

Abstract
One of the most common symptoms of Alzheimer's disease (AD) and related tauopathies is memory loss. The exact mechanisms leading to memory loss in tauopathies are not yet known; however, decreased translation due to ribosomal dysfunction has been implicated as a part of this process. Here we use a proteomics approach that incorporates subcellular fractionation and coimmunoprecipitation of tau from human AD and non-demented control brains to identify novel interactions between tau and the endoplasmic reticulum (ER). We show that ribosomes associate more closely with tau in AD than with tau in control brains, and that this abnormal association leads to a decrease in RNA translation. The aberrant tau–ribosome association also impaired synthesis of the synaptic protein PSD-95, suggesting that this phenomenon contributes to synaptic dysfunction. These findings provide novel information about tau-protein interactions in human brains, and they describe, for the first time, a dysfunctional consequence of tau–ribosome associations that directly alters protein synthesis.
SIGNIFICANCE STATEMENT Despite the identification of abnormal tau–ribosomal interactions in tauopathies >25 years ago, the consequences of this association remained elusive until now. Here, we show that pathological tau associates closely with ribosomes in AD brains, and that this interaction impairs protein synthesis. The overall result is a stark reduction of nascent proteins, including those that participate in synaptic plasticity, which is crucial for learning and memory. These data mechanistically link a common pathologic sign, such as the appearance of pathological tau inside brain cells, with cognitive impairments evident in virtually all tauopathies.
Keywords: Alzheimer, ribosome, RNA, tau, tauopathies, translation
Introduction
Aberrant accumulation of tau is associated with the etiology of >18 known neurodegenerative diseases collectively termed tauopathies. Alzheimer's disease (AD), the most common tauopathy, affects >36 million people world-wide (Alzheimer's Association, 2014). A common and early symptom in tauopathic patients is progressive memory loss. There is no cure for tauopathies, and current therapeutics stave off symptoms only temporarily. This is partly due to limited understanding of the molecular mechanisms linking tau and disease onset.
Synaptic function depends on constant protein synthesis; therefore, neurons are especially vulnerable to chronic attenuation of RNA translation (Moreno et al., 2012). Although, transient suppression of translation is an adaptive cellular response to endoplasmic reticulum (ER) stress (Harding et al., 1999), chronic inhibition of RNA translation contributes to the pathogenesis of multiple neurodegenerative disorders, including tauopathies (Vanderweyde et al., 2012; Abisambra et al., 2013b; Ash et al., 2014). Pronounced ribosomal deficiencies appear in regions where tau pathology is evident (Ding et al., 2005), yet the link between tau and ribosomal function has not been established.
Protein synthesis drives memory formation (Duvarci et al., 2008). The fact that progressive memory loss is common to virtually all tauopathies suggests that ribosomal dysfunction could be an underlying mechanism leading to clinical symptoms. Indeed, tau binds to ribosomes in the brain, and this interaction is enhanced in tauopathic brains (Papasozomenos and Binder, 1987; Piao et al., 2002).
Here we show that ribosomes associate with pathologic and nonpathological tau. Consequently, ribosomal function becomes impaired and global protein synthesis is reduced including the synaptic protein PSD-95. These data suggest that tau-mediated ribosomal dysfunction is a common pathogenic process that affects synapses leading to cognitive impairment.
Materials and Methods
Human brain samples.
Human samples were obtained from the University of Kentucky (UK) Alzheimer's Disease Center. Sample collection and experimental procedures involving human tissue were in compliance with the UK Institutional Review board. Samples from Brodmann areas 21/22 were used. AD tissues were scored as Braak V (female, 93-year-old), VI (male, 88-year-old), and VI (female, 80-year-old); samples from non-demented controls were Braak I (male, 79-year-old), II (female, 94-year-old), and II (female, 88-year-old). The average postmortem interval was 3 h.
Microsomes, coimmunoprecipitation, and liquid-chromatography tandem mass spectrometry.
