

Abstract
The medial prefrontal cortex (mPFC) serves executive functions that are impaired in neuropsychiatric disorders and pain. Underlying mechanisms remain to be determined. Here we advance the novel concept that metabotropic glutamate receptor 5 (mGluR5) fails to engage endocannabinoid (2-AG) signaling to overcome abnormal synaptic inhibition in pain, but restoring endocannabinoid signaling allows mGluR5 to increase mPFC output hence inhibit pain behaviors and mitigate cognitive deficits. Whole-cell patch-clamp recordings were made from layer V pyramidal cells in the infralimbic mPFC in rat brain slices. Electrical and optogenetic stimulations were used to analyze amygdala-driven mPFC activity. A selective mGluR5 activator (VU0360172) increased pyramidal output through an endocannabinoid-dependent mechanism because intracellular inhibition of the major 2-AG synthesizing enzyme diacylglycerol lipase or blockade of CB1 receptors abolished the facilitatory effect of VU0360172. In an arthritis pain model mGluR5 activation failed to overcome abnormal synaptic inhibition and increase pyramidal output. mGluR5 function was rescued by restoring 2-AG-CB1 signaling with a CB1 agonist (ACEA) or inhibitors of postsynaptic 2-AG hydrolyzing enzyme ABHD6 (intracellular WWL70) and monoacylglycerol lipase MGL (JZL184) or by blocking GABAergic inhibition with intracellular picrotoxin. CB1-mediated depolarization-induced suppression of synaptic inhibition (DSI) was also impaired in the pain model but could be restored by coapplication of VU0360172 and ACEA. Stereotaxic coadministration of VU0360172 and ACEA into the infralimbic, but not anterior cingulate, cortex mitigated decision-making deficits and pain behaviors of arthritic animals. The results suggest that rescue of impaired endocannabinoid-dependent mGluR5 function in the mPFC can restore mPFC output and cognitive functions and inhibit pain.
SIGNIFICANCE STATEMENT Dysfunctions in prefrontal cortical interactions with subcortical brain regions, such as the amygdala, are emerging as important players in neuropsychiatric disorders and pain. This study identifies a novel mechanism and rescue strategy for impaired medial prefrontal cortical function in an animal model of arthritis pain. Specifically, an integrative approach of optogenetics, pharmacology, electrophysiology, and behavior is used to advance the novel concept that a breakdown of metabotropic glutamate receptor subtype mGluR5 and endocannabinoid signaling in infralimbic pyramidal cells fails to control abnormal amygdala-driven synaptic inhibition in the arthritis pain model. Restoring endocannabinoid signaling allows mGluR5 activation to increase infralimbic output hence inhibit pain behaviors and mitigate pain-related cognitive deficits.
Keywords: amygdala, cannabinoids, mGluR, pain, plasticity, prefrontal cortex
Introduction
The medial prefrontal cortex (mPFC) serves executive functions, such as top-down cognitive control, and its infralimbic region interacts closely with the amygdala to suppress (“extinguish”) aversive behaviors (Likhtik et al., 2005; Herry et al., 2010; Pape and Pare, 2010; Sotres-Bayon and Quirk, 2010; Orsini and Maren, 2012; Marek et al., 2013). Fear extinction is associated with increased activity of infralimbic neurons (Milad and Quirk, 2002; Chang et al., 2010; Sepulveda-Orengo et al., 2013) through a mechanism that involves metabotropic glutamate receptor 5 (mGluR5; Fontanez-Nuin et al., 2011; Sepulveda-Orengo et al., 2013). Conversely, decreased infralimbic activity (Hefner et al., 2008; Chang and Maren, 2010; Kim et al., 2010; Sierra-Mercado et al., 2011; Wei et al., 2012) and inhibition or loss of infralimbic mGluR5 function (Xu et al., 2009; Fontanez-Nuin et al., 2011; Sepulveda-Orengo et al., 2013) have been linked to extinction deficits. Increasing infralimbic activity by blocking GABAergic inhibition rescued impaired extinction retrieval (Fitzgerald et al., 2014). Therefore, intact mGluR5 function in the infralimbic cortex is required for certain cognitive control processes, and so we hypothesized that it may also be important for pain control.
The present study addressed pain-related synaptic changes and mGluR5-mediated signaling in the infralimbic cortex, because clinical and preclinical evidence suggests that pain impairs mPFC-dependent cognitive functions, such as decision-making (Apkarian et al., 2004; Pais-Vieira et al., 2009; Ji et al., 2010; Moriarty et al., 2011). Work from our group (Ji et al., 2010; Ji and Neugebauer, 2011, 2014) and others (Metz et al., 2009; Zhang et al., 2015) showed mPFC dysfunction in models of inflammatory and neuropathic pain. We detected decreased pyramidal cell activity in the prelimbic and infralimbic regions of the mPFC, using extracellular single-unit recordings in anesthetized rats in a model of arthritis pain (Ji et al., 2010; Ji and Neugebauer, 2014), but synaptic and cellular mechanisms remain to be determined. Two recent studies reported prelimbic deactivation in a neuropathic pain model (Wang et al., 2015; Zhang et al., 2015). A novel rescue strategy to increase mPFC activity in an arthritis pain model was tested in our recent in vivo study (Ji and Neugebauer, 2014). Pharmacologic activation of mGluR5 and cannabinoid receptor CB1 produced the desired outcome on mPFC activity, but the mechanistic basis and behavioral consequences of this dual strategy remain to be determined. To the best of our knowledge, the present study is the first to show a breakdown of mGluR5-endocannabinoid signaling in the mPFC and beneficial effects of a rescue strategy on pain-related behaviors and cognitive functions.
mGluR5 belong to the group I family of G-protein coupled glutamate receptors which can activate the phospholipase C-diacylglycerol lipase α (DAGLα) pathway that leads to the formation of 2-arachidonoylglycerol (2-AG) endocannabinoids (Guindon and Hohmann, 2009; Di Marzo, 2011). mGluR5 in the mPFC is expressed mostly on postsynaptic elements (Muly et al., 2003). Activation of mGluR5 normally has excitatory effects on layer V pyramidal cells (Marek and Zhang, 2008; Fontanez-Nuin et al., 2011; Kiritoshi et al., 2013). Postsynaptically produced endocannabinoids act retrogradely on presynaptic CB1 receptors to inhibit excitatory or inhibitory synaptic transmission (Lovinger, 2008; Guindon and Hohmann, 2009; Kano et al., 2009; Di Marzo, 2011). In the mPFC, CB1 receptors are exclusively expressed in GABAergic interneurons (Marsicano and Lutz, 1999; Wedzony and Chocyk, 2009), axon terminals with CB1 receptors synapse on mPFC pyramidal cells expressing mGluR5 and DAGLα (Lafourcade et al., 2007), and CB1 activation can inhibit synaptic inhibition of pyramidal cells (Lin et al., 2008).
Therefore, we hypothesized that the 2-AG-CB1 system might be a useful target to control abnormal synaptic inhibition in a pain model and to restore mGluR5 function and mPFC output. We analyzed synaptic and cellular interactions of mGluR5 and endocannabinoid signaling using pharmacology, electrophysiology, optogenetics and behavior to show that mGluR5-driven endocannabinoid signaling at the basolateral amygdala (BLA)-mPFC synapse is impaired in an arthritis pain model but can be restored to remove abnormally enhanced feedforward inhibition, increase pyramidal output, and mitigate cognitive deficits and emotional pain responses.
Materials and Methods
Animals
Male Sprague-Dawley rats (120–320 g; Harlan Laboratories.) were housed in a temperature-controlled room under a 12 h light/dark cycle. Water and food were available ad libitum. Animals for brain slice physiology experiments were 5 to 10 weeks old (120–320 g); animals tested in the behavioral experiments were 7 to 10 weeks old (200–320 g). On the day of the experiment, rats were transferred from the animal facility and allowed to acclimate to the laboratory for at least 1 h. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at TTUHSC and conform to the guidelines of the International Association for the Study of Pain and of the National Institutes of Health (NIH).
Arthritis pain model
In some rats a mono-arthritis was induced in the left knee joint as described in detail previously (Neugebauer et al., 2007). A kaolin suspension (4%, 100 μl) was injected slowly into the joint cavity followed by repetitive flexions and extensions of the knee for 15 min. Next, a carrageenan solution (2%, 100 μl) was injected into the knee joint cavity, and the leg was flexed and extended for another 5 min. This treatment paradigm reliably leads to a localized inflammation confined to one knee joint within 1–3 h, persists for weeks, and is significantly associated with pain behaviors and activity changes in the peripheral and CNS (Neugebauer et al., 2007). Electrophysiological and behavioral experiments were performed 5–6 h after arthritis induction and data were compared with those obtained from normal naive rats. Our previous works showed that brain activity and behavior of normal rats was not different from rats that received intraarticular saline injection (Neugebauer et al., 2003) or needle insertion (Grégoire and Neugebauer, 2013), and so we used normal rats as a control group.
Electrophysiology
Brain slice preparation.
Brain slices containing the mPFC (3.2–2.7 mm anterior to bregma) were obtained from normal and arthritic rats as described before (Kiritoshi et al., 2013; Kiritoshi and Neugebauer, 2015). In some experiments, amygdala brain slices were used to verify the physiological effect of optogenetic activation of amygdala neurons expressing a light-sensitive channel (see Synaptic transmission and optogenetics). Brains were quickly removed and immersed in oxygenated ice-cold sucrose-based physiological solution containing the following (in mm): 87 NaCl, 75 sucrose, 25 glucose, 5 KCl, 21 MgCl2, 0.5 CaCl2, and 1.25 NaH2PO4 (Kasanetz et al., 2013). Coronal brain slices (400 μm) were prepared using a Vibratome (Series 1000 Plus). The mPFC slices were then incubated in oxygenated artificial CSF (ACSF) at room temperature (21°C) for at least 1 h before patch recordings. ACSF contained the following (in mm): 117 NaCl, 4.7 KCl, 1.2 NaH2PO4, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, and 11 glucose. A single brain slice was transferred to the recording chamber and submerged in ACSF (31 ± 1°C) superfusing the slice at ∼2 ml/min. Only one or two brain slices per animal were used. Only one neuron was recorded in each slice and a fresh slice was used for each new experimental protocol. Numbers in the text refer to the number of neurons tested for each parameter.
Patch-clamp recording.
