

Abstract
The p70 ribosomal S6 kinase (S6K) is a downstream substrate that is phosphorylated and activated by the mammalian target of rapamycin complex and regulates multiple cellular processes associated with fibrogenesis. Recent studies demonstrate that aberrant mTORC1-S6K signaling contributes to various pathological conditions, but a direct role in pulmonary fibroproliferation has not been established. Increased phosphorylation of the S6K pathway is detected immediately following transforming growth factor-α (TGF-α) expression in a transgenic model of progressive lung fibrosis. To test the hypothesis that the S6K directly regulates pulmonary fibroproliferative disease we determined the cellular sites of S6K phosphorylation during the induction of fibrosis in the TGF-α model and tested the efficacy of specific pharmacological inhibition of the S6K pathway to prevent and reverse fibrotic disease. Following TGF-α expression increased phosphorylation of the S6K was detected in the airway and alveolar epithelium and the mesenchyme of advanced subpleural fibrotic regions. Specific inhibition of the S6K with the small molecule inhibitor LY-2584702 decreased TGF-α and platelet-derived growth factor-β-induced proliferation of lung fibroblasts in vitro. Administration of S6K inhibitors to TGF-α mice prevented the development of extensive subpleural fibrosis and alterations in lung mechanics, and attenuated the increase in total lung hydroxyproline. S6K inhibition after fibrosis was established attenuated the progression of subpleural fibrosis. Together these studies demonstrate targeting the S6K pathway selectively modifies the progression of pulmonary fibrosis in the subpleural compartment of the lung.
Keywords: epidermal growth factor receptor, pleural fibrosis, mammalian target of rapamycin
lung fibrosis is a significant contributor to morbidity and mortality in a diverse spectrum of ailments, including interstitial lung diseases, idiopathic interstitial pneumonias as part of several systemic connective tissue and childhood interstitial lung disease syndromes, and in response to many types of lung injury, including radiation and chemotherapeutic drugs (13, 24, 39). Because lung fibrosis is heterogeneous in its clinical etiology, so too are the likely molecular pathogenic causes, and studies over the past three decades have identified several growth factors and cytokines associated with inducing and augmenting pulmonary fibrosis. Regardless of the underlying clinical disease and associated cytokines, common downstream pathological effector processes are seen in all pulmonary fibrotic disorders, which include the proliferation and differentiation of mesenchymal cells with increased extracellular matrix deposition into gas-exchanging regions of the lung.
Mesenchymal cell proliferation in the lung is predominantly mediated by growth factors, and there is a complex interface between these growth factors, their receptors, and the downstream signaling pathways that regulate cellular processes. Receptor tyrosine kinases (RTK) are the high-affinity cell surface receptors for many polypeptide growth factors and cytokines known to control lung fibrosis, including the epidermal growth factor receptor (EGFR), platelet-derived growth factor (PDGF), insulin-like growth factor, basic fibroblast growth factor (FGF), and vascular endothelial growth factor receptors (VEGF) (11, 21, 22). Following RTK activation, the phosphorylated tyrosine residues on the intracellular domains become docking sites for signaling molecules that activate multiple pathways, including the mitogen-activated protein kinase (MAPK), phosphatidylinositide 3-kinases (PI3K), and mammalian target of rapamycin (mTOR) pathways. A number of experimental studies have demonstrated that these pathways directly control cellular processes associated with fibrosis, including cell growth, proliferation, and protection from apoptosis (1, 49, 50). In addition, activation of the MAPK, PI3K, and mTOR pathways is elevated in human fibrotic disorders (2, 3, 18). Together, there is emerging evidence that there are relatively limited downstream converging signaling pathways that mediate fibroproliferation from heterogeneous upstream growth factors and cytokines.
The p70 ribosomal S6 kinase (S6K) is a mitogen and amino acid-sensitive serine-threonine kinase that is immediately downstream of TORC1, a critical component of the mTOR complex (44). There are two S6Ks (S6K1 and S6K2) that subsequently phosphorylate the 40S ribosomal protein S6 (34). In addition to TORC1, the S6K pathway has been shown to be directly or indirectly activated by kinases or signaling pathways known to regulate cell proliferation, differentiation, and apoptosis. These pathways include the 3-phosphoinositide-dependent protein kinase, which can directly phosphorylate S6K, the PI3K/Akt pathway, which activates the TORC1 pathway, and MAPK/Erk, which directly activates S6 using an mTOR-independent pathway through the p90 ribosomal S6 kinases (32, 40, 42, 45). In addition to S6K, TORC1 also phosphorylates the eukaryotic initiation factor (EIF)-4E-binding protein (4E-BPs), causing dissociation from EIF4E and allowing interaction between EIF4G1/EIF4G3 and EIF4E, leading to initiation of translation. TORC1 thus signals along parallel pathways to promote protein synthesis and control cell cycle regulation, cellular growth, and translation (18).
Emerging evidence suggests that aberrant mTORC1-S6K signaling contributes to various pathological conditions, including diabetes, obesity, organ hypertrophy, and cancer. There are limited data on the contribution for the S6K in mediating pulmonary remodeling. S6K is necessary for the proliferative responses of pulmonary artery adventitial fibroblasts in response to hypoxia (8). In cultured human airway smooth muscle cell activation of S6K is required for transforming growth factor-β (TGF-β) and endothelin-1-induced airway smooth muscle cell size enlargement (6). Recent data analyzing human lung tissue for phosphorylated S6 demonstrated increased expression in three of five idiopathic pulmonary fibrosis (IPF) samples compared with both controls and patients with chronic obstructive pulmonary disease by Western blot (36). Furthermore, immunohistochemical analysis for phosphorylated S6 on lung tissues taken at transplantation revealed weak expression in healthy lung tissue while the phosphorylated S6 was strongly expressed in myofibroblasts from IPF lung tissue (10). These findings indicate the S6K pathway is upregulated in at least a portion of patients with IPF and emphasize the importance to develop a greater understanding on the relevance of the S6K pathway in mediating fibroproliferative disease.
To further understand the biology of pulmonary fibroproliferation in vivo, our laboratory previously generated doxycycline (Dox)-regulatable transgenic mice overexpressing TGF-α under control of the lung epithelial-specific 2.3-kb rat Clara cell secretory protein (CCSP) gene promoter. When CCSP/TGF-α mice are administered Dox, progressive pulmonary fibrosis develops and is characterized by specific phenotypes that are observed in human fibrotic disease, including epithelial and mesenchymal proliferation with myofibroblast transformation, progressive migration of fibrotic lesions from the pleura into the interstitium, extracellular matrix deposition, severe restrictive changes in lung mechanics, and secondary pulmonary hypertension (15, 25). Hierarchical clustering revealed that transcriptional profiles of IPF were more similar to TGF-α induction than bleomycin injury, largely because of the extensive cytokine increase in bleomycin injury not detected in either IPF or the TGF-α model (14).
We have previously demonstrated increased phosphorylation of the S6K pathway but not the 4E-BPs immediately following TGF-α induction (23). Previous studies in the CCSP/TGF-α model confirmed that selective pharmacological inhibition of either the mTORC1, MAPK, or PI3K pathways inhibited progression of pulmonary fibrosis while also reducing phosphorylation of S6K (23, 27). Because the S6K pathway is downstream of several fibroproliferative receptor tyrosine kinase signaling pathways, including EGFR, we therefore hypothesized that the S6K was a crucial converging downstream pathway that could be pharmacologically targeted to regulate pulmonary fibroproliferative disease. To test this hypothesis we determined to identify cellular sites of S6K phosphorylation during the induction of fibrosis in the CCSP/TGF-α model and tested the efficacy of specific pharmacological inhibition of the S6K pathway in vitro and in vivo to prevent and reverse fibrotic disease.