Microsomes were isolated as previously described (Lopez et al., 2007) and modified for the brain (Abisambra et al., 2010). Coimmunoprecipitation (co-IP) was performed as previously described (Jinwal et al., 2012) using anti-Tau46 (Cell Signaling Technology) and anti-actin (Sigma-Aldrich). Protein complexes were identified using liquid-chromatography tandem mass spectrometry (LC-MS/MS) and UNIPROT as previously described (Meier et al., 2015).
In vitro translation assay.
1-Step IVT Kit (Thermo) was used with minor modifications: a black bottom 96-well plate was loaded with in vitro translation (IVT)-kit components and 10 ng of recombinant proteins. GFP (ex-482 nm/em-512 nm) was measured every 15 min in a BioTek Synergy HT at 30°C for 6 h. Each sample was run in triplicate and analyzed using GraphPad Prism (Student's t test).
Cell culture, primary neurons, and immunoblotting.
Cell maintenance, harvesting, and tau expression were performed as previously described (Abisambra et al., 2012, 2013b). P0–P1 primary neurons were obtained as previously described (Abisambra et al., 2013a). Samples were processed for immunoblotting as described earlier using BCA (Pierce) to estimate protein concentration, tris-glycine gels (Invitrogen), and PVDF membranes (Jones et al., 2011). Primary antibodies: anti-tau h-150 (1:1000; Santa Cruz Biotechnology), actin (1:1000, Sigma-Aldrich), RPL28 and RPP0 (1:1000, GeneTex), PHF1 (1:500), Hsp70 (1:1000, ENZO), PSD-95 (1:1000, Cell Signaling Technology), and puromycin (1:1000, EMD Millipore). Bands were detected using (Pierce ECL). Image analysis was performed using ImageJ. Bands of the protein of interest were normalized to a loading control. Statistical analysis was performed using Student's t test in GraphPad Prism.
Surface sensing of translation.
Surface sensing of translation (SUnSET) was performed as previously described (Schmidt et al., 2009) with minor modifications: cells were incubated with 10 μg/ml of puromycin in cell culture media for 1 h before harvest. Proteins were analyzed using immunoblots.
Immunofluorescence.
Immunofluorescence (IF) was performed as previously described (Abisambra et al., 2013b). Primary antibodies: T22 (1:100) and RPS6 (1:250; Santa Cruz Biotechnology). Tissues were also stained with Sudan black and Neurotrace (1:200). Slides with AD and control sections were stained omitting primary antibodies to establish nonspecific background signal.
Microscopy.
A Nikon Eclipse Ti laser-scanning confocal microscope was used to capture images. Fields analyzed using 40× and 100× objectives included areas of tau staining with morphologic distribution in agreement with Neurotrace labeling. All acquisition intensities, field sizes, and settings were kept consistent across all images. Images were prepared using the NIS Elements 4.20 (Nikon) and Photoshop Cs6 (Adobe) software and were based upon cells that most closely represented the group.
Quantitative real-time PCR.
Total RNA was extracted from rTg4510 tau transgenic and littermate control primary neuronal cultures using EZNA total RNA Kit II according to manufacture instructions (Omega Bio-tek, catalog #R6934-01). RNA was quantified using a BioTek spectrophotometer and cDNA was produced using SuperScript IV (Invitrogen). Quantitative real-time PCR was performed using TaqMan Gene Expression probes for PSD-95 and GAPDH using a ViiA 7 Real Time PCR System (Applied Biosystems). PSD-95 expression was evaluated by normalizing to GAPDH as an internal control. The real-time values for each sample were average and evaluated using the comparative CT method.
Results
We recently determined ER-bound proteins associate with tau (Abisambra et al., 2013b; Meier et al., 2015). To identify tau-associated ER proteins in AD, microsomes were isolated from non-demented control and AD brain tissues. Full-length tau (or actin as control) was co-IP from microsomes, and tau-associated peptides were identified using LC-MS/MS. Of the 216 identified proteins, 68.5% were unique in AD, 22.2% were unique in control, and 9.3% were common between both groups (Fig. 1A). Proteins were grouped into functional categories based on UNIPROT. A striking difference was that tau associated with more RNA-binding proteins in AD than in control (Fig. 1B,C).
Figure 1.