Whole-cell patch-clamp recordings were obtained from visually identified layer V pyramidal cells in the infralimbic mPFC of the right hemisphere (∼700 μm lateral to the interhemispheric fissure) using infrared DIC-IR videomicroscopy as described previously (Kiritoshi et al., 2013; Kiritoshi and Neugebauer, 2015). The right infralimbic cortex was targeted because right-hemispheric lateralization of pain-related plasticity has been reported for the amygdala (Carrasquillo and Gereau, 2008; Ji and Neugebauer, 2009), and this study focused on synaptic mechanisms of amygdala influences on the mPFC. To verify direct neuronal activation by optogenetic activation of light-sensitive channels in BLA neurons recordings were made in amygdala brain slices in control experiments (see synapitc transmission and optogenetic stimulation). Recording electrodes (3–5 MΩ tip resistance) were made from borosilicate glass and filled with intracellular solution containing the following (in mm): 122 K-gluconate, 5 NaCl, 0.3 CaCl2, 2 MgCl2, 1 EGTA, 10 HEPES, 5 Na2-ATP, and 0.4 Na3-GTP; pH was adjusted to 7.2–7.3 with KOH and osmolarity to 280 mOsm/kg with sucrose. For the recording of IPSCs, QX-314 (5 mm) was included in the internal solution. For the analysis of depolarization-induced suppression of synaptic inhibition (DSI), IPSCs were recorded using a high chloride intracellular solution (Kiritoshi et al., 2013) containing the following (in mm): 126 KCl, 10 NaCl, 1 MgCl2, 11 EGTA, 10 HEPES, 2 Mg-ATP, and 0.25 Na3-GTP (pH was adjusted to 7.2–7.3 with KOH and osmolarity to 280 mOsm/kg with sucrose). Data acquisition and analysis was done using a dual four-pole Bessel filter (Warner Instruments), low-noise Digidata 1322 interface (Axon Instruments, Molecular Devices), Axoclamp-2B amplifier (Axon Instruments, Molecular Devices), Pentium PC, and pClamp9 software (Axon Instruments). Headstage voltage was monitored continuously on an oscilloscope to ensure precise performance of the amplifier. If series resistance (monitored with pClamp9 software) changed >10%, the neuron was discarded.
Synaptic transmission and optogenetics.
Electrical and optogenetic stimulations were used to evoke EPSCs, IPSCs, EPSPs, and action potentials (E–S coupling) in pyramidal cells. Neurons were voltage-clamped at −70 or 0 mV for the study of EPSCs and IPSCs, respectively. The calculated equilibrium potential for chloride in this system was −68.99 mV (Nernst equation, pClamp9 software).
For focal electrical synaptic stimulation (150 μs square-wave pulses; using an S88 stimulator; Grass Technologies) a concentric bipolar stimulating electrode (David Kopf Instruments) was positioned in layer IV (500 μm from the medial surface of the slice) of the infralimbic cortex where our previous studies (Ji et al., 2010; Sun and Neugebauer, 2011; Kiritoshi et al., 2013) identified anterogradely labeled afferents from the BLA to the infralimbic and prelimbic regions of the mPFC following stereotaxic injections of a fluorescent tracer (DiI) into the BLA.
For optogenetic stimulation, a viral vector encoding channel rhodopsin 2 (ChR2) under the control of the CaMKII promoter (rAAV5/CaMKIIa-ChR2(H134R)-eYFP; courtesy of the Karl Deisseroth Laboratory, packaged by the vector core facility at the University of North Carolina, Chapel Hill) was injected stereotaxically into the right BLA, using the following stereotaxic coordinates: 2.3 mm posterior to bregma; 4.3 mm lateral to midline; depth, 7.0 mm. Animals were allowed to recover 4 weeks for viral expression before brain slices were obtained for electrophysiology (see Brain slice preparation). ChR2-expressing afferent fiber terminals from the BLA were activated optically in the mPFC by laser light pulses (5 ms, 0.1 Hz) generated by a blue laser (473 nm; Thorlabs) controlled by a Grass stimulator; they were delivered through the 40× objective of the microscope. Illumination area (0.24 mm2) was centered on the soma of the patched cell. Light power density was measured using an optical power meter (PM200, Thorlabs) placed under the objective. In control experiments, light activation of BLA neurons expressing ChR2 caused an inward current as predicted (Fig. 1C).
Figure 1.
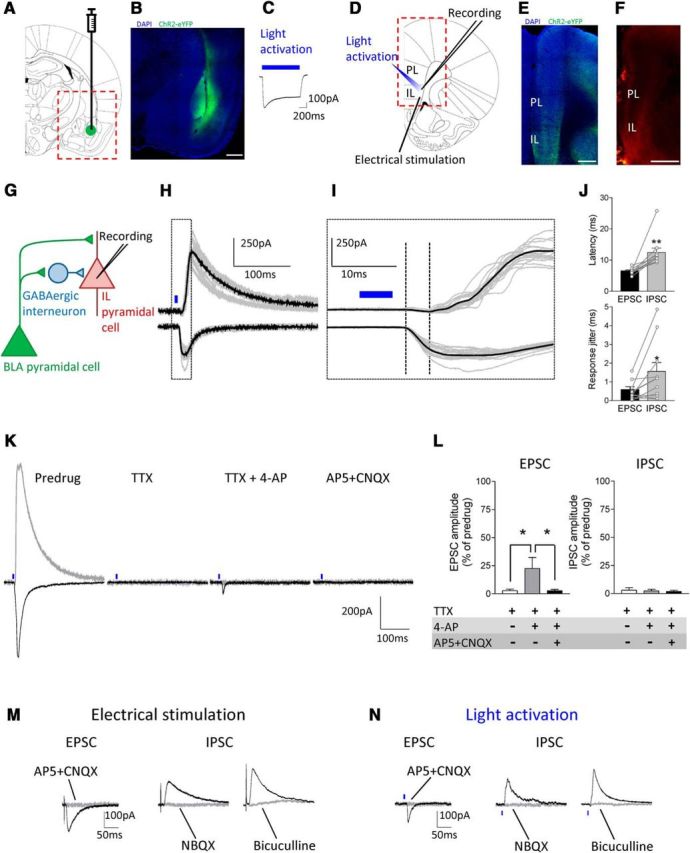
Optogenetic activation of BLA inputs to infralimbic mPFC pyramidal cells in brain slices from normal rats. A–C, Amygdala. A, Schematic representation of viral injection site into the BLA. B, Viral vector-mediated ChR2-eYFP expression in the BLA (green, ChR2-eYFP; blue, DAPI). Scale bar, 500 μm. C, ChR2-mediated current in a BLA pyramidal neuron evoked by a 1 s blue laser light pulse in an amygdala brain slice. D–F, mPFC. D, Schematic representation of recording and stimulation sites in an mPFC slice. E, ChR2-eYFP-expressing BLA fibers in an mPFC slice in experiments using optogenetic synaptic stimulation (green, ChR2-eYFP; blue, DAPI). Scale bar, 500 μm. F, Anterogradely labeled amygdala projections in the mPFC by DiI injection into the BLA for experiments using electrical synaptic stimulation. Scale bar, 1 mm. G, Hypothesized synaptic circuitry of BLA-driven monosynaptic excitatory and feedforward inhibitory inputs to mPFC neurons tested with optogenetic strategies. H–L, Synaptic responses of mPFC neurons generated by optogenetic activation of BLA axon terminals. H, Traces show individual examples of monosynaptic EPSCs (recorded at −70 mV) and polysynaptic IPSCs (at 0 mV) generated in an infralimbic layer V pyramidal cell by light activation (blue symbols) of ChR2-expressing BLA pyramidal cell axons. I, Enlarged view of the boxed area in H shows differences in latencies. J, Summary of onset latencies and jitter of light-evoked EPSC and IPSC. Each symbol shows one neuron. Bar histograms show mean ± SE (n = 11 neurons); *p < 0.05, **p < 0.01, compared with EPSCs, paired t test. K, L, Individual examples and summary of data showing synaptic nature of the optical-evoked responses. TTX (1 μm) abolished light-evoked EPSC and IPSC. 4-AP (1 mm) partially rescued EPSC but not IPSC. EPSC was blocked by AP5 (50 μm) and CNQX (20 μm). Summary of the effect of TTX, TTX + 4-AP, and TTX + 4-AP + AP5 + CNQX on light-evoked EPSC and IPSC in the same neurons (n = 5). *p < 0.05, repeated-measures ANOVA with Bonferroni posttests. M, N, Comparison of synaptic responses evoked by focal electrical (M) and optical (N) activation. Both techniques generated monosynaptic EPSCs that were blocked by AP5 (50 μm) and CNQX (20 μm) and glutamate-driven IPSCs that were blocked by NBQX (10 μm) or bicuculline (10 μm). Light intensity in the optogenetic experiments was set to evoke synaptic responses of submaximal amplitude (0.11–0.65 mW at brain slice; power density of 0.46–2.7 mW/mm2).
Behavior
Spinal reflexes.
Thresholds of hindlimb withdrawal reflexes evoked by mechanical stimulation of the knee joint were measured as described previously (Han et al., 2005; Ren et al., 2013; Medina et al., 2014). Mechanical stimuli of continuously increasing intensity were applied to the knee using a calibrated forceps equipped with a force transducer. Withdrawal threshold was defined as the minimum stimulus intensity that evoked a withdrawal reflex. Measurements were repeated two times at 5 min interval and the average was taken as the final value.
Vocalizations.
Audible and ultrasonic vocalizations were recorded and analyzed as described in detail previously (Han et al., 2005; Ren et al., 2013; Medina et al., 2014). Animals were briefly anesthetized with isoflurane (2%) and placed in a custom-designed recording chamber with openings for head and limbs. After habituation to the chamber, a calibrated forceps (see Spinal reflexes) was used to apply brief (15 s) mechanical stimuli of innocuous (300 g/30 mm2) and noxious (2000 g/30 mm2) intensities to the left knee joint. Audible (20 Hz to 16 kHz) and ultrasonic (25 ± 4 kHz) vocalizations were measured with a condenser microphone and a bat detector, respectively, placed at a fixed distance from the head. The sound detectors were connected to a filter and amplifier (UltraVox four-channel system; Noldus Information Technology). Vocalizations were recorded for 1 min, starting with the onset of the mechanical stimulus. Total durations of audible and ultrasonic vocalizations were analyzed using UltraVox 2.0 software (Noldus Information Technology). Experiments were performed in a shielded temperature- and light-controlled room, and appropriate filtering levels were used to avoid the recording of any background noise.
Gambling task.