METHODS
Transgenic mice.
CCSP-rtTA activator mice expressing the reverse tetracycline-responsive transactivator (rtTA) under control of the 2.3-kb rat CCSP gene promoter (47) were mated to conditional Dox-regulated transgenic mice containing the human TGF-α cDNA under the control of seven copies of the tetracycline operon [(TetO)7-cmv TGF-α] plus a minimal CMV promoter (16). Single transgenic CCSP-rtTA+/− mice (hereafter abbreviated CCSP/−) and bitransgenic CCSP-rtTA+/−/(TetO)7-cmv TGF-α+/− mice (hereafter abbreviated CCSP/TGF-α) were produced within the same litter by mating homozygous CCSP/− mice to hemizygous (TetO)7-cmv TGF-α+/− mice. All mice were derived from the FVB/NJ inbred strain. To induce TGF-α expression, Dox (Sigma, St. Louis, MO) was administered in the drinking water at a final concentration of 0.5 mg/ml and in food (62.5 mg/kg). Water was replaced three times per week. All mice were housed under specific pathogen-free conditions, and protocols were approved by the Institutional Animal Use and Care Committee of the Cincinnati Children's Hospital Research Foundation.
Lung histology, S6 immunostaining, and proliferation.
Lungs were inflation fixed using 4% paraformaldehyde and stained with trichrome as earlier described (25). Immunostaining for phosphorylated S6 (Ser235/236; Cell Signaling) used citrate/TBST antigen unmasking with antibody diluted 1:1,200. Secondary antibodies and DAB detection were performed as previously described (16). For immunohistochemical detection of proliferation, paraffin sections were immunostained with anti-mouse Ki-67 antigen (Dako, Glostrup, Denmark) with antibody diluted 1:500. The total numbers of Ki-67-staining nuclei were counted as well as the total number of nuclei in 10 randomly selected uniform fields (26.2 mm2) that encompassed alveolar, subpleural, and adventitial regions of the lung for each mouse. The proliferation index was determined by counting the total number of Ki-67-staining nuclei and dividing by the total number of nuclei in each field.
Fluorescence microscopy.
Lungs were harvested from CCSP/− and CCSP/TGF-α mice treated with Dox for 4 or 28 days, respectively, and embedded in OCT medium for cryosectioning (Tissue-Tek) at 6 μm in diameter. Sections were immunostained as described earlier (30). In brief, lung sections were blocked at room temperature for 1–2 h with 3% normal goat sera. Lung cells positive for phospho-S6 Ribosomal Protein were stained alone or colocalized with vimentin (clone 3B4; Millipore, Billerica, MA) and α-smooth muscle actin (SMA, Clone 1A4; Sigma Aldrich, St. Louis, MO) using a rabbit anti-phospho-S6 Ribosomal Protein (Ser235/236) antibody (Cell Signaling Technology). Appropriate secondary antibodies conjugated with either Alexa-Fluor 488, Alexa-Fluor 594, or Alexa 647 (Invitrogen, Grand Island, NY) were used to colocalize antigens by immunostaining. Confocal images were collected using a Nikon AIR-A1 laser-scanning confocal microscope (Melville, NY). Staining controls include lung sections stained only with the above conjugated secondary antibodies. Image exposures were held constant to reflect changes in staining intensities. Images were analyzed using Imaris (version 4.2.0; Bitplane, South Windsor, CT) and Adobe Photoshop (version 7.0).
Western blots.
Western blot analysis was performed on lung fibroblast lysates from culture and lung homogenates and quantified using the volume integration function on a PhosphorImager software Multigage (Fujifilm, Valhalla, NY) as previously described (12). Primary antibodies used include anti-GAPDH (Bethyl Labs, Montgomery, TX), total Akt [rabbit polyclonal antibody (pAb); Cell Signaling, Danvers, MA], phosphorylated Akt (Ser473) [rabbit monoclonal antibody (mAb); Cell Signaling], total p44/42 MAP kinase (Erk1/2) (rabbit pAb; Cell Signaling), phosphorylated p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (rabbit mAB; Cell Signaling), total and phosphorylated S6 (Ser235/236) (rabbit mAb; Cell Signaling), total and phosphorylated p70 S6 kinase (Thr389) (rabbit mAb; Cell Signaling), and total and phosphorylated 4E-BP1 (Thr37/46) (rabbit mAb; Cell Signaling).
Administration of S6K inhibitors in vivo.
The mTOR-S6K signaling networks have emerged as attractive targets for novel therapeutic strategies to treat cancer, resulting in the search for specific pharmacological inhibitors. LY-2584702 tosylate (hereafter referred to as LY-2584702; Eli Lilly, Indianapolis, IN) is an oral, potent, highly selective small-molecule ATP competitive inhibitor of p70S6 kinase that inhibits both S6K 1 and 2 isoforms. LY-2584702 is selective against 83 other kinases as determined by a ubiquitin kinase panel and 45 cell surface markers as determined by a CEREP minipanel. LY-2584702 demonstrated significant single-agent efficacy in glioblastoma and colon carcinoma xenograft models and is currently in advanced oncology trials (20, 48). Stock solutions of LY-2584702 were made fresh immediately before treatments. A stock vehicle of 15.6% cavasol (Wacker Chemical, Adrian, MI), 15.6% polysorbate 80 (Tween 80; Sigma-Aldrich) was made in sterile distilled water. The stock vehicle was first vortexed and then centrifuged (2,000 revolutions/min, 10 min). LY-2584702 was first suspended in polyethylene glycol (PEG-300; Sigma-Aldrich) and then added to the stock vehicle for a final concentration of 4 mg/ml. PEG-300 was added to the stock vehicle to administer to vehicle control mice. Mice were anesthetized (Isoflurane; Abbott Labs, Chicago, IL), and LY-2584702 or sterile vehicle was administered by gavage using a 20-gauge feeding catheter (Harvard Apparatus, Holliston, MA) in volumes ranging from 120 to 200 μl based on the weight of each mouse. Body weights for the mice were measured weekly, and dosing throughout the study was based on original baseline weights and not adjusted for weight changes. Mice were treated two times daily Monday through Saturday (∼7:00 A.M. and 7:00 P.M.) and one time on Sunday afternoon for up to four weeks.
Mouse fibroblast cultures.
Mouse primary fibroblasts were generated by culturing the lungs of CCSP/− mice for 10 days as described earlier (35). For signaling studies using Western blots, mouse primary fibroblasts were seeded at 0.5 million cells/well using 0.1% fetal bovine serum containing DMEM (Invitrogen) in 12-well plates. After 12 h of resting, fibroblasts were pretreated for 30 min with 0.1 and 0.5 μM of LY-2584702 in DMSO. Final concentration of DMSO in cultures was <0.1%, and no effect of DMSO was observed on signaling or viability of fibroblasts. Control or LY-2584702-added cells were treated with or without TGF-α (20 ng/ml) and PDGF-β (1 ng/ml; R&D Systems, Minneapolis, MN). After 30 min of treatment, media were removed, and cell lysates were prepared for Western blot analysis in RIPA buffer (Santa Cruz). Cell proliferation was determined by repeating the above experiments, allowing cells to incubate for 2 days, then counting cells with an automated cell counter (Nexcelom, Lawrence, MA). Because cell proliferation was greater when TGF-α and PDGF-β were added in combination than alone, we report the effects of the combination of growth factors on fibroblasts.
RNA preparation and real-time PCR.