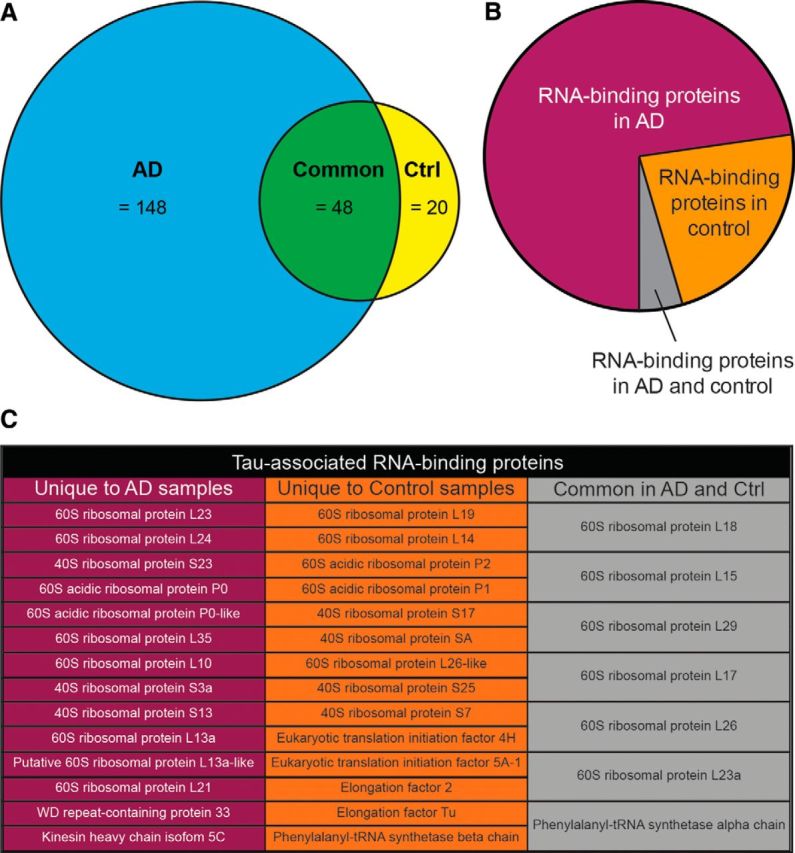
Tau associates with ER proteins differentially in AD versus control brains. A, Venn diagram showing the number of tau-associated ER proteins unique to AD brains (blue), unique to control brains (yellow), and common to both (green). A major group of proteins that was identified corresponded to RNA-binding proteins. B, Pie chart showing relative abundance of tau-associated, RNA-binding proteins identified in human AD and control brains by LC-MS/MS. C, List of proteins identified in B.
To further characterize the tau–ribosome association, we compared tau levels in AD and control subcellular fractions (Fig. 2A). Although tau levels were similar between the fractions lacking ribosomes, tau was significantly increased in the AD ribosomal fraction (Fig. 2A). We also detected a PHF1-positive smear in the AD ribosomes (Fig. 2B). PHF1 recognizes pS396/S404, which is associated with late stage tangles in AD (Greenberg et al., 1992).
Figure 2.

Pathological tau associates closely with ribosomes in AD brains. Representative immunoblots showing total tau (A) and PHF1-positive smear (B) enriched in AD brain ribosomal fraction. P0 and L28 (ribosomal proteins) and Hsp70 (cytosolic) confirm subcellular fractionation. C–E, Representative IF of AD brains showing overlap of oligomeric tau and rpS6 in human AD brain. Co-IF labeling of tau oligomers with T22 (red; C), rpS6 (green; D), and neurons (Neurotrace; blue). E, Merged inset image shows red and green overlap (arrowheads). F–H, Representative images of AD brains in which primary antibody incubation was omitted.
Recent studies show that oligomeric tau is highly toxic (Lasagna-Reeves et al., 2011), and it exhibits a prion-like behavior by propagating and seeding (Guo and Lee, 2011). We speculated that if aberrant tau–ribosome complexes were pathogenic, then tau oligomers would associate with ribosomes. To test this, we co-IF labeled tau oligomers and a ribosomal protein, rpS6, in AD and control brain sections (Fig. 2C–H). Confocal imaging revealed that tau oligomers and rpS6 signals overlapped (Fig. 2C–E), suggesting that oligomeric tau associates closely with ribosomes in AD.