Decision making was measured in a rodent gambling task (RGT) as described before (Ji et al., 2010; Sun and Neugebauer, 2011). The customized computerized system consisted of an octagonal arena connected to a runway with an automated guillotine door. The arena was partially divided by a Plexiglas panel, and each half of the arena contained one lever to release chocolate-coated food pellets (Research Diets) from an automated dispenser connected to a food cup. After a 5 s waiting period in the closed runway, the rat entered the arena and was given 20 s to explore the arena and choose and press one of two levers to receive a food pellet. Then, the animal was hand-removed from the arena and placed back in the runway for a new trial. When the animals had learned the association between lever presses and food delivery, they were subjected to the non-gambling phase (5 sessions; 1 per day), in which each of the two levers provided one food pellet upon pressing in 9 of 10 trials. In this phase, animals were excluded from the experiment if they showed preference for the lever on one side. The actual task (RGT) consisted of a single session of 90 consecutive trials, in which one lever continued to deliver in randomized order one pellet in 9 of 10 trials (low-risk lever), whereas the other lever was altered to return three pellets in only 3 of 10 trials (high-risk lever). The side of the high-risk lever remained the same throughout the 90 consecutive trials but was changed randomly between different animals to avoid any lateralization bias in the test environment. Preference index was calculated for each of 10 consecutive trials using the following formula: [(low-risk lever choices) − (high-risk lever choices)]/number of completed trials. Final preference index for the last 10 consecutive trials in a session was used for statistical analysis.
Drugs
The following drugs were used: mGluR5-positive allosteric modulator N-cyclobutyl-6-((3-fluorophenyl)ethynyl)nicotinamide hydrochloride (VU0360172, VU'172); CB1 receptor agonist N-(2-chloroethyl)-5Z,8Z,11Z,14Z-eicosatetraenamide (ACEA); CB1 receptor antagonist N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251); DAGL inhibitor N-formyl-l-leucine (1S)-1-[[(2S,3S)-3-hexyl-4-oxo-2-oxetanyl]methyl]dodecyl ester (tetrahydrolipstatin; THL); ABHD6 inhibitor N-methyl-N-[[3-(4-pyridinyl)phenyl]methyl]-4′-(aminocarbonyl)[1,1′-biphenyl]-4-yl carbamic acid ester (WWL70); MAGL inhibitor 4-[bis(1,3-benzodioxol-5-yl)hydroxymethyl]-1-piperidinecarboxylic acid 4-nitrophenyl ester (JZL184); TRPV1 receptor antagonist (2E)-N-(2,3-dihydro-1,4-benzodioxin-6-yl)-3-[4-(1,1-dimethylethyl)phenyl]-2-propenamide (AMG9810); NMDA receptor antagonist DL-2-amino-5-phosphonopentanoic acid (AP5); non-NMDA receptor antagonists 6-cyano-7-nitroquinoxaline-2,3-dione disodium salt (CNQX), and 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide disodium salt (NBQX); GABAA receptor antagonists bicuculline and picrotoxin; sodium channel blocker tetrodotoxin citrate (TTX); potassium channel blocker 4-aminopyridine (4-AP). THL was purchased from Sigma-Aldrich; all other drugs were purchased from Tocris Bioscience (R&D Systems). Selectivity and target concentrations have been established in the literature for VU'172 (Rodriguez et al., 2010), ACEA (Hillard et al., 1999), AM251 (Pertwee, 2010), THL (Bisogno et al., 2006; Zhang et al., 2011), WWL70 (Li et al., 2007; Marrs et al., 2010), JZL184 (Long et al., 2009; Marrs et al., 2010), and AMG9810 (Gavva et al., 2005). Drugs were prepared as stock solutions and diluted (1;1000) to their final concentration in ACSF on the day of the experiment. ACEA, a synthetic analog of anandamide, was supplied predissolved in anhydrous ethanol. AP5, CNQX, NBQX, bicuculline, TTX, and 4-AP were dissolved in water. VU'172, THL, WWL70, JZL184, and picrotoxin were dissolved in dimethyl sulfoxide. AM251 and AMG9810 were dissolved in ethanol.
Drug application
Brain slices.
Drugs were applied by gravity-driven superfusion of the brain slice in ACSF (∼2 ml/min). Solution flow into the recording chamber (1 ml volume) was controlled with a three-way stopcock. Drugs were applied for at least 15 min to establish equilibrium in the tissue. ACSF served as vehicle control in all experiments.
Drug application by microdialysis.
As described in detail previously (Ji et al., 2010; Sun and Neugebauer, 2011), a guide cannula was implanted stereotaxically the day before behavioral measurements with a stereotaxic apparatus (David Kopf Instruments). The animal was anesthetized with pentobarbital sodium (Nembutal, 50 mg/kg, i.p.), and the guide cannula was implanted on the dorsal margin of the right infralimbic mPFC, using the following coordinates (Paxinos and Watson, 1998): 3.2 mm rostral to bregma, 0.7 mm lateral to midline, depth 4.2 mm. The rationale for targeting the right infralimbic cortex is provided in Electrophysiology. For off-site control injections into the anterior cingulate cortex, the following coordinates were used for implanting the guide cannula: 1.6 mm rostral to bregma, 0.6 mm lateral to midline, depth 2.2–2.5 mm. The cannula was fixed to the skull with dental acrylic. Antibiotic ointment was applied to the exposed tissue to prevent infection. On the day of the experiment, a microdialysis probe (CMA/7) was inserted through the guide cannula so that the probe protruded by 1 mm. The probe was connected to an infusion pump (Harvard Apparatus) and perfused with ACSF (oxygenated and equilibrated to pH 7.4) for at least 1 h to establish equilibrium in the tissue. Drugs were dissolved in ACSF on the day of the experiment and applied by microdialysis at a rate of 5 μl/min for at least 20 min to establish equilibrium in the tissue.
Histology
The sites of the viral vector injection into the BLA (optogenetic experiments) and the position of microdialysis probes in the infralimbic mPFC (behavioral experiments) were verified histologically. The brains were fixed in 4% paraformaldehyde for 6–12 h and switched to 30% sucrose. Fifty micrometer sections were made on a freezing microtome and mounted on gelatin-coated slides. For the verification of ChR2-eYFP expression, sections were treated as described previously (Ji and Neugebauer, 2012) and mounted on slides using Vectashield mounting medium with DAPI (Vector Laboratories). Fluorescence images were acquired with a Nikon confocal microscope. For the verification of the position of the microdialysis probes, sections were stained with hematoxylin and eosin.
Statistical analysis
All averaged values are given as the mean ± SE. Statistical significance was accepted at the level p < 0.05. GraphPad Prism 3.0 software was used for all statistical analyses. Statistical analysis was performed on the raw data. Student's t test was used to compare two sets of data that had Gaussian distribution and similar variances. For multiple comparisons, ANOVA (repeated measures where appropriate) was used with Bonferroni post-tests as indicated in the text and figure legends.
Results
This study focusses on synaptic physiology and pharmacology in the infralimbic region (area 25) of the mPFC in rats. Although prefrontal cortical nomenclature is somewhat controversial and “infralimbic” is not a term used for primate research (for discussion, see Vogt and Paxinos, 2014), reference to “infralimbic mPFC” is well established in the rodent literature on fear extinction centered on interactions between mPFC and amygdala (Amir et al., 2011; Sierra-Mercado et al., 2011; Do-Monte et al., 2015). This nomenclature has also been applied to pain-related studies of mPFC function (Wang et al., 2015; Zhang et al., 2015) The present study and previous work are closely aligned with this body of literature and so we will use the term “infralimbic mPFC” that can be easily identified in rodent brain slice (Fig. 1).
Excitatory and inhibitory amygdala inputs to infralimbic cortex
The goal of this study was to determine pain-related synaptic changes in the infralimbic cortex and their modulation by mGluR5-endocannabinoid signaling to control pain-related behaviors and rescue cognitive functions. To do so we first measured excitatory and inhibitory synaptic transmission onto infralimbic pyramidal cells using electrical and optogenetic stimulations of fibers from the BLA. Our previous work suggested that enhanced activity in the amygdala leads to deactivation of prelimbic cortical pyramidal cells based on pharmacological data and electrical stimulation of anterogradely (from BLA) labeled fiber tracts (Ji et al., 2010). We also observed decreased activity (extracellularly recorded action potentials) in the infralimbic region (Ji and Neugebauer, 2014). We postulated that BLA pyramidal cells project directly to prelimbic and infralimbic pyramidal cells providing monosynaptic excitatory inputs but also target mPFC interneurons to generate feedforward inhibition of mPFC pyramidal cells (Ji et al., 2010; Kiritoshi et al., 2013). However, this hypothesis (Fig. 1G) remains to be tested directly, and only the availability of optogenetic tools now allows us to do so. Determining the identity of synaptic inputs is critical for the better understanding of disease mechanisms and drug actions in defined brain circuits.
First we established that selective optogenetic activation of axons from amygdala (BLA) projection neurons can reproduce the results we had found with electrical stimulation of fiber tracts containing anterogradely labeled BLA axons (Ji et al., 2010; Kiritoshi et al., 2013). Figure 1 shows that BLA inputs provide simultaneously direct monosynaptic excitation and glutamate-driven feedforward inhibition of infralimbic pyramidal cells. Light-sensitive ion channels (ChR2) were expressed in BLA neurons (Fig. 1B), following stereotaxic injection of a viral vector encoding ChR2 and yellow fluorescent protein (YFP) under the control of the CaMKII promoter (rAAV5/CaMKIIa-ChR2(H134R)-eYFP) into the BLA (Fig. 1A) as described before for the mPFC (Ji and Neugebauer, 2012). As a functional control, light activation (see Materials and Methods) through the microscope objective produced a ChR2-mediated inward current in BLA neurons recorded in amygdala brain slices (see individual example in Fig. 1C).
Light activation of ChR2-expressing BLA axon terminals in brain slices containing the mPFC (Fig. 1D,E) generated monosynaptic EPSCs and polysynaptic IPSCs in visually identified infralimbic layer V pyramidal cells (Fig. 1H–J). Compared with EPSCs, IPSCs occurred with significantly longer (Fig. 1J, top) and more variable (“jitter”; Fig. 1J, bottom) latencies (n = 11 neurons, p < 0.01 and 0.05, respectively; paired t tests). Light-activated responses were synaptically evoked, because they were blocked by the sodium channel blocker TTX (1 μm); subsequent addition of a potassium channel blocker (4-AP, 1 mm) partially rescued the EPSC but not IPSC, and the EPSC was blocked by glutamate receptor antagonists AP5 (50 μm) and CNQX (20 μm). Figure 1K shows an individual example and Figure 1L summarizes the data (n = 5 neurons; p < 0.05; F(1,8) = 1.98, repeated-measures ANOVA with Bonferroni posttests). Focal electrical stimulation (Fig. 1M) of fibers identified by anterograde labeling from the BLA (Fig. 1F) and light activation of ChR2-expressing BLA axons (Fig. 1N) evoked monosynaptic EPSCs that were blocked with AP5 and NBQX and glutamate receptor-driven IPSCs that were blocked by NBQX or bicuculline. Light intensity was set to evoke EPSCs and IPSCs of submaximal amplitude (9–50 mW at laser source, which translated into 0.11–0.65 mW at slice and power density of 0.46–2.7 mW/mm2). Importantly, there was no temperature change by the laser light measured in the recording chamber with maximum laser power (50 mW at laser source; 2.7 mW/mm2 under the objective). These experiments were done in brain slices from normal animals.
