

Abstract
Large cytoplasmic domains (CD) are a common feature among integral membrane proteins. In virtually all cases, these CD have a function (e.g., binding cytoskeleton or regulatory factors) separate from that of the membrane domain (MD). Strong associations between CD and MD are rare. Here we studied SLC4A11, a membrane transport protein of corneal endothelial cells, the mutations of which cause genetic corneal blindness. SLC4A11 has a 41-kDa CD and a 57-kDa integral MD. One disease-causing mutation in the CD, R125H, manifests a catalytic defect, suggesting a role of the CD in transport function. Expressed in HEK-293 cells without the CD, MD-SLC4A11 is retained in the endoplasmic reticulum, indicating a folding defect. Replacement of CD-SLC4A11 with green fluorescent protein did not rescue MD-SLC4A11, suggesting some specific role of CD-SLC4A11. Homology modeling revealed that the structure of CD-SLC4A11 is similar to that of the Cl−/HCO3− exchange protein AE1 (SLC4A1) CD. Fusion to CD-AE1 partially rescued MD-SLC4A11 to the cell surface, suggesting that the structure of CD-AE1 is similar to that of CD-SLC4A11. The CD-AE1-MD-SLC4a11 chimera, however, had no functional activity. We conclude that CD-SLC4A11 has an indispensable role in the transport function of SLC4A11. CD-SLC4A11 forms insoluble precipitates when expressed in bacteria, suggesting that the domain cannot fold properly when expressed alone. Consistent with a strong association between CD-SLC4A11 and MD-SLC4A11, these domains specifically associate when coexpressed in HEK-293 cells. We conclude that SLC4A11 is a rare integral membrane protein in which the CD has strong associations with the integral MD, which contributes to membrane transport function.
Keywords: corneal dystrophy, cytoplasmic domain, membrane domain, protein misfolding, SLC4A11
the integral membrane transport protein SLC4A11 is located in the water-transporting thin descending portion of the loop of Henle (14, 34) and basolaterally in the corneal endothelial layer, where it faces the corneal stroma (45). The SLC4A11 gene [Mendelian Inheritance in Man (MIM) no. 610206] encodes an 891-amino acid membrane protein, composed of an NH2-terminal 374-amino acid cytoplasmic domain (CD) and a 517-amino acid integral membrane domain (MD) with 14 transmembrane segments (Fig. 1B) (46). SLC4A11 is a member of the SLC4 family of bicarbonate transporters (3). Similar to anion exchanger 1 [AE1 (SLC4A1, band 3)], a well-studied protein in the SLC4 family, SLC4A11 exists as dimers (44).
Fig. 1.

Substrate translocation pore along the cytoplasmic domain (CD) and membrane domain (MD) of dimeric SLC4A11. A: SLC4A11 has a 374-amino acid NH2-terminal CD that may form an extension of the substrate translocation pore of the MD. Catalytically inactive mutants such as R125H may occlude the cytosolic pathway, interfering with transport function, but not folding, of the protein. Arrows indicate the possible substrate translocation pathway along the MD and CD of SLC4A11; R indicates residue R125 lining the pathway. B: topology model of SLC4A11 indicating start positions (arrows) of the MD constructs HA-353-MD, HA-347-MD, HA-329-MD, and HA-307-MD. FECD, Fuchs endothelial corneal dystrophy (FECD); CHED, congenital hereditary endothelial dystrophy (CHED) (1, 2, 11, 16, 21, 26, 27, 35, 37, 41, 42, 47, 48).
Although it belongs to the SLC4 family of bicarbonate transporters, SLC4A11 has no observed bicarbonate transport activity (19, 32). The plant SLC4A11 ortholog Bor1 facilitates borate transport (20). Human SLC4A11 was reported to be a Na+-coupled borate transporter (33), but this activity has not been confirmed (19, 32). Human SLC4A11 also has been reported to facilitate Na+-coupled OH− transport (19, 32), Na+-independent H+(OH−) transport (23), and H+-NH3 cotransport (50). We previously reported that human SLC4A11 facilitates transmembrane water movement driven by a hyposmotic extracellular environment (29, 45). SLC4A11 is the first protein shown to move water osmotically that does not belong to the major intrinsic protein family (45).
SLC4A11 mutations cause three posterior corneal dystrophies: 1) recessive congenital hereditary endothelial dystrophy (CHED; MIM no. 217700) (16, 21, 37, 42, 47); 2) recessive Harboyan syndrome (MIM no. 217400) (10), a combination of corneal dystrophy and perceptive deafness; and 3) dominant, late-onset Fuchs endothelial corneal dystrophy (FECD; MIM no. 136800) (10, 47, 48). These diseases are marked by fluid accumulation (edema) in the corneal stroma layer, which blurs vision. A urinary concentrating defect has been identified in mice lacking SLC4A11 (14), but renal deficits have not been reported in humans with SLC4A11 mutations.
Of the ∼60 SLC4A11 point mutations that have been identified (1, 2, 11, 16, 21, 24, 26, 27, 35, 37, 41, 42, 47, 48), 19 are located in the CD and the rest in the MD. Mutations are distributed, with no distinguishable pattern, throughout the protein (Fig. 1B). Most CHED, Harboyan syndrome, and FECD mutations result in intracellular SLC4A11 retention in the endoplasmic reticulum (ER), while a few, including SLC4A11-R125H, may be trafficked to the plasma membrane but are functionally inactive (29). Some ER-retained SLC4A11 mutants can be rescued to the plasma membrane surface, where they display functional activity (29).
One-third of SLC4A11 mutations map to the CD, indicating an important role in protein function; yet the role of the CD is unknown. Since the CD mutation R125H renders the protein inactive, a role of the CD in SLC4A11 membrane transport activity is suggested. Among SLC4 proteins, the two-domain (CD and MD) structure is entirely conserved. In the case of the red cell anion exchanger AE1 (band 3, SLC4A1), the CD are readily separated, and AE1 is functional in the absence of the CD (13, 39). Yet, important interactions between SLC4 CD and transport function are suggested by the role of AE2 (SLC4A2) CD in regulation of the rate of bicarbonate transport (51). Finally, the electrogenic Na+-HCO3− cotransporter NBCe1 (SLC4A4) has been proposed to have a substrate pathway through its CD. This pathway has been proposed to guide HCO3− to the MD, on the basis of structural modeling studies and a CD mutant (R298S), which affects the transport function, but not cell surface trafficking, of NBCe1 (9). In this study we set out to explore the role of CD-SLC4A11 in the structure and function of the protein.
MATERIALS AND METHODS
DNA constructs.
NH2-terminally hemagglutinin (HA) epitope-tagged splicing variant 2 of human SLC4A11 encoding an 891-amino acid protein (National Center for Biotechnology Information reference sequence NG_017072.1) in a eukaryotic expression construct (pSKL1) has been reported previously (29). The MD variants HA-353-MD (pSKL2), HA-347-MD (pSKL3), HA-329-MD (pSKL4), HA-307-MD (pSKL5), and Myc-307-MD-SLC4A11 (pSKL6) and the CD variants HA-CD (pSKL7) and Myc-CD-SLC4A11 (pSKL8) were prepared using a PCR-based strategy with pSKL1 as a template and primers (Table 1) that contain a 5′-NheI restriction site, an NH2-terminal HA or Myc tag, and a 3′-XhoI restriction site. PCR products were cloned into NheI/XhoI-digested pSKL1. The same strategy was employed to clone the 340-MD-AE1 (pSKL9) construct, except pPBAE1-oocyte was used as the template (5), with appropriate primers (Table 1). The AE1 cDNA corresponds to the human erythrocyte transcript, known as eAE1. His-tagged CD-SLC4A11 (CD-SLC4A11-His6, pCML1) was constructed by subcloning the insert (created by PCR using the forward and reverse primers 5′-AAGGAGATATACATATGAGCCAGGTCGGG-3′ and 5′-GGTGGTGGTGCTCGAGTCCTCCTCCTCCCTGGAAGTACAGGTTCTCTCCTCCTCCTTTCCCAATAATGCCATCAGTGAAGTCC-3′ and pSKL1 as the template) into the NdeI and XhoI sites of pET-12b using the In-Fusion cloning system and following the manufacturer's protocol. The primer incorporated a sequence encoding a tobacco etch virus protease cleavage site positioned 5′ to the His6 coding sequence in pET-21b. The QuikChange Lightning mutagenesis kit (Agilent) and the primers 5′-GTCGGAGGGCGTGGAGACCGTTGCACTCAGGAGGTCCAG-3′ and 5′-CTCCTGAGTGCAACGGTCTCCACGCCCTCCGACCTG-3′ were used to create a partially codon-optimized expression construct for CD-SLC4A11-His6 (pCML1). A glutathione-S-transferase (GST) fusion construct was created using pGEX-6P1 (GE Healthcare). The construct was modified to include a COOH-terminal tobacco etch virus cleavage site and a His10 tag. The BamHI/NotI sites in this modified construct, primers (5′-GGGATCCCCGGAATTCATGAGCCAGGTCGGAGG-3′ and 5′-TTTCCCAATAATGCCATCAGTGAAGTCC-3′), codon-optimized CD-SLC4A11-His6 as template, and the In-Fusion cloning kit were used to create the final construct (pCML2), which encoded CD-SLC4A11 with an NH2-terminal GST tag and a COOH-terminal His10 tag (GST-CD-SLC4A11-His10). The plasmids pSKL15 (encoding HA-CD-300-SLC4A11) and pSKL16 (encoding Myc-CD-300-SLC4A11), which include only SLC4A11 amino acids 1–300, were cloned using appropriate primers (Table 1), pSKL7 and pSKL8, respectively, as templates and the Q5 mutagenesis kit following the manufacturer's protocol.