The consequences of the tau–ribosome association are unknown. We hypothesized that the aberrant interaction between pathological tau species and ribosomes impairs translation. To test this, we measured the impact of tau on ribosomal function in three model systems. First, we tested the effect of tau oligomers on translation using a cell-free assay. We added recombinant tau oligomers or bovine serum albumin (BSA) as control with in vitro translation assay components and a GFP plasmid reporter. The rate of translation, measured by GFP, was significantly decreased in the presence of tau oligomers (Fig. 3A). To determine whether this effect was specific to tau oligomers, we tested the impact of oligomeric α-synuclein and insulin on translation (Fig. 3B). We found no significant change in GFP, suggesting that this effect is specific to tau.
Figure 3.

Pathological tau species decrease translation. A, IVT graph showing 32% reduction of translational output (after 6 h) in wells with tau oligomers compared with BSA control. B, Oligomeric α-synuclein and insulin did not affect translation. Immunoblots confirm the presence of oligomers in the translation assay; α-synuclein samples were denatured to yield monomers because high molecular weight oligomeric conformers are not detectable with anti-α-synuclein antibodies. C, Total and PHF1 tau are inversely proportional to the rate of protein synthesis. iHEK-Tau cells were stimulated with tetracycline to express tau for 4 d. Nascent proteins were tagged with puromycin. D, Cessation of tau expression rescues protein synthesis. Tau expression was induced with tetracycline for 24 or 96 h (lanes 1 and 2). At 96 h, puromycin levels decreased, whereas PHF1 increased. After continuous tau expression for 96 h (lanes 3 and 4), cells were washed and incubated with media lacking tetracycline, thereby halting tau expression for 24 or 96 h (lanes 3 and 4, respectively). E, Representative immunoblots showing the effect of wild-type (WT) and ΔK280 tau on protein synthesis. Changes in tau levels inversely correlated with puromycin. F, Quantification of E showing no significant difference in levels of protein synthesis between wild-type and ΔK280 mutant tau-expressing cells. G, SUnSET comparing the effect of P301L, ΔK280 tau, and WT in transiently transfected HEK293 cells. H, Quantification of G showing no significant difference between translation levels. I, Blot showing that overall translation (Puro) and PSD-95 are decreased in rTg4510 primary neurons. J, Quantification of I. Puromycin and PSD-95 were significantly decreased in rTg4510 neurons by 43 and 92%, respectively (*p < 0.05). K, Quantification of PSD-95 mRNA expression from rTg4510 neurons as measured by RT-PCR (**p = 0.0013).
We next measured the effect of pathological tau on the rate of translation in eukaryotic cells using SUnSET (Schmidt et al., 2009): a puromycin-based pulse assay. Puromycin, which is incorporated into recently translated proteins, was added to medium 1 h before harvesting cells. Nascent proteins, which appear as a smear, can then be quantified via immunoblots with anti-puromycin antibodies.
We used iHEK-Tau cells, an inducible HEK line that overexpresses wild-type human 4R0N tau upon addition of tetracycline (Abisambra et al., 2013b). Use of tetracycline allowed control over the start and overall duration of tau expression. We first performed a time course experiment in which tetracycline was added to iHEK-Tau cells over 72 h. We found that increased PHF1 and total tau levels correlated with decreased puromycin signal (Fig. 3C). We then performed a rescue experiment in which tau was expressed for 96 h and then tau expression was turned off for 24 or 96 h (Fig. 3D). We found that puromycin levels were rescued back to normal control once PHF1 levels were reduced.