Loss and rescue of endocannabinoid-dependent mGluR5 facilitation in an arthritis pain model
Next we studied the synaptic integration of excitatory and inhibitory inputs onto infralimbic pyramidal cells in brain slices from normal and arthritic rats (5 h postinduction) by measuring synaptically evoked spiking (E–S coupling) in current-clamp mode. The goal was to determine the role of mGluR5-endocannabinoid signaling under normal conditions and in a pain model. Effects of electrical (Fig. 2A–C) and optogenetic (Fig. 2D–F) stimulations were compared. Stimulation intensity was set to evoke three to four spikes in a series of 10 stimulations to allow the detection of any facilitatory effects. Measurements were made every 5 min before and during drug application. The focus was on mGluR5 because of its important role in cognitive control processes that involve amygdala-mPFC interactions such as fear extinction mGluR5 function (Xu et al., 2009; Fontanez-Nuin et al., 2011; Sepulveda-Orengo et al., 2013).
Figure 2.
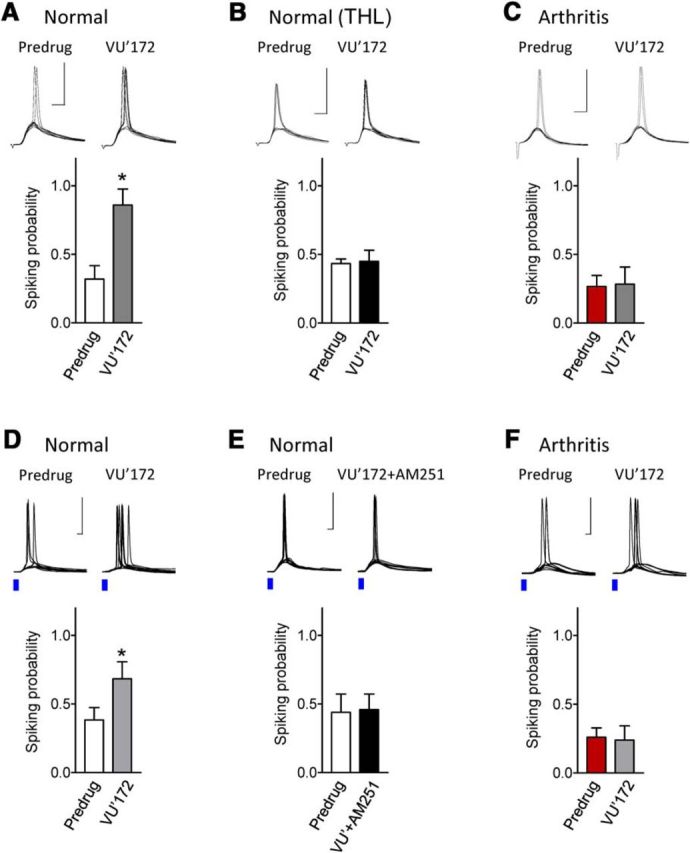
Impaired CB1 receptor-mediated facilitatory effects of mGluR5 in the arthritis pain model. Synaptically evoked spiking (E–S coupling) in infralimbic pyramidal cells using electrical (A–C) and optical (D–F) stimulations of BLA afferents (see Materials and Methods). Stimulation intensity was set to evoke three to four spikes in a series of 10 consecutive trials. Voltage traces show individual examples recorded in current-clamp mode at −60 mV. Scale bars, 50 mV, 5 ms. Bar histograms show number of spikes (mean ± SEM) evoked in a series of 10 trials before and during application of VU'172 (1 μm). A, In brain slices from normal rats, VU'172 (1 μm) alone increased spiking probability significantly (n = 5 neurons). B, In brain slices from normal rats VU'172 (1 μm) alone had no effect when THL (10 μm) was included in the patch-pipette (n = 6 neurons). C, In brain slices from arthritic rats (5 h postinduction) VU'172 (1 μm) alone had no effect (n = 6 neurons). D, In brain slices from normal rats, VU'172 (1 μm) alone increased spiking evoked by optical stimulation (blue laser light) of BLA afferents (n = 6 neurons). E, Coapplication of a CB1 receptor antagonist (AM251, 10 μm) blocked the effect of VU'172 (n = 5 neurons) in brain slices from normal rats. F, In brain slices from arthritic rats (5 h postinduction) VU'172 (1 μm) alone had no effect (n = 5 neurons). *p < 0.05 compared with predrug, paired t test.
A positive allosteric modulator for mGluR5 (VU'172, 1 μm) increased probability of spiking evoked by electrical stimulation (n = 5 neurons; Fig. 2A) or optical activation of BLA inputs (n = 6; Fig. 2D) significantly (p < 0.05 compared with predrug; paired t tests). The facilitatory effect of mGluR5 activation was blocked by intracellular application of a DAGL inhibitor (THL, 10 μm; n = 6; Fig. 2B) or by bath application of a CB1 receptor antagonist (AM251, 10 μm; n = 5; Fig. 2E), suggesting that intact endocannabinoid-CB1 signaling is required. Importantly, mGluR5 activation had no effect in brain slices from arthritic rats (5 h postinduction; electrical stimulation, n = 6 neurons; Fig. 2C; optical activation, n = 5; Fig. 2F).
The data suggest that modulation of infralimbic mPFC output (synaptically evoked spiking) by mGluR5 requires endocannbinoid-CB1 receptor signaling and is impaired in an arthritis pain model. Therefore, we tested the hypothesis that mGluR function can be rescued by increasing the availability of endocannabinoids and/or by CB1 receptor activation in brain slices from arthritic rats (5 h postinduction). A CB1 receptor agonist (ACEA, 10 nm) restored the facilitatory effect of VU'172 (1 μm) on synaptically evoked spiking (n = 14 neurons; p < 0.01, paired t test; Fig. 3A) using electrical or optical stimulation (see individual examples). Intracellular application of an inhibitor of postsynaptic 2-AG hydrolyzing enzyme ABHD6 (WWL70, 10 μm; n = 6 neurons) or perfusion of the brain slice with an inhibitor of monoacylglycerol lipase MGL (JZL184, 1 μm; n = 7 neurons) restored the facilitatory effects of VU'172 (p < 0.05 compared with predrug; paired t tests). The data support our hypothesis that endocannabinoids, particularly 2-AG, activating CB1 receptors can restore the facilitatory effect of mGluR5. Next, we addressed potential mechanisms.
Figure 3.

Activation of endocannabinoid-CB1 receptor signaling restores impaired facilitatory effects of mGluR5 in the arthritis pain model. Synaptically evoked spiking (E–S coupling) was measured in infralimbic pyramidal cells in brain slices from arthritic rats (same display as in Fig. 2). A, Coapplication of a CB1 receptor agonist (ACEA, 10 nm) restored the facilitatory effect of VU'172 (1 μm). Results obtained with electrical or optical stimulation (see individual examples) were not different, and so the data were pooled (bar histograms; n = 14 neurons). B, Including an inhibitor of postsynaptic 2-AG hydrolyzing enzyme ABHD6 (WWL70, 10 μm) in the patch pipette restored the facilitatory effect of VU'172 (n = 6 neurons). C, An inhibitor of monoacylglycerol lipase MGL (JZL184, 1 μm) restored the effect of VU'172 (n = 7 neurons). Slices were preincubated with JZL184 (1 μm) for 30 min, and recordings were performed in the continued presence of the inhibitor. D, Including a GABAA receptor antagonist (picrotoxin, 50 μm) in the patch-pipette also restored the effect of VU'172 (n = 5 neurons). *p < 0.05, **p < 0.01 compared with predrug; paired t tests.
Restoring control of increased synaptic inhibition in an arthritis pain model
Our previous work showed increased feedforward inhibition of prelimbic pyramidal cells in the arthritis pain model (Ji et al., 2010), which has been confirmed in a neuropathic pain model (Zhang et al., 2015), but this remains to be determined for the infralimbic cortex (Fig. 4). Here we tested the hypothesis that failure of 2-AG-CB1 receptors to control abnormal feedforward inhibition in the pain model would impair the ability of mGluR5 to drive infralimbic pyramidal cell output. As a starting point, and in support of our hypothesis, intracellular application of a GABAA receptor antagonist (picrotoxin, 50 μm) restored the effect of VU'172 in infralimbic pyramidal cells significantly (n = 5 neurons; p < 0.05 compared with predrug; paired t test; Fig. 3D).
Figure 4.
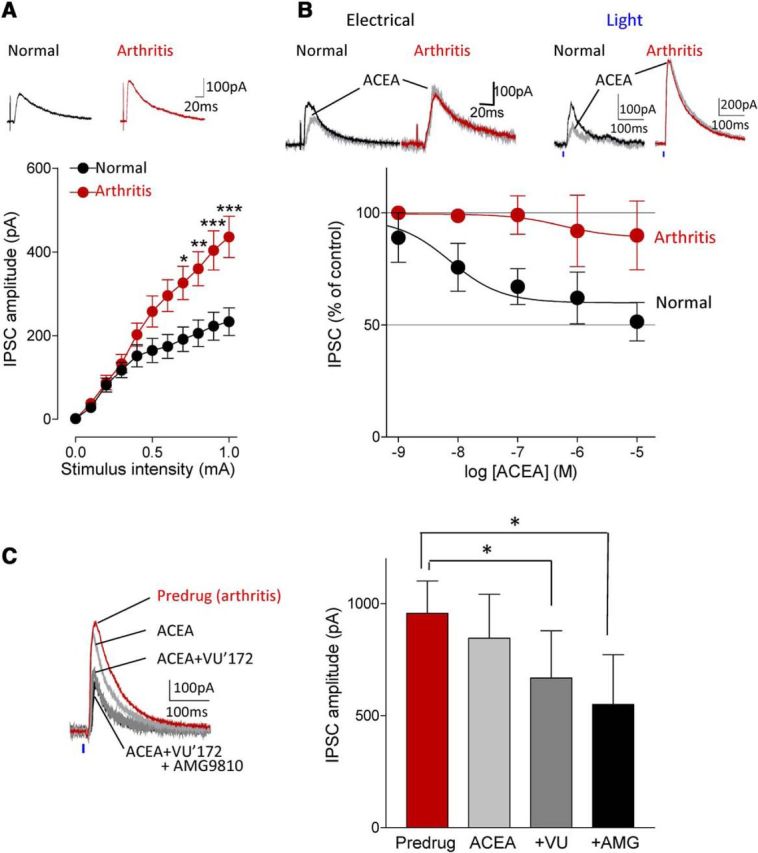
Coactivation of CB1 and mGluR5 inhibits enhanced inhibitory synaptic transmission in the arthritis pain model. A, Input-output functions of IPSCs recorded in slices from arthritic rats (n = 34 neurons) were significantly different (F(1,605) = 40.84, p < 0.0001, main effect of arthritis, two-way ANOVA) from normal controls (n = 23 neurons). IPCSs could be blocked with bicuculline or NBQX (data not shown). *,**,***p < 0.05–0.001, compared with normal (Bonferroni posttests). B, ACEA (10 nm) decreased IPSCs evoked by electrical or optical stimulation under normal condition but had no significant effect in the arthritis pain model. Concentration-response curves for ACEA under normal conditions (n = 4–8 neurons) and in the arthritis pain model (n = 3–8 neurons) were significantly different (p < 0.001; F(1,39) = 16.33, two-way ANOVA). C, Application of ACEA (10 nm) alone had no effect but addition of VU'172 (1 μm) decreased IPSCs in the pain model. The inhibitory effect of the combination persisted in the presence of a TRPV1 receptor antagonist AMG9810 (10 μm, n = 5 neurons; same neurons were tested with ACEA alone, ACEA and VU'172, and addition of AMG9810). *p < 0.05, F(1,12) = 56.23, repeated-measures ANOVA with Bonferroni posttests compared with predrug. A–C, Current traces show IPSCs (average of 8–10) evoked with electrical stimulation of 0.6 and 0.8 mA (A, B) and with optical activation of BLA terminals with 40 mW (at laser source; power density, 2.17 mW/mm2).