Table 1.
Oligonucleotides used in cloning of MD and CD variants of SLC4A11 and AE1
| Primer |
||
|---|---|---|
| Construct: Plasmid Name/Construct | Forward | Reverse |
| pSKL2 (HA-353-MD-SLC4A11) | 5′-CTAGCTAGCGCCACCATGTACCCATACGATGTTCCGGATTACGCTAGGTTCCCCTTGTACCCCTTG-3′ | 5′-CCGCTCGAGTCAAGGCCTGTGCTCAGCGTC-3′ |
| pSKL3 (HA-347-MD-SLC4A11) | 5′-CTAGCTAGCGCCACCATGTACCCATACGATGTTCCGGATTACGCTCGGGAGGACATCGCACGCAGG-3′ | 5′-CCGCTCGAGTCAAGGCCTGTGCTCAGCGTC-3′ |
| pSKL4 (HA-329-MD-SLC4A11) | 5′-CTAGCTAGCGCCACCATGTACCCATACGATGTTCCGGATTACGCTAGACACCCAGAGCCCCCAAAG-3′ | 5′-CCGCTCGAGTCAAGGCCTGTGCTCAGCGTC-3′ |
| pSKL5 (HA-307-MD-SLC4A11) | 5′-CTAGCTAGCGCCACCATGTACCCATACGATGTTCCGGATTACGCTGTGAGCCACGGTCCAGTGGCG-3′ | 5′-CCGCTCGAGTCAAGGCCTGTGCTCAGCGTC-3′ |
| pSKL6 (Myc-307-MD-SLC4A11) | 5′-CTAGCTAGCGCCACCATGGAACAAAAGCTAATTTCAGAAGAAGACCTAGTGAGCCACGGTCCAGTGGCG-3′ | 5′-CCGCTCGAGTCAAGGCCTGTGCTCAGCGTC-3′ |
| pSKL7 (HA-CD-SLC4A11) | 5′-CTAGCTAGCGCCACCATGTACCCATACGATGTTCCGGATTACGCTAGCCAGGTCGGGGGGCGGGGAGAC-3′ | 5′-CCGCTCGAGTCATTTCCCAATAATGCCATCAGT-3′ |
| pSKL8 (Myc-CD-SLC4A11) | 5′-CTAGCTAGCGCCACCATGGAACAAAAGCTAATTTCAGAAGAAGACCTAAGCCAGGTCGGGGGGCGGGGAGAC-3′ | 5′-CCGCTCGAGTCATTTCCCAATAATGCCATCAGT-3′ |
| pSKL9 (340-MD-AE1) | 5′-CTAGCTAGCGCCACCAGGGAGCTACTTCGAAGGC-3′ | 5′-CCGCTCGAGTCACACAGGCATGGCCAC-3′ |
| pSKL15 (HA-CD-300-SLC4A11) | 5′-TGACTCGAGGACTGTGCCTTCTAGTTGCCAGC-3′ | 5′-ATGCACCAAGGCCTCCTTG-3′ |
| pSKL16 (Myc-CD-300-SLC4A11) | 5′-TGACTCGAGGACTGTGCCTTCTAGTTGCCAGC-3′ | 5′-ATGCACCAAGGCCTCCTTG-3′ |
Underlined bases code for hemagglutinin (HA) or Myc epitope tag. MD, membrane domain; CD, cytosolic domain.
Constructs encoding the fusion proteins GFP-368-MD, GFP-307-MD, mNect-368-MD, and mNect-307-MD-SLC4A11 were created using a three-step megaprimer cloning strategy and the indicated oligonucleotides (Table 2). With primers 1 and 2 and peGFP-C1 (Clontech) used as the template for GFP-368-MD (pSKL10) and GFP-307-MD (pSKL11), the green fluorescent protein (GFP) cDNA was cloned. The resulting first PCR product had a 5′-NheI restriction site, a DNA sequence encoding for GFP, and the first 18 bp of MD-SLC4A11 DNA. This PCR product was purified and diluted 1:50 and used as a megaprimer for a second PCR, along with primers 1 and 3 (Table 2) and pSKL1 as the template. The second PCR product had a 5′-NheI restriction site, a DNA sequence encoding GFP, a HA tag and MD of SLC4A11 (starting at amino acid 368 or 308 of WT-SLC4A11), and a 3′-XhoI restriction site. NheI/XhoI-digested pSKL1 and the second PCR product were ligated, resulting in pSKL10 and pSKL11. Similarly, mNect-368-MD (pSKL12) and mNect-308-MD-SLC4A11 (pSKL13) were cloned using pDEJ6 (22), which encodes mNectarine (mNect), as the template. To construct CD-AE1-MD-SLC4A11 (pSKL14), a similar strategy was used, except pPBAE1-oocyte (5) was used as the template to clone CD-AE1 (the first PCR product) along with primers 1 and 2 (5′-CTAGCTAGCGCCACCATGTACCCATACGATGTTCCGGATTACGCTGAGGAGCTGCAGGAT-3′ and 5′-GCGTGCGATGTCCTCCCGGATGCCCTTCCCAAATGGCTTGGCAGGGCTGGACTGATAGCGCCTTCG-3′). With the purified and diluted first PCR product as the megaprimer, pSKL1 as the template, and primers 1 and 3 (5′-CCGCTCGAGTCAAGGCCTGTGCTCAGCGTC-3′), the pSKL14 fusion construct was created. Integrity of all the clones was confirmed by DNA sequencing (Institute for Biomolecular Design, Department of Biochemistry, University of Alberta).
Table 2.
Oligonucleotides used in cloning of GFP and mNectarine fusion variants of SLC4A11
| Primer Number |
|||
|---|---|---|---|
| Construct: Plasmid Name/Construct | 1 | 2 | 3 |
| pSKL10 (GFP-368-MD-SLC4A11)/pSKL11 (mNect-368-MD-SLC4A11) | 5′-CTAGCTAGCGCCACCATGGTGA GCAAGGGCGAGGAG-3′ | 5′-GCCCACAGCCTTGTTTTT AGCGTAATCCGGAACATCGTATGGGTA CTTGTACAGCTC-3′ | 5′-CCGCTCGAGTCAAGGCCTGT GCTCAGCGTC-3′ |
| pSKL12 (GFP-307-MD- SLC4A11)/pSKL13 (mNect-307- MD- SLC4A11) | 5′-CTAGCTAGCGCCACCATGGTGA GCAAGGGCGAGGAG-3′ | 5′-CACTGGACCGTGGCTCAC AGCGTAATCCGGAACATCGTATGGGTA CTTGTACAGCTC-3′ | 5′-CCGCTCGAGTCAAGGCCTGT GCTCAGCGTC-3′ |
Underlined bases code for HA epitope tag.
Cell culture and transfection.
cDNAs encoding SLC4A11 variants were transiently transfected using the calcium phosphate method (38). HEK-293 cells were grown at 37°C or 30°C in a 5% CO2-air environment in complete DMEM [DMEM supplemented with 5% (vol/vol) fetal bovine serum, 5% (vol/vol) calf serum, and 1% (vol/vol) penicillin-streptomycin-glutamine]. All experiments involving transiently transfected cells were carried out 40–48 h posttransfection.
Bacterial expression of CD-SLC4A11.
Escherichia coli were transformed with pCML1 or pCML2 and grown in lysogeny broth containing 0.1 mg/ml ampicillin. Cultures were grown at 37°C until absorbance at 600 nm reached 0.6–0.8. Protein expression was induced by addition of isopropyl-β-d-1-thiogalactopyranoside to a final concentration of 0.1 or 1 mM and incubation for 2–4 h at 37°C. Cells were harvested by centrifugation at 7,500 g for 10 min at 4°C. Bacterial pellets were lysed in extraction buffer [PBS: 140 mM NaCl, 3 mM KCl, 6.5 mM Na2HPO3, and 1.5 mM KH2PO3 (pH 7.4) containing 1 mM PMSF, 2 mM EDTA, 1 μg/ml DNase, and 1× Roche protease inhibitor cocktail] by sonication using a probe sonifier (model W185, Heat Systems Ultrasonics, Plainview, NY) with eight 10-s pulses while kept on ice. An aliquot of total lysate was removed, and the remaining lysate was centrifuged at 15,000 g to isolate the supernatant as the soluble fraction and the pellet as the insoluble fraction.
Immunoblots and densitometry.
