

Abstract
We used mRNA tagging to identify genes expressed in the intestine of C. elegans. Animals expressing an epitope-tagged protein that binds the poly-A tail of mRNAs (FLAG::PAB-1) from an intestine-specific promoter (ges-1) were used to immunoprecipitate FLAG::PAB-1/mRNA complexes from the intestine. A total of 1938 intestine-expressed genes (P<0.001) were identified using DNA microarrays. First, we compared the intestine-expressed genes with those expressed in the muscle and germline, and identified 510 genes enriched in all three tissues and 624 intestine-, 230 muscle- and 1135 germ line-enriched genes. Second, we showed that the 1938 intestine-expressed genes were physically clustered on the chromosomes, suggesting that the order of genes in the genome is influenced by the effect of chromatin domains on gene expression. Furthermore, the commonly expressed genes showed more chromosomal clustering than the tissue-enriched genes, suggesting that chromatin domains may influence housekeeping genes more than tissue-specific genes. Third, in order to gain further insight into the regulation of intestinal gene expression, we searched for regulatory motifs. This analysis found that the promoters of the intestine genes were enriched for the GATA transcription factor consensus binding sequence. We experimentally verified these results by showing that the GATA motif is required in cis and that GATA transcription factors are required in trans for expression of these intestinal genes.
Keywords: C. elegans, Intestine, Gene expression, GATA transcriptional regulation, Chromosomal clustering
INTRODUCTION
C. elegans is an excellent model organism with which to study development at the systems level using functional genomics. First, the simple anatomy of C. elegans is extremely well characterized and consists of five main tissues: neurons, muscle, skin, a digestive tract and a germline. The body plan is known at the level of single cells and the complete lineage of the 959 somatic cells has been shown to be essentially invariant (Sulston and Horvitz, 1977; Sulston et al., 1983). Second, there are exceptionally good genomic resources with which to study development at the systems level. The complete genome sequence of C. elegans has been determined, DNA microarrays and an RNAi library containing nearly every gene in the genome are readily available (Jiang et al., 2001; Kamath and Ahringer, 2003), and protein interactions have been studied on a large scale (Li et al., 2004).
In order to refine our understanding of development from cellular to molecular resolution, our aim is to define most or all of the genes expressed in each of the major tissue types. A global developmental profile of gene expression in C. elegans will elucidate the genes expressed in specific tissues and in all tissues, expand our understanding of tissue differentiation, and lead to insights in regulation of tissue-specific gene expression.
The small size of C. elegans (1 mm in length) makes it impractical to measure gene expression directly by dissecting tissues. One approach used to identify genes expressed in a cell lineage or tissue is mRNA tagging (Roy et al., 2002). Genes expressed in the body wall muscle were identified by expressing an epitope-tagged protein that binds poly-A tails on messenger RNA (poly-A binding protein or PAB-1) from the muscle-specific promoter for the gene myo-3. Epitope-tagged PAB-1 in the muscle was crosslinked to mRNA in that tissue, the PAB-1/mRNA complexes were enriched by immunoprecipitation with an antibody to the epitope, and DNA microarrays were used to identify 1354 muscle-expressed genes (Roy et al., 2002).
In order to extend tissue profiling in C. elegans, we have employed mRNA tagging to identify genes expressed in the intestine. C. elegans is a filter feeder with a digestive system composed of three main parts: pharynx, intestine and rectum. The pharynx concentrates and processes food before passing it to the intestine. The intestine is a tube that twists 180° along its length and is composed of twenty epithelial cells with a layer of microvilli that surround a lumen (White, 1988). The 14 posterior-most intestinal cells undergo nuclear division at the beginning of the L1 larval stage and become binucleate. All 20 cells undergo endoreduplications of their DNA at each larval stage, making the adult intestinal nuclei 32-ploid (Hedgecock and White, 1985). The intestine secretes digestive enzymes into the lumen, absorbs processed nutrients, functions as a storage organ with granules packed with lipids, proteins or carbohydrates, and nurtures germ cells by producing yolk proteins that are transported to the oocytes (Kimble and Sharrock, 1983). The third part of the digestive tract, the rectum, is composed of endothelial and muscle cells.
