

Abstract
Arginylation is a posttranslational modification that plays a global role in mammals. Mice lacking the enzyme arginyltransferase in skeletal muscles exhibit reduced contractile forces that have been linked to a reduction in myosin cross-bridge formation. The role of arginylation in passive skeletal myofibril forces has never been investigated. In this study, we used single sarcomere and myofibril measurements and observed that lack of arginylation leads to a pronounced reduction in passive forces in skeletal muscles. Mass spectrometry indicated that skeletal muscle titin, the protein primarily linked to passive force generation, is arginylated on five sites located within the A band, an important area for protein-protein interactions. We propose a mechanism for passive force regulation by arginylation through modulation of protein-protein binding between the titin molecule and the thick filament. Key points are as follows: 1) active and passive forces were decreased in myofibrils and single sarcomeres isolated from muscles lacking arginyl-tRNA-protein transferase (ATE1). 2) Mass spectrometry revealed five sites for arginylation within titin molecules. All sites are located within the A-band portion of titin, an important region for protein-protein interactions. 3) Our data suggest that arginylation of titin is required for proper passive force development in skeletal muscles.
Keywords: arginylation, titin, posttranslational modification, passive force, sarcomere, myofibril
arginylation is a posttranslational modification with physiological importance for muscles (13). Mediated by the enzyme arginyl-tRNA-protein transferase (ATE1), arginylation transfers arginine from tRNA onto the NH2 terminus of proteins, as well as to side chains of aspartate and glutamate residues in intact protein chains (2, 35). Posttranslationally added Arg also serves as target sites for Arg methylation (29), one of the most common types of methylation in mammals and a key process for the proper function of different proteins and the performance of a broad number of regulatory pathways (6, 34).
Several studies have shown that arginylation is essential for the proper functioning of different proteins, cells and organisms (14, 29, 39). In mammals, lack of ATE1 leads to embryonic lethality due to severe defects in cardiovascular development (15, 26). Cardiac muscle-specific ATE1-knockout (ATE1-KO) mice survive to adulthood, but the muscle structure and contractility are impaired, leading to dilated cardiomyopathy and high rates of lethality after 6 mo of age (14). Recently, our group showed that skeletal muscle-specific Ate1 KO (Ckmm-ATE1) results in a reduced active force in skeletal muscles by modifying the number of myosin cross-bridges attached to actin (3), suggesting that arginylation plays a key role in muscle regulation.
Titin is a giant multifunctional protein (3–4 MDa) present in sarcomeres of striated muscles (21, 37). Spanning the half-sarcomere, titin's NH2 terminus and COOH terminus are connected to the Z disk and M band, respectively. Titin is divided into distinct functional domains. The I-band portion of titin is mainly responsible for passive tension due to sarcomere length (SL) changes. Titin A band is largely composed of fibronectin-type III (Fn3)-like and immunoglobulin-like (Ig) domains, arranged in well-ordered super-repeats. The A-band topology is highly conserved among different isoforms of titin, an important feature for protein-protein interactions that occur along its structure and play key roles in muscle functions. These interactions include myosin, myosin binding protein C (MyBP-C), and myomesin (32) and are important for maintaining sarcomere structure and mechanics.
In this study, we investigated the role of arginylation in passive force generation and titin function in skeletal muscles using a mouse line with KO of ATE1 driven by skeletal muscle-specific creatine kinase M promoter (Ckmm-ATE1 mice). Using single sarcomeres and myofibrils, we found that lack of arginylation results in a pronounced reduction in the passive force. Furthermore, mass spectrometry revealed five arginylated sites within titin's A band, located in important areas for protein-protein interactions. These results led us to propose a mechanism for arginylation-dependent regulation of titin's function in the passive force generation through modulation of titin's interactions with key proteins of the sarcomere.
MATERIALS AND METHODS
Mice.
Skeletal muscle ATE1-KO mice were obtained by crossing Ate1-floxed mouse line generated by targeted insertion of loxP sites upstream and downstream of the critical region of the Ate1 gene with the Ckmm-Cre mouse line commercially available from The Jackson Laboratory, as previously described (3). After crossing, Cre recombinase expression driven by skeletal muscle-specific creatine kinase (Ckmm) promoter drives the deletion of Ate1 in striated muscle in the mice that are homozygous for the floxed allele and express at least one copy of the Cre transgene. Throughout the data collection 34 male mice of ∼1 yr of age were used. All animal procedures used in this study were approved by a committee for animal care at the University of Pennsylvania.
Myofibrils preparation and experiments.
Small muscle bundles from mouse soleus were dissected from wild-type (WT; 4 animals) and Ckmm-ATE1 mice (4 animals), rinsed in rigor solution (in mM: 50 Tris, 100 NaCl, 2 KCl, 2 MgCl2, and 10 EGTA, and pH 7.4), and tied to wooden sticks. The samples were stored in rigor solution/glycerol (50:50) solution for 15 h in −20°C and then transferred to a fresh rigor solution/glycerol (50:50) solution containing a cocktail of protease inhibitors (Roche Diagnostics) for at least 7 days before experiments. On the day of the experiment, small muscle pieces were cut and defrosted in rigor solution at 4°C for 1 h. These pieces were then homogenized in rigor solution using the following sequence: twice for 5 s at 8,000 rpm, twice for 3 s at 15,000 rpm, and once for 1 s at 21,000 rpm (Advanced Homogenizing System 250; VWR). The homogenate was transferred to an experimental temperature-controlled bath (10°C), where rigor solution was slowly replaced to relaxing solution (in mM: 70 KCl, 20 imidazole, 5 MgCl2, 5 ATP, 14.5 creatine phosphate, 0.015 CaCl2, 7 EGTA, and pH 7.4). Under high magnification (×60), the contrast between the dark A bands and the light I bands provided a dark-light intensity pattern, representing the striation pattern produced by the sarcomeres, which allowed for measurement of the SL during the experiments. A myofibril was chosen for mechanical measurements based on its striation pattern.