Bacterial or HEK-293 cell lysates were prepared in SDS-PAGE sample buffer [10% (vol/vol) glycerol, 2% (wt/vol) SDS, 0.5% (wt/vol) bromophenol blue, and 75 mM Tris (pH 6.8)] containing cOmplete protease inhibitor cocktail. Lysates were made to 1% (vol/vol) 2-mercaptoethanol and heated for 5 min at 65°C, and insoluble material was removed by centrifugation at 16,000 g for 10 min. Samples were resolved by SDS-PAGE on 7.5%, 10%, or 12% (wt/vol) acrylamide gels (28). Immunoblots were processed as described elsewhere (46). Mouse anti-HA, mouse anti-GAPDH, mouse anti-Myc, mouse anti-His, mouse anti-AE1, or mouse anti-GST was used at 1:2,000, 1:5,000, 1:2,000, 1:2,500, 1:5,000, or 1:5,000 dilution, respectively, in TBS-TM [5% skim milk powder in TBS-T: 0.1% (vol/vol) Tween 20, 0.15 M NaCl, and 50 mM Tris·HCl (pH 7.5)]. After incubation with sheep anti-mouse HRP-conjugated secondary antibody at 1:5,000 dilution, immunoblots were developed using Millipore Luminata Crescendo Western HRP reagent and visualized using a biomolecular imager (GE ImageQuant LAS4000, GE Healthcare). Quantitative densitometric analyses were performed using ImageQuant TL 8.1 software.
Cell surface processing assays.
Cell surface processing assays were performed as described elsewhere (44). Transfected cells were rinsed with 4°C PBS, washed with 4°C borate buffer [in mM: 154 NaCl, 7.2 KCl, 1.8 CaCl2, and 10 mM boric acid (pH 9.0)], and labeled with sulfo-NHS-SS-biotin (0.5 mg/ml). Cells were washed three times at 4°C with quenching buffer [192 mM glycine and 25 mM Tris (pH 8.3)] and then solubilized in 500 μl of IPB buffer [1% (vol/vol) IGEPAL CA-630, 5 mM EDTA, 150 mM NaCl, 0.5% (wt/vol) sodium deoxycholate, and 10 mM Tris (pH 7.5)] containing cOmplete protease inhibitor. For each sample, half of the total recovered supernatant (T) was retained for later SDS-PAGE analysis. The remaining half of the recovered supernatants was combined with 100 μl of 50% suspension of immobilized streptavidin-Sepharose resin, and the supernatant was collected [unbound protein (U)]. The T and U fractions of each sample were processed for SDS-PAGE analysis and immunoblotting as described above. After blot densitometry, the following formula was used to calculate the percentage of biotinylated protein: (T − U)/T × 100.
Immunoprecipitation.
Immunoprecipitation experiments were performed as described elsewhere (29). HEK-293 cell lysates were prepared in IPB buffer containing cOmplete protease inhibitor cocktail and incubated at 4°C for 20 min. Cell lysates were centrifuged at 13,200 g for 20 min at 4°C, and protein concentration was determined by bicinchoninic acid assay (40). A 50-μg aliquot of total protein (T) was set aside, and 300 μg of the remaining lysate were incubated at 4°C overnight with 1.5 μl of rabbit polyclonal anti-Myc or rabbit nonimmune serum along with 35 μl of Dynabeads Protein G resin. After 18 h, the resin was washed three times with 4°C IPB buffer containing cOmplete protease inhibitors, resuspended in 40 μl of SDS-PAGE sample buffer, and heated at 65°C for 5 min. Samples were then processed for immunoblotting.
Enzymatic deglycosylation.
HEK-293 cell lysates were prepared in IPB buffer as described above, and total protein concentration was measured by bicinchoninic acid assay (48). Samples (25 μg protein) were heated for 5 min at 65°C and then incubated with 5 μl (2,500 units) of N-glycosidase F (PNGase F) or 3 μl of endoglycosidase H (Endo H, 1,500 units) for 2 h at 37°C. SDS-PAGE sample buffer was added to stop the reaction, and the samples were processed for immunoblotting.
Perfluorooctanoic acid-PAGE.
Samples were processed as described elsewhere (36, 44). All steps after homogenization of the cells were performed at 4°C. Transfected HEK-293 cells were homogenized in PBS (pH 7.4) containing protease inhibitors and 1% (wt/vol) IGEPAL CA-630 for 20 min on ice. Samples were centrifuged at 16,000 g for 10 min at 4°C. Lysates (50 μg protein) were combined with perfluorooctanoic acid (PFO)-PAGE sample buffer [0.8% (wt/vol) NaPFO, 20% (vol/vol) glycerol, 50 mM DTT, 0.005% (wt/vol) bromophenol blue, and 100 mM Tris (pH 8.0)] or SDS-PAGE sample buffer and incubated for 15 min at 4°C. Samples were resolved on 10% Tris-glycine gels without SDS. Electrophoresis was performed at 4°C using PFO running buffer [192 mM glycine, 0.5% (wt/vol) NaPFO, and 25 mM Tris (pH 8.5)]. Immunoblotting was performed as described above.
Cross-linking of SLC4A11.
Samples were processed as described elsewhere (44). Briefly, transfected HEK-293 cells were washed with PBS and incubated for 30 min at 20°C in PBS containing 2 mM dithiobis(succinimidyl propionate) (DTSP), a membrane-permeant, thiol-cleavable cross-linker. To stop cross-linking reactions, the cells were incubated with quenching buffer [192 mM glycine and 25 mM Tris (pH 8.3)] for 15 min. Cells were then rinsed with PBS and lysed in IPB buffer containing cOmplete protease inhibitor cocktail, and equal amounts of the samples were prepared with or without 50 mM DTT and then incubated for 30 min at 37°C. Lysates (50 μg of protein) were combined with SDS-PAGE sample buffer containing cOmplete protease inhibitor cocktail and heated for 5 min at 65°C. Samples were resolved on 7.5% or 10% SDS-polyacrylamide gels.
Assays of osmotically driven water flux.
HEK-293 cells were grown on poly-l-lysine-coated 25-mm round glass coverslips and cotransfected with cDNA encoding enhanced GFP (eGFP; peGFP-C1 vector, Clontech) and pcDNA 3.1 (empty vector) or the indicated SLC4A11 plasmid constructs in a 1:8 molar ratio (29, 45). After 48 h, coverslips were mounted in a 35-mm-diameter cell chamber (Attofluor, Molecular Probes) and washed with PBS. The chamber was mounted on the stage of a spinning-disk confocal microscope (Wave FX, Quorum Technologies, Guelph, ON, Canada) with a scanning head (model CSU10, Yokogawa). The microscope was equipped with a motorized xy stage with piezo focus drive (MS-4000 xyz automated stage, ASI Imaging). During experiments, the chamber was perfused at 3.5 ml/min with isotonic MBSS buffer [in mM: 90 NaCl, 5.4 KCl, 0.4 MgCl2, 0.4 MgSO4, 3.3 NaHCO3, 2 CaCl2, 5.5 glucose, 100 d-mannitol, and 10 HEPES (pH 7.4, 300 mosmol/kg)] and then with hypotonic MBSS buffer [the same composition as isotonic buffer, but without d-mannitol (pH 7.4, 200 mosmol/kg)]. Osmolarity of the solutions was verified previously using an osmometer (model 3D3, Advanced Instruments). Data were acquired with an electron-multiplying CCD camera system (model C9100-13, Hamamatsu) and a ×20 objective during excitation with a laser (Spectral Applied Research, Richmond Hill, ON, Canada) at 491 nm. eGFP fluorescence, collected though a dichroic cube (Quorum Technologies) at 520- to 540-nm wavelengths, was acquired at 1 point/s for 4–6 min. Quantitative image analysis for each HEK-293 cell in a selected region of interest was performed using Volocity 6.0 software (PerkinElmer, Guelph, ON, Canada). After the switch to hypotonic medium, the rate of fluorescence change was determined from the initial 15 s of linear fluorescence change.
Statistical analysis.
Numerical values are means ± SE. Statistical analyses were performed using Prism software (GraphPad Prism 6). Groups were compared with one-way ANOVA and unpaired t-test; P < 0.05 was considered significant.
RESULTS
Role of the SLC4A11 CD.
Most disease-causing point mutations of SLC4A11 protein lead to misfolding and retention in the ER, thereby impairing plasma membrane transport activity (29). By contrast, the disease mutation R125H in the SLC4A11 CD (Fig. 1) is a catalytic mutant (45). This led us to consider how mutations in the CD affect SLC4A11 transport function. Studies of the Na+-HCO3− cotransporter NBCe1 (SLC4A4) suggest that this SLC4 family member has a CD pore through which substrate translocates (9). As a SLC4 family member, the function of SLC4A11 might be similar to that of NBCe1. A substrate translocation pathway might pass through the CD, enabling access to the SLC4A11 MD (Fig. 1A), such that mutation of R125 would impair transport activity (Fig. 1A).
To explore the importance of CD-SLC4A11 in the protein's function, we sought to assess the activity of the MD alone. We prepared four truncation mutants of CD-SLC4A11, HA-353-MD, HA-347-MD, HA-329-MD, and HA-307-MD-SLC4A11, where an NH2-terminal HA epitope tag was added to MD sequences starting at the indicated residue of the SLC4A11 CD (Fig. 1B). After expression in HEK-293 cells (Fig. 2A), densitometry revealed that HA-353-MD, HA-347-MD, and HA-329-MD-SLC4A11 accumulated to only 5%, 10%, and 15%, respectively, of the level of WT-SLC4A11 (Fig. 2B). Only HA-307-MD-SLC4A11 had significant expression (97 ± 11% of WT-SLC4A11), suggesting an important role of the M307-R329 region in conferring SLC4A11 stability. These data show that MD-SLC4A11 requires the CD for protein accumulation.
