

Abstract
Homeostatic regulation is essential for the maintenance of synaptic strength within the physiological range. The current study is the first to demonstrate that both induction and reversal of homeostatic upregulation of synaptic vesicle release can occur within seconds of blocking or unblocking acetylcholine receptors at the mouse neuromuscular junction. Our data suggest that the homeostatic upregulation of release is due to Ca2+-dependent increase in the size of the readily releasable pool (RRP). Blocking vesicle refilling prevented upregulation of quantal content (QC), while leaving baseline release relatively unaffected. This suggested that the upregulation of QC was due to mobilization of a distinct pool of vesicles that were rapidly recycled and thus were dependent on continued vesicle refilling. We term this pool the “homeostatic reserve pool.” A detailed analysis of the time course of vesicle release triggered by a presynaptic action potential suggests that the homeostatic reserve pool of vesicles is normally released more slowly than other vesicles, but the rate of their release becomes similar to that of the major pool during homeostatic upregulation of QC. Remarkably, instead of finding a generalized increase in the recruitment of vesicles into RRP, we identified a distinct homeostatic reserve pool of vesicles that appear to only participate in synchronized release following homeostatic upregulation of QC. Once this small pool of vesicles is depleted by the block of vesicle refilling, homeostatic upregulation of QC is no longer observed. This is the first identification of the population of vesicles responsible for the blockade-induced upregulation of release previously described.
SIGNIFICANCE STATEMENT The current study is the first to demonstrate that both the induction and reversal of homeostatic upregulation of synaptic vesicle release can occur within seconds. Our data suggest that homeostatic upregulation of release is due to Ca2+-dependent priming/docking of a small homeostatic reserve pool of vesicles that normally have slow-release kinetics. Following priming, the reserve pool of vesicles is released synchronously with the normal readily releasable pool of synaptic vesicles. This is the first description of this unique pool of synaptic vesicles.
Keywords: acetylcholine receptor, mouse, neuromuscular junction, quantal content, synapse
Introduction
The ways in which activity regulates synaptic strength have been extensively studied, but are still only partially understood. There are a series of changes in synaptic function following manipulations of activity that appear to function to maintain the excitability of networks within certain boundaries. This type of synaptic plasticity is known as homeostatic regulation, and its cardinal feature is that changes in synaptic strength serve to oppose perturbation of synaptic function. Homeostatic regulation of synaptic function appears to involve several distinct mechanisms, including changes in the number of vesicles released [quantal content (QC)], and changes in the response to an individual quantum (quantal size) (Rich and Wenner, 2007; Turrigiano, 2012; Müller et al., 2015).
Some of the first descriptions of what would now be termed “homeostatic synaptic plasticity” were close to 35 years ago at mammalian neuromuscular junction (NMJ), where partial block of acetylcholine receptors (AChRs) triggers a compensatory increase in QC (Katz and Miledi, 1978; Cull-Candy et al., 1980; Wilson, 1982; Plomp et al., 1992; Tian et al., 1994; Wang et al., 2010b).
The majority of studies suggest that the induction of homeostatic regulation of QC occurs within 5–10 min at both the mammalian and Drosophila NMJ (Wilson, 1982; Tian et al., 1994; Frank et al., 2006; Weyhersmüller et al., 2011), while one study suggested it takes hours (Plomp et al., 1992). No study has been performed to determine the time course of the reversibility of homeostatic regulation of QC. The time course of reversibility may place limits on mechanisms that underlie the homeostatic regulation of QC.
Homeostatic regulation of QC has been most extensively studied at the Drosophila NMJ. Blocking postsynaptic receptors at the Drosophila NMJ has been shown to trigger upregulation of QC that is absent in flies with a point mutations in the α1 subunit of Cav2.1 Ca2+ channels (Frank et al., 2006, 2009) and is accompanied by an increase of presynaptic Ca2+ influx (Müller and Davis, 2012). A mechanism contributing to the increase in QC is an increase in the readily releasable pool (RRP) of synaptic vesicles, and this increase is blocked by mutations of RIM (rab3 interacting molecule; Müller et al., 2012) and RIM binding protein (Müller et al., 2015). Together, the studies in Drosophila suggest that both the upregulation of presynaptic Ca2+ entry and an increase in the RRP contribute to the upregulation of QC. The characteristics of the pool of vesicles recruited to increase the RRP during homeostatic upregulation of QC remains unknown.
In the current study, we set out to determine the time course, reversibility, and characteristics of the pool of synaptic vesicles recruited during homeostatic regulation of QC at the mammalian NMJ. We identified a distinct homeostatic reserve pool of vesicles that are rapidly and reversibly recruited during homeostatic upregulation of QC.
Materials and Methods
All procedures involving animals were approved by the Wright State Laboratory Animal Care and Use Committee.
Several strains of mice were used. In most experiments, mice expressing the yellow fluorescent protein (YFP) transgene driven by the Thy-1 promoter (Feng et al., 2000) were used. The presence of YFP in motor terminal allowed for easier identification of NMJs. In some experiments, where indicated, unaffected sibling mice of the ClCn1adr-mto2J strain (The Jackson Laboratory) were used (ClC mice). We have used this strain previously for studies of homeostatic regulation of QC (Wang et al., 2004, 2005, 2010b). We found no difference in homeostatic upregulation of QC between the two strains. GCamp2 and GCamp3 mice were obtained from The Jackson Laboratory (B6;CBA-Tg(Thy1-GCaMP2.2c)8Gfng/J, stock #017892; and B6;CBA-Tg(Thy1-GCaMP3)6Gfng/J, stock #017893). In addition to genotyping, each muscle was checked for Gcamp2/Gcamp3 fluorescence before recording. The experiment procedures used to measure QC in the mouse tibialis anterior (TA) muscle have been extensively described in previous publications (Wang et al., 2004, 2005, 2010b). Briefly, 2- to 3-month-old mice of either sex were killed using CO2 inhalation, and the TA muscle was removed and pinned in a Sylgard plated dish. After staining with 10 μm 4-(4-diethylaminostyryl)-N-methylpyridinium iodide (Invitrogen), NMJs were visualized using an epifluorescence microscope, and were perfused at a speed of 3–6 ml/min with an external solution containing the following (in mm): 118 NaCl, 0.7 Mg2SO4, 2 CaCl2, 3.5 KCl, 26.2 NaHCO3, 1.7 NaH2PO4, and 5.5 glucose, pH 7.3–7.4 (at 20–22°C), equilibrated with 95% O2 and 5% CO2. Endplate currents (EPCs) were recorded using two-electrode voltage clamp, and the nerve branch of the TA muscle was stimulated via an extracellular tungsten electrode (FHC).