The myofibrils were attached between an atomic force cantilever (AFC; model ATEC-CONTPt; Nanosensors; stiffness: 50.01 ± 0.6 or 34.0 ± 0.02 nN/μm) and a rigid glass microneedle using micromanipulators. A laser beam was directed through an optical periscope onto the AFC and reflected back into a photo-quadrant detector (18). When the cantilever was pulled by the myofibril during contractions or stretches, the displacement was detected by changes in laser diffraction. Since the AFC stiffness (K) is known, force (F) can be calculated based on the cantilever displacement (Δd) during contraction (F = K × Δd). A computer-controlled, multichannel fluidic system connected to a double-barrelled pipette was used for activation/deactivation of the myofibrils by changing between channels containing activating or relaxing solution.
The myofibrils were adjusted to an average SL of ∼2.4 μm and then activated to produce maximal isometric force. After relaxation, the myofibril was adjusted again to the initial SL and passively stretched three times by ∼25% of its initial length [WT 2.39 ± 0.001 μm (n = 13) and Ckmm-ATE1 2.40 ± 0.003 μm (n = 12)] at 3 μm/s. Between each step, the myofibrils were held isometric for 25 s to reach a steady state (see Fig. 2B). Force and average SL were recorded throughout the protocol.
Fig. 2.

Custom AFM data reveal impairment in passive force in Ckmm-arginyl-tRNA-protein transferase (ATE1) myofibrils. A: typical traces obtained during contractions produced by wild-type (WT; blue line) and Ckmm-ATE1 myofibrils (grey line) and corresponding sarcomere length (SL) traces during activation and relaxation. B: typical passive force traces during consecutive stretches produced by WT (blue lines) and Ckmm-ATE1 (gray lines) myofibrils. C: means ± SE of peak and steady-state passive forces passive forces produced during consecutive stretches. D: rates of force decay in WT (blue) and Ckmm-ATE1 (gray) produced by myofibrils after the stretches. The Ckmm-ATE1 myofibrils presented a faster rate of force decay compared with WT myofibrils. Inset: example of normalized viscoelastic transients of Ckmm-ATE1 and WT myofibrils.
Single sarcomeres preparation and experiments.
The experimental setup for mechanical experimentation of sarcomeres consisted of a newly designed chamber that allows the use of custom-made needles that transversally pierce the sarcomeres, in parallel to the Z lines. This system provides a high signal:noise ratio during mechanical measurements performed with sarcomeres and a clear visualization of the sarcomere structure. Capillary glass needles that were used to grab the sarcomeres and for force measurements were pulled using a needle/pipette puller (Kopf Model 720; David Kopf Instruments, Tujunga, CA). Needle stiffness was calculated by a cross-bending method using an atomic force microscope (AFM) cantilever (ATEC-CONTPt-20; Nanosensors; stiffness: 42.8 nN/μm). The AFM stiffness was measured using scanning electron microscopy. The same cantilever was used to measure the stiffness of all needles used during the experiments. Briefly, the needle tip and the flat surface of the cantilever were brought together and moved horizontally using a piezo motor. The cantilever/needle displacement was measured and subtracted from the free cantilever displacement. Each displacement was multiplied by the cantilever stiffness and the average needle stiffness used. One thin needle and one thick needle were used in each experiment (stiffness ranging from 50 to 100 nN/μm and 500 to 1,000 nN/μm, respectively). The pair of needles was matched at a ratio of at least 0.1 (deflectable needle stiffness/inflexible needle stiffness).
Sarcomeres were isolated from myofibrils of WT (n = 6 animals) and Ckmm-ATE1 (n = 6 animals) soleus muscles using two microneedles with stiffness of 43.44 ± 2.89 nN/μm (deflectable) and >700 nN/μm (rigid), respectively. During these experiments, myofibrils were imaged in a phase-contrast inverted microscope (Eclipse TE2000-U; Nikon) at ×150 magnification (Plan Fluor ×100 oil objective plus ×1.5 built-in microscope magnification). The myofibrils were maintained in relaxing solution at 10°C. A myofibril image was taken for measurements of its cross-sectional area, and three measurements of the A-band diameter (in parallel with the M line) were averaged for force normalization. One sarcomere in a selected myofibril was isolated by piercing it with the microneedles in an area externally adjacent to the Z lines (Fig. 1B). The sarcomere was slightly lifted from the coverslip and its length adjusted to ∼2.2 μm. At this length the sarcomere is slack and does not shorten further at resting conditions (slack length). At the slack length, sarcomeres are not expected to produce passive force (Fig. 3A) (22, 25). During mechanical measurements, the sarcomeres were stretched in several steps of 300 nm using a piezo motor connected to the rigid needle. The sarcomeres were maintained at a steady length for 10 s (see Fig. 3B). Forces obtained during this period were used to create passive force-length relations (see Fig. 3C). All experimental procedures were recorded and analyzed using ABSnake plugin for ImageJ software (National Institutes of Health).
Fig. 1.

Schematics of the myofibril and single sarcomere systems. A: myofibril attached between the atomic force microscopy (AFM) cantilever and a glass microneedle. In red, the laser hits the cantilever and is reflected into the photodiode array (18). Bottom: myofibril before and after stretch. Scale bar = 10 μm. B: sarcomere attached between 2 glass microneedles. The L-shaped needle at left is flexible, whereas the needle at right is not flexible. Bottom: procedure for isolating the sarcomere from the myofibrils. Scale bar = 3 μm.
Fig. 3.