Fig. 2.

Expression and plasma membrane localization of SLC4A11 CD truncation mutants. HEK-293 cells were transfected with cDNA encoding hemagglutinin (HA) epitope-tagged WT-SLC4A11 and CD truncation mutants. A: cell lysates (50 μg protein) were loaded on SDS-polyacrylamide gels and processed for immunoblotting with anti-HA (α-HA) and anti-GAPDH (α-GAPDH) antibodies. Arrows show band corresponding to HA-353-MD-SLC4A11. B: densitometry of expression of SLC4A11 variants, normalized for GAPDH abundance and standardized to WT-SLC4A11. C: transfected cells were labeled with membrane-impermeant sulfo-NHS-SS-biotin. Cell lysates were divided into two equal fractions, and one was incubated with streptavidin-Sepharose resin to remove biotinylated proteins. Unbound fraction (U) and total cell lysate (T) were processed for immunoblots using anti-HA antibody. D: fraction of SLC4A11 and GAPDH labeled by sulfo-NHS-SS-biotin (SNSB). Values are means ± SE (n = 3). Dashed line indicates level of biotinylated GAPDH, which is the background level of the assay. *P < 0.05 vs. WT-SLC4A11 and 307-MD. #P < 0.05 vs. GAPDH.
To assess the state of protein maturation of the CD truncation mutants, we used glycosidase enzymes. The migration position of fully deglycosylated protein was established upon treatment with PNG F (data not shown). Core glycosylated protein, with immature glycosylation associated with ER location, is sensitive to cleavage by Endo H, whereas mature glycosylated chains are insensitive. Consistent with post-ER (mature) glycosylation of WT-SLC4A11, the protein was resistant to Endo H, but its carbohydrate was cleaved by PNG F (data not shown). In contrast, the CD truncation mutants were sensitive to Endo H to varying degrees (data not shown). This suggests that the mutants predominantly fail to mature. HA-329-MD-SLC4A11, however, shows some resistance to Endo H, consistent with partial maturation (data not shown).
To test the cellular localization of CD truncation mutants, we also carried out cell surface biotinylation assays (46) (Fig. 2C). The fraction of biotin-labeled (plasma membrane-localized) protein was calculated relative to WT-SLC4A11 (Fig. 2D). None of these mutants localized to the cell surface significantly. As a result, the functional activity of the MD alone could not be assessed, as functional assays require cell surface localization.
GFP and mNectarine fusion proteins to cover a hydrophobic surface.
A possible explanation for the instability of the isolated MD-SLC4A11 is that the MD must be associated with the CD, perhaps to satisfy hydrophobic bonding requirements. To test this notion, we created two fusion proteins, GFP and mNectarine (a variant of red fluorescent protein) (22), fused at the NH2 terminus to varying lengths of MD-SLC4A11. GFP is dimeric and might induce dimerization when fused to MD-SLC4A11, while mNectarine is monomeric (18, 22, 43). We considered the possibility that the essential role of CD-SLC4A11 might be facilitation of SLC4A11 dimerization, a function that might be complemented by GFP, but not by mNectarine.
GFP-368-MD, mNect-368-MD, GFP-307-MD, and mNect-307-MD-SLC4A11 were expressed in HEK-293 cells, and their expression was assessed on immunoblots (Fig. 3A). GFP-368-MD and mNect-368-MD accumulated to <5% of the level of WT-SLC4A11, while accumulation of GFP-307-MD and mNect-307-MD was ∼15% of WT levels (Fig. 3B). Cell surface processing assays revealed that plasma membrane surface trafficking was insignificant for the four fusion variants (Figs. 3, C and D). Fusion of GFP or mNectarine failed to rescue the protein stability and trafficking defects of MD-SLC4A11. Thus, neither a surface covering the SLC4A11 MD nor forced dimerization of the CD (with dimeric GFP) suffices to induce rescue of the MD to the cell surface.
Fig. 3.

Expression and plasma membrane localization of SLC4A11 MD chimeras. Green fluorescent protein (GFP) and mNectarine (mNect, a modified form of red fluorescent protein), which exist as dimers and monomers, respectively, were fused to the NH2 terminus of MD-SLC4A11 at amino acid 368 (GFP-368-MD and mNect-368-MD) or amino acid 307 (GFP-307-MD and mNect-307-MD). A: HEK-293 cells were transfected with cDNA encoding HA-tagged WT-SLC4A11 and the indicated chimeras. Cell lysates (50 μg of protein) were loaded on SDS-polyacrylamide gels and processed for immunoblotting with anti-HA (α-HA) and anti-GAPDH (α-GAPDH) antibodies. Arrows indicate faint bands. B: densitometry of expression of SLC4A11 variants normalized for GAPDH abundance and standardized to WT-SLC4A11. C: HEK-293 cells were transfected with cDNA encoding HA-tagged SLC4A11 chimeras and subjected to cell surface biotinylation assays. Unbound fraction (U) and total cell lysate (T) were probed on immunoblots with indicated antibodies. D: fraction of SLC4A11 and GAPDH labeled by sulfo-NHS-SS-biotin. Values are means ± SE (n = 3). Dashed line indicates level of biotinylated GAPDH, which is the background level of the assay. *P < 0.05 vs. WT-SLC4A11. #P < 0.05 vs. GAPDH.
CD-AE1 and MD-SLC4A11 chimera.
Since fusion of GFP and mNectarine did not stabilize MD-SLC4A11, we hypothesized that the MD requires a protein with a structural fold similar to that of CD-SLC4A11. To identify such a protein, we submitted the domain's sequence (amino acids 1–370) to Protein Homology/analogY Recognition Engine V 2.0 (Phyre2), a protein structure prediction server (25). The highest-probability structure was based on AE1 (SLC4A1), a Cl−/HCO3− exchanger in the SLC4 family with SLC4A11. The homology model of CD-SLC4A11 based on AE1 crystal structure (49) had four β-sheets and several α-helices (Fig. 4A). Only the SLC4A11 R125-P341 region had a structural alignment with 100% confidence to the AE1 template (Fig. 4B), while the remainder of SLC4A11 was modeled with <20% confidence (Fig. 4C, highlighted in gray). Considering the amino acid sequence identity of just 18% between human AE1 and SLC4A11 CD, the structural alignment confidence of 100% from R125 to P341 is remarkable. The Q301-P331 region of SLC4A11 (Fig. 4, B and C, red) corresponds to the dimeric interface, CD-AE1 F314-R344 region in the AE1 crystal structure (49). The catalytically inactive mutant R125H (highlighted in yellow) maps to the core of the predicted structure, and it is possible that it could line a substrate translocation pathway. We cannot, however, be confident of this assumption, since there is low confidence in the structural alignment with AE1 in SLC4A11 amino acids 1–124 and 341–371.
Fig. 4.
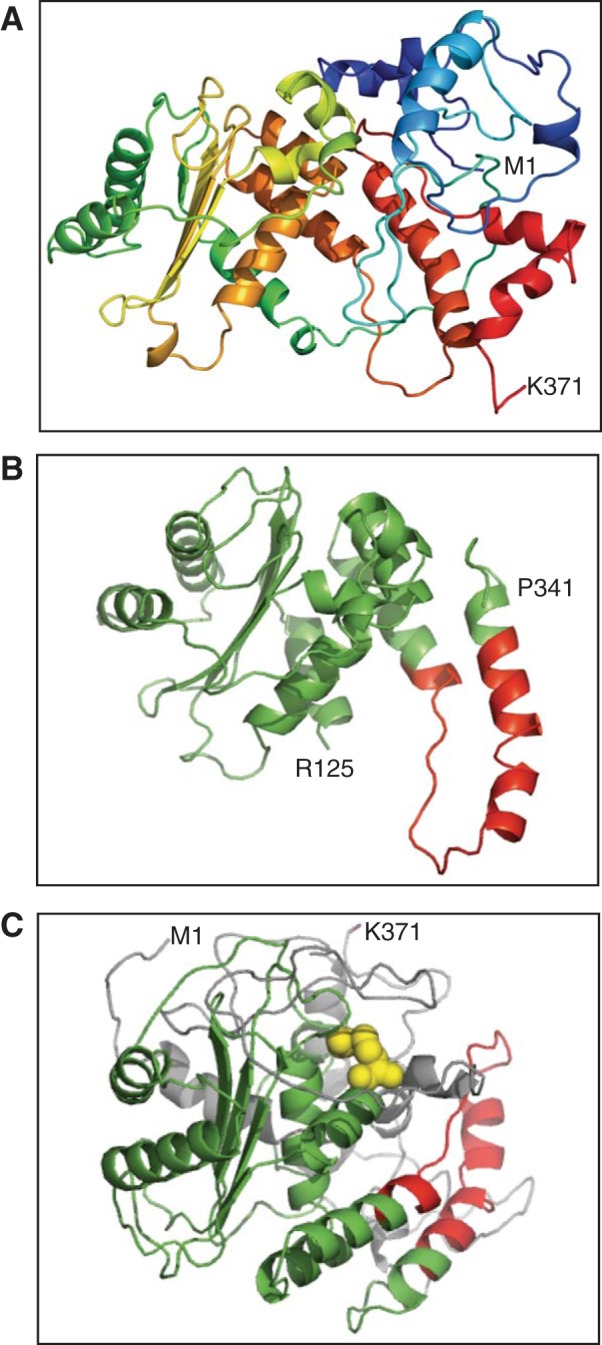
Homology model of SLC4A11 CD. A: homology model of CD-SLC4A11 (M1-K371 region) was prepared using Protein Homology/analogY Recognition Engine V 2.0 (Phyre2) software and the structure of CD-AE1 (M1-F379 region) as template (49). B: homology model (R125-P341 region) of CD-SLC4A11 based on structural alignment with 100% confidence on CD-AE1. Red color indicates SLC4A11 (Q301-P331 region) corresponding to the AE1 dimerization motif (49). C: homology model of CD-SLC4A11, with R125 highlighted as space filling the model in yellow and dimeric interface in red. Residues with high (100% confidence) probability of correct structural prediction in Phyre2 are shown as green and red; low-confidence (∼18%) structural fitting in Phyre2 is shown as gray.