Muscle fibers were crushed on both ends of the TA, away from the endplate band to eliminate contractions upon nerve stimulation, and the holding potential was set at −45 mV. In some experiments [where α-bungarotoxin (BTX) was used], a more negative holding potential was required to detect miniature EPCs (mEPCs) due to block of postsynaptic AChRs. In these experiments, muscle fibers were not crushed, and contraction was prevented by the addition of 1–3 μm μ-conotoxin GIIIB (Peptide Institute, Inc.) into the external solution to inhibit muscle Na+ channels. We have shown that the QC obtained from preparations treated with both methods is identical (Wang et al., 2004, 2005).
Drugs were applied either by adding them into the perfusate or by pressure puff delivery to individual NMJs with a picospritzer II (20 psi). α-BTX was purchased from Sigma-Aldrich and applied at a dose of 1 mg/ml for 2 min. d-Tubocurarine (d-TC) was purchased from Sigma-Aldrich, and bath applied at a dose of 0.1 μm and puffed at a concentration of 3 μm. 3,4-Diaminopyridine (DAP) was purchased from Sigma-Aldrich. For bar graphs of QC before and after block, EPC amplitude was determined from an average of 10 EPCs evoked at 0.5 Hz using the protocol shown in Figure 1B, while mEPC amplitude was determined from an average obtained from at least 30 mEPCs recorded over a 1 or 2 min period (depending on the frequency of mEPCs).
Figure 1.

Upregulation of QC triggered by blocking AChRs is rapidly reversible. A, QC was increased by partially blocking AChRs with two antagonists: a, QC was sampled at a muscle holding potential of −70 mV from six TA muscles from ClC mice before and 1 h after the muscle was briefly exposed to α-BTX (1 mg/ml, 2 min) to irreversibly block AChRs. The QC was 80.5 ± 6.3 before and 133.8 ± 5.1 after partial block of AChRs (p < 0.01), a 66% increase; b, QC was sampled at −70 mV from three TA muscles from YFP mice before, after incubation with 0.1 μm d-TC for 1 h, and after washout. QC increased from 63.2 ± 5.8 to 98.4 ± 5.9 (p < 0.01), a 56% increase. Muscle contraction was prevented by the addition of 1 μm μ-conotoxin in the bathing solution. The numbers inside each bar indicate the number of fibers recorded. B, Recording protocol used in C (and other experiments wherever indicated). Two EPCs at 0.5 Hz and 20 s of mEPCs were alternately recorded, and the averaged values were used to calculate the corresponding QC and construct the plot shown in C. EPCs and mEPCs are scaled differently so that they both fit on the same trace. C, Average EPC (up triangle), mEPC (down triangle), and QC (square) were plotted over time during infusion and washout of D-TC from a TA muscle preparation voltage clamped at -45 mV. The bathing solution was switched to a solution containing 0.1 μm d-TC, where indicated. D, Averaged traces of EPCs and mEPCs taken from the record shown in C. The time they were recorded is indicated by corresponding numbers. E, Averaged QC taken before and 3 min after d-TC infusion. Data from 19 endplates were pooled and averaged together. QC was increased from 76.5 ± 3.6 to 113.3 ± 6.9 by d-TC infusion (p < 0.01, paired t test). F–G, Average EPC, mEPC, and QC are plotted over time during a brief block of AChRs via the application of d-TC from a wide-bore patch pipette. Current traces shown in G correspond to the filled symbols in plot F. Immediately after 3 μm d-TC was pressure puffed (20 psi, 1 s) to the endplate being voltage clamped, both EPCs and mEPCs were not detectable. As the washout continued, first the EPC, then the mEPC became detectable. QC was obtained as soon as mEPCs could be accurately detected. QC was maximal as soon as it could be calculated and then declined to baseline over 2–3 min as d-TC was washed out.
A point-by-point deconvolution of EPCs was performed using a method detailed in studies by Rich et al. (2002) and Wang et al. (2010a). The averaged EPC was deconvolved with the fitted mEPC, which was obtained by best fitting onset-aligned and averaged mEPC records using the least-squares method.
A nested ANOVA (SYSTAT; Systat Software) was used for comparing the effect of drugs and other experimental manipulations. Plots and curve fittings were made using SigmaPlot software (Systat Software). The averaged results are expressed as the mean ± SEM. p Values <0.05 and <0.01 are denoted by one and two stars, respectively.
Results
Upregulation of QC following block of acetylcholine receptors is rapid and reversible
We and others (Plomp et al., 1992; Wang et al., 2010b) have previously shown that long-term blocking of AChRs in vivo with α-BTX triggers an increase in QC at the mouse NMJ. It has been suggested previously (Plomp et al., 1992) that this increase in QC following application of BTX takes >3 h to develop. In contrast, it has been reported by that the block of AChRs in vitro using d-TC triggers an increase in QC within 5–10 min (Wilson and Thomsen, 1992; Tian et al., 1997). The difference in time course between the reported effects of d-TC and BTX raises the possibility that different mechanisms underlie the increase in QC following application of these different AChRs blockers.
We re-examined whether both toxins were able to trigger increased QC within 1–2 h in vitro. Partial blockade of AChRs with either d-TC or BTX for 1–2 h in vitro triggered similar increases in QC (Fig. 1A). After the α-BTX exposure, QC was increased by 66% (p < 0.01). Under similar conditions with a similar degree of block of AChRs, d-TC induced a 56% increase (p < 0.01). These data are consistent with a similar time course of induction of increased QC following the application of either BTX or d-TC.