Single sarcomeres isolated from Ckmm-ATE1 produced lower passive force than WT. A: 4 steps of a single sarcomere being stretched from slack length to a long length. The inflexible needle is at right and the deflectable needle at left. B: examples of 6 experiments showing passive force development in WT (blue circles) and Ckmm-ATE1 (gray triangles) myofibrils. C: cloud plot showing passive forces produced by WT and Ckmm-ATE1 isolated sarcomeres. Each point corresponds to a plateau area from the raw data [WT (n = 23) and Ckmm-ATE1 (n = 25)].
Mass spectrometry.
Identification of arginylated proteins by mass spectrometry was performed in isolated myofibrils from soleus muscles of both WT and Ckmm-ATE1 as described previously (39, 42).
Electrophoresis and phosphorylation assays.
Skeletal muscle tissues (WT = 4 and KO = 4) were weighed and pulverized in liquid nitrogen using a mortar and pestle, until a fine powder was obtained. Tissues were primed at −20°C for a minimum of 20 min and then solubilized in 8 M urea buffer containing the following (in mol/l): 8 urea, 2 thiourea, 0.05 Tris·HCl, 0.075 dithiothreitol with 3% SDS, and 0.03% bromophenol blue, pH 6.8; and 50% glycerol with protease inhibitors including the following (in mmol/l): 0.04 E64, 0.16 leupeptin, and 0.2 PMSF. Solubilization was performed for 10 min at 60°C, after which the tissue was centrifuged at room temperature. Finally, the samples were aliquoted into smaller volumes, flash frozen in liquid nitrogen, and stored at −80°C.
Titin isoform N2A was resolved using gradient SDS-PAGE (40). The solubilized samples were electrophoresed on 2–7% gradient gel run at 60 V for 1 h and then at 90 V for 1 h (Criterion 26-well gel system; Bio-Rad, Hercules CA). The gels were stained using Coomassie brilliant blue (Acros Organics) and scanned (Epson 800; Epson, Long Beach CA). For analysis of titin phosphorylation by PKCα [Serine 26 (Ser11878, UnitprotKB: Q8WZ42) and Serine 170 (Ser12022, UnitprotKB: Q8WZ42)], solubilized samples were run in a 0.8% agarose, vertical gel electrophoresis chamber (38). Gels were run at 15 mA for 2 h 50 min and transferred onto PVDF membrane (Immobilon-FL; cat. no. IPFL00010; Millipore; 0.45 μm) using a semidry transfer unit (Trans-Blot Cell; Bio-Rad) for 2 h 30 min. Blots were stained with Ponceau-S (cat. no. P7170-1L; Sigma) for visualization of total protein transferred. Blots were probed with phospho-specific rabbit polyclonal antibodies against phosphorylated S26 and S170 of the PEVK sequence of the skeletal titin isoform (8). Membranes were labeled with secondary antibodies conjugated with fluorescent dyes with infrared excitation spectra (CF680; cat. no. 20067-1; goat anti-rabbit; Biotium, Hayward CA). Blots were scanned using Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln NE), and the images were analyzed using LI-COR software. Ponceau-S scans were analyzed in One-D scan to normalize phosphorylation signal to total protein loading.
Electron microscopy.
Small pieces of soleus muscle from Ckmm-ATE1 (n = 7) and WT (n = 6) mice were fixed for 3 days at 4°C in 2.5% Glutaraldehyde in 0.1 M sodium cacodylate buffer and 4% sucrose. The samples were postfixed with 1% aqueous OsO4 plus 1.5% aqueous potassium ferrocyanide for 2 h at 4°C and dehydrated by incubation in increasing concentrations of acetone for 15 min each in 30, 50, 70, 80, and 90% and 3× 100% ethanol and embedded in Epon. Thin sections of both cross- and longitudinal sections were obtained using an ultramicrotome and stained using 1% uranyl acetate. The sections were overlaid onto Formvar-coated grids for electron microscopy. Filament counting was made in several randomly chosen areas of 0.01 μm2 of perpendicular cross-sectional cuts using ImageJ software.
Statistics.
For the experiments with myofibrils a two-way ANOVA for repeated measures was used to compare peak forces, steady-state forces, and rates of force decay at different SLs. When significant changes were observed, multiple comparisons were performed with Student-Newman-Keuls tests. A level of significance of P ≤ 0.05 was set for all analyses. All values are presented as means ± SE.
RESULTS
Passive force development is impaired in Ckmm-ATE1 myofibrils.
Isolated myofibrils from age-matched WT and Ckmm-ATE1 mice were first activated with calcium (pCa 4.5) to evaluate their active force production. Consistent with our previous observations (3), Ckmm-ATE1 sarcomeres produced less active force than WT (Fig. 2A).
When a myofibril was stretched at low Ca2+ concentration, the passive forces increased during and after the stretches. There were two components of force increase: a fast transient component related to viscoelastic properties and a steady component, in which the force reached a quasi-steady state and persisted while the myofibril was held isometric. This behavior is similar to data published in previous studies (20, 36). Both viscoelastic and steady-state forces developed by Ckmm-ATE1 myofibrils were lower than the force developed by WT myofibrils (Fig. 2, B and C). The average SL at the end of all the stretches was significantly increased in Ckmm-ATE1 mice, suggesting a larger compliance than in WT muscles.
To assess the viscoelastic behavior of the myofibrils, we calculated the force decrease and the rates of force decay between consecutive stretches, after the peak force was achieved. Although the relative magnitudes of force decrease among stretches were not different, the rates of force decay were faster in KO than in WT (Fig. 2D). The faster force decay suggests that Ckmm-ATE1 myofibrils are unable to sustain stable levels of passive force for prolonged periods of time.
Passive force is impaired in Ckmm-ATE1 sarcomeres.