On the basis of the structural alignment (Fig. 4), amino acids 1–354 of human erythrocyte CD-AE1 and amino acids 341–891 of MD-SLC4A11 were fused to create a chimera (CD-AE1-MD-SLC4A11). Expression and localization of the chimera were assessed in HEK-293 cells (Fig. 5, A and C). The expression level of CD-AE1-MD-SLC4A11 was not significantly different from that of WT-SLC4A11 (Fig. 5B). Cell surface trafficking was 69 ± 10% for WT-SLC4A11 and 25 ± 5% for CD-AE1-MD-SLC4A11, and both cell surface trafficking levels were significantly different from the background level (Fig. 5D).
Fig. 5.

Fusion to AE1 CD restores MD-SLC4A11 localization but not function. HEK-293 cells were transfected with cDNA encoding HA epitope-tagged WT-SLC4A11 and CD-AE1-MD-SLC4A11 chimera (AE1 residues 1–354 fused to SLC4A11 residues 341–891). A: cell lysates (50 μg of protein) were immunoblotted with anti-HA (α-HA) and anti-GAPDH (α-GAPDH) antibodies. B: densitometry of expression, normalized for GAPDH abundance and standardized to WT-SLC4A11. C: transfected cells were labeled with membrane-impermeant sulfo-NHS-SS-biotin for cell surface biotinylation assays, and cell lysates were probed on immunoblots. D: fraction of WT-SLC4A11, CD-AE1-MD-SLC4A11, and GAPDH labeled by sulfo-NHS-SS-biotin. Values are means ± SE (n = 3). Dashed line indicates level of biotinylated GAPDH, which represents the background level for the assay. E: HEK-293 cells, grown on coverslips, were transiently cotransfected with cDNA encoding GFP and vector or SLC4A11 chimera. Cells were perfused with isosmotic and hyposmotic media. F, fluorescence. F: rate of fluorescence change, which represents water flux. Values are means ± SE from 3 independent experiments with 8–10 cells measured per coverslip. *P < 0.05 vs. GAPDH. #P < 0.05 vs. CD-AE1-MD-SLC4A11. P̂ < 0.05 vs. WT-SLC4A11. NS, not significant (P > 0.05).
Since CD-AE1-MD-SLC4A11 was expressed at the cell surface at ∼40% of WT-SLC4A11 levels, we were able to assess its water flux activity. Cell swelling assays were performed in HEK-293 cells (45) coexpressing GFP and either WT-SLC4A11 or CD-AE1-MD-SLC4A11. Cytoplasmic GFP concentration was monitored during the switch from isotonic to hypotonic solution (Fig. 5E). The rate of fluorescence decrease, representing dilution of GFP in the cytoplasm, is a surrogate for water flux activity. Water flux in CD-AE1-MD-SLC4A11-expressing cells was indistinguishable from that in vector-transfected cells (Fig. 5F), while water flux in WT-SLC4A11 cells was significant, as reported elsewhere (45). Overall, the CD-AE1-MD-SLC4A11 chimera shows that a protein with a similar structural fold is required to stabilize MD-SLC4A11, allowing it to traffic to the cell surface. The absence of CD-SLC4A11, however, impairs water flux activity.
Bacterial overexpression of CD-SLC4A11.
Understanding the structure of CD-SLC4A11 might rationalize the CD mutants that cause corneal dystrophies. We thus expressed CD-SLC4A11 (amino acids 1–368) in E. coli to provide material for structural studies. In E. coli, the native sequence of CD-SLC4A11 was expressed at low levels that were barely detectable on immunoblots (Fig. 6A). We thus optimized codon usage for E. coli, replacing rare E. coli codons in the SLC4A11 cDNA to stabilize the nascent polypeptide. E. coli extracts from codon-optimized CD-SLC4A11 revealed a band of the expected size (∼40 kDa) on immunoblots (Fig. 6A). As the expression level was low, we fused GST to the NH2 terminus of CD-SLC4A11 (GST-CD-SLC4A11-His10) and expressed in E. coli. A 72-kDa band on Coomassie blue-stained SDS-polyacrylamide gels, consistent with the size of GST-CD-SLC4A11-His10, increased in intensity upon induction (Fig. 6B). Supernatants and pellets of E. coli cell lysates were assessed on Coomassie blue-stained gels and immunoblots. The majority of CD-SLC4A11 was found in the pellet (Fig. 6C), not in the supernatant, whether expressed alone or fused to GST. This suggests that CD-SLC4A11 is poorly soluble, even when fused to a large soluble tag such as GST.
Fig. 6.

Bacterial expression of SLC4A11 CD. A: native human or codon-optimized (for Escherichia coli) cDNA encoding CD-SLC4A11 (amino acids 1–368), tagged with 6× histidine (CD-SLC4A11-His6), were transformed into E. coli and grown in lysogeny broth. After 2–4 h of induction with isopropyl-β-d-1-thiogalactopyranoside (IPTG), cultures were centrifuged, sonicated, and processed for immunoblotting with anti-His (α-His) antibody. B and C: cDNA, encoding GST fused to the NH2 terminus of SLC4A11 CD and tagged with 10× His (GST-CD-SLC4A11-His10), was transformed into E. coli. After induction with IPTG, cultures were collected and sonicated. Total cell lysate (T) was centrifuged, and supernatant (S) and pellet (P) fractions were collected. Samples were subjected to SDS-PAGE and stained with Coomassie blue dye (B) or immunoblotted and probed with anti-GST antibody (C).
CD-SLC4A11 and MD-SLC4A11 strongly associate.
Removal of the CD destabilized MD-SLC4A11. Similarly, CD-SLC4A11 is poorly soluble when expressed in bacteria without MD-SLC4A11. We hypothesized that the CD and MD might strongly associate in the SLC4A11 native structure, such that each domain was unstable unless positioned against its complementary surface. We assessed a possible interaction between Myc epitope-tagged CD-SLC4A11 (Myc-CD-SLC4A11) and HA epitope-tagged MD-SLC4A11 (HA-307-MD-SLC4A11). When coexpressed in HEK-293 cells, HA-307-MD-SLC4A11 and Myc-CD-SLC4A11 coimmunoprecipitated (Fig. 7A), indicating that the domains associate. Providing further validation, we found that when the epitope tags were reversed, HA-CD-SLC4A11 coimmunoprecipitated with Myc-307-MD-SLC4A11 (Fig. 7B).
Fig. 7.

Association between SLC4A11 CD and MD. HEK-293 cells were cotransfected with cDNA encoding Myc-tagged CD-SLC4A11 and HA-tagged 307-MD-SLC4A11 (A), Myc-tagged 307-MD-SLC4A11 and HA-tagged CD-SLC4A11 (B), or Myc-tagged CD-SLC4A11 and HA-tagged 340-MD-AE1 (C). Cell lysates (300 μg of total protein) were immunoprecipitated with anti-Myc antibody (IP) or nonimmune serum (NI). Immunoprecipitates (both IP and NI) and 50 μg of total protein cell lysate (T) were probed on immunoblots with anti-HA (α-HA) and anti-AE1 (α-AE1) antibody.
Interaction between the CD and MD was specific, as Myc-CD-SLC4A11 did not associate with MD-AE1 (340-MD-AE1) (Fig. 7C). No signal was found in any of the samples when nonimmune serum was used for immunoprecipitation, indicating the specificity of the antibody. These immunoprecipitation data provide additional support for a strong association between SLC4A11 domains.
When the MD-AE1 was coexpressed as two separate halves, the two complementary fragments associated and were able to transport anions (15). Since CD-SLC4A11 and MD-SLC4A11 associated, we wanted to assess their ability to reconstitute a water flux. Since trafficking to the cell surface is required for water flux assays, using a cell surface biotinylation technique, we first measured the ability of the coexpressed fragments to traffic to the cell surface (Fig. 8). The level of biotinylation of MD-SLC4A11 was barely higher than that of cytosolic GAPDH (Fig. 8). Thus, while MD-SLC4A11 and CD-SLC4A11 associate in coimmunoprecipitates, virtually none of the complex traffics to the cell surface. It was therefore not possible to assess the function of the assembled complementary fragments.