To determine the reversibility of the upregulation of QC, we tracked changes in QC in individual fibers during reversible block of AChRs with d-TC. To follow QC over time in individual fibers, two evoked EPCs were recorded every 20 s using a stimulation rate of 0.5 Hz. Between the recordings of EPCs, spontaneous mEPCs were recorded. By dividing the average EPC amplitude by the average mEPC amplitude of mEPCs collected during the following 20 s, we were able to determine QC every 22 s to follow changes over time (Fig. 1B). Individual muscle fibers were voltage clamped for up to 1 h as we repeatedly switched the solution between one containing d-TC and one lacking d-TC. During bath infusion of d-TC, mEPC amplitude was reduced over minutes and during this time there was an increase in QC (Fig. 1C,D). Experiments using the same protocol were repeated in 19 endplates from six mice. When measured before d-TC application and at the peak of QC increase during d-TC infusion, QC was increased by 48% (p < 0.01, paired t test; Fig. 1E), an increase similar to that induced by >1 h of either α-BTX or d-TC. The increase in QC paralleled the reduction in mEPC amplitude, and could repeatedly be induced and reversed in individual fibers within minutes.
To determine the speed of induction and reversibility of changes in QC as accurately as possible, we briefly puffed d-TC onto the NMJ being recorded using a wide-bore patch pipette (Fig. 1F,G). Immediately following the application of 3 μm d-TC, mEPC amplitudes fell below the level of detection. As d-TC was washed out over several minutes, mEPCs became detectable, and at that time QC was increased. As the smallest mEPCs fall into the noise, we likely underestimated the magnitude of the increase in QC. These data suggest that QC is increased within 90 s of the application of d-TC, and this increase was reversed as rapidly as we could wash out d-TC. There was no lag in the decrease in QC, such that QC decreased in parallel with the recovery of mEPC amplitude. We conclude that reversal of the increase in QC occurs within seconds. High-dose d-TC puff tests were repeatedly performed in three endplates, and the results were consistent. These data are the first demonstration that the increase in QC triggered by block of neurotransmitter receptors is both rapidly inducible and rapidly reversible.
Upregulation of QC following block of acetylcholine receptors is Ca2+ dependent
When either external Ca2+ level was lowered or external Mg2+ level was raised, blocking AChRs in the short term did not cause QC to increase (Fig. 2A). Manipulating the external Ca2+ or Mg2+ level could be preventing the upregulation of QC by blocking signaling in either the postsynaptic muscle fiber or via disruption of a Ca2+-dependent process in the presynaptic terminal. For example, a significant amount of Ca2+ enters skeletal muscle through AChRs (Scuka and Mozrzymas, 1992), and that Ca2+ entry might play a key role in regulating a retrograde signal from muscle that triggers the upregulation of QC.
Figure 2.

The homeostatic upregulation of QC is Ca2+ dependent. All experiments were performed using the protocol shown in Figure 1B. The dose of d-TC for all infusions was 0.1 μm. A, Averaged QC taken before and at the peak of d-TC infusion in variety of Ca2+ and Mg2+ combinations as indicated below each pair of bars (in mm). The numbers above the bars indicate the number of fibers recorded. Significant QC increase by d-TC (paired t test, from left to right: p = 0.49, p = 0.64, p = 0.34, p = 0.17, p = 0.20, p < 0.01, p < 0.01, p < 0.01) was seen in 1 Ca/0.7 Mg, 2 Ca/0.7 Mg, and 4 Ca/0.7 Mg groups (numbers represent concentrations in mm). B, QC upregulation is attenuated in transgenic mice expressing GCamp in motor neurons. QC values averaged from GCamp2 wild-type mice (n = 16) and transgenic (n = 27) mice, and GCamp3 wild-type (n = 7) and transgenic (n = 25) mice before and after the infusion of d-TC 0.1 μm are shown in the left panel. Although d-TC caused significant QC increases in both wild-type and transgenic groups, the percentage increases in the transgenic groups were significantly attenuated, as shown in the right panel. C, The frequency of mEPCs, reported as the number of events per 20 s, was measured from 27 endplates before and after the increase in QC induced by the infusion d-TC (paired t test, p = 0.67). D, Plot of an experiment in which a QC increase induced by 0.1 μm d-TC infusion was absent in a low-Ca2+ (2 mm Ca2+/8 mm Mg2+) solution. Upon the addition of 40 μm 3,4-DAP, the baseline QC increased from 10 to 80. Following the addition of d-TC, QC increased further to 120. E, Representative EPC and mEPC traces taken from the corresponding numbers in D. F, Data were collected in 2 mm Ca2+/8 mm Mg2+ in the presence (n = 4, p < 0.01) and absence (n = 19, p = 0.34) of 40 μm 3,4-DAP.
To selectively manipulate presynaptic Ca2+ levels, we used transgenic mice in which GCamp2 and GCamp3 were driven by the Thy1 promoter (Chen et al., 2012). We hypothesized that the GCamp Ca2+ indicator dyes would partially buffer the presynaptic Ca2+ signal (Tian et al., 2009; Faas et al., 2011). Baseline QC was not reduced in GCamp transgenic mice. However, in both lines, transgenic mice exhibited attenuated upregulation of QC triggered by partial AChR block (Fig. 2B). The attenuation of homeostatic synaptic plasticity in these mice is consistent with the possibility that the effect of manipulation of external Ca2+ or Mg2+ in wild-type mice is due to effects on Ca2+ in the presynaptic nerve terminal rather than the muscle.
Presynaptic voltage-gated Ca2+ influx during the action potential is critical
The Ca2+ dependence of the homeostatic increase in QC could derive from either an increase in basal levels of Ca2+ in the nerve terminal or an increase in Ca2+ influx during the action potential. If there was an increase in basal Ca2+, we would expect to see an increase in mEPC frequency (Angleson and Betz, 2001). However, increased QC was not paralleled by an increase in mEPC frequency (Fig. 2C). This suggested that the Ca2+ dependence of the increase in QC was most likely due to increased Ca2+ entry during the action potential rather than an increase in resting Ca2+ level.
We tested the role of voltage-gated Ca2+ entry into the presynaptic terminal by application of 3,4-DAP, which enhances Ca2+ entry during action potentials by blocking presynaptic voltage-gated K+ channels (Thomsen and Wilson, 1983). The addition of 3,4-DAP increased baseline QC under conditions of low external Ca2+ back to normal levels and restored the d-TC-induced increase in QC (Fig. 2D,F). These data suggest the Ca2+ dependence of the increase in QC derives from Ca2+ entry through presynaptic voltage-gated Ca2+ channels during the action potential.