Isolated sarcomeres were stretched in several steps of 300 nm. Just after the initial stretch, passive force dropped quickly, due to the viscoelastic properties of titin. Forces obtained during the elastic period were used to create passive force-length relations (Fig. 3C). Ckmm-ATE1 sarcomeres developed lower passive forces than WT sarcomeres [WT (n = 23) and Ckmm-ATE1 (n = 25)]. The difference in force between the two muscles started at SL of ∼2.75 μm and increased with longer SLs (Fig. 3C). The passive forces obtained with single sarcomere measurements matched remarkably well the forces produced by the myofibrils. Thus passive force reduction in the absence of arginylation is present at the single sarcomere level and may result from impairments in the properties of individual titin molecules and/or its interaction with other sarcomeric proteins.
The A band of titin contains five sites for arginylation.
The reduction in the passive sarcomere forces seen in myofibrils lacking Ate1 suggests that arginylation may directly regulate the key proteins involved in passive force generation. To test whether any of these proteins are arginylated in vivo, we analyzed myofibrils isolated from WT and Ckmm-ATE1 myofibrils using mass spectrometry. This analysis revealed five arginylated sites on titin. All of these sites are located within the A band (Fig. 4), and four of them are located in Fn3 domains. The first site was a glutamic acid (E14609) localized in the first Fn3 domain (A2) at the super-repeat 1, just after a transition area between I band and A band. The second and third sites (D19156 and D19159) were aspartic acids located in the same Fn3 domain at the super-repeat 7 (A48). The fourth site, also an aspartic acid (D27727), was located in an Fn3 domain (A135) at the super-repeat 15. Both super-repeats 7 and 15 are located at the C-zone area of the A band, where the binding sites to MyBPCs are found.
Fig. 4.

Representation of titin domains along the A band of the sarcomere. A: 5 sites for arginylation in titin N2A located in the A band (NP_035782.3). Blue rectangles are Ig-domains and white rectangles are Fn3 domains. Bottom: in red rectangles the domains were sites for arginylation were found: E1609 at A2 in the super-repeat 1, E19156 and D19159 at A48 in the super-repeat 7, D27727 at A135 in the super-repeat 15, and D32535 located in an interdomain connecting 2 Ig domains (*monomethylated sites). B: sequence alignments were performed according to the position of the Fn3 domain in the super-repeat (23). Domains were we found sites for arginylation are highlighted in green and the corresponding site is in bold. E14609 at A2 and D27727 at A135 are highly conserved among their Fn3 family.
To assess the functionality of titin arginylation at these sites, we performed sequence alignment of the Fn3 domains according to their position within the super-repeat (23) and found that two arginylated sites (E14609 and D27727) are highly conserved among the Fn3 domains (Fig. 4B). The fifth site is a glutamic acid (E32535) located at the P-Zone, in an interdomain connecting two Ig domains (M4–M5), in between the titin kinase domain and the COOH terminus. Three of these five sites (E14609, D19156, and E32535) were found monomethylated, suggesting that arginylation at these positions is relatively stable.
To test whether arginylation also affects the abundance or isoform composition of titin, we analyzed WT and Ckmm-ATE1 samples by gel electrophoresis. We found no difference in band mobility between the titin isoforms in WT and Ckmm-ATE1 (Fig. 5A). Moreover, no difference was found in the abundance of N2A-titin and in the titin degradation product T2. We also investigated a possible effect of arginylation on titin phosphorylation and observed a lack of difference between the N2A titin in the two muscles (Fig. 5B). Finally, back phosphorylation assays using 32P showed no difference for PKA activity between the two muscles. Phospho-specific antibodies (pS26 and pS170) that positively recognize PKCα phosphorylation in the PEVK area revealed similar fluorescence intensity.
Fig. 5.

Arginylation does not affect titin abundance and phosphorylation status. A: SDS-PAGE gels showing a lack of changes in titin mobility in WT and CKmm-ATE1 myofibrils. The optical density of the degradation product T2 was not increased, indicating a lack of protein degradation in the Ckmm-ATE1 skeletal muscles. B: phosphorylation assays evaluating phosphorylation of both residues PS26 and PS170 of the spring-like segment PEVK in WT and Ckmm-ATE1 myofibrils. MHC, myosin heavy chain.
Electron microscopy.
The ultrastructure of Ckmm-ATE1 sarcomeres showed a normal appearance (Fig. 6B). A ratio of six titin molecules per myosin filament is expected for these muscles, and therefore a reduction in the number of myosin filaments could account for the reduction in passive force. To test this possibility, we performed cross-sectional imaging of the muscles to evaluate filament arrangement and counting (Fig. 6A). The lattice arrangement appeared normal and no difference was found in the density of myosin filaments [WT 12.3 ± 0.427 myosin filaments/0.01 μm2 (n = 41) and Ckmm-ATE1 12.8 ± 0.35 myosin filaments/0.01 μm2 (n = 53)]. Overall, our data show that the reduction in passive force on CKmm-Ate1 myofibrils is not due to reduction in protein content, protein degradation, nor changes in titin phosphorylation conditions.
Fig. 6.