The regulatory network of transcription factors that direct the differentiation of the intestine has been studied in detail. The P1 cell differentiates into the EMS and P2 cells by SKN-1-dependent activation of med-1 and med-2 in the EMS cell and PIE-1-dependent blocking of SKN-1 in the P2 cell (Maduro et al., 2001). EMS divides into the E and MS cells. end-1 and end-3 are the direct targets of MED-1 and MED-2, and are consequently expressed in the E cell (Maduro et al., 2001). END-1 and END-3 induce the expression of elt-2 and elt-7, leading to the activation of downstream targets that differentiate the E cell into the 20 intestinal cells (Fukushige et al., 1998; Maduro and Rothman, 2002). elt-2 and elt-7 expression is maintained into adulthood by an autoregulatory loop, propagating intestinal cell identity (Maduro and Rothman, 2002).
The organogenesis of the C. elegans intestine has been detailed at the cellular level (Leung et al., 1999). It includes cytoplasmic polarization of cells in the intestinal primordium, intercalation of specific sets of cells, generation of an extracellular cavity within the primordium, and adherens junction formation. The adherens junctions present an ideal model with which to investigate epithelial cell polarity and several proteins involved in the process have been identified such as PAR-3, PAR-6, PKC-3, SMA-1, ERM-1, LET-413, DLG-1, AJM-1 and others (Knust and Bossinger, 2002). A molecular profile of the intestine would help to identify more genes involved in cell polarity and its development.
By generating a profile of gene expression in the C. elegans intestine, we have identified the molecules that define intestinal function. The list of intestine-expressed genes includes genes of known and unknown function. The genes with known functions provide insight into mechanisms and pathways used in diverse intestinal functions, such as epithelial cell polarity, digestion, and resistance to pathogens and toxicity. The intestinal expression of genes with previously unknown function implies a role in intestinal processes.
A genome-wide profile of intestinal gene expression can also be used to elucidate the regulatory networks that maintain intestinal differentiation. We have defined intestine-specific target genes and transcription factors. We searched for DNA sequence motifs enriched in the promoters of the intestine-enriched genes that might function as cis-acting regulatory motifs. This analysis allowed us to generate a first draft of the intestinal regulatory network by linking intestinal transcription factors to their targets via DNA motifs in their promoters.
In addition to identifying muscle-expressed genes, Roy et al. (Roy et al., 2002) were able to show that these genes are positionally clustered on the chromosomes. We have shown that intestine-expressed genes are also located in chromosomal clusters. Interestingly, we found a strong bias for chromosomal clustering in the housekeeping rather than the intestine-enriched genes, suggesting a role of chromatin organization in the regulation of gene expression.
MATERIALS AND METHODS
Molecular biology
Plasmid pPJCSK1 containing Pges-1::FLAG::PAB-1 was constructed (contact authors for details). A transgenic strain expressing Pges-1::FLAG::PAB-1 was made by microinjecting (Mello and Fire, 1995) and integrating the plasmid using γ-irradiation (3300 rads) resulting in strain SD1084.
mRNA tagging
The mRNA-tagging protocol was carried out as described by Roy et al. (Roy et al., 2002) with modifications (contact authors for details). RNA was linearly amplified as previously described (Wang et al., 2000). The DNA microarrays, probe preparation and microarray hybridizations were carried out as previously described (Roy et al., 2002).
Data analysis
All raw data can be found and downloaded from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo, GSE2626) or the Stanford Microarray Database (http://genome-www.stanford.edu/microarray). To identify genes that are significantly enriched by mRNA tagging, we first normalized the total amount of Cy3 and Cy5 signal to each other in each hybridization. We measured the ratio of the signals from the co-immunoprecipitated mRNA (Cy5) to total RNA in the cell extract (Cy3), and calculated the percentile rank for each gene relative to all genes in each hybridization. The mean percentile rank was determined from eight repeats of the mRNA-tagging experiment. Student’s t-test was used to determine which genes showed a mean enrichment significantly greater than the median enrichment for all genes (P<0.001).