Blocking AChRs increases RRP size
We found previously that BTX treatment in vivo increased the number of releasable vesicles (the binomial parameter n) rather than increasing the probability of release (the binomial parameter p; Wang et al., 2010b). We examined whether partial block of AChRs increased n by increasing the size of RRP (Rosenmund and Stevens, 1996). The size of RRP can be estimated by measuring cumulative QC evoked by high-frequency stimulations (Schneggenburger et al., 1999; Liu et al., 2011), assuming that the release probability and the rate of replenishment are constant, and therefore depression during high-frequency stimulation is mainly due to the reduction of vesicles in the RRP. In the example shown in Figure 3, the increase in QC measured using 0.5 Hz stimulation (Fig. 3A) was paralleled by an increase in the calculated RRP size from 289 to 469, as estimated from the y-intercept of the linear fit of data plotted in Figure 3D. An average increase of 245 quanta (from 280.3 ± 24.1 to 524.5 ± 40.4) was obtained from eight individual NMJs (Fig. 3E). The increase in RRP size was not accompanied by an increase in the steady-state QC during 100 Hz stimulation (Fig. 3C), such that the slope of the data plotted in Figure 3D was unaltered. These data suggest that the block of AChRs selectively increases the RRP, while leaving the rate of vesicle mobilization unaltered.
Figure 3.

Blocking AChRs increases the RRP size. A, QC from an individual NMJ was monitored during the infusion of d-TC, as previously described. Two 100 Hz trains were recorded where indicated by the numbers. B, Fifty EPCs recorded in response to 100 Hz stimulation at times 1 (baseline, top) and times 2 (in the presence of d-TC, bottom). Averaged mEPC traces before train stimulation at each time point are shown in the right panel. C, Plot of QC, which was calculated from the example shown in A and B. D, The cumulative QC plot was constructed from the plot of QCs shown in C, and the RRP size was estimated by the intercepts of linear regression of the linear portion of the cumulative QC plot (last 20 points). Upon d-TC blockade, RRP was increased to 468.9 from an initial value of 288.7, while the slope was unchanged (34.7 vs 34.8). E, Averaged results from eight individual NMJs show that upon AChRs blockade, RRP was increased (from 280.3 ± 24.1 to 524.5 ± 40.4; p < 0.01). The steady-state QC was unchanged by d-TC infusion (average QC from the last 20 pulses was 29.0 ± 1.9 at baseline and 27.9 ± 1.9 after d-TC infusion; p = 0.13). 1st QC, First QC of the train (p < 0.01); P-QC, QC when EPC amplitude has plateaued.
Deconvolution of EPCs reveals an increase in synchronized release following partial block of AChRs
One change that might cause an increase in the RRP is an increase in the amount of Ca2+ entering through Ca2+ channels. We have previously used deconvolution of EPCs with mEPCs to demonstrate that with increased Ca2+ entry there is an increase in the peak rate of vesicle release (Wang et al., 2010a). We term this wave of rapidly released vesicles “synchronously released vesicles” (Fig. 4A). In addition to synchronously released vesicles, there is a continued release of vesicles at a lower rate that we term “late release” (Fig. 4A). As external Ca2+ is increased, there is a greater increase in late release than in synchronous release (Wang et al., 2010a). This leads to an increase in the ratio of late to total release (Fig. 4A–E).
Figure 4.
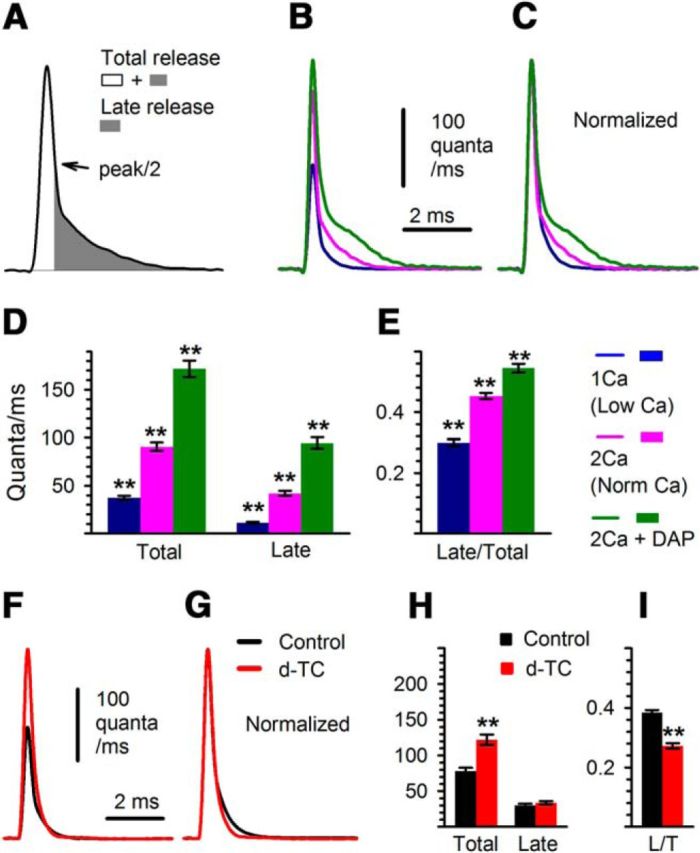
Changes in release kinetics following d-TC are distinct from the changes triggered by increased Ca2+ entry. A, Measurements of total release and late release (defined as the integral from half of the peak on the decay phase to the end of the decay, shaded area) are illustrated. B, Deconvolution traces, obtained at 1 mm (n = 29, blue) and 2 mm (n = 46, magenta) Ca2+ and 0.7 mm Mg2+, and at 2 mm Ca2+ and 0.7 mm Mg2+ with the addition of 10 μm 3,4-DAP (n = 26, green). The superimposed deconvolution waveforms shown represent average traces for each condition. C, Shown are the waveforms in B normalized so that the ratio of late to peak release can be compared. D, E, Bar graphs of total and late release, and the ratio of total to late release. All differences are statistically significant (p < 0.01). F, G, Averaged deconvolution traces obtained from 41 NMJs before (black trace) and after (red trace) AChRs were partially blocked by d-TC are superimposed (F) and peak normalized (G). H, I, Corresponding statistics of measurements are shown (Total, p < 0.01; Late, p = 0.33; ratio of Late to Total (L/T) release, p < 0.01).