Fig. 8.

Plasma membrane localization of coexpressed MD-SLC4A11 and CD-SLC4A11. HEK-293 cells were transfected with cDNA encoding HA epitope-tagged 307-MD-SLC4A11 and Myc epitope-tagged CD-SLC4A11. A: transfected cells were labeled with membrane-impermeant sulfo-NHS-SS-biotin. Cell lysates were divided into two equal fractions, and one was incubated with streptavidin-Sepharose resin to remove biotinylated proteins. Unbound fraction (U) and total cell lysate (T) were processed for immunoblots using anti-HA or anti-Myc antibody. B: fraction of 307-MD-SLC4A11 and GAPDH labeled with sulfo-NHS-SS-biotin. Values are means ± SE (n = 3). Dashed line indicates level of biotinylated cytosolic GAPDH, which is the background level of the assay.
CD-SLC4A11 dimerizes independently of the MD.
WT-SLC4A11 is dimeric (44), raising the following question: Does the CD dimerize independently of the MD? We expressed CD-SLC4A11 in HEK-293 cells, as the protein was largely insoluble when expressed in E. coli. Nondenaturing gel electrophoresis (PFO-PAGE) experiments were performed to examine CD-SLC4A11 oligomeric structure. HA-CD-SLC4A11 migrated as a single band at 43 kDa when treated with SDS, which disrupts protein-protein interactions, consistent with a monomer (Fig. 9A). In contrast, bands at 43 and ∼85 kDa were observed in cells treated with nondenaturing PFO (Fig. 9A). The upper band at ∼85 kDa corresponds to the predicted molecular weight of dimeric CD-SLC4A11.
Fig. 9.

SLC4A11 CD are dimeric. A: HEK-293 cells, transfected with cDNA encoding NH2-terminally HA-tagged CD-SLC4A11, were solubilized in PBS containing 0.5% (vol/vol) nonionic IGEPAL detergent and incubated for 30 min at 4°C. Samples were combined with SDS sample buffer (SDS) or perfluorooctanoic acid (PFO) sample buffer, subjected to PFO-PAGE, and probed on immunoblots with anti-HA antibody (α-HA). B and C: HEK-293 cells, transfected with cDNA encoding HA-tagged full-length WT-SLC4A11 (B) or CD-SLC4A11 (C) were incubated with dithiobis(succinimidyl propionate) and then solubilized. Equal amounts of samples were treated with DTT (+) or left untreated (−) and probed on immunoblots. D and E: HEK-293 cells were cotransfected with cDNAs encoding HA-CD-SLC4A11 and Myc-CD-SLC4A11 (D) or HA-CD-300-SLC4A11 (SLC4A11 amino acids 1–300) and Myc-CD-300-SLC4A11 (E). Cells were solubilized 48 h posttransfection, and a 50-μg total protein aliquot (T) was set aside. Lysate (300 μg of protein) was immunoprecipitated with rabbit polyclonal anti-Myc (IP) or rabbit nonimmune serum (NI). Samples were probed on immunoblots.
To assess further CD-SLC4A11 oligomeric structure, we performed cross-linking experiments with the membrane-permeable, thiol-cleavable cross-linker DTSP. We first tested full-length WT-SLC4A11, which has been established to be dimeric (44). In the absence of reducing agents, DTSP-treated WT-SLC4A11 migrates as a predominant 200-kDa band and a lower-abundance 100-kDa band. This is consistent with cross-linked dimeric SLC4A11 (∼200 kDa) and non-cross-linked SLC4A11 (∼100 kDa) running as a monomer in SDS (Fig. 9B). In confirmation of this idea, DTT treatment shifted the protein to a ∼100-kDa species, consistent with a monomer. DTSP-treated CD-SLC4A11 migrated at ∼85 kDa, corresponding to a dimeric cross-linked protein (Fig. 9C). DTT led to the appearance of a band at 43 kDa, corresponding to monomeric CD-SLC4A11 (Fig. 9C). Some signal above 85 kDa in the CD-SLC4A11 lane may have arisen because of aggregation.
Coimmunoprecipitation was used as a final approach to assess CD-SLC4A11 oligomerization. Myc and HA epitope-tagged versions of CD-SLC4A11 were coexpressed in HEK-293 cells. Immunoprecipitation assays revealed association of the Myc- and HA-tagged CD-SLC4A11 (Fig. 9D). No signal was found when nonimmune serum was used to pull down the protein, indicating specificity of the coimmunoprecipitation.
We next explored which region of CD-SLC4A11 is responsible for the domain's dimerization. Consideration of the AE1 dimerization region and the CD-SLC4A11 homology model (Fig. 4) suggested that the Q301-P331 region might form the SLC4A11 dimeric interface. We thus coexpressed HA and Myc epitope-tagged versions of SLC4A11 amino acids 1–300. Immunoprecipitation studies show association between HA-CD-300-SLC4A11 and Myc-CD-300-SLC4A11 (Fig. 9E). Taken together, these experiments show that the CD-SLC4A11 dimerizes independently of the MD. Furthermore, the CD-SLC4A11 region Q301-K370 is not essential for CD dimerization.
DISCUSSION
We found that SLC4A11 CD is essential for stability of the MD. Furthermore, SLC4A11 CD is required for the membrane transport function of SLC4A11. Modeling studies and the ability of CD-AE1 to complement CD-SLC4A11 in trafficking of SLC4A11 indicate that MD-AE1 and CD-SLC4A11 have very similar structures. Finally, SLC4A11 CD and MD are strongly associated, and the CD dimerizes on its own. About one-third of corneal dystrophy-causing mutations are located in SLC4A11 CD, and this work provides the first insights into the role of the CD in the function of SLC4A11. Together, our findings support the urgent need for additional insight into the structure and function of solute carrier proteins such as SLC4A11 (8).
Role of the CD in MD-SLC4A11 stabilization.
CD-SLC4A11 has a key role in stabilizing the full-length protein. About one-third of all identified SLC4A11 point mutations map to the CD, and many of these result in a molecular phenotype marked by retention of the protein in the ER (29, 41, 44). This suggests a key role of the CD, permitting trafficking of SLC4A11 to the cell surface. Point mutations could, however, cause CD-SLC4A11 to misfold, resulting in surveillance by the ER quality-control apparatus.
Experiments presented here indicate that the role of CD-SLC4A11 is more than permissive to exit from the ER. Truncation of the CD led to progressive dysfunction, such that the greater the truncation of the CD, the less the MD was able to accumulate. This is consistent with ER-associated degradation of proteins recognized as misfolded, leading to reduced levels of steady-state accumulation. All the CD-SLC4A11 truncation mutants failed to process to the cell surface. Point mutations that cause CD-SLC4A11 to be recognized as misfolded inhibit SLC4A11 MD maturation, as does removal of the domain.
What is the role of SLC4A11 CD in MD stabilization? As discussed above, SLC4A11 MD is unstable (accumulates to a low level and traffics poorly to the cell surface) when expressed alone, but we also found that bacterially overexpressed CD was poorly soluble, even with a large, soluble tag. These observations would be unified if the two SLC4A11 domains strongly associate in the mature full-length protein. Expression of the two domains independently would thus expose surfaces that are normally buried, likely hydrophobic, and prone to aggregation. In support of this model, coexpressed SLC4A11 CD and MD associated in immunoprecipitation assays.
Our data support a specific role of CD-SLC4A11 in stabilizing the MD. The idea that SLC4A11 MD has a surface that must be covered suggests that any properly folded domain could complement the function. In contrast, we found that fusion of two different fluorescent proteins failed to rescue the processing defect of SLC4A11 MD. This indicates that maturation of SLC4A11 requires close association with a specific structure.
Structural modeling revealed for the first time evidence that SLC4A11 CD and AE1 CD have regions with significant structural similarity. In support of this idea, AE1 CD rescued SLC4A11 MD trafficking to the cell surface. This suggests that AE1 has structural features sufficiently similar to SLC4A11's domain that the chimera permits appropriate folding of the MD, enabling escape from the cell's quality-control apparatus.
Another possible obligatory role of CD-SLC4A11 is assistance with dimerization, since the full-length protein is dimeric (44). However, we found that fusion of GFP, a dimeric protein, to SLC4A11 failed to rescue SLC4A11 MD trafficking to the same degree as mNectarine, a monomeric fluorescent protein. While AE1 CD is dimeric (49), the failure of GFP to rescue the MD indicates that SLC4A11 CD contributes more than dimerization to the protein's stability. Further argument against a requisite role of the CD in protein dimerization involves AE1, another member of the SLC4 family. AE1 CD and MD are independently dimeric.
AE1 does, however, provide interesting contrasts with the data we have presented for SLC4A11. AE1 (SLC4A1) and SLC4A11 belong to the same protein family and, thus, likely share a common fold of their MD. AE1 MD can, however, be stably expressed without the CD (6), and CD-AE1 is sufficiently stable by itself that it has been overexpressed and crystallized (49). The two AE1 domains can be readily proteolytically cleaved and separated under mild conditions, indicating weak interactions between the domains (13). Our data thus indicate that while there are structural similarities between SLC4A11 and AE1, there are major differences in the way the proteins' two domains associate.