To assess the effects of p70S6K inhibition on lung collagen gene expression, RNA was obtained from whole lung homogenates from CCSP/TGF-α treated with and without LY-2584702 following both 2 days or 4 wk of Dox. Whole lung homogenates were lysed with buffer RLT (Qiagen Sciences, Valencia, CA), and total RNA was extracted using the RNeasy Mini Kit from Qiagen (Qiagen Sciences) as described previously (29). Extracted RNA was used in real-time PCR (RT-PCR) assays performed with the CFX384 Touch Real-Time PCR detection system (Bio-Rad, Hercules, CA). The relative quantities of mRNA for several genes were determined using iTaq universal SYBR green supermix (Bio-Rad) or TaqMan gene expression master mix (Applied Biosystems, Life Technologies, Grand Island, NY). Target gene transcripts in each sample were normalized to hypoxanthine guanine phosphoribosyltransferase, 18s rRNA, or b-actin and expressed as a relative increase or decrease compared with control. Real-time primer sequences used for mouse collagens were as follows: type I, α1 (Col1α1) 5′-AGACATGTTCAGCTTTGTGGAC (forward), 5′-GCAGCTGACTTCAGGGATG (reverse); type III, α1 (Col3α1) 5′-TCCCCTGGAATCTGTGAATC (forward), 5′-TGAGTCGAATTGGGGAGAAT (reverse); type V, α1 (Col5α1) 5′-CTACATCCGTGCCCTGGT (forward), 5′-CCAGCACCGTCTTCTGGTAG (reverse).
Measurements of subpleural thickness, pulmonary mechanics, and total lung collagen.
The subpleural thickness was measured by histomorphometric measurement of Mason trichrome-stained lung sections using the measured distance function of MetaMorph as previously described (31). Three random lung areas are photographed per mouse using a Leica DM2700 M brightfield microscope (Leica Microsystems, Buffalo Grove, IL). High-magnification images (X20) were captured with a 3CCD color video camera and analyzed using MetaMorph imaging software (version 6.2; Molecular Devices, Sunnyvale, CA). Ten subpleural measurements per photograph are then taken for a total of 30 measurements/mouse. Lung mechanics were assessed on mice with a computerized Flexi Vent system (SCIREQ, Montreal, Canada), as previously described (19, 43). Total lung collagen was determined by measuring hydroxyproline levels as previously described (26, 31).
Statistics.
All data were analyzed with Prism (version 5; GraphPad). Unpaired t-test and one-way ANOVA with Tukey's Multiple Comparison posttest are used to compare different experimental groups, and data were considered statistically significant for P values <0.05. A Fisher's exact test was used to compare the difference in proportion of death rates in experimental groups.
RESULTS
Activation of S6K in the lung cells during TGF-α-induced pulmonary fibrosis.
We previously demonstrated TGF-α expression increases phosphorylation of S6K as measured by Western blot (23). To investigate the distribution of phosphorylated S6K (P-S6K) in the fibrotic lesions of the lung, immunohistochemistry and immunofluorescence staining with anti-S6K antibody was performed on lung sections from CCSP/TGF-α transgenic mice after 4 days on Dox when fibrosis is developing, and at 4 wk when fibrosis is established. At 4 days of Dox intense P-S6K immunostaining is detected primarily in airway epithelial cells, and the early adventitial fibrotic regions with infrequent staining in alveolar epithelial cells and the subpleural region (Fig. 1, A and B). By 4 wk on Dox airway epithelial staining persists although less intense when compared with 4 days of Dox. Strong immunohistochemical staining for P-S6K was detected in the fibrotic subpleural regions, with minimal staining in the peribronchial and perivascular adventitial regions (Fig. 1, A and B). Immunofluorescence for P-S6K following 4 wk of Dox demonstrates minimal staining in CCSP/− controls and confirms strong staining in the fibrotic lesions of the lung pleura of CCSP/TGF-α transgenic mice (Fig. 1C) with minimal staining in the adventitial fibrotic regions (data not shown). Colocalization of P-S6K with α-SMA and vimentin was detected in the subpleural fibrotic regions (Fig. 1D). Colocalization of Ki-67 with α-SMA was not identified (data not shown), supporting our previous studies that proliferating cells in fibrotic subpleural regions following TGF-α overexpression are predominantly fibroblasts (17, 28) and similar to in vitro studies that demonstrate TGF-β-mediated myofibroblast differentiation and proliferation of the NRK fibroblasts are mutually exclusive cellular responses to TGF-β (9). Together, these studies support a strong activation of P-S6K in the airway epithelial cells, with selective activation in the mesenchymal cells in the subpleural fibrotic regions in the advanced fibrotic lesions.
Fig. 1.

Phosphorylated p70 ribosomal S6 kinase (S6K) expression in the lung following transforming growth factor (TGF)-α overexpression. Clara cell secretory protein (CCSP)/TGF-α mice were administered doxycycline (Dox) to induce TGF-α expression. Immunostaining for phosphorylated S6 was performed on the lung sections of TGF-α mice administered Dox for 4 days or 4 wk comparing the subpleural compartment (A) with the airway/adventitial area (B). Insets demonstrate higher magnification of cells shown on bottom. C: immunofluorescence for phosphorylated S6 (red) on the 4-wk sections in the parenchyma of CCSP/− controls and CCSP/TGF-α mice. Insets demonstrate higher magnification of cells along the subpleural surface. D: immunofluorescence for phosphorylated S6 (red), α-smooth muscle actin (SMA), or vimentin (green) and colocalization (white) on the subpleural fibrotic regions for CCSP/TGF-α mice following 4 wk of Dox reveal colocalization of pS6 with mesenchymal cells. Staining is representative of individual sections from 4–8 mice/group.
LY-2584702 selectively inhibits TGF-α-induced phosphorylation of S6K and proliferation in fibroblasts.
To determine if LY-2584702 selectively inhibits S6K activation in fibroblasts, primary lung fibroblasts were isolated from FVB/N CCSP/− mice and stimulated with recombinant human TGF-α and PDGF-β. Cell signaling intermediates were then measured from fibroblast cell lysates without and with incubation of the S6K pharmacological inhibitor LY-2584702. TGF-α and PDGF-β added [alone (data not shown) or in combination] to fibroblasts directly induced phosphorylation of S6 and the PI3K pathway, evident by phosphorylation of Akt (Fig. 2A). Incubation of fibroblasts with LY-2584702 selectively prevented phosphorylation of S6 in a dose-dependent pattern while not effecting phosphorylation of Akt. LY-2584702 did not affect baseline proliferation of fibroblasts, but prevented TGF-α- and PDGFβ-stimulated proliferation (Fig. 2B).
Fig. 2.
