prevent an acute increase in endothelial permeability due to VEGF observed in this and previous studies. Somewhat higher concentrations of ascorbate also blocked permeability increases due to direct treatment with H2O2, again suggesting that ascorbate could buffer ROS/RNS in vivo and that any culture studies of endothelial barrier function should include physiologic ascorbate concentrations in the culture medium.
The increase in endothelial barrier permeability induced by VEGF was associated with a modest but significant increase in ROS/RNS generation that was prevented by ascorbate loading. It was accompanied by a 25% decrease in intracellular ascorbate over 90 minutes of incubation. This suggests that ascorbate is both sensitive to such stress and that its ability to block VEGF-induced ROS/RNS might contribute to its prevention of VEGF-induced increases in barrier permeability. At the concentrations measured in the current studies, ascorbate can neutralize intracellular superoxide [29] as well as its highly reactive downstream product peroxynitrite [30]. This primary antioxidant scavenging activity of ascorbate may thus account for a portion of its effect to decrease VEGF-induced increases in endothelial barrier permeability.
We also found that blockade of eNOS with L-NAME (but not D-NAME) prevented VEGF-induced increases in endothelial barrier permeability, as it did in most [12, 13, 31], but not all [3] previous studies. This supports the notion that the rapid increase in endothelial barrier leakage due to VEGF requires NO. Activation of eNOS by VEGF is thought to be due to VEGF-induced increases in intracellular calcium [32, 33]. However, it appears that in increasing eNOS activity, VEGF uncouples the enzyme, resulting in increased superoxide and peroxynitrite generation.
We previously showed that the vitamin tightens the endothelial permeability barrier in both the basal [16] and thrombin-activated state [15] by preserving NO. That it may also do so with VEGF is plausible, especially since increasing tetrahydrobiopterin by providing the cells with its precursor sepiapterin also blocked acute VEGF-induced increases in barrier permeability. Thus ascorbate could block VEGF-induced barrier opening both by scavenging ROS/RNS and by preserving NO itself.
The ability of ascorbate to prevent vascular leak due to VEGF could have clinical implications. For example, macular edema in diabetics with retinopathy is treated by injecting anti-VEGF antibodies into the vitreous humor of the eye [34]. Since ascorbate is severely depleted in the vitreous humor of diabetics with poor glycemic control [35] who are also likely to have macular edema [34], it is possible that its repletion could help to prevent or reverse macular edema due to VEGF, providing a safe and effective adjunct to more invasive therapies.
Acknowledgments
This work was supported by National Institutes of Health grant DK050435 and by the Cell Culture Core of the Vanderbilt Diabetes Research and Training Center (DK020593).
Abbreviations
- D-NAME
Nω-nitro-D-arginine methyl ester hydrochloride
- eNOS
endothelial nitric oxide synthase
- Hepes
N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid
- KRH
Krebs-Ringer Hepes
- L-NAME
Nω-nitro-L-arginine methyl ester hydrochloride
- NO
nitric oxide
- ROS/RNS
reactive oxygen and nitrogen species
- VEGF
vascular endothelial growth factor
References
- 1.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 2.Roberts WG, Palade GE. Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J Cell Sci. 1995;108(Pt 6):2369–2379. doi: 10.1242/jcs.108.6.2369. [DOI] [PubMed] [Google Scholar]
- 3.Marumo T, Noll T, Schini-Kerth VB, Harley EA, Duhault J, Piper HM, Busse R. Significance of nitric oxide and peroxynitrite in permeability changes of the retinal microvascular endothelial cell monolayer induced by vascular endothelial growth factor. J Vasc Res. 1999;36:510–515. doi: 10.1159/000025694. [DOI] [PubMed] [Google Scholar]