Role of SLC4A11 CD in membrane transport activity.
As discussed above, the CD has an important and specific role in MD folding, stability, and trafficking. There is additional evidence that the domain has a role in the membrane transport functions of SLC4A11. The SLC4A11 R125H mutation causes CHED (16) and resides in the CD (46). R125H-SLC4A11 targets normally to the cell surface but does not support the water flux activity in cells expressing WT-SLC4A11 (45). We assume that if the mutation affected the folding of SLC4A11, the ER quality-control apparatus would retain the protein in the ER. How can a CD mutation affect membrane transport function?
One possibility is that the SLC4A11 CD contains a pathway through which transported substrates traverse (Fig. 1A). Such pathways are well established in some ion channels, for example, the voltage-gated K+ channel (30, 31). Similarly, the CFTR Cl− channel was recently proposed to have “fenestrations” in its CD, guiding substrate Cl− to the transmembrane pore (12). Closer to SLC4A11, evidence suggests that the electrogenic Na+-HCO3− cotransporter NBCe1 (SLC4A4) has a channel pathway through its CD (9).
Amino acid sequence alignment with ClustalW2 identifies the residue corresponding to NBCe1 R298 as SLC4A11 R270. Interestingly, this site is flanked by two CHED mutations, A269V and T271M, suggesting a conserved sensitivity to causing disease. Furthermore, R298 is located in a largely solvent inaccessible polar subsurface pocket and forms a salt bridge (9). In the SLC4A11 homology model, we did not find such a pocket for R125 (data not shown). Salt bridges, predicted using Visual Molecular Dynamics software (17), were not close to R125. This could, however, be due to the low confidence in the homology structure in the SLC4A11 amino acid 1–125 region.
Our data are, however, consistent with a role of CD-SLC4A11 in membrane transport function. Coimmunoprecipitations revealed association between CD-SLC4A11 and MD-SLC4A11, as one would expect if the CD had a pathway guiding substrate to the MD. More importantly, partial cell surface trafficking was restored to MD-SLC4A11 by fusion of CD-AE1. AE1-SLC4A11 fusion, however, did not support water flux activity. This strongly suggests that CD-SLC4A11 contributes to the membrane transport activity of the protein. Lack of functional activity in the CD-AE1-MD-SLC4A11 chimera could be due to 1) absence of the substrate translocation pathway in CD-AE1 [consistent with this idea, a substrate access tunnel in the CD is not an important feature of AE1 (39)] or 2) absence of specific residues required for SLC4A11 substrate translocation. Our data are unable to reveal the contribution of the CD to MD function, but providing a pathway for substrate funneling to the MD, as proposed for NBCe1, is a possibility. Nevertheless, all these experiments show that the CD is critical to the cell surface trafficking and membrane transport function of SLC4A11.
Dimerization of SLC4A11 CD.
Using three different methods, we found that CD-SLC4A11 dimerizes independently of the MD. AE1 MD and CD dimerize independently of each other (4, 7, 49). Unfortunately, we could not test the oligomerization of SLC4A11 MD, as it was unstable and does not reach the cell surface. Dimerization of CD-SLC4A11 raises the possibility that the dimeric unit performs a key function in SLC4A11 not supported by the individual CD monomers. For example, the proposed cytosolic substrate access pore might pass through the dimeric interface.
We explored the role in dimerization of the SLC4A11 region homologous to the AE1 dimerization motif. Deletion of the SLC4A11 Q301-K370 region yielded a dimeric CD. While the Q301-P331 region might play a role in SLC4A11 dimerization, it was clearly dispensable. Interestingly, however, the SLC4A11 Q301-P331 region has an important role in the protein's structure. Truncation of SLC4A11 amino acids 1–329 led to a substantial loss of protein expression, which was rescued when amino acids 307–328 were added (Fig. 2B). The overlap of amino acids 307–328 with the predicted dimerization region is hard to overlook, but our data do not show an essential role of the region in dimerization. Unfortunately, the region's structure is poorly modeled by homology to AE1. Perhaps SLC4A11 amino acids 307–328 interact with the MD?
Conclusions.
We explored the role of the SLC4A11 CD to rationalize disease-causing mutations. We conclude that 1) the SLC4A11 CD is strongly associated with and required for stability of the MD, 2) the CD is essential for SLC4A11 water flux activity (possibly forming a substrate access pathway in the cytosol), 3) SLC4A11 CD and AE1 CD share a common core structure, and 4) SLC4A11 CD dimerizes independently of the MD.
GRANTS
Operating support was provided by the Canadian Institutes of Health Research. S. K. Loganathan was supported by a graduate studentship from the Natural Sciences and Engineering Research Council-supported International Research Training Group in Membrane Biology.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.K.L. and J.R.C. developed the concept and designed the research; S.K.L. and C.M.L. performed the experiments; S.K.L. and C.M.L. analyzed the data; S.K.L. and J.R.C. interpreted the results of the experiments; S.K.L., C.M.L., and J.R.C. prepared the figures; S.K.L. and J.R.C. drafted the manuscript; S.K.L. and J.R.C. edited and revised the manuscript; S.K.L., C.M.L., and J.R.C. approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Reinhart Reithmeier, Katherine Badior, and Darpan Malhotra for helpful comments on the manuscript.
REFERENCES
- 1.Aldahmesh MA, Khan AO, Meyer BF, Alkuraya FS. Mutational spectrum of SLC4A11 in autosomal recessive CHED in Saudi Arabia. Invest Ophthalmol Vis Sci 50: 4142–4145, 2009. [DOI] [PubMed] [Google Scholar]
- 2.Aldave AJ, Yellore VS, Bourla N, Momi RS, Khan MA, Salem AK, Rayner SA, Glasgow BJ, Kurtz I. Autosomal recessive CHED associated with novel compound heterozygous mutations in SLC4A11. Cornea 26: 896–900, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Alka K, Casey JR. Bicarbonate transport in health and disease. IUBMB Life 66: 596–615, 2014. [DOI] [PubMed] [Google Scholar]
- 4.Basu A, Mazor S, Casey JR. Measurement of distances within a concatamer of the plasma membrane Cl−/HCO3− exchanger, AE1. Biochemistry 49: 9226–9240, 2010. [DOI] [PubMed] [Google Scholar]
- 5.Bonar P, Schneider HP, Becker HM, Deitmer JW, Casey JR. Three-dimensional model for the human Cl−/HCO3− exchanger, AE1, by homology to the E. coli ClC protein. J Mol Biol 425: 2591–2608, 2013. [DOI] [PubMed] [Google Scholar]
- 6.Bonar PT, Casey JR. Over-expression and purification of functional human AE1, expressed in Saccharomyces cerevisiae suitable for protein crystallography. Protein Exp Purif 74: 106–115, 2010. [DOI] [PubMed] [Google Scholar]
- 7.Casey JR, Reithmeier RAF. Analysis of the oligomeric state of band 3, the anion transport protein of the human erythrocyte membrane, by size exclusion high performance liquid chromatography: oligomeric stability and origin of heterogeneity. J Biol Chem 266: 15726–15737, 1991. [PubMed] [Google Scholar]
- 8.Cesar-Razquin A, Snijder B, Frappier-Brinton T, Isserlin R, Gyimesi G, Bai X, Reithmeier RA, Hepworth D, Hediger MA, Edwards AM, Superti-Furga G. A call for systematic research on solute carriers. Cell 162: 478–487, 2015. [DOI] [PubMed] [Google Scholar]
- 9.Chang MH, Dipiero J, Sonnichsen FD, Romero MF. Entry to “HCO3− tunnel” revealed by SLC4A4 human mutation and structural model. J Biol Chem 283: 18402–18410, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desir J, Abramowicz M. Congenital hereditary endothelial dystrophy with progressive sensorineural deafness (Harboyan syndrome). Orphanet J Rare Dis 3: 28–36, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desir J, Moya G, Reish O, Van Regemorter N, Deconinck H, David KL, Meire FM, Abramowicz M. Borate transporter SLC4A11 mutations cause both Harboyan syndrome and non-syndromic corneal endothelial dystrophy. J Med Genet 44: 322–326, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Hiani Y, Linsdell P. Functional architecture of the cytoplasmic entrance to the cystic fibrosis transmembrane conductance regulator chloride channel pore. J Biol Chem 290: 15855–15865, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grinstein S, Ship S, Rothstein A. Anion transport in relation to proteolytic dissection of band 3 protein. Biochim Biophys Acta 507: 294–304, 1978. [DOI] [PubMed] [Google Scholar]