Because mutations on tau are associated with risk for many tauopathies, we sought to determine whether mutant tau variants inhibited protein synthesis. To this end, we performed SUnSET in iHEK cells that overexpress the disease-associated ΔK280 tau (Rosso et al., 2003). We found that expression of WT or ΔK280 tau decreased protein synthesis (Fig. 3E), and that this reduction was not significantly different between the two cell lines (Fig. 3F). We then compared translation levels between mutant forms of tau by transiently transfecting HEK293 cells with WT, ΔK280, or P301L tau plasmids. We found no significant difference in protein synthesis levels between WT, ΔK280, or P301L-expressing cells (Fig. 3G,H). These data suggest that tau expression and accumulation of hyperphosphorylated tau impairs protein synthesis.
To determine the impact of pathological tau species in a more neurologically relevant model, we measured changes in the rate of translation in primary neurons derived from rTg4510 tau transgenic and control mice using SUnSET. The rTg4510 model overexpresses human P301L mutant tau (Santacruz et al., 2005). At 14 DIV, puromycin was added to the medium and primary neurons were harvested. Puromycin-tagged proteins were decreased by 43% in rTg4510 neurons (*p = 0.011) compared with control (Fig. 3I,J). We then investigated whether a more direct correlate of synaptic integrity could be affected by pathological tau in this model. To this end, we measured the levels of the PSD-95 and found that it was reduced by 92% in rTg4510 neurons (*p = 0.049; Fig. 3I,J). We hypothesized that tau-mediated disruption of RNA translation would result in increased mRNA levels. We performed RT-PCR and found that PSD-95 cDNA was increased by twofold in rTg4510 primary neurons compared with controls (Fig. 3K). These data suggest that P301L tau impairs RNA translation, and that this pathological mechanism directly affects translation without impairing gene expression.
Discussion
This study couples cellular and biochemical approaches with proteomics to expand the understanding of the ER-specific tau interactome. The unbiased proteomics approach (Fig. 1) identified 216 tau-associated ER proteins; many of which were unreported. The largest change corresponded to an increase in the association of ribosomal proteins with tau in AD. Using three in vitro models, we show that protein synthesis was significantly decreased as a consequence of the aberrant tau–ribosome association. Although it has been previously reported that tau interacted with ribosomes (Papasozomenos and Binder, 1987; Nelson et al., 1993), the functional consequences of this interaction was unknown until now. We have identified a dysfunctional consequence of tau–ribosome association that impairs protein synthesis, providing the first steps to understanding the mechanism delineating cognitive decline symptoms in tauopathic patients (Fig. 4).
Figure 4.

Schematic representation showing the consequences of pathological tau association with ribosomes. Under normal conditions, tau promotes tubulin polymerization and stabilizes microtubules. De novo protein synthesis, and in particular nascent neurotransmitter production is necessary for normal learning and memory. Our data suggest that in tauopathic brains, tau adopts aberrant conformations that associate with ribosomes. This interaction reduces nascent protein synthesis. Abrogation of new proteins impairs memory. This mechanism links the most common symptom of tauopathies.
LC-MS/MS results suggest that many other ER proteins (216 total) associate with tau. The use of mass spectrometry for identification of protein-protein interactions is limited and only provides a “first pass” suggestion. For example, whether these interactions are direct, indirect, or false-positives need to be investigated further. As shown in Figure 2, we validated the interaction between tau and ribosomes, and the validation of other interactions is currently underway. When comparing the RNA-binding proteins that associate with tau, we did not find elongation or initiation factors associating with tau in AD samples (Fig. 1C). These data suggest that the association of pathological tau with ribosomes abrogates the recruitment of translation factors to the tau–ribosome complex, and this could lead to reduced translation.
To avoid false-positive results from the LC-MS/MS, we implemented rigorous exclusion criteria (Meier et al., 2015), which omitted well known tau-associated proteins, but increased confidence in these interactions. Our co-IP focused on identifying mature, and not nascent, tau. We used a tau antibody (Tau46) that recognizes the carboxy-terminal tau sequence (404–441). As such this approach obviates caspase-cleaved tau, which is cleaved at D421 and is implicated in the formation of neurofibrillary tangles in AD (Rissman et al., 2004). Future efforts to identify cleaved tau-associated ER proteins might determine novel pathologic mechanisms. Nonetheless, our current list does provide further information about the diverse interactions of tau and other ER proteins.