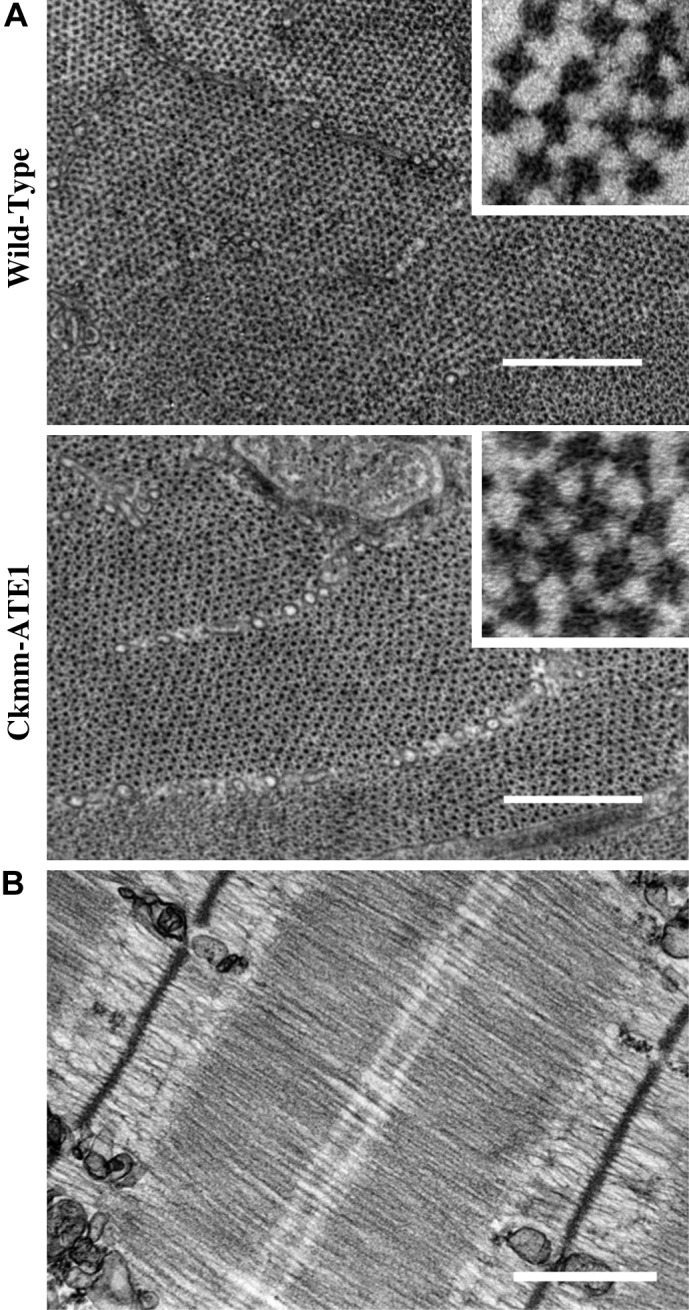
Ckmm-ATE1 presented normal hexagonal arrangement of filaments. A: electron micrographs of cross-sectional views in WT and Ckmm-ATE1 myofibrils. Insets show a closer view of the filaments (area = 0.01 μm2). B: sarcomere structure appeared normal in Ckmm-ATE1 mice. Scale bars = 500 nm.
DISCUSSION
Our study is the first to demonstrate that titin N2A isoform is arginylated in vivo and that passive force is reduced in skeletal muscles lacking ATE1. Passive force is essential for muscle function, and titin is the main protein responsible for passive force development within muscle cells. Our data show that arginylation does not alter the ratio between intact titin and T2 or the abundance of titin N2A (Fig. 5A). In skeletal muscles, titin stiffness is driven by the extension of the serially linked Ig-like domains and the PEVK (proline, glutamatic acid, valine, and lysine residue rich segment) spring-like segment. After the SL of 2.7 μm, PEVK is the main responsible for passive force development (31). Posttranslational phosphorylation of titin spring-segments is known to affect titin stiffness; nevertheless, we found that arginylation does not affect titin through an indirect change in PEVK phosphorylation status (Fig. 5B). Finally, we observed that muscles from Ckmm-ATE1 present a normal ultrastructure and there is no reduction in the amount of titin compared with WT (Figs. 5A and 6). Thus our result strongly suggests that arginylaton directly affects titin functioning.
N2A titin presents four spring-like segments in its I band: proximal Ig domain, N2A, PEVK, and the distal Ig domain. The synchronic work of these segments controls passive tension inside each sarcomere. During stretch, there are two components for the development of passive force, a fast component and a slow component (Fig. 1B). The fast component is mainly attributed to a viscous drag originated from the interactions between titin and actin filaments (20, 43). Our data show a lower peak force in Ckmm-ATE1 myofibrils, which may be related to the titin-actin interaction. Sites for arginylation have been identified in skeletal muscle actin (3), and it was demonstrated that arginylation regulates actin polymerization in vivo (3, 28). Hence, arginylation of actin might influence passive force viscoelastic development, although it is not likely to account for the decrease in passive force observed at the steady-state level.
After the fast viscoelastic component, the force drops exponentially towards an elastic quasi-steady-state component. We observed a reduction in the elastic portion of the passive force component in myofibrils and single sarcomeres, suggesting that the mechanical properties of titin are also affected by arginylation. All sites for arginylation were located in the A band of titin, which is not commonly associated with the elasticity of these molecules. Since the architecture of the A-band region of titin is well structured, any modification could play a role regulating passive forces.
During myofibrillogenesis, the association between titin and myosin starts very early, and titin works as a molecular scaffold for myosin creating a very large overlap between both proteins (11, 33). In a previous study, cardiac myofibrils lacking arginylation were structurally defective before the development of any specific phenotype (14, 15, 27). It is tempting to speculate that protein arginylation may occur before the incorporation of titin and myosin into the sarcomere, which could result in a poor anchorage among A-band proteins. We observed previously that myosin and MyBP-C are arginylated (3). Hence, we cannot exclude the possibility passive force weakness observed in the KO mice is related to impairment in the interaction of myosin with titin, leading to a systematic abnormal anchoring of titin in the A band. It has been hypothesized that, in normal muscle fibers, an extensive stretch may dislodge titin anchorage on the thick filament, preventing forces from rising (36). In our experiments, passive force of Ckmm-ATE1 reaches a plateau after 3.0 μm (Fig. 3A), and its myofibrils are more compliant than WT (Fig. 2C). Further studies are necessary to evaluate whether arginylation is required to avoid early dislodgment of titin anchorage from thick filament when the sarcomeres undergo stretch.