Analysis of inhibitory synaptic transmission onto infralimbic pyramidal cells showed for the first time significantly increased IPSCs in brain slices from arthritic rats (n = 34 neurons) compared with normal controls (n = 23 neurons; p < 0.001; F(1,605) = 40.84, two-way ANOVA; Fig. 4A). IPCSs were blocked by bicuculline or NBQX as shown in Figure 1M and N, which is generally accepted as evidence for the concept of glutamate-driven feedforward inhibition. Activation of CB1 receptors with ACEA (10 nm) decreased synaptic inhibition concentration-dependently under normal conditions (n = 4–8 neurons) but had no significant effect in the arthritis pain model (n = 3–8 neurons; Fig. 4B). The difference was significant (p < 0.001; F(1,39) = 16.33, two-way ANOVA), and was observed with electrical stimulation and with optical activation of afferent fibers from the BLA, and so the data were pooled for the concentration-response analysis (Fig. 4B).
Because the combination of VU'172 and ACEA increased synaptically evoked spiking (Fig. 3) and our previous study showed that VU'172 can inhibit inhibitory transmission in the infralimbic cortex under normal conditions through a CB1 receptor-dependent mechanism (Kiritoshi et al., 2013), we tested the effect of ACEA combined with VU'172 on inhibitory transmission in the arthritis model (Fig. 4C). These studies were done using optical activation of BLA axon terminals in the infralimbic cortex in slices from arthritic rats. Coapplication of ACEA (10 nm) and VU'172 (1 μm) decreased IPSCs in infralimbic pyramidal cells significantly (p < 0.05; F(1,12) = 56.23; repeated-measures ANOVA with Bonferroni post-tests) whereas ACEA alone had no significant effect (Fig. 4B, see data). Importantly, the inhibitory effect of VU'172 combined with ACEA was not mediated through TRPV1 receptors, which have been implicated in some actions of endocannabinoids, because a TRPV1 receptor antagonist (AMG9810, 10 μm) did not block the inhibitory effect of the combination (n = 5 neurons; same neurons were tested with ACEA alone, ACEA and VU'172, and addition of AMG9810; Fig. 4C).
The data so far suggest that effects of mGluR5 and CB1 activation are lost in an arthritis pain model but can be restored by a combination strategy that makes endocannabinoids available to mGluR5 and directly activates CB1; the underlying mechanism involves restoring the ability of mGluR5-CB1 interactions to control increased BLA-driven synaptic inhibition of infralimbic pyramidal cells. A more direct way to assess endocannabinoid function and control of synaptic inhibition is the analysis of DSI (Kano et al., 2009). DSI involves postsynaptic calcium influx following depolarization, activation of specific 2-AG synthesizing (ie, DAGLα) enzymes, synthesis and release of 2-AG, and retrograde activation of CB1 receptors on the presynaptic terminal to inhibit transmitter release (Lovinger, 2008; Kano et al., 2009; Di Marzo, 2011; Rivera et al., 2014).
In agreement with our previous studies (Kiritoshi et al., 2013) DSI could be demonstrated in infralimbic pyramidal cells in brain slices from normal animals. A brief (4 s) depolarization decreased IPSCs recorded at −70 mV with a high chloride internal solution in brain slices from normal animals (n = 5 neurons; Fig. 5A,E). VU'172 (1 μm) prolonged the duration of DSI in (n = 5 neurons). For the recording of DSI, the stimulation electrode was placed close to the recording pipette (450–650 μm from the recording pipette) to evoke monosynaptic IPSC in the presence of AP5 and CNQX. In brain slices from arthritic rats, DSI was not detected (Fig. 5B–E). Application of VU'172 (1 μm, n = 5 neurons; Fig. 5B) or ACEA (10 nm, n = 6 neurons; Fig. 5C) alone partially rescued DSI in the arthritis pain model. Coapplication of VU'172 with ACEA fully restored DSI in slices from arthritic rats (n = 6 neurons; Fig. 5D,E).
Figure 5.
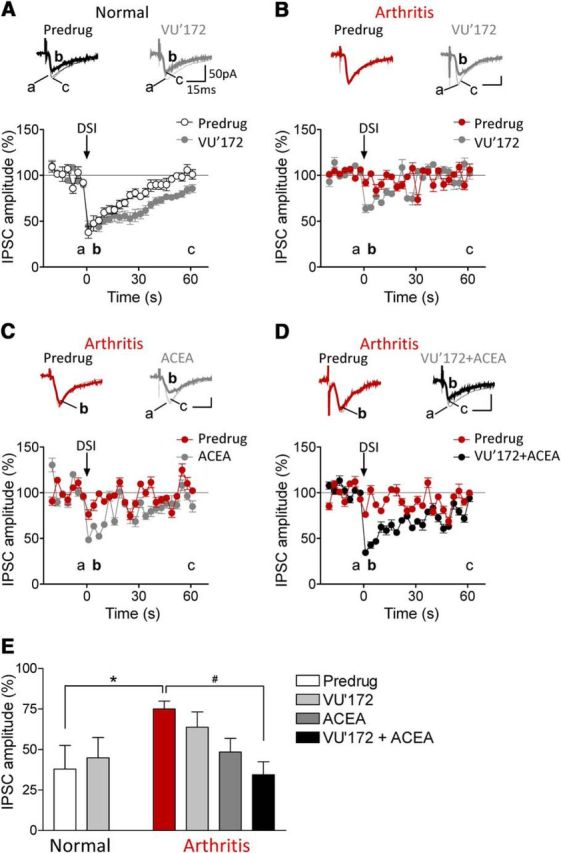
Coactivation of CB1 and mGluR5 restores DSI in the arthritis pain model. IPSCs were recorded at −70 mV with a high chloride internal solution. Brief (4 s) depolarization of pyramidal cells decreased IPSCs (=DSI). A–D, Current traces show IPSCs (average of 8–10) before (a) and 1–3 s (b), and 60 s after depolarization (c). Graphs show IPSC amplitudes normalized to pre-DSI control values averaged for the sample of neurons (mean ± SE). A, VU'172 (1 μm) prolonged the duration of DSI in slices from normal rats (n = 5 neurons). B–D, DSI could not be induced in arthritis. Application of VU'172 (B; n = 5 neurons) or ACEA (C, 10 nm, n = 6 neurons) alone partially rescued DSI in the arthritis pain model. D, Coapplication of VU'172 with ACEA fully restored DSI in arthritis (n = 6 neurons). E, Bar histogram summarizes the data shown in A–D. Magnitude of DSI at 1 s after depolarization is shown as IPSC amplitude normalized to pre-DSI values. *p < 0.05, one-way ANOVA with Bonferroni post-tests.
Together the results suggest that CB1-mediated control of synaptic inhibition is impaired in the arthritis pain model but can be restored by increasing mGluR5-2-AG signaling. Next, we evaluated behavioral consequences of the rescue strategy of combined mGluR5 and CB1 activation in the infralimbic mPFC.
Beneficial effects of mGluR5 and CB1 coactivation on arthritis pain-related behaviors
Audible and ultrasonic vocalizations evoked by compression of the knee with different intensities were measured before and during stereotaxic application of VU'172 (100 μm, concentration in microdialysis probe) and ACEA (10 μm) or ACSF (vehicle control) into the infralimbic mPFC by microdialysis for 20 min (Fig. 6). The combined application of VU'172 and ACEA, which increased pyramidal cell output (Fig. 3), inhibited spinal withdrawal reflexes (Fig. 6A) and audible and ultrasonic vocalizations (Fig. 6B,C) that were increased in the arthritis pain model (5–6 h postinduction). Vocalizations were evoked by brief (15 s) mechanical compression of the knee joint. Drug effects were significant (n = 6 rats, p < 0.05; Bonferroni post-tests). Importantly, stereotaxic injections of VU'172 and ACEA into the anterior cingulate cortex (ACC; area 24b) had no significant effect on hindlimb withdrawal thresholds and vocalizations of arthritic rats (n = 6 for each test; p > 0.05, paired t tests; Fig. 7).
Figure 6.

Coactivation of CB1 and mGluR5 in infralimbic mPFC inhibits pain-related behaviors. VU'172 (100 μm) and ACEA (10 μm) or ACSF (vehicle control) were applied stereotaxically into the infralimbic mPFC by microdialysis for 20 min. Note that numbers refer to concentration in the microdialysis probe. Tissue concentrations are estimated to be 100 times lower (see Materials and Methods). A, Mechanical thresholds for hindlimb withdrawal reflexes were measured by compressing the knee joint with a calibrated forceps (n = 6 rats). ***p < 0.001, compared with normal; #p < 0.05, compared with predrug in arthritis (Bonferroni posttests). B, C, Duration of audible and ultrasonic vocalizations evoked by compression of the knee with a calibrated forceps for 15 s (n = 6 rats). **p < 0.01, ***p < 0.001, compared with normal; #p < 0.05, compared with predrug in arthritis (Bonferroni posttests). D, Results of rodent gambling task (see Materials and Methods). Preference index for “high-risk” (3 food pellets in 3/10 trials) or “low-risk” (1 food pellet in 9/10 trials) choices of levers providing food rewards was calculated and averaged for every 10 consecutive trials (session of 90 trials; see Materials and Methods). Negative preference indicates high-risk decision making. Normal rats (n = 6 rats) changed their strategy to prefer low-risk lever, but arthritic rats (n = 8 rats) did not. Intra-mPFC administration of VU'172 and ACEA had no effect in normal rats (n = 5 rats) but restored decision making (ability to switch preference) in arthritic rats (n = 5 rats). E, Final preference index was calculated for the last 10 trials of each session for statistical analysis. ns, p > 0.05 compared with normal vehicle, ***p < 0.001, compared with normal vehicle, ###p < 0.001 compared with arthritis vehicle (Bonferroni posttests). F, Histologic verification of drug application sites. Diagrams show coronal brain slices. Numbers indicate distance from the bregma. Symbols show the positions of the microdialysis probes in the mPFC of normal (○; n = 17) and arthritic animals (•; n = 19).