It has been proposed that homeostatic upregulation of QC in Drosophila may be due to the broadening of presynaptic action potentials such that there is prolongation of presynaptic Ca2+ entry (Younger et al., 2013). We examined the effect of broadening the presynaptic action potential on release using 10 μm 3,4-DAP to block presynaptic K channels (Thomsen and Wilson, 1983) and found that late release was increased to a greater degree than peak release (Fig. 4B–E). Thus, both the elevation of extracellular Ca2+ and the widening of the action potential trigger an increase in the ratio of late to total release.
We next examined the change in release kinetics triggered during homeostatic upregulation of QC. The changes in the kinetics of release following the application of d-TC were distinct from those induced by either increasing the external Ca2+ level or widening the presynaptic action potential. Instead of triggering an increase in the ratio of late to total release, partial block of AChRs selectively increased the peak rate of release such that the ratio of late to total release was decreased (Fig. 4F–I). The marked difference between the effects of elevated external Ca2+ level and partial block of AChRs suggest that Ca2+ entry is not increased following homeostatic upregulation of release at the mouse NMJ. This agrees with data in Figure 2 showing no homeostatic upregulation of QC when external Ca2+ is reduced. It further suggests there is no broadening of the presynaptic action potential at the mouse NMJ. We conclude that adequate Ca2+ entry plays a permissive role in homeostatic upregulation of QC, but there is no increase in Ca2+ entry.
Inhibition of vesicle recycling does not block the initial d-TC-induced increase in QC, but blocks subsequent upregulation of QC
We wished to determine how the RRP is increased to cause the increase in QC. Partial block of AChRs might increase the size of the RRP by increasing vesicle mobilization/recycling. One way to increase vesicle mobilization is to rapidly endocytose membrane and refill vesicles with neurotransmitter. Another is to move vesicles from a reserve pool. If rapid vesicle recycling and refilling is the mechanism underlying the increase in the RRP, blocking vesicle refilling should prevent the homeostatic increase in QC. To examine this possibility, we interrupted vesicle refilling by applying 5 μm vesamicol, a vesicular ACh transporter inhibitor (Van der Kloot, 2003). At a concentration of 5 μm, vesamicol was found to have no detectable postsynaptic effect (Enomoto, 1988). It does not alter the quantal size of preformed vesicles (Van der Kloot, 2003) and does not disturb synaptic vesicle recycling (Parsons et al., 1999), but it completely abolishes refilling (Bahr and Parsons, 1986). As expected, vesamicol caused a gradual decrease in QC over time as vesicles were released, but not refilled (Fig. 5A). Vesamicol did not block the initial response of NMJ to the infusion of d-TC (Fig. 5A). However, subsequent infusions of d-TC no longer caused an increase in QC in both the NMJ recorded from during the initial infusion of d-TC as well as all other NMJs from the same muscle that were exposed to the initial infusion of d-TC. This finding was repeated in 10 muscles. These data suggest that the pool of vesicles mobilized during homeostatic upregulation of QC was almost completely depleted following the first application of d-TC, at a time when the pool of vesicles normally released was reduced by <40%. We conclude that the pool of vesicles mobilized following homeostatic upregulation of QC is distinct from the pool of vesicles normally released. We term this novel pool of vesicles the “homeostatic reserve pool.”
Figure 5.
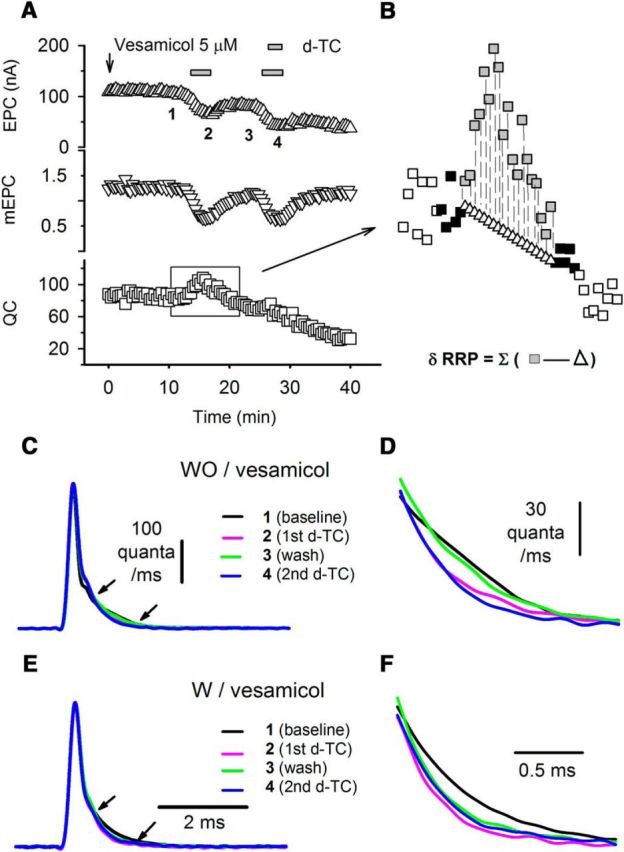
Vesamicol does not block the initial d-TC-induced increase in QC, but blocks the subsequent upregulation of QC. A, An example of the action of vesamicol. Averaged EPC (upward triangle), mEPC (downward triangle), and QC (square) values are plotted against time. Vesamicol 5 μm was added into the bathing solution where indicated by the arrow and was present in the bathing solution for the rest of the recording. Low-dose d-TC (0.1 μm) was infused and washed out twice, as indicated during the recording. QC was increased only during the first d-TC infusion. No clear increase in QC could be detected for the subsequent infusion of d-TC. B, Estimation of pool size. The method used to calculate the size of the vesicle pool recruited by infusion of d-TC is indicated by the expanded area of the square region of the QC plot. In the expanded plot, filled squares indicate QC at time points used to generate the linear fit of the decay in QC. Open triangles indicate the estimate for what QC would have been without infusion of d-TC at each time point. Gray squares indicate the QC actually measured. Open squares are QCs that were measured, but not used for calculations. C, Deconvolution traces derived from the experiment shown in Figure 1C each represent the average of five traces from the times indicated by the corresponding numbers in Figure 1C. The traces are shown superimposed with the peak normalized. D, Zoomed plot of the region in C indicated by the arrows shows differences in late release before and after each infusion of d-TC. In comparison, E and F show averaged deconvolution traces, derived from the experiment shown in A, processed in the same manner as in C and D, in a situation where vesicle recycling was blocked by the infusion of vesamicol.