- 4.Hippenstiel S, Krull M, Ikemann A, Risau W, Clauss M, Suttorp N. VEGF induces hyperpermeability by a direct action on endothelial cells. Am J Physiol. 1998;274:L678–L684. doi: 10.1152/ajplung.1998.274.5.L678. [DOI] [PubMed] [Google Scholar]
- 5.Wang W, Merrill MJ, Borchardt RT. Vascular endothelial growth factor affects permeability of brain microvessel endothelial cells in vitro. Am J Physiol. 1996;271:C1973–C1980. doi: 10.1152/ajpcell.1996.271.6.C1973. [DOI] [PubMed] [Google Scholar]
- 6.Esser S, Wolburg K, Wolburg H, Breier G, Kurzchalia T, Risau W. Vascular endothelial growth factor induces endothelial fenestrations in vitro. J Cell Biol. 1998;140:947–959. doi: 10.1083/jcb.140.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng Y, Venema VJ, Venema RC, Tsai N, Behzadian MA, Caldwell RB. VEGF-induced permeability increase is mediated by caveolae. Invest Ophthalmol Vis Sci. 1999;40:157–167. [PubMed] [Google Scholar]
- 8.Chen J, Braet F, Brodsky S, Weinstein T, Romanov V, Noiri E, Goligorsky MS. VEGF-induced mobilization of caveolae and increase in permeability of endothelial cells. American Journal of Physiology: Cell Physiology. 2002;282:C1053–C1063. doi: 10.1152/ajpcell.00292.2001. [DOI] [PubMed] [Google Scholar]
- 9.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. 2006;8:1223–1234. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 10.Murakami T, Felinski EA, Antonetti DA. Occludin phosphorylation and ubiquitination regulate tight junction trafficking and vascular endothelial growth factor-induced permeability. J Biol Chem. 2009;284:21036–21046. doi: 10.1074/jbc.M109.016766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han J, Shuvaev VV, Muzykantov VR. Catalase and superoxide dismutase conjugated with platelet-endothelial cell adhesion molecule antibody distinctly alleviate abnormal endothelial permeability caused by exogenous reactive oxygen species and vascular endothelial growth factor. J Pharmacol Exp Ther. 2011;338:82–91. doi: 10.1124/jpet.111.180620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murohara T, Horowitz JR, Silver M, Tsurumi Y, Chen D, Sullivan A, Isner JM. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation. 1998;97:99–107. doi: 10.1161/01.cir.97.1.99. [DOI] [PubMed] [Google Scholar]
- 13.Breslin JW, Pappas PJ, Cerveira JJ, Hobson RW, Duran WN. VEGF increases endothelial permeability by separate signaling pathways involving ERK-1/2 and nitric oxide. Am J Physiol Heart Circ Physiol. 2003;284:H92–H100. doi: 10.1152/ajpheart.00330.2002. [DOI] [PubMed] [Google Scholar]
- 14.May JM, Qu ZC. Ascorbic acid prevents increased endothelial permeability caused by oxidized low density lipoprotein. Free Radic Res. 2010;44:1359–1368. doi: 10.3109/10715762.2010.508496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parker WH, Qu ZC, May JM. Intracellular ascorbate prevents endothelial barrier permeabilization by thrombin. J Biol Chem. 2015;290:21486–21497. doi: 10.1074/jbc.M115.662098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.May JM, Qu ZC. Nitric oxide mediates tightening of the endothelial barrier by ascorbic acid. Biochem Biophys Res Commun. 2011;404:701–705. doi: 10.1016/j.bbrc.2010.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heller R, Unbehaun A, Schellenberg B, Mayer B, Werner-Felmayer G, Werner ER. L-ascorbic acid potentiates endothelial nitric oxide synthesis via a chemical stabilization of tetrahydrobiopterin. Journal of Biological Chemistry. 2001;276:40–47. doi: 10.1074/jbc.M004392200. [DOI] [PubMed] [Google Scholar]
- 18.Moy AB, Bodmer JE, Blackwell K, Shasby S, Shasby DM. cAMP protects endothelial barrier function independent of inhibiting MLC20-dependent tension development. Am J Physiol. 1998;274:L1024–L1029. doi: 10.1152/ajplung.1998.274.6.L1024. [DOI] [PubMed] [Google Scholar]
- 19.Stelzner TJ, Weil JV, O'Brien RF. Role of cyclic adenosine monophosphate in the induction of endothelial barrier properties. J Cell Physiol. 1989;139:157–166. doi: 10.1002/jcp.1041390122. [DOI] [PubMed] [Google Scholar]