Figure 7.

Coactivation of CB1 and mGluR5 in anterior cingulate cortex has no effect on pain-related behaviors. VU'172 (100 μm) and ACEA (10 μm) or ACSF (vehicle control) were applied stereotaxically into the ACC (area 24b) by microdialysis for 20 min (Fig. 6; see Materials and Methods). A, Mechanical thresholds for hindlimb withdrawal reflexes evoked by compressing the knee joint with a calibrated forceps (n = 6 rats). B, C, Duration of audible and ultrasonic vocalizations evoked by compression of the knee with a calibrated forceps for 15 s (n = 6 rats). D, Histologic verification of drug application sites. Diagrams show coronal brain slices. Number indicates distance from the bregma.
Our previous studies showed that amygdala hyperactivity (Ji et al., 2010) or mPFC deactivation (Sun and Neugebauer, 2011) impaired reward-based decision-making in a rodent gambling task model. Results from the present study confirm that arthritic rats (n = 8; 5–6 h postinduction) fail to switch strategies and persist in preferring the “high-risk” lever that provides three chocolate-coated food pellets in only 3 of 10 trials in a series of 90 consecutive trials, reflected in a negative preference index (Fig. 6D). In contrast, normal animals (n = 6 rats) switch from preferring the high-risk lever initially to preferring the low-risk lever that provides one food reward consistently in 9 of 10 trials. In these animals, ACSF was administered as vehicle control. Stereotaxic coapplication of VU'172 (100 μm, concentration in microdialysis probe) and ACEA (10 μm) into the infralimbic mPFC had no effect in normal rats (n = 5 rats) but restored decision making in arthritic rats (n = 5) so that they were able to switch strategies like normal rats. For statistical analysis, the final preference index was calculated (average of final 10 trials of each session). Differences were significant (F(3,20) = 15.15 ANOVA) for arthritic rats compared with normal rats (p < 0.001 Bonferroni posttests) and for the effect of VU'172 plus ACEA in arthritic rats compared with ACSF control in arthritic rats (p < 0.001).
The data suggest that pharmacological activation of infralimbic output with a combination of mGluR5 and CB1 activators inhibits pain-related behaviors and restores decision making in the arthritis pain model.
Discussion
Using patch-clamp slice physiology, pharmacology, optogenetics, and behavior this study advances the novel concept that mGluR5 fails to engage endocannabinoid (2-AG) signaling to overcome abnormal synaptic inhibition of the infralimbic mPFC pyramidal cells in an arthritis pain model; restoring endocannabinoid signaling allows mGluR5 to increase mPFC output hence inhibit pain behaviors and mitigate cognitive deficits. We report several conceptually and technologically innovative findings on pain-related cortical dysfunction and rescue strategies. To the best of our knowledge, this is the first demonstration of direct excitatory and feedforward synaptic inputs from the BLA to infralimbic mPFC using an optogenetic approach. Our previous studies used electrical stimulation of fibers labeled with a fluorescent tracer injected into the BLA (Ji et al., 2010; Sun and Neugebauer, 2011; Kiritoshi et al., 2013), but activation of additional fibers from cortical and extracortical sources could not be excluded entirely. Here we show that optical activation of light-sensitive channels (ChR2) expressed in BLA pyramidal cells using the CaMKII promotor reproduces our results with electrical stimulation. This is not trivial because it is important to understand the source of information to the cortex and the site(s) of drug actions. This study shows that it is indeed input from the BLA that undergoes maladaptive changes in the arthritis pain model.
The results provide several novel insights into pain-related changes and function of the infralimbic mPFC (use of this nomenclature is explained in Results). We found enhanced feedforward inhibition and loss of DSI in the arthritis pain model. Our previous work linked enhanced feedforward inhibition in the prelimbic cortex to cortical deactivation (Ji et al., 2010; Sun and Neugebauer, 2011). Differential roles have been suggested for prelimbic and infralimbic cortices (Sierra-Mercado et al., 2011; Mendoza et al., 2015) and so it is important to analyze and compare pain-related changes in these regions. We focused on the infralimbic cortex for a number of reasons. The infralimbic region of the mPFC inhibits amygdala output to “extinguish” aversive behaviors (Likhtik et al., 2005; Sah and Westbrook, 2008; Herry et al., 2010; Kim and Richardson, 2010; Pape and Pare, 2010; Sotres-Bayon and Quirk, 2010; Orsini and Maren, 2012). Decreased infralimbic activity has been implicated in extinction deficits (Hefner et al., 2008; Chang and Maren, 2010; Kim et al., 2010; Sierra-Mercado et al., 2011). Extinction deficits have been proposed as a mechanism of the persistence of pain and its negative affective dimension (Apkarian et al., 2009), and failure to activate the mPFC has been linked to visceral hypersensitivity in patients (Mayer et al., 2005).
Importantly, our data link the dramatic increase in inhibition of infralimbic pyramidal cells mechanistically to dysfunction of mGluR5-2-AG signaling. In the rodent mPFC, CB1 is exclusively expressed in GABAergic interneurons (Marsicano and Lutz, 1999; Wedzony and Chocyk, 2009) and presynaptic CB1 receptors face postsynaptic mGluR5 on pyramidal cells (Lafourcade et al., 2007). mGluR5 can couple to 2-AG synthesis (Katona and Freund, 2008; Di Marzo, 2011) and to retrograde endocannabinoid signaling involving presynaptic CB1 receptors to depress inhibitory transmission (Freund et al., 2003; Lovinger, 2008; Kano et al., 2009). Our data show that this is the case in infralimbic pyramidal cells because the facilitatory effects of mGluR5 can be blocked with an inhibitor of 2-AG synthesis or a CB1 receptor antagonist (Fig. 2), suggesting an important interaction of mGluR5 and CB1 activation in the regulation of pyramidal cell output measured as BLA-driven synaptically evoked spiking. However, this interaction fails in the pain model where the facilitatory effects of a selective mGluR5 activator (VU'172; Rodriguez et al., 2010) on pyramidal output are lost and so is the ability of a selective CB1 agonist (ACEA; Hillard et al., 1999) to control synaptic inhibition.
The following results support the conclusion that impaired mGluR5-2-AG signaling fails to control synaptic inhibition, resulting in decreased pyramidal output. In the arthritis pain model, activation of mGluR5 failed to increase evoked spiking, but the facilitatory effect was restored by increasing the availability of endocannabinoids in the postsynaptic pyramidal cell with an inhibitor of postsynaptic 2-AG hydrolyzing enzyme ABHD6 (WWL70, in the patch pipette) or an inhibitor of monoacylglycerol lipase MGL (JZL184). Activating CB1 exogenously with ACEA or removing abnormal synaptic inhibition of pyramidal cells with picrotoxin included in the patch-pipette also restored the effect of mGluR5 activation on pyramidal output. The combined activation of mGluR5 and CB1 controlled abnormally enhanced feedforward inhibition and restored endocannabinoid-mediated presynaptic inhibition of synaptic inhibition (DSI). The data suggest a lack of available 2-AG rather than of functional CB1 receptors. The data also argue against impaired release of endocannabinoids, because increasing availability of 2-AG in the postsynaptic cell restored endocannabinoid-dependent facilitation of pyramidal output by mGluR5.
The importance of intact interactions between mGluR5 and the endocannabinoid system for pain modulation was shown recently in the dorsolateral periaqueductal gray (Gregg et al., 2012). Footshock produced stress-induced antinociception by activating mGluR5 and mobilizing 2-AG through a mechanism that required postsynaptic diacylglycerol lipase activity and presynaptic CB1 receptors (Gregg et al., 2012). Our behavioral data suggest that intact mGluR5-2-AG interaction is also required in the infralimbic mPFC for pain control. Coactivation of mGluR5 and CB1 in the infralimbic, but not anterior cingulate, cortex inhibited pain responses and restored decision-making in the arthritis pain model. Together with the electrophysiological results, our data suggest that restoring infralimbic output is a powerful pain control mechanism and strategy, which is consistent the finding that consolidation of fear extinction depends on mGluR5 activation in the infralimbic cortex (Fontanez-Nuin et al., 2011). In contrast to mGluR5, other mGluRs would be less suitable targets to enhance pyramidal output. Our previous work showed that mGluR1 activates feedforward inhibition of mPFC pyramidal cells (Sun and Neugebauer, 2011), whereas group II mGluRs inhibit direct excitatory transmission as well as feedforward inhibition onto pyramidal cells, but their net effect is decreased pyramidal cell output (Kiritoshi and Neugebauer, 2015).
The concept of activating mPFC to inhibit pain behaviors has recently been addressed in an elegant study showing that optogenetic activation of the prelimbic cortex produced strong antinociceptive effects in a neuropathic pain model (Lee et al., 2015). That study implicated projections to the nucleus accumbens in the antinociceptive effects of prelimbic mPFC activation (Lee et al., 2015). Although the present study did not determine the involvement of subcortical structures and pathways, we showed previously that pharmacological activation of the infralimbic cortex can inhibit activity of amygdala output neurons (Ji and Neugebauer, 2014). This is consistent with a key role of the infralimbic cortex in certain aspects of behavioral extinction through inhibition of amygdala output (Maren and Quirk, 2004; Likhtik et al., 2005; Akirav and Maroun, 2007; Sah and Westbrook, 2008; Herry et al., 2010; Kim and Richardson, 2010; Pape and Pare, 2010; Sotres-Bayon and Quirk, 2010). Pain-related enhanced feedforward inhibition and decreased neuronal activity have been found in the prelimbic (Ji et al., 2010; Ji and Neugebauer, 2011) and infralimbic (this study) cortex. A recent study from another group (Zhang et al., 2015) confirmed enhanced feedforward inhibition in the prelimbic mPFC in a neuropathic pain model and showed that optogenetic inhibition of GABAergic interneurons decreased pain responses in freely moving mice (Zhang et al., 2015). Although the relative contribution of different cortico-subcortical loops to pain control remains to be determined, a unifying picture is emerging that removing abnormal inhibition and increasing mPFC output can inhibit pain.