Although tau mutations are not typically associated with risk for AD, there are 48 tau mutations that are associated with onset of other tauopathies (for review, see Zhang et al., 2015). The defining pathologic hallmark of tangles in AD is hyperphosphorylated tau (Grundke-Iqbal et al., 1986). Interestingly, we found that protein synthesis was impaired equally by WT and two disease-associated mutant tau variants: P301L and ΔK280. P301L tau is most commonly associated with frontotemporal dementia and Parkinsonism linked to chromosome 17 (FTDP-17; Clark et al., 1998). Expression of this form of tau in the rTg4510 transgenic mice leads to earlier onset and robust neurofibrillary tangle formation (Santacruz et al., 2005). The ΔK280 tau mutant is also associated with FTDP-17 (Rosso et al., 2003), as well as AD (Momeni et al., 2009). The presence of this mutation decreases tau's ability to bind to microtubules (Rizzu et al., 1999), and leads to increased levels of tau aggregation (Barghorn et al., 2000). Our findings indicate that tau-mediated impairment of protein synthesis could be a common mechanism of neuronal dysfunction between tauopathies (Fig. 4).
Our data support the hypothesis that pathological tau specifically reduces ribosomal function, which could lead to memory alterations in tauopathies. We also found that PSD-95, a synaptic protein that participates in learning and memory (Migaud et al., 1998), was decreased in rTg4510 neurons (Fig. 3I,J). Cognitive impairment seen tauopathy models could be attributed not only to reduced protein synthesis, but also to targeted decrease of synaptic proteins.
Interestingly, oligomers of other proteins (α-synuclein and insulin) did not alter ribosomal function (Fig. 3B). This finding suggests that there is a mechanism of ribosomal downregulation that specifically implicates tau, but not all other oligomeric and pathologically altered proteins. However, it is possible that proteins involved in the pathogenic mechanisms of other neurodegenerative disorders such as Aβ, poly-glutamines, and TDP-43 could also inhibit de novo protein synthesis. Experiments to test the effects of these oligomers are currently underway.
Characterization of this ribosome-directed molecular mechanism of tauopathies could provide novel therapeutic opportunities. For instance, therapeutic strategies aiming to uncouple pathological tau from the ribosome might restore RNA translation and prove effective to treat AD and other tauopathies. To do this, specific ribosomal proteins that associate with tau need to be identified as well as the discreet regions where tau binds to these proteins. This would guide the proof-of-concept use of peptides resembling these amino acid stretches to outcompete tau from associating with the ribosome and thereby restore protein synthesis.
This work suggests a direct effect of tau on translation by its association with ribosomes; however, there is also an indirect relationship between tau and translation. One of these is the chronic activation of the protein kinase RNA-like endoplasmic reticulum kinase (PERK). Accumulation of tau impairs ER-associated degradation, which then activates the unfolded protein response and subsequently the PERK pathway (Abisambra et al., 2013b). The prolonged activation of the PERK pathway leads to a reduction in RNA translation through phosphorylation of the initiation factor eIF2α (Marciniak et al., 2006). This alteration could be a cumulative result of tau's direct and indirect effects on translation. Our study provides further evidence that tau's involvement in disease is multi-faceted, that pathological tau heavily affects translation of vital proteins, and that the tau–ribosome complex could serve as a key therapeutic target for tauopathies.
Footnotes
The University of Kentucky Alzheimer's Disease Center (UK-ADC) and its Neuropathology Core is supported by NIH/NIA P30 AG028383; the University of Kentucky Proteomics Core is partially supported by grants from the NIH/NIGMS (P20GM103486) and the NIH/NCI (P30CA177558); this work was also supported in part by NIH R01NS077284 (H.Z.); the LC-MS/MS instrument was acquired with a High-End Instrumentation Grant S10RR029127 (H.Z.) from the National Institutes of Health; NIH/NINDS P30NS051220 supported the maintenance of the microscopy core used for imaging; J.F.A., D.L., and S.M. were supported by NIH/NINDS 1R01 NS091329-01, Alzheimer's Association NIRG-14-322441, NIH/NCATS 5UL1TR000117-04, NIH/NIGMS 5P30GM110787, GlaxoSmithKline, Department of Defense AZ140097, UK-ADC Pilot Award 8.1 supported by NIH/NIA P30 AG028383, the University of Kentucky Epilepsy Center and NIH/NIMHD L32 MD009205-01. We thank Dr Pedro Vera and the Lexington, VA Medical Center for support, Dr Peter Davies for his generous contribution of the PHF1 antibody used for immunofluorescent staining, Dr Chad Dickey for developing and sharing the iHEK cell lines and the tau plasmids, Ela Patel and Sonya Anderson with their assistance accessing the human brain tissue, Dr Fred Schmitt for insightful discussions and crucial intellectual contributions to this study, and the generous contribution of oligomers and T22 antibodies from Dr Rakez Kayed and Urmi Sengupta.