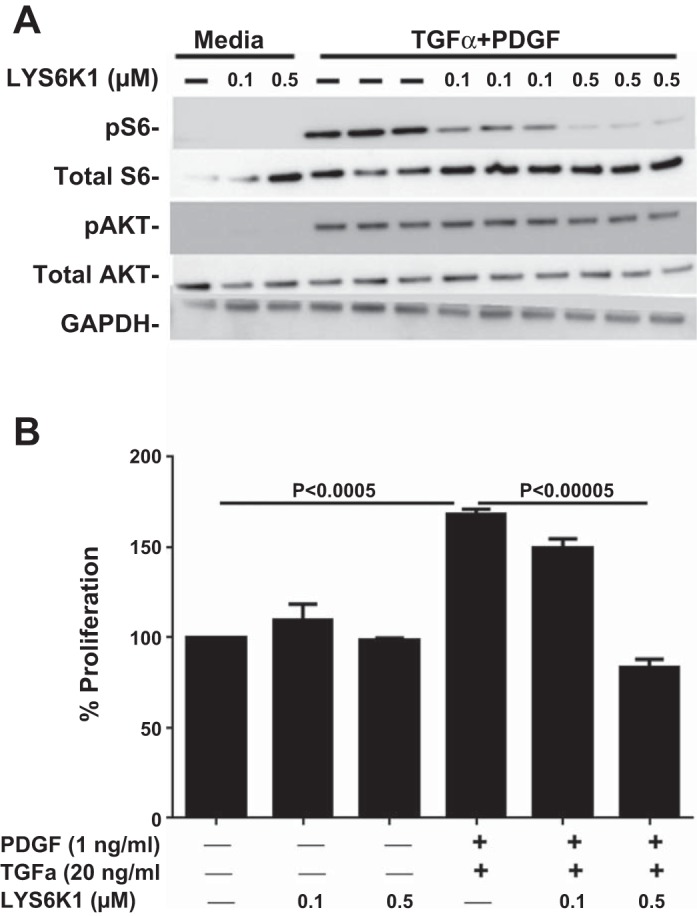
LY-2584702 selectively inhibits phosphorylation of S6 and proliferation of primary mouse lung fibroblasts. Primary mouse lung fibroblasts were isolated and stimulated with TGF-α and platelet-derived growth factor (PDGF)-β. A: Western blot analysis of phosphorylated S6 from fibroblasts treated without and with LY-2584702. B: proliferation assay for fibroblasts treated without and with LY-2584702. Data are means ± SE, and statistical differences shown were performed using ANOVA. Experiments were repeated in duplicate.
LY-2584702 prevents TGF-α-induced phosphorylation of S6K and lung cell proliferation but does not alter collagen gene transcription in vivo.
CCSP/TGF-α mice were administered Dox for 2 days to induce TGF-α expression. Phosphorylated S6 (Ser235/236), as measured by Western blot analysis, more than doubled compared with Dox-treated CCSP/− control mice. LY-2584702 treatment at doses of 25 or 50 mg/kg two times per day prevented increases in P-S6 in lung homogenates of CCSP/TGF-α mice (Fig. 3, A and B). To determine if modulation of S6 phosphorylation is associated with physiological changes of proliferation, we measured total cell proliferation with Ki-67 staining in paraffin lung sections of CCSP/TGF-α mice. Following 2 days of Dox administration, proliferation more than doubled in CCSP/TGF-α mice treated with vehicle compared with controls. Treatment of CCSP/TGF-α mice with 25 or 50 mg/kg of LY-2584702 two times per day prevented TGF-α-induced proliferation (Fig. 3C). Ki-67 staining cells morphologically were airway epithelial cells and mesenchymal cells (data not shown), consistent with previous observations in the CCSP/TGF-α model (16, 17, 28). TGF-α induction for 2 days increased the expression of mouse lung collagen transcripts in CCSP/TGF-α mice treated with vehicle compared with controls (2.1-fold for Col1α1, 1.8-fold for Col3α1, and 1.5-fold for Col5α1), but neither dose of LY-2584702 treatment prevented the increases in these collagen transcript levels (data not shown). Together these studies demonstrate that LY-2584702 prevented TGF-α-induced phosphorylation of S6 and cellular proliferation in vivo. Because both doses of LY-2584702 reduced parameters to control levels, consequential in vivo studies were performed using 25 mg/kg two times a day.
Fig. 3.
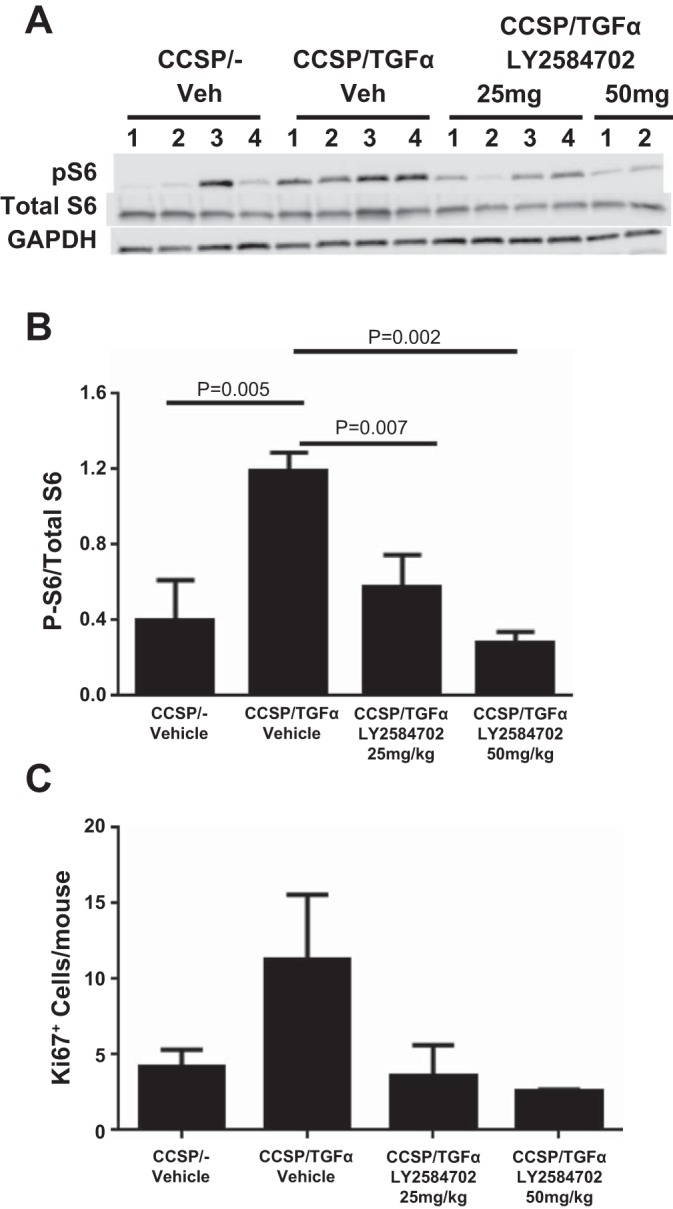
LY-2584702 inhibits phosphorylation of S6 and prevents total lung cell proliferation but does not inhibit increased collagen matrix transcription in vivo. CCSP/TGF-α mice were administered Dox for 2 days to induce TGF-α expression. A: Western blot analysis of phosphorylated S6 from lung homogenates of mice treated with vehicle or LY-2584702. Mice were treated with LY-2584702 at doses of 25 or 50 mg/kg, 2 times/day. B: quantification of ratio of the intensity of phosphorylated S6 over total S6. C: total lung cell proliferation measured by Ki-67 staining in paraffin lung sections of mice treated with vehicle or LY-2584702. Data are means ± SE, and statistical differences shown were performed using an unpaired t-test.
LY-2584702 prevents TGF-α-induced pulmonary fibrosis.