- 20.Yamada Y, Furumichi T, Furui H, Yokoi T, Ito T, Yamauchi K, Yokota M, Hayashi H, Saito H. Roles of calcium, cyclic nucleotides, and protein kinase C in regulation of endothelial permeability. Arteriosclerosis. 1990;10:410–420. doi: 10.1161/01.atv.10.3.410. [DOI] [PubMed] [Google Scholar]
- 21.May JM, Qu ZC, Mendiratta S. Protection and recycling of α-tocopherol in human erythrocytes by intracellular ascorbic acid. Arch Biochem Biophys. 1998;349:281–289. doi: 10.1006/abbi.1997.0473. [DOI] [PubMed] [Google Scholar]
- 22.Hardy TA, May JM. Coordinate regulation of L-arginine uptake and nitric oxide synthase activity in cultured endothelial cells. Free Radic Biol Med. 2002;32:122–131. doi: 10.1016/s0891-5849(01)00781-x. [DOI] [PubMed] [Google Scholar]
- 23.Jones W, Li X, Perriott LM, Whitesell RR, May JM. Uptake, recycling, and antioxidant functions of α-lipoic acid in endothelial cells. Free Radic Biol Med. 2002;33:83–93. doi: 10.1016/s0891-5849(02)00862-6. [DOI] [PubMed] [Google Scholar]
- 24.May JM, Qu ZC, Qiao H. Transfer of ascorbic acid across the vascular endothelium: mechanism and self-regulation. Am J Physiol Cell Physiol. 2009;297:C169–C178. doi: 10.1152/ajpcell.00674.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siflinger-Birnboim A, del Vecchio PJ, Cooper JA, Blumenstock FA, Shepard JM, Malik AB. Molecular sieving characteristics of the cultured endothelial monolayer. J Cell Physiol. 1987;132:111–117. doi: 10.1002/jcp.1041320115. [DOI] [PubMed] [Google Scholar]
- 26.Utoguchi N, Ikeda K, Saeki K, Oka N, Mizuguchi H, Kubo K, Nakagawa S, Mayumi T. Ascorbic acid stimulates barrier function of cultured endothelial cell monolayer. J Cell Physiol. 1995;163:393–399. doi: 10.1002/jcp.1041630219. [DOI] [PubMed] [Google Scholar]
- 27.Bergsten P, Amitai G, Kehrl J, Dhariwal KR, Klein HG, Levine M. Millimolar concentrations of ascorbic acid in purified human mononuclear leukocytes. Depletion and reaccumulation. J Biol Chem. 1990;265:2584–2587. [PubMed] [Google Scholar]
- 28.May JM, Qu ZC. Ascorbic acid efflux and re-uptake in endothelial cells: maintenance of intracellular ascorbate. Mol Cell Biochem. 2009;325:79–88. doi: 10.1007/s11010-008-0022-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackson TS, Xu AM, Vita JA, Keaney JF., Jr Ascorbate prevents the interaction of superoxide and nitric oxide only at very high physiological concentrations. Circulation Research. 1998;83:916–922. doi: 10.1161/01.res.83.9.916. [DOI] [PubMed] [Google Scholar]
- 30.Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols - Implications for uncoupling endothelial nitric-oxide synthase. Journal of Biological Chemistry. 2003;278:22546–22554. doi: 10.1074/jbc.M302227200. [DOI] [PubMed] [Google Scholar]
- 31.El-Remessy AB, Al-Shabrawey M, Platt DH, Bartoli M, Behzadian MA, Ghaly N, Tsai N, Motamed K, Caldwell RB. Peroxynitrite mediates VEGF's angiogenic signal and function via a nitration-independent mechanism in endothelial cells. FASEB J. 2007;21:2528–2539. doi: 10.1096/fj.06-7854com. [DOI] [PubMed] [Google Scholar]
- 32.Brock TA, Dvorak HF, Senger DR. Tumor-secreted vascular permeability factor increases cytosolic Ca2+ and von Willebrand factor release in human endothelial cells. Am J Pathol. 1991;138:213–21. [PMC free article] [PubMed] [Google Scholar]
- 33.Hamdollah Zadeh MA, Glass CA, Magnussen A, Hancox JC, Bates DO. VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation. 2008;15:605–14. doi: 10.1080/10739680802220323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abbate M, Cravedi P, Iliev I, Remuzzi G, Ruggenenti P. Prevention and treatment of diabetic retinopathy: evidence from clinical trials and perspectives. Curr Diabetes Rev. 2011;7:190–200. doi: 10.2174/157339911795843168. [DOI] [PubMed] [Google Scholar]
- 35.Barba I, Garcia-Ramirez M, Hernandez C, Alonso MA, Masmiquel L, Garcia-Dorado D, Simo R. Metabolic fingerprints of proliferative diabetic retinopathy: an 1H-NMR-based metabonomic approach using vitreous humor. Invest Ophthalmol Vis Sci. 2010;51:4416–4421. doi: 10.1167/iovs.10-5348. [DOI] [PubMed] [Google Scholar]