Some technical aspects need to be considered. We used selective compounds at concentrations that are well established in the literature. ACEA, a synthetic analog of anandamide, is one of the most selective agonists for CB1 (Hillard et al., 1999). Although high concentrations of ACEA (>10 μm) can activate TRPV1 in the brain (Casarotto et al., 2012), we used a concentration of 10 nm in the brain slice experiments and 10 μm in the microdialysis probe in the behavioral studies where the concentration achieved in the tissue is at least 100-fold lower due to the concentration gradient across the microdialysis membrane and diffusion in the tissue. Importantly, the effects of ACEA and VU0360172 persisted in the presence of a TRPV1 receptor antagonist (AMG9810; Fig. 4). Further, CB1 and TRPV1 receptors have opposing effects in the prefrontal cortex (Rubino et al., 2008; Giordano et al., 2012) and TRPV1 activation counteracts endocannabinoid-mediated retrograde inhibition of GABAergic transmission in the striatum (Di Marzo, 2011). We did not test for CB2 involvement because it is CB1 that mediates DSI; CB2 produces opposite effects in the mPFC and CB2 activation reduces firing of mPFC pyramidal cells through calcium-activated chloride channels (den Boon et al., 2012). VU0360172 (VU'172) is one of the most potent and selective mGluR5-positive allosteric modulators (Rodriguez et al., 2010). We showed previously that the effects of VU'172 on synaptically evoked spiking were inhibited by a selective negative allosteric modulator of mGluR5 (MTEP) (Kiritoshi et al., 2013). Microdialysis was chosen for drug delivery in the behavioral experiments because it provides steady-state drug levels without a volume effect (Stiller et al., 2003). Drug injection into the anterior cingulate cortex as placement control had no effect. The distance between injections sites into infralimbic and anterior cingulate cortex was <2 mm, suggesting that drugs did not spread beyond a distance of 1 mm around the tip of the microdialysis probe. We cannot rule out the possibility of some drug diffusion from the infralimbic injection site into the adjacent prelimbic cortex. However, such spread would have also occurred with the injection into the anterior cingulate region, which had no effect. Another issue relates to optogenetic activation and comparability across animals and brain slices, which depend on viral vector-mediated expression levels. To control for any variability, we selected intensities that produce similar levels of synaptically evoked spiking and results obtained with optogenetic activation matched those with electrical synaptic stimulation.
In conclusion, breakdown of mGluR5-endocannabinoid signaling at BLA synaptic inputs to infralimbic mPFC fails to control abnormal synaptic inhibition of infralimbic pyramidal cells in an arthritis pain model. Restoring endocannabinoid signaling allows mGluR5 activation to increase infralimbic output hence inhibit pain behaviors and mitigate cognitive deficits.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke (NIH/NINDS) Grants NS081121 and NS038261.
The authors declare no competing financial interests.
References
- Akirav I, Maroun M. The role of the medial prefrontal cortex-amygdala circuit in stress effects on the extinction of fear. Neural Plast. 2007;2007:30873. doi: 10.1155/2007/30873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir A, Amano T, Pare D. Physiological identification and infralimbic responsiveness of rat intercalated amygdala neurons. J Neurophysiol. 2011;105:3054–3066. doi: 10.1152/jn.00136.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apkarian AV, Sosa Y, Krauss BR, Thomas PS, Fredrickson BE, Levy RE, Harden RN, Chialvo DR. Chronic pain patients are impaired on an emotional decision-making task. Pain. 2004;108:129–136. doi: 10.1016/j.pain.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Apkarian AV, Baliki MN, Geha PY. Towards a theory of chronic pain. Prog Neurobiol. 2009;87:81–97. doi: 10.1016/j.pneurobio.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Cascio MG, Saha B, Mahadevan A, Urbani P, Minassi A, Appendino G, Saturnino C, Martin B, Razdan R, Di Marzo V. Development of the first potent and specific inhibitors of endocannabinoid biosynthesis. Biochim Biophys Acta. 2006;1761:205–212. doi: 10.1016/j.bbalip.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW., 4th Hemispheric lateralization of a molecular signal for pain modulation in the amygdala. Mol Pain. 2008;4:24. doi: 10.1186/1744-8069-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarotto PC, Terzian AL, Aguiar DC, Zangrossi H, Guimarães FS, Wotjak CT, Moreira FA. Opposing roles for cannabinoid receptor type-1 (CB) and transient receptor potential vanilloid type-1 channel (TRPV1) on the modulation of panic-like responses in rats. Neuropsychopharmacology. 2012;37:478–486. doi: 10.1038/npp.2011.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Maren S. Strain difference in the effect of infralimbic cortex lesions on fear extinction in rats. Behav Neurosci. 2010;124:391–397. doi: 10.1037/a0019479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Berke JD, Maren S. Single-unit activity in the medial prefrontal cortex during immediate and delayed extinction of fear in rats. PLoS One. 2010;5:e11971. doi: 10.1371/journal.pone.0011971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boon FS, Chameau P, Schaafsma-Zhao Q, van Aken W, Bari M, Oddi S, Kruse CG, Maccarrone M, Wadman WJ, Werkman TR. Excitability of prefrontal cortical pyramidal neurons is modulated by activation of intracellular type-2 cannabinoid receptors. Proc Natl Acad Sci U S A. 2012;109:3534–3539. doi: 10.1073/pnas.1118167109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V. Endocannabinoid signaling in the brain: biosynthetic mechanisms in the limelight. Nat Neurosci. 2011;14:9–15. doi: 10.1038/nn.2720. [DOI] [PubMed] [Google Scholar]
- Do-Monte FH, Manzano-Nieves G, Quiñones-Laracuente K, Ramos-Medina L, Quirk GJ. Revisiting the role of infralimbic cortex in fear extinction with optogenetics. J Neurosci. 2015;35:3607–3615. doi: 10.1523/JNEUROSCI.3137-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald PJ, Whittle N, Flynn SM, Graybeal C, Pinard CR, Gunduz-Cinar O, Kravitz AV, Singewald N, Holmes A. Prefrontal single-unit firing associated with deficient extinction in mice. Neurobiol Learn Mem. 2014;113:69–81. doi: 10.1016/j.nlm.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanez-Nuin DE, Santini E, Quirk GJ, Porter JT. Memory for fear extinction requires mGluR5-mediated activation of infralimbic neurons. Cereb Cortex. 2011;21:727–735. doi: 10.1093/cercor/bhq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Gavva NR, Tamir R, Qu Y, Klionsky L, Zhang TJ, Immke D, Wang J, Zhu D, Vanderah TW, Porreca F, Doherty EM, Norman MH, Wild KD, Bannon AW, Louis JC, Treanor JJ. AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther. 2005;313:474–484. doi: 10.1124/jpet.104.079855. [DOI] [PubMed] [Google Scholar]
- Giordano C, Cristino L, Luongo L, Siniscalco D, Petrosino S, Piscitelli F, Marabese I, Gatta L, Rossi F, Imperatore R, Palazzo E, de Novellis V, Di M, Di Marzo V, Maione S. TRPV1-dependent and -independent alterations in the limbic cortex of neuropathic mice: impact on glial caspases and pain perception. Cereb Cortex. 2012;22:2495–2518. doi: 10.1093/cercor/bhr328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg LC, Jung KM, Spradley JM, Nyilas R, Suplita RL, 2nd, Zimmer A, Watanabe M, Mackie K, Katona I, Piomelli D, Hohmann AG. Activation of type 5 metabotropic glutamate receptors and diacylglycerol lipase-alpha initiates 2-arachidonoylglycerol formation and endocannabinoid-mediated analgesia. J Neurosci. 2012;32:9457–9468. doi: 10.1523/JNEUROSCI.0013-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grégoire S, Neugebauer V. 5-HT2CR blockade in the amygdala conveys analgesic efficacy to SSRIs in a rat model of arthritis pain. Mol Pain. 2013;9:41. doi: 10.1186/1744-8069-9-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, Hohmann AG. The endocannabinoid system and pain. CNS Neurol Disord Drug Targets. 2009;8:403–421. doi: 10.2174/187152709789824660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Bird GC, Li W, Neugebauer V. Computerized analysis of audible and ultrasonic vocalizations of rats as a standardized measure of pain-related behavior. 2005:261–269. doi: 10.1016/j.jneumeth.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Hefner K, Whittle N, Juhasz J, Norcross M, Karlsson RM, Saksida LM, Bussey TJ, Singewald N, Holmes A. Impaired fear extinction learning and cortico-amygdala circuit abnormalities in a common genetic mouse strain. J Neurosci. 2008;28:8074–8085. doi: 10.1523/JNEUROSCI.4904-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ferraguti F, Singewald N, Letzkus JJ, Ehrlich I, Lüthi A. Neuronal circuits of fear extinction. Eur J Neurosci. 2010;31:599–612. doi: 10.1111/j.1460-9568.2010.07101.x. [DOI] [PubMed] [Google Scholar]
- Hillard CJ, Manna S, Greenberg MJ, DiCamelli R, Ross RA, Stevenson LA, Murphy V, Pertwee RG, Campbell WB. Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1) J Pharmacol Exp Ther. 1999;289:1427–1433. [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Hemispheric lateralization of pain processing by amygdala neurons. J Neurophysiol. 2009;102:2253–2264. doi: 10.1152/jn.00166.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Pain-related deactivation of medial prefrontal cortical neurons involves mGluR1 and GABA(A) receptors. J Neurophysiol. 2011;106:2642–2652. doi: 10.1152/jn.00461.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Modulation of medial prefrontal cortical activity using in vivo recordings and optogenetics. Mol Brain. 2012;5:36–45. doi: 10.1186/1756-6606-5-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. CB1 augments mGluR5 function in medial prefrontal cortical neurons to inhibit amygdala hyperactivity in an arthritis pain model. Eur J Neurosci. 2014;39:455–466. doi: 10.1111/ejn.12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Sun H, Fu Y, Li Z, Pais-Vieira M, Galhardo V, Neugebauer V. Cognitive impairment in pain through amygdala-driven prefrontal cortical deactivation. J Neurosci. 2010;30:5451–5464. doi: 10.1523/JNEUROSCI.0225-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Kasanetz F, Lafourcade M, Deroche-Gamonet V, Revest JM, Berson N, Balado E, Fiancette JF, Renault P, Piazza PV, Manzoni OJ. Prefrontal synaptic markers of cocaine addiction-like behavior in rats. Mol Psychiatry. 2013;18:729–737. doi: 10.1038/mp.2012.59. [DOI] [PubMed] [Google Scholar]