To determine if S6K inhibition prevented the physiological consequences of progressive lung fibrosis, CCSP/TGF-α mice were administered Dox to induce TGF-α expression and concomitantly treated with either vehicle or LY-2584702 (25 mg/kg) two times per day for 4 wk, and fibrosis endpoints were compared with CCSP/− nonfibrotic controls. Induction of TGF-α caused extensive subpleural, perivascular, and peribronchial fibrosis on lung histology. CCSP/TGF-α mice treated with LY-2584702 display a significant decrease in the subpleural fibrosis compared with vehicle-treated CCSP/TGF-α mice (Fig. 4A). However, LY-2584702 treatment had only a minimal effect on the development of fibrotic lesions in the perivascular and peribronchial regions of the lung. Morphometric measurement of subpleural thickness confirmed near complete inhibition of increased subpleural thickness in CCSP/TGF-α mice on Dox treated with LY-2584702 (Fig. 4B). Total lung collagen levels as determined by hydroxyproline were significantly reduced in LY-2584702-treated CCSP/TGF-α mice but remained significantly elevated compared with nonfibrotic controls (Fig. 4C). In agreement with the 2-day prevention study, TGF-α expression for 4 wk increased the expression of mouse lung collagen transcripts in CCSP/TGF-α mice treated with vehicle compared with controls (6.1-fold for Col1α1, 5.4-fold for Col3α1, and 5.2-fold for Col5α1), but LY-2584702 treatment did not prevent the increases in these collagen transcript levels (data not shown), consistent with a posttranscriptional role of the S6K pathway in collagen deposition. Increases in airway elastance were significantly prevented in CCSP/TGF-α mice treated with LY-2584702 with no statistical changes in elastance and lung compliance (Fig. 5). Together these studies demonstrate that S6K inhibition administered at the time of TGF-α overexpression prevented collagen accumulation primarily in the lung pleura along with the accompanying selected alterations in lung mechanics.
Fig. 4.

LY-2584702 prevents TGF-α-induced pulmonary fibrosis. CCSP/TGF-α mice were administered Dox for 4 wk to induce pulmonary fibrosis and treated with vehicle or LY-2584702 (25 mg/kg, 2 times/day). A: representative photomicrographs of lung tissues stained with Masson trichrome staining. Panel on top centers on the peribronchial and perivascular areas and the panel on bottom the subpleural regions of the lung. B: morphometric quantification of the subpleural thickness measured from lung sections. C: total lung collagen of right lung measured with hydroxyproline. Data are means ± SE (n = 8–11 mice in each group), and statistical differences shown were performed using ANOVA.
Fig. 5.

LY-2584702 prevents TGF-α-induced changes in lung mechanics. CCSP/TGF-α mice were administered Dox for 4 wk to induce pulmonary fibrosis and airway resistance, airway elastance, and lung compliance measured in mice treated with vehicle or LY-2584702. Data are means ± SE (n = 8–11 mice for each group), and statistical differences shown were performed using an unpaired t-test.
LY-2584702 inhibition attenuates the progression of established subpleural fibrosis.
To determine whether S6K inhibition influences the progression of established fibrosis, following 4 wk of Dox treatment when fibrosis is already manifest, CCSP/TGF-α mice were treated with LY-2584702 (25 mg/kg, 2 times/day) while remaining on Dox for an additional 4 wk (8 wk total). Controls included CCSP/− and CCSP/TGF-α mice treated with vehicle while remaining on Dox an additional 4 wk. No deaths occurred in the 10 CCSP/− controls during the 8-wk study. For the CCSP/TGF-α mice, 5 out of 22 mice treated with vehicle died, whereas 7 of 22 mice treated with LY-2584702 died (Fig. 6A). The death rates in CCSP/TGF-α mice were not statistically significant between groups. Body weights of CCSP/TGF-α mice treated with either vehicle or LY-2584702 decreased significantly from CCSP/− vehicle controls beginning at week 5 and decreased almost 25% from baseline following 8 wk of Dox (Fig. 6A). There were no statistical differences in body weights at any point between vehicle- or LY-2584702-treated CCSP/TGF-α mice.
Fig. 6.

LY-2584702 inhibition as a rescue therapy minimally alters the progression of established pulmonary fibrosis. CCSP/TGF-α mice were administered Dox for 8 wk; 5 wk into Dox, mice were treated with either vehicle or LY-2584702 (25 mg/kg, 2 times/day). A: changes in body weight of CCSP/− controls and CCSP/TGF-α mice treated with either vehicle or LY-2584702. “X” represent mouse deaths in CCSP/TGF-α mice treated with either vehicle (red) or LY-2584702 (green). B: representative low-power photomicrographs of lung tissues stained with Masson trichrome staining. C: morphometric quantification of the subpleural thickness measured from lung sections. D: total lung collagen of the right lung measured with hydroxyproline. Data are means ± SE (n = 8–22 mice in each group), and statistical differences shown were performed using ANOVA.
CCSP/TGF-α mice treated with Dox and vehicle for 8 wk demonstrated marked subpleural thickening with advanced perivascular and peribronchial fibrosis affecting large and small vessels and airways (Fig. 6B). CCSP/TGF-α mice treated with LY-2584702 demonstrated significantly reduced subpleural fibrosis as quantified with morphometry compared with vehicle-treated mice. However, LY-2584702 treatment had no effect on perivascular and peribronchial fibrosis, similar to vehicle-treated mice (Fig. 6, B and C). Total lung collagen was increased fourfold in CCSP/TGF-α mice treated with vehicle and was unchanged by LY-2584702 treatment (Fig. 6D). Lung mechanics were unaltered in CCSP/TGF-α mice treated with LY-2584702 compared with CCSP/TGF-α mice treated with vehicle (Fig. 7).
Fig. 7.

LY-2584702 inhibition as a rescue therapy does not alter changes in lung mechanics. CCSP/TGF-α mice were administered Dox for 8 wk; 5 wk into Dox, mice were treated with either vehicle or LY-2584702, and airway resistance, airway elastance, and lung compliance were measured. Data are means ± SE (n = 8–17 mice for each group), and statistical differences shown were performed using ANOVA.
Because LY-2584702 treatment was only minimally effective in altering the progression of fibrosis as a rescue therapy, we measured phosphorylation of signaling intermediates from other fibrogenic pathways to determine if LY-2584702 treatment altered their activation. We did not detect differences in Akt or Erk phosphorylation patterns on Western blot of lung homogenates between vehicle- and LY-2584702-treated CCSP/TGF-α mice (data not shown), suggesting the PI3K or MAPK pathways were not altered following LY-2584702 treatment. Overexpression of TGF-α alone had no significant effect on phosphorylation of 4E-BP1, a direct substrate of mTORC1. However, there was a significant increase of 4E-BP1 phosphorylation in the lung homogenates of CCSP/TGF-α mice treated with LY-2584702 compared with vehicle-treated mice (Fig. 8A). In addition, LY-2584702-treated mice demonstrated increased phosphorylation of S6K1 at Thr389 (Fig. 8B). Together, these findings suggest LY-2584702 administered at doses effective to prevent phosphorylation of S6 at Ser235/236 may lead to a shunting toward increased activation of the 4E-BP1 pathway.
Fig. 8.

LY-2584702 inhibition increases phosphorylation of 4E-binding protein (4E-BP1) and p70S6K. CCSP/TGF-α mice were administered Dox for 2 days to induce TGF-α expression. A: Western blot analysis of lung homogenates from mice treated with vehicle or LY-2584702 (25 or 50 mg/kg, 2 times/day) for phosphorylated 4E-BP1 at the Thr37/46 sites (A) and p70S6K at the Thr389 site (B). Total p70S6K is the band on top (70 kDa). Right, quantification of ratio of the intensity of phosphorylated over total for each intermediate. Data are means ± SE, and statistical differences shown were performed using ANOVA (A) and an unpaired t-test (B).
DISCUSSION
Significant findings from this study include demonstration that epithelial overexpression of TGF-α in vivo resulted in increased phosphorylation of the S6 in the airway and alveolar epithelium as well in the mesenchyme of advanced subpleural fibrotic regions. LY-2584702 treatment in vivo administered at the initiation of TGF-α overexpression prevented the development of extensive subpleural fibrosis and increases in airway elastance and attenuated lung collagen accumulation. Together, these experiments demonstrate a direct role for the S6K pathway in mediating fibroproliferative pathological disease processes in the lung.