Titin's A-band topology is extremely conserved among different titin isoforms. The majority of the A-band structure is highly organized in 17 super-repeats: six 7-domains super-repeats {[Ig-(fn3)2-Ig-(fn3)3]n} and eleven 11-domains super-repeats {[Ig-(fn3)2-Ig-(fn3)3]n} (17, 23). Five sites for arginylation were found by mass spectrometry, three of which are monomethylated, suggesting that these sites may be relatively stable and likely carry important physiological roles (Fig. 4A). The A band is an important area for interaction between titin and many other proteins, especially with myosin and MyBP-C. Four arginylated sites are located on Fn3 domains. The first site (E14609) is monomethylated and located at the beginning of the thick filament on the first Fn3 (A2) domain of titin. Interestingly, this residue is conserved in all second position Fn3 domains of the short super-repeats. The super-repeat 1 follows an important region of transition between the I band and A band. In fact, there is an arginylated site in the same titin's Fn3 domain in cardiac muscles (L7960) that also produces low passive forces (14).
The second and third (E19156 and D19159, respectively) arginylated sites are located on the same Fn3 domain (A48). The fact that two sites are located closely to each other and that one is monomethylated suggests an important site for the titin. A48 is part of the super-repeat 7, which is the first long super-repeat of the C zone, where MyBPC is present. The fourth site (D27727) is also an Fn3 domain (A135) located at the C zone, on the long super-repeat 15. This is a surface residue located at the 28th position of the sequence alignment. It has been reported that D28 is a highly conserved residue among all Fn3 domains and might play a role in the structural flexibility of Fn3 domains (23). Mutations in the exon 335 that express A131 to A136 have been linked to patients with dilated cardiomyopathy, leading to truncated titin in cardiac muscles but not in skeletal muscles (7). The function of the Fn3 domains is still not fully understood. Previous studies indicate that Fn3 domains bind to different parts of myosin, especially to the myosin subfragment 1 (S1) and to the light meromyosin (9, 16, 23, 30).
The M band of the sarcomere has a high concentration of different types of proteins, forming a very important region of protein interactions and strong structural support for the sarcomere. Titin's serine/threonine type kinase domain and its surroundings interact with a variety of important proteins like MuRF1, FHL2, and myomesin. The fifth arginylation site (D32535) is located at the M band, in an interdomain connecting two Ig-domains (M4 and M5). The Ig-domain M4 is a possible site for binding of myomesin on titin. Interestingly, besides L7960, two other sites for arginylation were reported in cardiac N2B isoform. One of them is placed in an Ig domain (A72) at the super-repeat 9 and the second one is a monomethylated site in the kinase domain (V1503 and C24818, respectively). The kinase domain is important for mechanical load sensing and mutation in an arginine residue in the kinase domain has been linked to hereditary myopathy in humans (19).
Alternative mechanisms.
Although titin and myosin contain several arginylated sites, other sarcomeric proteins may play a role in the impairment of passive force observed in this study. One such protein is MyBPC, which is arginylated in the elastic M domain (3, 4, 10, 12). It is unlikely that this site plays a role in the passive force, since the M domain is associated with interactions between MyBPC and actin, whereas the binding site to titin and to the thick filament in MyBPC is at COOH-terminal domain.
The fifth arginylated site within titin molecule is located in close proximity where myomesin binds on titin. Myomesin is a protein found in all striated muscles, being responsible for interconnecting titin and myosin (24, 41). The bridge formed by myomesin is important for stress sensing and for the structural integrity of the sarcomere (1, 24, 41). However, the sarcomere M-band senses mechanical stress only during sarcomere contraction, which makes this interaction between titin and myomesin less likely to account for our results (1).
Another possibility is that α-actinin may play a role in the reduction of passive force in KO mice. α-Actinin is a highly conserved actin-binding protein that cross-links actin filaments from adjacent sarcomeres on the Z disks. Four spectrin-like repeats form the α-actinin rod, an important area for protein-protein binding and Z-disk assembly and function (44). In skeletal muscles, there are three arginylated residues located within the α-actinin rod, at the second spectrin-like repeat, which is essential for α-actinin binding to titin (3, 5, 44). Hence, an impairment of this interaction could change titin spring-properties at the Z disk. More research is needed to investigate the potential role of α-actinin in the regulation of passive force in association with arginylation.
In conclusion, myofibrils and sarcomeres isolated from skeletal muscles lacking protein arginylation presented a reduction in passive force, without apparent structural defects, changes in isoform or phosphorylation status of titin. These findings strengthen the idea of posttranslational arginylation as a regulator of force production in striated muscles.
GRANTS
F. S. Leite is a recipient of a Conselho Nacional de Desenvolvimento Cientifico e Tecnologico (CNPq) scholarship, Brazil. A. S. Cornachione is a recipient of a Fundacao de Amparo a Pesquisa do Estado de Sao Paulo (FAPESP) fellowship (2013/07104-6), Brazil. This study was funded by Natural Sciences and Engineering Research Council (NSERC) of Canada (to D. E. Rassier) and National Institutes of Health Grants GM-104003 and GM117984 (to A. S. Kashina) and 5R01-AR-053897 (to H. Granzier).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
F.S.L., A.K., J.R.Y., A.S.K., and D.E.R. conception and design of research; F.S.L., F.C.M., A.S.C., Y.-S.C., N.A.L., X.H., and C.S. performed experiments; F.S.L., F.C.M., J.R.Y., and A.S.K. analyzed data; F.S.L., H.G., A.S.K., and D.E.R. interpreted results of experiments; F.S.L., F.C.M., and A.K. prepared figures; F.S.L., F.C.M., A.K., and D.E.R. drafted manuscript; F.S.L., H.G., A.S.K., and D.E.R. edited and revised manuscript; F.S.L., F.C.M., A.K., A.S.C., Y.-S.C., N.A.L., X.H., C.S., J.R.Y., H.G., A.S.K., and D.E.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Clara Franzini-Armstrong for assistance during preparation of soleus muscle for electron microscopy. We also thank the Facility for Electron Microscopy Research, McGill University.