The authors declare no competing financial interests.
References
- Abisambra JF, Fiorelli T, Padmanabhan J, Neame P, Wefes I, Potter H. LDLR expression and localization are altered in mouse and human cell culture models of Alzheimer's disease. PloS One. 2010;5:e8556. doi: 10.1371/journal.pone.0008556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abisambra JF, Jinwal UK, Suntharalingam A, Arulselvam K, Brady S, Cockman M, Jin Y, Zhang B, Dickey CA. DnaJA1 antagonizes constitutive Hsp70-mediated stabilization of tau. J Mol Biol. 2012;421:653–661. doi: 10.1016/j.jmb.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abisambra J, Jinwal UK, Miyata Y, Rogers J, Blair L, Li X, Seguin SP, Wang L, Jin Y, Bacon J, Brady S, Cockman M, Guidi C, Zhang J, Koren J, Young ZT, Atkins CA, Zhang B, Lawson LY, Weeber EJ, et al. Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol Psychiatry. 2013a;74:367–374. doi: 10.1016/j.biopsych.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abisambra JF, Jinwal UK, Blair LJ, O'Leary JC, 3rd, Li Q, Brady S, Wang L, Guidi CE, Zhang B, Nordhues BA, Cockman M, Suntharalingham A, Li P, Jin Y, Atkins CA, Dickey CA. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J Neurosci. 2013b;33:9498–9507. doi: 10.1523/JNEUROSCI.5397-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer's Association. 2014 Alzheimer's disease facts and figures. Alzheimers Dement. 2014;10:e47–e92. doi: 10.1016/j.jalz.2014.02.001. [DOI] [PubMed] [Google Scholar]
- Ash PE, Vanderweyde TE, Youmans KL, Apicco DJ, Wolozin B. Pathological stress granules in Alzheimer's disease. Brain Res. 2014;1584:52–58. doi: 10.1016/j.brainres.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghorn S, Zheng-Fischhöfer Q, Ackmann M, Biernat J, von Bergen M, Mandelkow EM, Mandelkow E. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry. 2000;39:11714–11721. doi: 10.1021/bi000850r. [DOI] [PubMed] [Google Scholar]
- Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, Li D, Payami H, Awert F, Markopoulou K, Andreadis A, D'Souza I, Lee VM, Reed L, Trojanowski JQ, Zhukareva V, Bird T, Schellenberg G, Wilhelmsen KC. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci U S A. 1998;95:13103–13107. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Markesbery WR, Chen Q, Li F, Keller JN. Ribosome dysfunction is an early event in Alzheimer's disease. J Neurosci. 2005;25:9171–9175. doi: 10.1523/JNEUROSCI.3040-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvarci S, Nader K, LeDoux JE. De novo mRNA synthesis is required for both consolidation and reconsolidation of fear memories in the amygdala. Learn Mem. 2008;15:747–755. doi: 10.1101/lm.1027208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SG, Davies P, Schein JD, Binder LI. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem. 1992;267:564–569. [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Lee VM. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286:15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Jinwal UK, Abisambra JF, Zhang J, Dharia S, O'Leary JC, Patel T, Braswell K, Jani T, Gestwicki JE, Dickey CA. Cdc37/Hsp90 protein complex disruption triggers an autophagic clearance cascade for TDP-43 protein. J Biol Chem. 2012;287:24814–24820. doi: 10.1074/jbc.M112.367268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JR, Lebar MD, Jinwal UK, Abisambra JF, Koren J, 3rd, Blair L, O'Leary JC, Davey Z, Trotter J, Johnson AG, Weeber E, Eckman CB, Baker BJ, Dickey CA. The diarylheptanoid (+)-aR,11S-myricanol and two flavones from bayberry (Myrica cerifera) destabilize the microtubule-associated protein tau. J Nat Prod. 2011;74:38–44. doi: 10.1021/np100572z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011;6:39. doi: 10.1186/1750-1326-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez D, Abisambra Socarrás JF, Bedi M, Ness GC. Activation of the hepatic LDL receptor promoter by thyroid hormone. Biochim Biophys Acta. 2007;1771:1216–1225. doi: 10.1016/j.bbalip.