The known biological effects of S6K include a number cellular processes found in fibrosis, including protein synthesis, cell cycle progression and cellular growth, cytoskeleton organization, and cell proliferation (46). Specific targeted inhibition of the S6K with LY-2584702 prevented both TGF-α/PDGFβ-induced proliferation of lung fibroblasts in vitro and TGF-α-driven proliferation in the CCSP/TGF-α model, further supporting the S6K as a key modulator in the proliferative drive during fibrogenesis. While LY-2584702 treatment prevented the physiological effects of 4 wk of TGF-α overexpression, RT-PCR for lung collagens from whole lung homogenates of CCSP/TGF-α mice were not reduced in LY-2584702-treated mice. Previous studies demonstrate that the S6K pathway regulates protein translation (32), and these findings support a posttranscriptional regulation of collagen deposition through the S6K pathway in the CCSP/TGF-α lung.
Immunohistochemistry for phosphorylated S6 demonstrated a direct correlation in vivo between the pulmonary regions of increased S6K phosphorylation and sensitivity to LY-2584702 treatment for inhibition of collagen deposition. Specifically, the lung pleura exhibited intense immunostaining for phosphorylated S6K and were responsive to LY-2584702 with almost complete inhibition of subpleural thickening following TGF-α overexpression. In contrast, the adventitial areas only rarely immunostained for phosphorylated S6K, and the degree of matrix deposition was unaffected by LY-2584702 treatment and likely accounts for the incomplete prevention of lung hydroxyproline increases in the prevention studies. This compartmentalization effect of S6K inhibition further supports the phenotypic and functional heterogeneity of mesenchymal cells in the lung. Evidence of lung fibroblast heterogeneity has been well described with regard to morphology, proliferative capacity, surface marker expression, collagen type and production, intracellular metabolic pathways, and cytokine production (5, 7, 38). In neonatal calves the pulmonary artery adventitia consists of multiple fibroblast subpopulations where only selective subpopulations expand under chronic hypoxic conditions (5). In IPF, fibroblastic foci were found to be part of an interconnected matrix, but comprised of polyclonal fibroblasts (4). Together, these findings support the concept that persistent local pathological stimuli, such as overexpression of growth factors, leads to a selective activation whereby fibroblast subpopulations with distinct fibroproliferative capabilities emerge and contribute to the disease process (5). In the current study, overexpression of epithelial TGF-α caused the expansion of mesenchymal cells in both subpleural and adventitial regions, but likely through distinctive signaling pathways, thereby further supporting the heterogeneity of fibrotic lesions.
When LY-2584702 was administered to mice as a rescue therapy after fibrosis was already embedded, the degree of subpleural thickness was significantly reduced compared with vehicle-treated CCSP/TGF-α mice. The subpleural thickness measurements for the rescue-treated mice (69 ± 6 μm; Fig. 6C) were increased compared with measurements from vehicle-treated CCSP/TGF-α mice following 4 wk of Dox (51 ± 13 μm; Fig. 4B), but these differences were not statistically significant. Because the 4 wk of Dox values represent the approximate degree of subpleural fibrosis already present at the beginning of the rescue treatment, these comparisons suggest that S6K inhibition significantly attenuated the progression of the subpleural disease. The strong immunostaining for S6K phosphorylation in the pleura combined with the effective inhibition of S6K to prevent and diminish the progression of subpleural fibrosis in the CCSP/TGF-α model suggest there may be translational relevance for human lung diseases where there is a prominent subpleural component. In the TGF-α model we do not detect expansion of mesothelial cells as determined with calretinin staining (unpublished data), and our findings support further investigation of the role of the S6K pathway in plural fibrosis models.
In contrast to the effects on the lung pleura, LY-2584702 rescue therapy had no effect on the increases in total lung collagen, alterations in lung mechanics, or reduction in total body weight when compared with vehicle-treated CCSP/TGF-α mice following 8 wk of Dox. These findings differ from our previous study where mTORC1 inhibition with rapamycin administered to CCSP/TGF-α mice as a rescue therapy after 4 wk of Dox stabilized declining body weights, increases in total lung collagen, and alterations in lung mechanics (23). Explanations for why fibrosis progressed despite effective inhibition of S6K phosphorylation are premised upon the interaction and overlap of S6K with other regulatory processes. The S6K is known to serve as a negative regulator of Akt phosphorylation, and S6K inhibition could release feedback inhibition of the PI3K pathway as has been demonstrated in several neoplastic cell culture lines (45). However, we did not detect increased phosphorylation of Akt in either lung fibroblasts in vitro or in total lung homogenates in vivo by Western blot following LY-2584702 treatment, suggesting the PI3K pathway was not upregulated. Activation of the S6K is a complex process requiring multiple signaling inputs involving phosphorylation at several sites. Treatment of CCSP/TGF-α mice with LY-2584702 inhibited phosphorylation of the S6K substrate S6 yet also enhanced phosphorylation of S6K at the Thr389 site. The precise mechanisms for this seemingly paradoxical finding are not clear. However, another specific inhibitor of the S6K, PF-4708671, also led to a significant dose-dependent enhancement of S6K1 phosphorylation at Thr389 in vitro that was ablated by inactivation of mTORC1 (37). The results from this investigation suggest there is a feedback pathway whereby the S6K regulates its own phosphorylation via mTORC1. Our findings further support this hypothesis by demonstrating that inhibition of S6K in vivo can lead to increased phosphorylation of S6K at selected catalytic sites.
In addition to S6K, the mTORC1 complex also phosphorylates 4E-BP1, a protein important for cap-dependent translation. Overexpression of TGF-α in our model did not directly lead to detectable changes in 4E-BP phosphorylation; however, administration of LY-2584702 in vivo was associated with a dramatic increase in 4E-BP phosphorylation. Therefore, one possible mechanism for the ineffective reversal of fibrosis by LY-2584702 is the increased phosphorylation of S6K at Thr389 and a shunt toward increased activation the 4E-BP pathway. However, LY-2584702 treatment was largely effective in preventing the onset of fibrosis even while 4E-BP was already upregulated, thereby suggesting that there are other mechanisms or pathways driving fibroproliferation.
A more likely explanation for the ineffectiveness of LY-2584702 in reversing disease is that the S6K pathway is too narrow a singular target to effectively prevent progression of disease after fibrosis is extensive and rampant. Following persistent activation of an initial key fibroproliferative pathway, which is EGFR signaling in the CCSP/TGF-α model, there is likely amplification over time of multiple related pathways that also promote fibroproliferation to some extent. We have previously demonstrated that pharmacological inhibition of either the PI3K or MAPK signaling pathways alone in the CCSP/TGF-α model prevented the development of lung fibrosis, but were less effective in resolving ongoing disease when inhibitors were administered as a rescue treatment (25, 30). In contrast, combined inhibition of both PI3K and MAPK pathways provided significantly improved reversal of ongoing disease than inhibition of separate pathways (27). The superior resolution of this combined approach suggests that each pathway stimulates distinctive downstream fibrogenic intermediates. Because previous investigations have demonstrated that both PI3K and MAPK signaling intermediates can ultimately activate the S6K pathway, we hypothesized that the fibroproliferative cellular processes of both pathways could converge through the S6K and thereby provide a more selective target to reverse progressive disease. However, the incomplete resolution of LY-2584702 treatment compared with combined PI3K-MAPK treatment supports activation of non-S6K-mediated pathways stimulating fibroproliferation.