Present address of A. S. Cornachione: Dept. of Pathology, Faculty of Medicine of Ribeirão Preto, University of São Paulo, Vila Monte Alegre, 14049900 Ribeirão Preto, São Paulo, Brazil.
REFERENCES
- 1.Agarkova I, Perriard JC. The M-band: an elastic web that crosslinks thick filaments in the center of the sarcomere. Trends Cell Biol 15: 477–485, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Balzi E, Choder M, Chen WN, Varshavsky A, Goffeau A. Cloning and functional analysis of the arginyl-tRNA-protein transferase gene ATE1 of Saccharomyces cerevisiae. J Biol Chem 265: 7464–7471, 1990. [PubMed] [Google Scholar]
- 3.Cornachione AS, Leite FS, Wang J, Leu NA, Kalganov A, Volgin D, Han X, Xu T, Cheng YS, Yates JR 3rd, Rassier DE, Kashina A. Arginylation of myosin heavy chain regulates skeletal muscle strength. Cell reports 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Craig R, Lee KH, Mun JY, Torre I, Luther PK. Structure, sarcomeric organization, and thin filament binding of cardiac myosin-binding protein-C. Pflügers Arch 466: 425–431, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Djinovic-Carugo K, Young P, Gautel M, Saraste M. Structure of the alpha-actinin rod: molecular basis for cross-linking of actin filaments. Cell 98: 537–546, 1999. [DOI] [PubMed] [Google Scholar]
- 6.Donlin LT, Andresen C, Just S, Rudensky E, Pappas CT, Kruger M, Jacobs EY, Unger A, Zieseniss A, Dobenecker MW, Voelkel T, Chait BT, Gregorio CC, Rottbauer W, Tarakhovsky A, Linke WA. Smyd2 controls cytoplasmic lysine methylation of Hsp90 and myofilament organization. Genes Dev 26: 114–119, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerull B, Atherton J, Geupel A, Sasse-Klaassen S, Heuser A, Frenneaux M, McNabb M, Granzier H, Labeit S, Thierfelder L. Identification of a novel frameshift mutation in the giant muscle filament titin in a large Australian family with dilated cardiomyopathy. J Mol Med 84: 478–483, 2006. [DOI] [PubMed] [Google Scholar]
- 8.Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H. PKC phosphorylation of titin's PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res 105: 631–638, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Houmeida A, Holt J, Tskhovrebova L, Trinick J. Studies of the interaction between titin and myosin. J Cell Biol 131: 1471–1481, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howarth JW, Ramisetti S, Nolan K, Sadayappan S, Rosevear PR. Structural insight into unique cardiac myosin-binding protein-C motif: a partially folded domain. J Biol Chem 287: 8254–8262, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Isaacs WB, Kim IS, Struve A, Fulton AB. Association of titin and myosin heavy chain in developing skeletal muscle. Proc Natl Acad Sci USA 89: 7496–7500, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karsai A, Kellermayer MS, Harris SP. Mechanical unfolding of cardiac myosin binding protein-C by atomic force microscopy. Biophys J 101: 1968–1977, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kashina A. Protein arginylation, a global biological regulator that targets actin cytoskeleton and the muscle. Anat Rec (Hoboken) 297: 1630–1636, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurosaka S, Leu NA, Pavlov I, Han X, Ribeiro PA, Xu T, Bunte R, Saha S, Wang J, Cornachione A, Mai W, Yates JR 3rd, Rassier DE, Kashina A. Arginylation regulates myofibrils to maintain heart function and prevent dilated cardiomyopathy. J Mol Cell Cardiol 53: 333–341, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwon YT, Kashina AS, Davydov IV, Hu RG, An JY, Seo JW, Du F, Varshavsky A. An essential role of N-terminal arginylation in cardiovascular development. Science 297: 96–99, 2002. [DOI] [PubMed] [Google Scholar]
- 16.Labeit S, Gautel M, Lakey A, Trinick J. Towards a molecular understanding of titin. EMBO J 11: 1711–1716, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Labeit S, Kolmerer B. Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science 270: 293–296, 1995. [DOI] [PubMed] [Google Scholar]
- 18.Labuda A, Brastaviceanu T, Pavlov I, Paul W, Rassier DE. Optical detection system for probing cantilever deflections parallel to a sample surface. Rev Sci Instrum 82: 013701, 2011. [DOI] [PubMed] [Google Scholar]
- 19.Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, Rostkova E, Kristensen J, Brandmeier B, Franzen G, Hedberg B, Gunnarsson LG, Hughes SM, Marchand S, Sejersen T, Richard I, Edstrom L, Ehler E, Udd B, Gautel M. The kinase domain of titin controls muscle gene expression and protein turnover. Science 308: 1599–1603, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Linke WA, Leake MC. Multiple sources of passive stress relaxation in muscle fibres. Phys Med Biol 49: 3613–3627, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Maruyama K, Kimura S, Ohashi K, Kuwano Y. Connectin, an elastic protein of muscle. Identification of “titin” with connectin. J Biochem 89: 701–709, 1981. [DOI] [PubMed] [Google Scholar]
- 22.Minozzo FC, Baroni BM, Correa JA, Vaz MA, Rassier DE. Force produced after stretch in sarcomeres and half-sarcomeres isolated from skeletal muscles. Sci Rep 3: 2320, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muhle-Goll C, Habeck M, Cazorla O, Nilges M, Labeit S, Granzier H. Structural and functional studies of titin's fn3 modules reveal conserved surface patterns and binding to myosin S1–a possible role in the Frank-Starling mechanism of the heart. J Mol Biol 313: 431–447, 2001. [DOI] [PubMed] [Google Scholar]