- 14.Groeger N, Froehlich H, Maier H, Olbrich A, Kostin S, Braun T, Boettger T. Slc4a11 prevents osmotic imbalance leading to corneal endothelial dystrophy, deafness, and polyuria. J Biol Chem 285: 14467–14474, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Groves JD, Wang L, Tanner MJ. Complementation studies with co-expressed fragments of human red cell band 3 (AE1): the assembly of the anion-transport domain in Xenopus oocytes and a cell-free translation system. Biochem J 332: 161–171, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hemadevi B, Veitia RA, Srinivasan M, Arunkumar J, Prajna NV, Lesaffre C, Sundaresan P. Identification of mutations in the SLC4A11 gene in patients with recessive congenital hereditary endothelial dystrophy. Arch Ophthalmol 126: 700–708, 2008. [DOI] [PubMed] [Google Scholar]
- 17.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph 14: 33–38, 1996. [DOI] [PubMed] [Google Scholar]
- 18.Ilagan RP, Rhoades E, Gruber DF, Kao HT, Pieribone VA, Regan L. A new bright green-emitting fluorescent protein-engineered monomeric and dimeric forms. FEBS J 277: 1967–1978, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jalimarada SS, Ogando DG, Vithana EN, Bonanno JA. Ion transport function of SLC4A11 in corneal endothelium. Invest Ophthalmol Vis Sci 54: 4330–4340, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jennings ML, Howren TR, Cui J, Winters MJ, Hannigan R. Transport and regulatory characteristics of the yeast bicarbonate transporter homolog Bor1p. Am J Physiol Cell Physiol 293: C468–C476, 2007. [DOI] [PubMed] [Google Scholar]
- 21.Jiao X, Sultana A, Garg P, Ramamurthy B, Vemuganti GK, Gangopadhyay N, Hejtmancik JF, Kannabiran C. Autosomal recessive corneal endothelial dystrophy (CHED2) is associated with mutations in SLC4A11. J Med Genet 44: 64–68, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson DE, Ai HW, Wong P, Young JD, Campbell RE, Casey JR. Red fluorescent protein pH biosensor to detect concentrative nucleoside transport. J Biol Chem 284: 20499–20511, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kao L, Azimov R, Abuladze N, Newman D, Kurtz I. Human SLC4A11-C functions as a DIDS-stimulatable H+(OH−) permeation pathway: partial correction of R109H mutant transport. Am J Physiol Cell Physiol 308: C176–C188, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaul H, Suman M, Khan Z, Ullah MI, Ashfaq UA, Idrees S. Missense mutation in SLC4A11 in two Pakistani families affected with congenital hereditary endothelial dystrophy (CHED2). Clin Exp Optom. In press. [DOI] [PubMed] [Google Scholar]
- 25.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4: 363–371, 2009. [DOI] [PubMed] [Google Scholar]
- 26.Kodaganur SG, Kapoor S, Veerappa AM, Tontanahal SJ, Sarda A, Yathish S, Prakash DR, Kumar A. Mutation analysis of the SLC4A11 gene in Indian families with congenital hereditary endothelial dystrophy 2 and a review of the literature. Mol Vis 19: 1694–1706, 2013. [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar A, Bhattacharjee S, Prakash DR, Sadanand CS. Genetic analysis of two Indian families affected with congenital hereditary endothelial dystrophy: two novel mutations in SLC4A11. Mol Vis 13: 39–46, 2007. [PMC free article] [PubMed] [Google Scholar]
- 28.Laemmli UK. Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature 227: 680–685, 1970. [DOI] [PubMed] [Google Scholar]
- 29.Loganathan SK, Casey JR. Corneal dystrophy-causing SLC4A11 mutants: suitability for folding-correction therapy. Hum Mutat 35: 1082–1091, 2014. [DOI] [PubMed] [Google Scholar]
- 30.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent shaker family K+ channel. Science 309: 897–903, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science 309: 903–908, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Ogando DG, Jalimarada SS, Zhang W, Vithana EN, Bonanno JA. SLC4A11 is an EIPA-sensitive Na+ permeable pHi regulator. Am J Physiol Cell Physiol 305: C716–C727, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park M, Li Q, Shcheynikov N, Zeng W, Muallem S. NaBC1 is a ubiquitous electrogenic Na+-coupled borate transporter essential for cellular boron homeostasis and cell growth and proliferation. Mol Cell 16: 331–341, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Praetorius J, Kim YH, Bouzinova EV, Frische S, Rojek A, Aalkjaer C, Nielsen S. NBCn1 is a basolateral Na+-HCO3− cotransporter in rat kidney inner medullary collecting ducts. Am J Physiol Renal Physiol 286: F903–F912, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Puangsricharern V, Yeetong P, Charumalai C, Suphapeetiporn K, Shotelersuk V. Two novel mutations including a large deletion of the SLC4A11 gene causing autosomal recessive hereditary endothelial dystrophy. Br J Ophthalmol 98: 1460–1462, 2014. [DOI] [PubMed] [Google Scholar]
- 36.Ramjeesingh M, Huan LJ, Garami E, Bear CE. Novel method for evaluation of the oligomeric structure of membrane proteins. Biochem J 342: 119–123, 1999. [PMC free article] [PubMed] [Google Scholar]
- 37.Ramprasad VL, Ebenezer ND, Aung T, Rajagopal R, Yong VH, Tuft SJ, Viswanathan D, El-Ashry MF, Liskova P, Tan DT, Bhattacharya SS, Kumaramanickavel G, Vithana EN. Novel SLC4A11 mutations in patients with recessive congenital hereditary endothelial dystrophy (CHED2). Hum Mutat 28: 522–523, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Ruetz S, Lindsey AE, Ward CL, Kopito RR. Functional activation of plasma membrane anion exchangers occurs in a pre-Golgi compartment. J Cell Biol 121: 37–48, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shnitsar V, Li J, Li X, Calmettes C, Basu A, Casey JR, Moraes TF, Reithmeier RA. A substrate access tunnel in the cytosolic domain is not an essential feature of the solute carrier 4 (SLC4) family of bicarbonate transporters. J Biol Chem 288: 33848–33860, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem 150: 76–85, 1985. [DOI] [PubMed] [Google Scholar]
- 41.Soumittra N, Loganathan SK, Madhavan D, Ramprasad VL, Arokiasamy T, Sumathi S, Karthiyayini T, Rachapalli SR, Kumaramanickavel G, Casey JR, Rajagopal R. Biosynthetic and functional defects in newly identified SLC4A11 mutants and absence of COL8A2 mutations in Fuchs endothelial corneal dystrophy. J Hum Genet 59: 444–453, 2014. [DOI] [PubMed] [Google Scholar]
- 42.Sultana A, Garg P, Ramamurthy B, Vemuganti GK, Kannabiran C. Mutational spectrum of the SLC4A11 gene in autosomal recessive congenital hereditary endothelial dystrophy. Mol Vis 13: 1327–1332, 2007. [PubMed] [Google Scholar]
- 43.Tsien RY. The green fluorescent protein. Annu Rev Biochem 67: 509–544, 1998. [DOI] [PubMed] [Google Scholar]
- 44.Vilas GL, Loganathan S, Quon A, Sundaresan P, Vithana EN, Casey JR. Oligomerization of SLC4A11 protein and the severity of FECD and CHED2 corneal dystrophies caused by SLC4A11 mutations. Hum Mutat 33: 419–428, 2012. [DOI] [PubMed] [Google Scholar]
- 45.Vilas GL, Loganathan SK, Liu J, Riau AK, Young JD, Mehta JS, Vithana EN, Casey JR. Transmembrane water-flux through SLC4A11: a route defective in genetic corneal diseases. Hum Mol Genet 22: 4579–4590, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vilas GL, Morgan PE, Loganathan S, Quon A, Casey JR. Biochemical framework for SLC4A11, the plasma membrane protein defective in corneal dystrophies. Biochemistry 50: 2157–2169, 2011. [DOI] [PubMed] [Google Scholar]
- 47.Vithana EN, Morgan P, Sundaresan P, Ebenezer ND, Tan DT, Mohamed MD, Anand S, Khine KO, Venkataraman D, Yong VH, Salto-Tellez M, Venkatraman A, Guo K, Hemadevi B, Srinivasan M, Prajna V, Khine M, Casey JR, Inglehearn CF, Aung T. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2). Nat Genet 38: 755–757, 2006. [DOI] [PubMed] [Google Scholar]
- 48.Vithana EN, Morgan PE, Ramprasad V, Tan DT, Yong VH, Venkataraman D, Venkatraman A, Yam GH, Nagasamy S, Law RW, Rajagopal R, Pang CP, Kumaramanickevel G, Casey JR, Aung T. SLC4A11 mutations in Fuchs endothelial corneal dystrophy (FECD). Hum Mol Genet 17: 656–666, 2008. [DOI] [PubMed] [Google Scholar]
- 49.Zhang D, Kiyatkin A, Bolin JT, Low PS. Crystallographic structure and functional interpretation of the cytoplasmic domain of erythrocyte membrane band 3. Blood 96: 2925–2933, 2000. [PubMed] [Google Scholar]
- 50.Zhang W, Ogando DG, Bonanno JA, Obukhov AG. Human SLC4A11 is a novel NH3:H+ co-transporter. J Biol Chem 290: 12871–12876, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Chernova MN, Stuart-Tilley AK, Jiang L, Alper SL. The cytoplasmic and transmembrane domains of AE2 both contribute to regulation of anion exchange by pH. J Biol Chem 271: 5741–5749, 1996. [DOI] [PubMed] [Google Scholar]