Recent clinical trials in patients with IPF further support the concept that multiple signaling pathways may mediate the maintenance and progression of fibrotic lesions. IPF patients randomized to receive the mTOR inhibitor everolimus unexpectedly demonstrated a more rapid disease progression and a trend toward an inferior survival benefit compared with placebo-treated patients (33). Similarly, surfactant protein C knockout mice (Sftpc−/−) treated with the mTOR inhibitor rapamycin before or after exposure to intratracheal bleomycin demonstrated decreased survival, worse lung function, increased expression of profibrotic Th2 cytokines, and reduced expression of INF-γ compared with Sftpc−/− mice not receiving rapamycin (29). The mechanisms for the worse outcomes with mTOR inhibition are not known but may be related to release of negative feedback loops or shunting from mTOR1 to other fibrogenic signaling pathways. In contrast, a recent trial in IPF patients randomized to receive nintedanib, an intracellular inhibitor that targets multiple tyrosine kinases, including the VEGF, FGF, and PDGF receptors, demonstrated significantly reduced decline in the forced vital capacity compared with placebo-treated patients (40, 41). Together these findings support the benefits of targeting multiple fibrogenic signaling pathways to treat advanced disease while single pathway inhibition may be ineffective in treating advanced fibrosis, and in some scenarios, could exacerbate disease.
Over the past decade a number of pharmacologically specific and effective signaling pathway inhibitors have emerged for treating human disease, primarily in oncology. Applying these successful treatments to target selective upregulated pathways in fibrotic lung disease is feasible. The current study confirms that the S6K pathway can be targeted pharmacologically in vivo to modify lung fibrosis, especially in the subpleural compartment of the lung. The findings from this study also highlight the challenges of reversing fibrosis when treatment is initiated after disease is extensive and progressive. The clinical heterogeneity of human fibrotic lung disease likely reflects the interpretation that multiple pathways mediate fibroproliferation, each of which may need to be targeted to ultimately successfully modify disease progression.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant P50-HL-107159 and a Cincinnati Children's Hospital Innovation Award (W. D. Hardie).
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: S.K.M., G.T., and W.D.H. conception and design of research; S.K.M., R.E., C.D., S.S., A.S., and W.D.H. performed experiments; S.K.M. and W.D.H. analyzed data; S.K.M. and W.D.H. interpreted results of experiments; S.K.M. and W.D.H. prepared figures; S.K.M. and W.D.H. drafted manuscript; S.K.M., G.T., R.E., C.D., S.S., A.S., and W.D.H. approved final version of manuscript; W.D.H. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank Dr. Sandaruwan Geeganage from Eli Lilly for assistance in procuring LY-2584702 and advice on solubilizing the drug and treating mice in vivo, Dr. Nanhua Zhang from pulmonary medicine at the Cincinnati Children's Hospital Medical Center (CCHMC) for statistical assistance in powering the study and statistical analysis of the endpoints, Dr. Steve Glasser from Pulmonary Biology at CCHMC for review of the manuscript, and the division of Veterinary Services at CCHMC for careful and expert care of the mice used in this study.
REFERENCES
- 1.Antoniou KM, Margaritopoulos GA, Soufla G, Symvoulakis E, Vassalou E, Lymbouridou R, Samara KD, Kappou D, Spandidos DA, Siafakas NM. Expression analysis of Akt and MAPK signaling pathways in lung tissue of patients with idiopathic pulmonary fibrosis (IPF). J Recept Signal Trans Res 30: 262–269, 2010. [DOI] [PubMed] [Google Scholar]
- 2.Cantley LC. The phosphoinositide 3-kinase pathway. Science 296: 1655–1657, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Chung EJ, Brown AP, Asano H, Mandler M, Burgan WE, Carter D, Camphausen K, Citrin D. In vitro and in vivo radiosensitization with AZD6244 (ARRY-142886), an inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2 kinase. Clin Cancer Res 15: 3050–3057, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cool CD, Groshong SD, Rai PR, Henson PM, Stewart JS, Brown KK. Fibroblast foci are not discrete sites of lung injury or repair: the fibroblast reticulum. Am J Respir Crit Care Med 174: 654–658, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das M, Dempsey EC, Reeves JT, Stenmark KR. Selective expansion of fibroblast subpopulations from pulmonary artery adventitia in response to hypoxia. Am J Physiol Lung Cell Mol Physiol 282: L976–L986, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Deng H, Hershenson MB, Lei J, Bitar KN, Fingar DC, Solway J, Bentley JK. p70 Ribosomal S6 kinase is required for airway smooth muscle cell size enlargement but not increased contractile protein expression. Am J Respir Cell Mol Biol 42: 744–752, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Derdak S, Penney DP, Keng P, Felch ME, Brown D, Phipps RP. Differential collagen and fibronectin production by Thy 1+ and Thy 1− lung fibroblast subpopulations. Am J Physiol Lung Cell Mol Physiol 263: L283–L290, 1992. [DOI] [PubMed] [Google Scholar]
- 8.Gerasimovskaya EV, Tucker DA, Stenmark KR. Activation of phosphatidylinositol 3-kinase, Akt, and mammalian target of rapamycin is necessary for hypoxia-induced pulmonary artery adventitial fibroblast proliferation. J Appl Physiol 98: 722–731, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Grotendorst GR, Rahmanie H, Duncan MR. Combinatorial signaling pathways determine fibroblast proliferation and myofibroblast differentiation. FASEB 18: 469–479, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Gui YS, Wang L, Tian X, Li X, Ma A, Zhou W, Zeng N, Zhang J, Cai B, Zhang H, Chen JY, Xu KF. mTOR overactivation and compromised autophagy in the pathogenesis of pulmonary fibrosis. PloS One 10: e0138625, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamada N, Kuwano K, Yamada M, Hagimoto N, Hiasa K, Egashira K, Nakashima N, Maeyama T, Yoshimi M, Nakanishi Y. Anti-vascular endothelial growth factor gene therapy attenuates lung injury and fibrosis in mice. J Immunol 175: 1224–1231, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Hardie WD, Davidson C, Ikegami M, Leikauf GD, Le Cras TD, Prestridge A, Whitsett JA, Korfhagen TR. EGF receptor tyrosine kinase inhibitors diminish transforming growth factor-alpha-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 294: L1217–L1225, 2008. [DOI] [PubMed] [Google Scholar]
- 13.Hardie WD, Glasser SW, Hagood JS. Emerging concepts in the pathogenesis of lung fibrosis. Am J Pathol 175: 3–16, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hardie WD, Korfhagen TR, Sartor MA, Prestridge A, Medvedovic M, Le Cras TD, Ikegami M, Wesselkamper SC, Davidson C, Dietsch M, Nichols W, Whitsett JA, Leikauf GD. Genomic profile of matrix and vasculature remodeling in TGF-α induced pulmonary fibrosis. Am J Respir Cell Mol Biol 37: 309–321, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardie WD, Korfhagen TR, Sartor MA, Prestridge A, Medvedovic M, Le Cras TD, Ikegami M, Wesselkamper SC, Davidson C, Dietsch M, Nichols W, Whitsett JA, Leikauf GD. Genomic profile of matrix and vasculature remodeling in TGF-alpha induced pulmonary fibrosis. Am J Respir Cell Mol Biol 37: 309–321, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hardie WD, Le Cras TD, Jiang K, Tichelaar JW, Azhar M, Korfhagen TR. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 286: L741–L749, 2004. [DOI] [PubMed] [Google Scholar]