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Marciniak SJ, Garcia-Bonilla L, Hu J, Harding HP, Ron D. Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J Cell Biol. 2006;172:201–209. doi: 10.1083/jcb.200508099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier S, Bell M, Lyons DN, Ingram A, Chen J, Gensel JC, Zhu H, Nelson PT, Abisambra JF. Identification of novel tau interactions with endoplasmic reticulum proteins in Alzheimer's brain. J Alzheimers Dis. 2015;48:687–702. doi: 10.3233/JAD-150298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O'Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- Momeni P, Pittman A, Lashley T, Vandrovcova J, Malzer E, Luk C, Hulette C, Lees A, Revesz T, Hardy J, de Silva R. Clinical and pathological features of an Alzheimer's disease patient with the MAPT delta K280 mutation. Neurobiol Aging. 2009;30:388–393. doi: 10.1016/j.neurobiolaging.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, Barrett DA, Tsaytler P, Bertolotti A, Willis AE, Bushell M, Mallucci GR. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature. 2012;485:507–511. doi: 10.1038/nature11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Marton L, Saper CB. Alz-50 immunohistochemistry in the normal sheep striatum: a light and electron microscope study. Brain Res. 1993;600:285–297. doi: 10.1016/0006-8993(93)91385-6. [DOI] [PubMed] [Google Scholar]
- Papasozomenos SC, Binder LI. Phosphorylation determines two distinct species of tau in the central nervous system. Cell Motil Cytoskeleton. 1987;8:210–226. doi: 10.1002/cm.970080303. [DOI] [PubMed] [Google Scholar]
- Piao YS, Hayashi S, Wakabayashi K, Kakita A, Aida I, Yamada M, Takahashi H. Cerebellar cortical tau pathology in progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. 2002;103:469–474. doi: 10.1007/s00401-001-0488-2. [DOI] [PubMed] [Google Scholar]
- Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, LaFerla FM, Rohn TT, Cotman CW. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004;114:121–130. doi: 10.1172/JCI200420640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzu P, Van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A, Niermeijer MF, Hillebrand M, Ravid R, Oostra BA, Goedert M, van Duijn CM, Heutink P. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in The Netherlands. Am J Hum Genet. 1999;64:414–421. doi: 10.1086/302256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosso SM, Donker Kaat L, Baks T, Joosse M, de Koning I, Pijnenburg Y, de Jong D, Dooijes D, Kamphorst W, Ravid R, Niermeijer MF, Verheij F, Kremer HP, Scheltens P, van Duijn CM, Heutink P, van Swieten JC. Frontotemporal dementia in The Netherlands: patient characteristics and prevalence estimates from a population-based study. Brain. 2003;126:2016–2022. doi: 10.1093/brain/awg204. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- Vanderweyde T, Yu H, Varnum M, Liu-Yesucevitz L, Citro A, Ikezu T, Duff K, Wolozin B. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci. 2012;32:8270–8283. doi: 10.1523/JNEUROSCI.1592-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CC, Xing A, Tan MS, Tan L, Yu JT. The role of MAPT in neurodegenerative diseases: genetics, mechanisms and therapy. Mol Neurobiol. 2015 doi: 10.1007/s12035-015-9415-8. Advance online publication. Retrieved Sep. 12, 2015. [DOI] [PubMed] [Google Scholar]