- 24.Obermann WM, Gautel M, Weber K, Furst DO. Molecular structure of the sarcomeric M band: mapping of titin and myosin binding domains in myomesin and the identification of a potential regulatory phosphorylation site in myomesin. EMBO J 16: 211–220, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pavlov I, Novinger R, Rassier DE. The mechanical behavior of individual sarcomeres of myofibrils isolated from rabbit psoas muscle. Am J Physiol Cell Physiol 297: C1211–C1219, 2009. [DOI] [PubMed] [Google Scholar]
- 26.Rai R, Wong CC, Xu T, Leu NA, Dong DW, Guo C, McLaughlin KJ, Yates JR 3rd, Kashina A. Arginyltransferase regulates alpha cardiac actin function, myofibril formation and contractility during heart development. Development 135: 3881–3889, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ribeiro PA, Ribeiro JP, Minozzo FC, Pavlov I, Leu NA, Kurosaka S, Kashina A, Rassier DE. Contractility of myofibrils from the heart and diaphragm muscles measured with atomic force cantilevers: effects of heart-specific deletion of arginyl-tRNA-protein transferase. Int J Cardiol 168: 3564–3571, 2013. [DOI] [PubMed] [Google Scholar]
- 28.Saha S, Mundia MM, Zhang F, Demers RW, Korobova F, Svitkina T, Perieteanu AA, Dawson JF, Kashina A. Arginylation regulates intracellular actin polymer level by modulating actin properties and binding of capping and severing proteins. Mol Biol Cell 21: 1350–1361, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saha S, Wong CC, Xu T, Namgoong S, Zebroski H, Yates JR 3rd, Kashina A. Arginylation and methylation double up to regulate nuclear proteins and nuclear architecture in vivo. Chem Biol 18: 1369–1378, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soteriou A, Gamage M, Trinick J. A survey of interactions made by the giant protein titin. J Cell Sci 104: 119–123, 1993. [DOI] [PubMed] [Google Scholar]
- 31.Trombitas K, Greaser M, Labeit S, Jin JP, Kellermayer M, Helmes M, Granzier H. Titin extensibility in situ: entropic elasticity of permanently folded and permanently unfolded molecular segments. J Cell Biol 140: 853–859, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tskhovrebova L, Trinick J. Flexibility and extensibility in the titin molecule: analysis of electron microscope data. J Mol Biol 310: 755–771, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Van der Ven PF, Ehler E, Perriard JC, Furst DO. Thick filament assembly occurs after the formation of a cytoskeletal scaffold. J Muscle Res Cell Motil 20: 569–579, 1999. [DOI] [PubMed] [Google Scholar]
- 34.Voelkel T, Andresen C, Unger A, Just S, Rottbauer W, Linke WA. Lysine methyltransferase Smyd2 regulates Hsp90-mediated protection of the sarcomeric titin springs and cardiac function. Biochim Biophys Acta 1833: 812–822, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Han X, Wong CC, Cheng H, Aslanian A, Xu T, Leavis P, Roder H, Hedstrom L, Yates JR 3rd, Kashina A. Arginyltransferase ATE1 catalyzes midchain arginylation of proteins at side chain carboxylates in vivo. Chem Biol 21: 331–337, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang K, McCarter R, Wright J, Beverly J, Ramirez-Mitchell R. Regulation of skeletal muscle stiffness and elasticity by titin isoforms: a test of the segmental extension model of resting tension. Proc Natl Acad Sci USA 88: 7101–7105, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang K, McClure J, Tu A. Titin: major myofibrillar components of striated muscle. Proc Natl Acad Sci USA 76: 3698–3702, 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Warren CM, Krzesinski PR, Greaser ML. Vertical agarose gel electrophoresis and electroblotting of high-molecular-weight proteins. Electrophoresis 24: 1695–1702, 2003. [DOI] [PubMed] [Google Scholar]
- 39.Wong CC, Xu T, Rai R, Bailey AO, Yates JR 3rd, Wolf YI, Zebroski H, Kashina A. Global analysis of posttranslational protein arginylation. PLoS Biol 5: e258, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu Y, Bell SP, Trombitas K, Witt CC, Labeit S, LeWinter MM, Granzier H. Changes in titin isoform expression in pacing-induced cardiac failure give rise to increased passive muscle stiffness. Circulation 106: 1384–1389, 2002. [DOI] [PubMed] [Google Scholar]
- 41.Xiao S, Grater F. Molecular basis of the mechanical hierarchy in myomesin dimers for sarcomere integrity. Biophys J 107: 965–973, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu T, Wong CC, Kashina A, Yates JR 3rd. Identification of N-terminally arginylated proteins and peptides by mass spectrometry. Nat Protoc 4: 325–332, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamasaki R, Berri M, Wu Y, Trombitas K, McNabb M, Kellermayer MS, Witt C, Labeit D, Labeit S, Greaser M, Granzier H. Titin-actin interaction in mouse myocardium: passive tension modulation and its regulation by calcium/S100A1. Biophys J 81: 2297–2313, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Young P, Ferguson C, Banuelos S, Gautel M. Molecular structure of the sarcomeric Z-disk: two types of titin interactions lead to an asymmetrical sorting of alpha-actinin. EMBO J 17: 1614–1624, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]