To generate tissue-specific and common gene lists, we used the P values from Student’s t-tests used to calculate significant enrichment in muscle, intestine, and germline. The P values for muscle-expressed genes were calculated as described by Roy et al. (Roy et al., 2002) and as described above for intestine-expressed genes. The P values for germ line-enriched genes were calculated from a Student’s t-test between log ratios for four repeats of wild-type and glp-4 animals at both the L4 and adult stages as described by Reinke et al. (Reinke et al., 2004). Commonly-expressed genes had a P value of less than 0.05 in the muscle, intestine and germline DNA microarray experiments. Tissue-enriched genes had a P value of less than 0.01 in the DNA microarray experiment involving the tissue of interest and greater than 0.5 in the experiments concerning the other two tissues. Analysis of chromosomal clustering was carried out as described by Roy et al. (Roy et al., 2002).
Motif search
The CompareProspector algorithm was used to search for regulatory motifs as described by Liu et al. (Liu et al., 2004). The Pearson correlation coefficient was calculated for every pair of genes in expression data from 979 C. elegans DNA microarray experiments (Stuart et al., 2003).
Construction of GFP reporters
The promoter::GFP constructs for gst-42, elo-6, D2030.5, ZK970.2, C25E10.8 and B0218.8 were obtained from D. Dupuy (Dupuy et al., 2004). Transgenic strains expressing GFP from the promoter of each gene were made by microinjecting pha-1(e2123) animals with promoter::GFP (50 g/μg) and pha-1(+) (pC1, 100 g/μl, a gift from A. Fire), generating an extrachromosomal array. The resulting strains for each gene were SD1245 and SD1246 for D2030.5; SD1144 for elo-6; SD1145, SD1242 and SD1243 for gst-42; SD1149 for ZK970.2; SD1147 for C25E10.8; and SD1146 for B0218.8.
Site-directed mutagenesis
One TGATAA site in the promoters of D2030.5, gst-42, and elo-6 was changed to GGTACC, a KpnI restriction site used as a diagnostic for mutagenesis, and confirmed by sequencing (contact authors for details). The mutated promoter::GFP constructs were used to generate transgenic strains as previously described. Strains with extrachromosomal arrays expressing GFP from the mutated promoters were generated for elo-6 (SD1228, SD1229 and SD1230), D2030.5 (SD1159, SD1160 and SD1161) and gst-42 (SD1162 and SD1163).
GATA transcription factor RNAi
NGM agar with 1 mM IPTG and 25 μg / μL carbenicillin was seeded with bacteria expressing dsRNA for each targeted gene (Kamath and Ahringer, 2003), as well as a negative control with bacteria expressing empty vector. Twenty L4 animals were picked onto each plate, transferred to a fresh RNAi plate after 2 days, and analyzed for GFP expression after 1 day. Four promoter::GFP lines were treated with RNAi to unc-22 as a negative control and all four showed no significant change in GFP expression.
Imaging and quantification of GFP expression for mutagenesis and RNAi
Twenty animals for each strain were analyzed for GFP expression using a Zeiss Axioplan microscope equipped with a CCD camera. Comparison of all images was carried out on the same day with the same microscope settings. The color images were converted to 8-bit images using ImageJ software (Rasband, 2004) and the measure tool was used to measure pixel intensity of each worm.
RESULTS
Intestine-enriched genes identified by mRNA tagging
To gain insights into the regulatory networks and the underlying cellular pathways that define intestinal functions, we used mRNA tagging to perform a genome-wide scan for genes expressed in this tissue. We generated a strain that expresses FLAG-tagged poly Abinding protein from the intestine-specific promoter for gut esterase-1 (ges-1). We verified that Pges-1::FLAG::PAB-1 is expressed specifically in the intestine by visualizing its expression with immunohistochemistry using anti-FLAG antibody conjugated to alkaline phosphatase (Fig. 1).
Fig. 1. ges-1p::FLAG::PAB-1 is expressed exclusively in the intestine.