The finding that the homeostatic reserve pool of vesicles was completely depleted during the initial infusion of d-TC allowed us to estimate the size of the pool. We took into account the gradual reduction in QC induced by vesamicol by linearly fitting the reduction in QC (Fig. 5B). We used the fitted QC to estimate what the QC would have been in the absence of d-TC and subtracted this from the observed QC at each time point. By summing the “additional” vesicles released during each EPC in the presence of d-TC, we calculated the size of the recruited pool and obtained an estimate of 316.0 ± 38.5 (n = 10 NMJs). This estimate agrees reasonably well with the estimated increase of 244 in RRP size obtained by high-frequency repetitive stimulation (Fig. 3) and thus suggests that the recruitment of the homeostatic reserve pool can fully account for the changes we found in the response to repetitive stimulation following partial block of AChRs.
We wished to further characterize the homeostatic reserve pool. Homeostatic upregulation of QC is absent in a solution containing a low external Ca2+ level (Fig. 2). Late release of vesicles is also absent in a solution containing a low external Ca2+ level (Fig. 4). This correlation led to the hypothesis that the homeostatic reserve pool normally participates primarily in late release and is recruited into the synchronously released pool to cause upregulation of QC. Supporting this was the finding that homeostatic upregulation of QC is paralleled by a rapidly reversible decrease in the percentage of vesicles released late (Fig. 5C,D). If homeostatic upregulation of QC is due to the recruitment of a distinct homeostatic reserve pool of vesicles, vesamicol should lead to a permanent decrease in late release as it prevents refilling of the homeostatic reserve pool. As shown in Figure 5, E and F, the decrease in late release triggered by d-TC was maintained following washout of d-TC in the presence of vesamicol. Our interpretation is that the pool of vesicles that normally participate in late release was completely depleted during homeostatic upregulation of QC in the presence of vesamicol. We conclude that the homeostatic reserve pool is distinct from the pool of vesicles that are synchronously released and normally have slower release kinetics.
Discussion
We provide evidence that homeostatic upregulation of QC is mediated by a rapid and reversible recruitment of a small pool of synaptic vesicles that we term the homeostatic reserve pool. We demonstrate that adequate Ca2+ entry during action potentials is necessary for the recruitment of this pool of vesicles during homeostatic upregulation of QC. However, our data suggest that there is no upregulation of Ca2+ entry during action potentials during homeostatic upregulation of QC. Inhibition of vesicle refilling as well as repetitive stimulation indicate that the homeostatic reserve pool of vesicle is small (∼300) and can be rapidly depleted. Deconvolution of EPCs to determine release kinetics suggests that the homeostatic reserve pool normally participates primarily in late release, and its recruitment is accompanied by an increase in its rate of vesicle release.
Mobilization of vesicles from a small, unique pool underlies homeostatic upregulation of QC at the mouse NMJ
Several findings in the current study suggest the homeostatic reserve pool is distinct from the normal RRP vesicles. First, the homeostatic reserve pool is more dependent on vesicle recycling than the pool normally participating in release. This conclusion is supported by experiments using vesamicol to prevent the refilling of recycled vesicles as well as the finding that during repetitive stimulation the homeostatic increase in QC is eliminated. Our interpretation is that this pool of vesicles dose not recycle fast enough to maintain the homeostatic upregulation of QC during repetitive stimulation. Second, deconvolution suggests that the homeostatic reserve pool is normally released later than the normal pool of vesicles. Recruitment of these vesicles appears to be accompanied by a selective increase in their rate of release such that release becomes more synchronous. Third, the homeostatic reserve pool is more sensitive to the reduction of extracellular Ca2+ than the normal pool of vesicles. This is suggested by the elimination of homeostatic upregulation of QC when extracellular Ca2+ is reduced.
Putting the above findings together led us to propose the model in Figure 6. In low Ca2+ the homeostatic reserve pool is functionally absent due to a lack of Ca2+-dependent vesicle mobilization. Thus, when AChRs are blocked, there is no homeostatic upregulation of QC. As the external Ca2+ level is increased and sufficient Ca2+ entry is ensured, the homeostatic reserve pool is mobilized, but is primarily limited to participating in late release. Following partial block of AChRs in normal Ca2+, the kinetics of release of the homeostatic reserve pool speed up to become similar to the synchronously released pool of vesicles. Sufficient Ca2+ entry during action potentials appears necessary to allow for Ca2+-dependent priming and/or recycling (Ceccarelli and Hurlbut, 1980; Sakaba and Neher, 2001) of the homeostatic reserve pool. The increased synchronization of release is both rapidly induced and rapidly reversed as AChRs are blocked and unblocked. The rapid reversal stems from the small size of the homeostatic reserve pool [300 of a total of 174,000 vesicles per mouse NMJ (Schofield and Marshall, 1980)], such that vesicles in that pool that are primed for synchronous release are rapidly depleted following washout of AChR blockers.
Figure 6.

Blocking postsynaptic AChRs mobilizes a homeostatic reserve pool of vesicles at the mouse NMJ. Illustration of single action potential evoked synaptic vesicle release (top diagram) and its corresponding release profile obtained by deconvolution of evoked current (bottom). When extracellular Ca2+ entry is low, the only pool of vesicles participating in release is the synchronously released pool of vesicles (blue vesicles). Both release probability and the number of releasable vesicles are low. With the elevation of extracellular Ca2+ level, there is an increase in the pool of synchronously released vesicles. The time course of release of these vesicles is indicated by vertical blue lines in the deconvolution profile. In normal Ca2+ levels, there is also recruitment of the homeostatic reserve pool (red vesicles), but most vesicles in this pool are released slowly, after the peak of vesicle release is over (red dotted arrow in diagram, vertical red lines in deconvolution profile). Following partial block of AChRs, the homeostatic reserve pool becomes primed and is released as rapidly as the synchronously released pool (superimposed red and blue vertical lines, deconvolution profile). The homeostatic reserve pool is small and must be continuously replenished through vesicle recycling (red arrow).