- Katona I, Freund TF. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat Med. 2008;14:923–930. doi: 10.1038/nm.f.1869. [DOI] [PubMed] [Google Scholar]
- Kim JH, Richardson R. New findings on extinction of conditioned fear early in development: theoretical and clinical implications. Biol Psychiatry. 2010;67:297–303. doi: 10.1016/j.biopsych.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Kim SC, Jo YS, Kim IH, Kim H, Choi JS. Lack of medial prefrontal cortex activation underlies the immediate extinction deficit. J Neurosci. 2010;30:832–837. doi: 10.1523/JNEUROSCI.4145-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiritoshi T, Neugebauer V. Group II mGluRs modulate baseline and arthritis pain-related synaptic transmission in the rat medial prefrontal cortex. Neuropharmacology. 2015;95:388–394. doi: 10.1016/j.neuropharm.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiritoshi T, Sun H, Ren W, Stauffer SR, Lindsley CW, Conn PJ, Neugebauer V. Modulation of pyramidal cell output in the medial prefrontal cortex by mGluR5 interacting with CB1. Neuropharmacology. 2013;66:170–178. doi: 10.1016/j.neuropharm.2012.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafourcade M, Elezgarai I, Mato S, Bakiri Y, Grandes P, Manzoni OJ. Molecular components and functions of the endocannabinoid system in mouse prefrontal cortex. PLoS One. 2007;2:e709. doi: 10.1371/journal.pone.0000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Manders TR, Eberle SE, Su C, D'amour J, Yang R, Lin HY, Deisseroth K, Froemke RC, Wang J. Activation of corticostriatal circuitry relieves chronic neuropathic pain. J Neurosci. 2015;35:5247–5259. doi: 10.1523/JNEUROSCI.3494-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Blankman JL, Cravatt BF. A functional proteomic strategy to discover inhibitors for uncharacterized hydrolases. J Am Chem Soc. 2007;129:9594–9595. doi: 10.1021/ja073650c. [DOI] [PubMed] [Google Scholar]
- Likhtik E, Pelletier JG, Paz R, Paré D. Prefrontal control of the amygdala. J Neurosci. 2005;25:7429–7437. doi: 10.1523/JNEUROSCI.2314-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Mao SC, Chen PS, Gean PW. Chronic cannabinoid administration in vivo compromises extinction of fear memory. Learn Mem. 2008;15:876–884. doi: 10.1101/lm.1081908. [DOI] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavón FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM. Presynaptic modulation by endocannabinoids. Handb Exp Pharmacol. 2008;184:435–477. doi: 10.1007/978-3-540-74805-2_14. [DOI] [PubMed] [Google Scholar]
- Marek GJ, Zhang C. Activation of metabotropic glutamate 5 (mGlu5) receptors induces spontaneous excitatory synaptic currents in layer V pyramidal cells of the rat prefrontal cortex. Neurosci Lett. 2008;442:239–243. doi: 10.1016/j.neulet.2008.06.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek R, Strobel C, Bredy TW, Sah P. The amygdala and medial prefrontal cortex: partners in the fear circuit. J Physiol. 2013;591:2381–2391. doi: 10.1113/jphysiol.2012.248575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S, Quirk GJ. Neuronal signalling of fear memory. Nat Rev Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, Bodor AL, Muccioli GG, Hu SS, Woodruff G, Fung S, Lafourcade M, Alexander JP, Long JZ, Li W, Xu C, Möller T, Mackie K, Manzoni OJ, Cravatt BF, Stella N. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. Eur J Neurosci. 1999;11:4213–4225. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- Mayer EA, Berman S, Suyenobu B, Labus J, Mandelkern MA, Naliboff BD, Chang L. Differences in brain responses to visceral pain between patients with irritable bowel syndrome and ulcerative colitis. Pain. 2005;115:398–409. doi: 10.1016/j.pain.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Medina G, Ji G, Grégoire S, Neugebauer V. Nasal application of neuropeptide S inhibits arthritis pain-related behaviors through an action in the amygdala. Mol Pain. 2014;10:32. doi: 10.1186/1744-8069-10-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza J, Sanio C, Chaudhri N. Inactivating the infralimbic but not prelimbic medial prefrontal cortex facilitates the extinction of appetitive Pavlovian conditioning in Long–Evans rats. Neurobiol Learn Mem. 2015;118:198–208. doi: 10.1016/j.nlm.2014.12.006. [DOI] [PubMed] [Google Scholar]
- Metz AE, Yau HJ, Centeno MV, Apkarian AV, Martina M. Morphological and functional reorganization of rat medial prefrontal cortex in neuropathic pain. Proc Natl Acad Sci U S A. 2009;106:2423–2428. doi: 10.1073/pnas.0809897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Moriarty O, McGuire BE, Finn DP. The effect of pain on cognitive function: a review of clinical and preclinical research. Prog Neurobiol. 2011;93:385–404. doi: 10.1016/j.pneurobio.2011.01.002. [DOI] [PubMed] [Google Scholar]
- Muly EC, Maddox M, Smith Y. Distribution of mGluR1alpha and mGluR5 immunolabeling in primate prefrontal cortex. J Comp Neurol. 2003;467:521–535. doi: 10.1002/cne.10937. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Bhave G, Gereau RW., 4th Synaptic plasticity in the amygdala in a model of arthritic pain: differential roles of metabotropic glutamate receptors 1 and 5. J Neurosci. 2003;23:52–63. doi: 10.1523/JNEUROSCI.23-01-00052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Han JS, Adwanikar H, Fu Y, Ji G. Techniques for assessing knee joint pain in arthritis. Mol Pain. 2007;3:8–20. doi: 10.1186/1744-8069-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini CA, Maren S. Neural and cellular mechanisms of fear and extinction memory formation. Neurosci Biobehav Rev. 2012;36:1773–1802. doi: 10.1016/j.neubiorev.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pais-Vieira M, Mendes-Pinto MM, Lima D, Galhardo V. Cognitive impairment of prefrontal-dependent decision-making in rats after the onset of chronic pain. Neuroscience. 2009;161:671–679. doi: 10.1016/j.neuroscience.2009.04.011. [DOI] [PubMed] [Google Scholar]
- Pape HC, Pare D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev. 2010;90:419–463. doi: 10.1152/physrev.00037.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. Receptors and channels targeted by synthetic cannabinoid receptor agonists and antagonists. Curr Med Chem. 2010;17:1360–1381. doi: 10.2174/092986710790980050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W, Kiritoshi T, Grégoire S, Ji G, Guerrini R, Calo G, Neugebauer V. Neuropeptide S: a novel regulator of pain-related amygdala plasticity and behaviors. J Neurophysiol. 2013;110:1765–1781. doi: 10.1152/jn.00874.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera P, Arrabal S, Cifuentes M, Grondona JM, Pérez-Martín M, Rubio L, Vargas A, Serrano A, Pavón FJ, Suárez J, Rodríguez de FF. Localization of the cannabinoid CB1 receptor and the 2-AG synthesizing (DAGLalpha) and degrading (MAGL, FAAH) enzymes in cells expressing the Ca(2+)-binding proteins calbindin, calretinin, and parvalbumin in the adult rat hippocampus. Front Neuroanat. 2014;8:56. doi: 10.3389/fnana.2014.00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, Blatt TN, Jadhav S, Menon UN, Vinson PN, Rook JM, Stauffer SR, Niswender CM, Lindsley CW, Weaver CD, Conn PJ. Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol Pharmacol. 2010;78:1105–1123. doi: 10.1124/mol.110.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino T, Realini N, Castiglioni C, Guidali C, Viganó D, Marras E, Petrosino S, Perletti G, Maccarrone M, Di Marzo V, Parolaro D. Role in anxiety behavior of the endocannabinoid system in the prefrontal cortex. Cereb Cortex. 2008;18:1292–1301. doi: 10.1093/cercor/bhm161. [DOI] [PubMed] [Google Scholar]
- Sah P, Westbrook RF. Behavioural neuroscience: the circuit of fear. Nature. 2008;454:589–590. doi: 10.1038/454589a. [DOI] [PubMed] [Google Scholar]
- Sepulveda-Orengo MT, Lopez AV, Soler-Cedeño O, Porter JT. Fear extinction induces mGluR5-mediated synaptic and intrinsic plasticity in infralimbic neurons. J Neurosci. 2013;33:7184–7193. doi: 10.1523/JNEUROSCI.5198-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology. 2011;36:529–538. doi: 10.1038/npp.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotres-Bayon F, Quirk GJ. Prefrontal control of fear: more than just extinction. Curr Opin Neurobiol. 2010;20:231–235. doi: 10.1016/j.conb.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiller CO, Taylor BK, Linderoth B, Gustafsson H, Warsame Afrah A, Brodin E. Microdialysis in pain research. Adv Drug Deliv Rev. 2003;55:1065–1079. doi: 10.1016/S0169-409X(03)00104-2. [DOI] [PubMed] [Google Scholar]
- Sun H, Neugebauer V. mGluR1, but not mGluR5, activates feed-forward inhibition in the medial prefrontal cortex to impair decision making. J Neurophysiol. 2011;106:960–973. doi: 10.1152/jn.00762.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt BA, Paxinos G. Cytoarchitecture of mouse and rat cingulate cortex with human homologies. Brain Struct Funct. 2014;219:185–192. doi: 10.1007/s00429-012-0493-3. [DOI] [PubMed] [Google Scholar]
- Wang GQ, Cen C, Li C, Cao S, Wang N, Zhou Z, Liu XM, Xu Y, Tian NX, Zhang Y, Wang J, Wang LP, Wang Y. Deactivation of excitatory neurons in the prelimbic cortex via Cdk5 promotes pain sensation and anxiety. Nat Commun. 2015;6:7660. doi: 10.1038/ncomms8660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedzony K, Chocyk A. Cannabinoid CB1 receptors in rat medial prefrontal cortex are colocalized with calbindin- but not parvalbumin- and calretinin-positive GABA-ergic neurons. Pharmacol Rep. 2009;61:1000–1007. doi: 10.1016/S1734-1140(09)70161-6. [DOI] [PubMed] [Google Scholar]
- Wei W, Coelho CM, Li X, Marek R, Yan S, Anderson S, Meyers D, Mukherjee C, Sbardella G, Castellano S, Milite C, Rotili D, Mai A, Cole PA, Sah P, Kobor MS, Bredy TW. p300/CBP-associated factor selectively regulates the extinction of conditioned fear. J Neurosci. 2012;32:11930–11941. doi: 10.1523/JNEUROSCI.0178-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhu Y, Contractor A, Heinemann SF. mGluR5 has a critical role in inhibitory learning. J Neurosci. 2009;29:3676–3684. doi: 10.1523/JNEUROSCI.5716-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wang M, Bisogno T, Di Marzo V, Alger BE. Endocannabinoids generated by Ca2+ or by metabotropic glutamate receptors appear to arise from different pools of diacylglycerol lipase. PLoS One. 2011;6:e16305. doi: 10.1371/journal.pone.0016305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Gadotti VM, Chen L, Souza IA, Stemkowski PL, Zamponi GW. Role of prelimbic GABAergic circuits in sensory and emotional aspects of neuropathic pain. Cell Rep. 2015;12:752–759. doi: 10.1016/j.celrep.2015.07.001. [DOI] [PubMed] [Google Scholar]