- 17.Hardie WD, Piljan-Gentle A, Dunlavy MR, Ikegami M, Korfhagen TR. Dose-dependent lung remodeling in transgenic mice expressing transforming growth factor-alpha. Am J Physiol Lung Cell Mol Physiol 281: L1088–L1094, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Hartford CM, Ratain MJ. Rapamycin: something old, something new, sometimes borrowed and now renewed. Clin Pharmacol Ther 82: 381–388, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Hokuto I, Ikegami M, Yoshida M, Takeda K, Akira S, Perl AK, Hull WM, Wert SE, Whitsett JA. Stat-3 is required for pulmonary homeostasis during hyperoxia. J Clin Invest 113: 28–37, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hollebecque A, Houede N, Cohen EE, Massard C, Italiano A, Westwood P, Bumgardner W, Miller J, Brail LH, Benhadji KA, Soria JC. A phase Ib trial of LY2584702 tosylate, a p70 S6 inhibitor, in combination with erlotinib or everolimus in patients with solid tumours. Eur J Cancer 50: 876–884, 2014. [DOI] [PubMed] [Google Scholar]
- 21.Hoyle GW, Li J, Finkelstein JB, Eisenberg T, Liu JY, Lasky JA, Athas G, Morris GF, Brody AR. Emphysematous lesions, inflammation, and fibrosis in the lungs of transgenic mice overexpressing platelet-derived growth factor. Am J Pathol 154: 1763–1775, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inoue Y, King TE Jr, Barker E, Daniloff E, Newman LS. Basic fibroblast growth factor and its receptors in idiopathic pulmonary fibrosis and lymphangioleiomyomatosis. Am J Respir Crit Care Med 166: 765–773, 2002. [DOI] [PubMed] [Google Scholar]
- 23.Korfhagen TR, Le Cras TD, Davidson CR, Schmidt SM, Ikegami M, Whitsett JA, Hardie WD. Rapamycin prevents transforming growth factor-alpha-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 41: 562–572, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuwano K. Epithelial cell apoptosis and lung remodeling. Cell Mol Immunol 4: 419–429, 2007. [PubMed] [Google Scholar]
- 25.Le Cras TD, Korfhagen TR, Davidson C, Schmidt S, Fenchel M, Ikegami M, Whitsett JA, Hardie WD. Inhibition of PI3K by PX-866 prevents transforming growth factor-alpha-induced pulmonary fibrosis. Am J Pathol 176: 679–686, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madala SK, Edukulla R, Davis KR, Schmidt S, Davidson C, Kitzmiller JA, Hardie WD, Korfhagen TR. Resistin-like molecule alpha1 (Fizz1) recruits lung dendritic cells without causing pulmonary fibrosis. Respir Res 13: 51, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madala SK, Edukulla R, Phatak M, Schmidt S, Davidson C, Acciani TH, Korfhagen TR, Medvedovic M, Lecras TD, Wagner K, Hardie WD. Dual targeting of MEK and PI3K pathways attenuates established and progressive pulmonary fibrosis. PloS One 9: e86536, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madala SK, Edukulla R, Schmidt S, Davidson C, Ikegami M, Hardie WD. Bone marrow-derived stromal cells are invasive and hyperproliferative and alter transforming growth factor-alpha-induced pulmonary fibrosis. Am J Respir Cell Mol Biol 50: 777–786, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madala SK, Maxfield MD, Davidson CR, Schmidt SM, Garry D, Ikegami M, Hardie WD, Glasser SW. Rapamycin regulates bleomycin-induced lung damage in SP-C-deficient mice. Pulmo Med 2011: 653524, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Madala SK, Schmidt S, Davidson C, Ikegami M, Wert S, Hardie WD. MEK-ERK pathway modulation ameliorates pulmonary fibrosis associated with epidermal growth factor receptor activation. Am J Respir Cell Mol Biol 46: 380–388, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Madala SK, Schmidt S, Davidson C, Ikegami M, Wert S, Hardie WD. MEK-ERK pathway modulation ameliorates pulmonary fibrosis associated with epidermal growth factor receptor activation. Am J Respir Cell Mol Biol 46: 380–388, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J 441: 1–21, 2012. [DOI] [PubMed] [Google Scholar]
- 33.Malouf MA, Hopkins P, Snell G, Glanville AR. An investigator-driven study of everolimus in surgical lung biopsy confirmed idiopathic pulmonary fibrosis. Respirology 16: 776–783, 2011. [DOI] [PubMed] [Google Scholar]
- 34.McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Dorfman AL, Longnus S, Pende M, Martin KA, Blenis J, Thomas G, Izumo S. Deletion of ribosomal S6 kinases does not attenuate pathological, physiological, or insulin-like growth factor 1 receptor-phosphoinositide 3-kinase-induced cardiac hypertrophy. Mol Cell Biol 24: 6231–6240, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moore BB, Kolodsick JE, Thannickal VJ, Cooke K, Moore TA, Hogaboam C, Wilke CA, Toews GB. CCR2-mediated recruitment of fibrocytes to the alveolar space after fibrotic injury. Am J Pathol 166: 675–684, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, Rosas IO, Morse D. Autophagy in idiopathic pulmonary fibrosis. PloS One 7: e41394, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pearce LR, Alton GR, Richter DT, Kath JC, Lingardo L, Chapman J, Hwang C, Alessi DR. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem J 431: 245–255, 2010. [DOI] [PubMed] [Google Scholar]
- 38.Phipps RP, Penney DP, Keng P, Quill H, Paxhia A, Derdak S, Felch ME. Characterization of two major populations of lung fibroblasts: distinguishing morphology and discordant display of Thy 1 and class II MHC. Am J Respir Cell Mol Biol 1: 65–74, 1989. [DOI] [PubMed] [Google Scholar]
- 39.Pinzani M. PDGF and signal transduction in hepatic stellate cells. Front Biosci 7: d1720–d1726, 2002. [DOI] [PubMed] [Google Scholar]
- 40.Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, Thomas G. Phosphorylation and activation of p70s6k by PDK1. Science 279: 707–710, 1998. [DOI] [PubMed] [Google Scholar]
- 41.Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker-Herceg R, Disse B, Collard HR. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370: 2071–2082, 2014. [DOI] [PubMed] [Google Scholar]
- 42.Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N, Blenis J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem 282: 14056–14064, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schuessler TF, Bates JH. A computer-controlled research ventilator for small animals: design and evaluation. IEEE Trans Biomed Eng 42: 860–866, 1995. [DOI] [PubMed] [Google Scholar]
- 44.Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J 17: 6649–6659, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith J, Rozengurt E. Different patterns of Akt and ERK feedback activation in response to rapamycin, active-site mTOR inhibitors and metformin in pancreatic cancer cells. PloS One 8: e57289, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tavares MR, Pavan IC, Amaral CL, Meneguello L, Luchessi AD, Simabuco FM. The S6K protein family in health and disease. Life Sci 131: 1–10, 2015. [DOI] [PubMed] [Google Scholar]
- 47.Tichelaar JW, Lu W, Whitsett JA. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J Biol Chem 275: 11858–11864, 2000. [DOI] [PubMed] [Google Scholar]
- 48.Tolcher A, Goldman J, Patnaik A, Papadopoulos KP, Westwood P, Kelly CS, Bumgardner W, Sams L, Geeganage S, Wang T, Capen AR, Huang J, Joseph S, Miller J, Benhadji KA, Brail LH, Rosen LS. A phase I trial of LY2584702 tosylate, a p70 S6 kinase inhibitor, in patients with advanced solid tumours. Eur J Cancer 50: 867–875, 2014. [DOI] [PubMed] [Google Scholar]
- 49.White ES, Thannickal VJ, Carskadon SL, Dickie EG, Livant DL, Markwart S, Toews GB, Arenberg DA. Integrin alpha4beta1 regulates migration across basement membranes by lung fibroblasts: a role for phosphatase and tensin homologue deleted on chromosome 10. Am J Respir Crit Care Med 168: 436–442, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xia H, Diebold D, Nho R, Perlman D, Kleidon J, Kahm J, Avdulov S, Peterson M, Nerva J, Bitterman P, Henke C. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med 205: 1659–1672, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]