Comparison of homeostatic regulation at the mouse and Drosophila NMJ
Homeostatic upregulation of QC has been extensively studied at the Drosophila NMJ. There are a number of features shared by homeostatic regulation at mouse and Drosophila NMJs. In both, there is upregulation of QC following partial block of postsynaptic receptors, with a strong correlation between the degree of block and the upregulation of QC (Plomp et al., 1994; Frank et al., 2006). In neither synapse is there a change in the rate of spontaneous mEPCs (Frank et al., 2006; our study), such that it appears unlikely that there is an increase in resting presynaptic Ca2+ level. In addition, in both systems the increase in QC is due to an increase in the RRP (Wang et al., 2010b; Müller et al., 2012, 2015; our study) and is not sustained during repetitive stimulation, suggesting a special dependence on rapid vesicle replenishment (Wilson, 1982; Tian et al., 1994; Frank et al., 2006; our study). These findings suggest that the homeostatic regulation of QC is a similar process in the mouse and Drosophila.
However, our study suggests there are several fundamental differences between the mouse and Drosophila. (1) In the mouse, upregulation of QC occurred as rapidly as we could measure it. In Drosophila, there is a several minute lag between the block of receptors and the upregulation of QC (Frank et al., 2006). (2) When external Ca2+ concentration is reduced, there is no increase in QC following either short-term block (Tian et al., 1994; Wilson et al., 1995; our study) or long-term block (Gallant, 1982; Plomp et al., 1994) of AChRs at the mammalian NMJ. In Drosophila, the increase in QC occurs at both low and normal extracellular Ca2+ levels (Frank et al., 2006; Müller et al., 2015). (3) Finally, in Drosophila the upregulation in QC is accompanied by an increase in presynaptic Ca2+ entry (Müller and Davis, 2012). In the mouse, we find no evidence to suggest the upregulation of Ca2+ entry. If there was an increase in Ca2+ entry, the increase should be sufficient to trigger an increase in QC independent of vesicle mobilization as the relationship between external Ca2+ and QC is most dramatic when external Ca2+ is low (Wang et al., 2010a). Our deconvolution analysis provides further evidence that there is no increase in presynaptic Ca2+ entry at the mouse NMJ. These differences suggest fundamental differences in homeostatic mechanisms between the two model synapses.
Time course and reversibility
Ours is the first study of the reversibility of homeostatic regulation of QC. We found that both the induction and reversal of homeostatic regulation of QC occurred in parallel with the block of AChRs. These data suggest that homeostatic upregulation and downregulation of QC occur faster than the 22 s sampling rate of QC that we were able to achieve. These findings have implications for underlying mechanisms. First, the signal that triggers the upregulation in QC must be induced rapidly. Second, the signal must have a half-life of <22 s. Finally, the signal needs to be able to be turned on and off repeatedly within minutes.
Our data suggest that homeostatic upregulation of QC is due to Ca2+-dependent priming/docking of a small homeostatic reserve pool of vesicles that normally have slow-release kinetics. Following activation of the homeostatic reserve pool, it is released synchronously with the normal RRP of synaptic vesicles. Once this small pool of vesicles is depleted, homeostatic upregulation of QC does not occur.
Footnotes
This work was supported by National Institutes of Health Grant P01-NS-057228 (M.M.R.) and Wright State University Emerging Science Grant 229118 (X.W.). We thank Kathrin Engisch and Peter Wenner for helpful comments.
The authors declare no competing financial interests.
References
- Angleson JK, Betz WJ. Intraterminal Ca(2+) and spontaneous transmitter release at the frog neuromuscular junction. J Neurophysiol. 2001;85:287–294. doi: 10.1152/jn.2001.85.1.287. [DOI] [PubMed] [Google Scholar]
- Bahr BA, Parsons SM. Demonstration of a receptor in Torpedo synaptic vesicles for the acetylcholine storage blocker L-trans-2-(4-phenyl[3,4–3H]-piperidino) cyclohexanol. Proc Natl Acad Sci U S A. 1986;83:2267–2270. doi: 10.1073/pnas.83.7.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli B, Hurlbut WP. Ca2+-dependent recycling of synaptic vesicles at the frog neuromuscular junction. J Cell Biol. 1980;87:297–303. doi: 10.1083/jcb.87.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Cichon J, Wang W, Qiu L, Lee SJ, Campbell NR, Destefino N, Goard MJ, Fu Z, Yasuda R, Looger LL, Arenkiel BR, Gan WB, Feng G. Imaging neural activity using Thy1-GCaMP transgenic mice. Neuron. 2012;76:297–308. doi: 10.1016/j.neuron.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy SG, Miledi R, Trautmann A, Uchitel OD. On the release of transmitter at normal, myasthenia gravis and myasthenic syndrome affected human end-plates. J Physiol. 1980;299:621–638. doi: 10.1113/jphysiol.1980.sp013145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto K. Post- and presynaptic effects of vesamicol (AH5183) on the frog neuromuscular junction. Eur J Pharmacol. 1988;147:209–215. doi: 10.1016/0014-2999(88)90779-0. [DOI] [PubMed] [Google Scholar]
- Faas GC, Raghavachari S, Lisman JE, Mody I. Calmodulin as a direct detector of Ca2+ signals. Nat Neurosci. 2011;14:301–304. doi: 10.1038/nn.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/S0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Pielage J, Davis GW. A presynaptic homeostatic signaling system composed of the Eph receptor, ephexin, Cdc42, and CaV2.1 calcium channels. Neuron. 2009;61:556–569. doi: 10.1016/j.neuron.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallant PE. The relationship between anti-acetylcholine receptor antibody levels and neuromuscular function in chronically myasthenic rats. J Neurol Sci. 1982;54:129–141. doi: 10.1016/0022-510X(82)90225-8. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. A re-examination of curare action at the motor endplate. Proc R Soc Lond B Biol Sci. 1978;203:119–133. doi: 10.1098/rspb.1978.0096. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sugiura Y, Lin W. The role of synaptobrevin1/VAMP1 in Ca2+-triggered neurotransmitter release at the mouse neuromuscular junction. J Physiol. 2011;589:1603–1618. doi: 10.1113/jphysiol.2010.201939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Davis GW. Transsynaptic control of presynaptic Ca(2)(+) influx achieves homeostatic potentiation of neurotransmitter release. Curr Biol. 2012;22:1102–1108. doi: 10.1016/j.cub.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Liu KS, Sigrist SJ, Davis GW. RIM controls homeostatic plasticity through modulation of the readily releasable vesicle pool. J Neurosci. 2012;32:16574–16585. doi: 10.1523/JNEUROSCI.0981-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Genç Ö, Davis GW. RIM-binding protein links synaptic homeostasis to the stabilization and replenishment of high release probability vesicles. Neuron. 2015;85:1056–1069. doi: 10.1016/j.neuron.2015.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons RL, Calupca MA, Merriam LA, Prior C. Empty synaptic vesicles recycle and undergo exocytosis at vesamicol-treated motor nerve terminals. J Neurophysiol. 1999;81:2696–2700. doi: 10.1152/jn.1999.81.6.2696. [DOI] [PubMed] [Google Scholar]
- Plomp JJ, van Kempen GT, Molenaar PC. Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J Physiol. 1992;458:487–499. doi: 10.1113/jphysiol.1992.sp019429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp JJ, van Kempen GT, Molenaar PC. The upregulation of acetylcholine release at endplates of α-bungarotoxin-treated rats: its dependency on calcium. J Physiol. 1994;478:125–136. doi: 10.1113/jphysiol.1994.sp020236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich MM, Wenner P. Sensing and expressing homeostatic synaptic plasticity. Trends Neurosci. 2007;30:119–125. doi: 10.1016/j.tins.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Rich MM, Wang X, Cope TC, Pinter MJ. Reduced neuromuscular quantal content with normal synaptic release time course and depression in canine motor neuron disease. J Neurophysiol. 2002;88:3305–3314. doi: 10.1152/jn.00271.2002. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/S0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron. 2001;32:1119–1131. doi: 10.1016/S0896-6273(01)00543-8. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/S0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Schofield GG, Marshall IG. Neuromuscular transmission in the athymic nude mouse. J Neurol Sci. 1980;48:21–34. doi: 10.1016/0022-510X(80)90147-1. [DOI] [PubMed] [Google Scholar]
- Scuka M, Mozrzymas JW. Postsynaptic potentiation and desensitization at the vertebrate end-plate receptors. Prog Neurobiol. 1992;38:19–33. doi: 10.1016/0301-0082(92)90033-B. [DOI] [PubMed] [Google Scholar]
- Thomsen RH, Wilson DF. Effects of 4-aminopyridine and 3,4-diaminopyridine on transmitter release at the neuromuscular junction. J Pharmacol Exp Ther. 1983;227:260–265. [PubMed] [Google Scholar]
- Tian L, Prior C, Dempster J, Marshall IG. Nicotinic antagonist-produced frequency-dependent changes in acetylcholine release from rat motor nerve terminals. J Physiol. 1994;476:517–529. doi: 10.1113/jphysiol.1994.sp020151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Prior C, Dempster J, Marshall IG. Hexamethonium- and methyllycaconitine-induced changes in acetylcholine release from rat motor nerve terminals. Br J Pharmacol. 1997;122:1025–1034. doi: 10.1038/sj.bjp.0701481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harbor Perspect Biol. 2012;4:a005736. doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Kloot W. Loading and recycling of synaptic vesicles in the Torpedo electric organ and the vertebrate neuromuscular junction. Prog Neurobiol. 2003;71:269–303. doi: 10.1016/j.pneurobio.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Wang X, Engisch KL, Li Y, Pinter MJ, Cope TC, Rich MM. Decreased synaptic activity shifts the calcium dependence of release at the mammalian neuromuscular junction in vivo. J Neurosci. 2004;24:10687–10692. doi: 10.1523/JNEUROSCI.2755-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li Y, Engisch KL, Nakanishi ST, Dodson SE, Miller GW, Cope TC, Pinter MJ, Rich MM. Activity-dependent presynaptic regulation of quantal size at the mammalian neuromuscular junction in vivo. J Neurosci. 2005;25:343–351. doi: 10.1523/JNEUROSCI.3252-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Pinter MJ, Rich MM. Ca2+ dependence of the binomial parameters p and n at the mouse neuromuscular junction. J Neurophysiol. 2010a;103:659–666. doi: 10.1152/jn.00708.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Q, Engisch KL, Rich MM. Activity-dependent regulation of the binomial parameters p and n at the mouse neuromuscular junction in vivo. J Neurophysiol. 2010b;104:2352–2358. doi: 10.1152/jn.00460.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyhersmüller A, Hallermann S, Wagner N, Eilers J. Rapid active zone remodeling during synaptic plasticity. J Neurosci. 2011;31:6041–6052. doi: 10.1523/JNEUROSCI.6698-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DF. Influence of presynaptic receptors on neuromuscular transmission in rat. Am J Physiol. 1982;242:C366–C372. doi: 10.1152/ajpcell.1982.242.5.C366. [DOI] [PubMed] [Google Scholar]
- Wilson DF, Thomsen RH. Effects of hexamethonium on transmitter release from the rat phrenic nerve. Neurosci Lett. 1992;143:79–82. doi: 10.1016/0304-3940(92)90237-2. [DOI] [PubMed] [Google Scholar]
- Wilson DF, West AE, Lin Y. Inhibitory action of nicotinic antagonists on transmitter release at the neuromuscular junction of the rat. Neurosci Lett. 1995;186:29–32. doi: 10.1016/0304-3940(95)11274-Z. [DOI] [PubMed] [Google Scholar]
- Younger MA, Müller M, Tong A, Pym EC, Davis GW. A presynaptic ENaC channel drives homeostatic plasticity. Neuron. 2013;79:1183–1196. doi: 10.1016/j.neuron.2013.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]