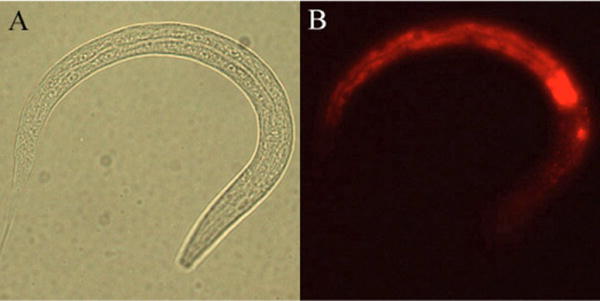
Immunohistochemistry of strain expressing Pges-1::FLAG::PAB-1 with anti-FLAG antibodies shows intestinal expression in fourth larval stage animals. (A) Bright-field image. (B) Antibody immunoflourescence.
To obtain a profile of gene expression in the fully differentiated intestine, we made extracts from a synchronous population of animals in the fourth larval stage. We crosslinked polyadenylated mRNA to FLAG::PAB-1, and enriched for mRNAs expressed in the intestine using anti-FLAG monoclonal antibodies for immunoprecipitation. Endogenous PAB-1 bound to mRNA in the rest of the worm does not have the FLAG tag and should not be immunoprecipitated with the FLAG antibody. The mRNA was extracted from FLAG::PAB-1/mRNA in the intestinal precipitate and used to prepare cDNA labeled with Cy5. mRNA from whole worm lysate was isolated from the same extract (before immunoprecipitation with α-FLAG antibody) and used to prepare cDNA labeled with Cy3. These two samples were hybridized to DNA microarrays representing ~94% of the genes in the C. elegans genome (Jiang et al., 2001). The mRNA-tagging experiment was repeated eight times to gain enough statistical power to distinguish enriched from unenriched mRNAs.
Genes expressed in the intestine are expected to have higher Cy5/Cy3 ratios than genes not expressed in the intestine. Therefore, we assigned a percentile rank for the Cy5/Cy3 ratios for each gene in a DNA microarray hybridization. We averaged the ranks for each gene across the eight samples, and ordered the list from the gene showing the highest (1.000) to the lowest enrichment (0.000). We used a Student’s t-test to identify genes that were significantly enriched above the median enrichment of 0.467, resulting in a list of 1938 genes enriched by mRNA tagging (P<0.001) (Fig. 2, see Table S1 in the supplementary material).
Fig. 2. Genes expressed in the intestine by mRNA tagging.
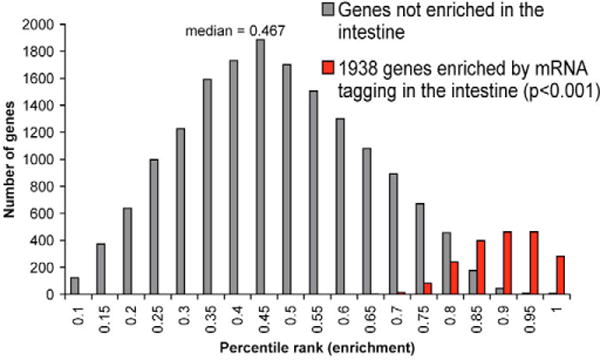
Histogram of the average percentile rank of enrichment after intestinal mRNA tagging. The x-axis shows the average percentile rank of enrichment, and the y-axis shows the number of genes.
We performed several controls to verify that the 1938 genes identified by mRNA tagging truly represent genes expressed in the intestine. First, 1938 enriched genes is much greater than the 18 genes expected by chance (at P<0.001) out of 18,345 genes on the DNA microarrays. Second, we generated a list from published literature of 80 genes that are not expressed in the intestine and showed that only one (1.25%) is in the list of 1938 genes (see Table S2 in the supplementary material). Third, the intestine gene list contains 271 genes whose expression pattern has been previously studied (see Table S3 in the supplementary material). Of these, 190 (70%) are expressed in the intestine. For many of the remaining 84 genes (30%), previous studies may have focused on expression in specific cells or tissues and may not have scored expression in the intestine. The fraction of genes from the list of 1938 mRNA tagged genes expressed in the intestine (at least 70%) is much higher than the fraction expressed in the intestine from a random set of genes. When 51 genes are chosen at random, only 13 (25%) are expressed in the intestine (Roy et al., 2002).
We analyzed the anatomical expression of six of the 1938 intestine-enriched genes (elo-6, gst-42, D2030.5, ZK970.2, C25E10.8 and B0218.8) by observing expression of GFP reporters (Fig. 3). These genes were selected because their expression profile had not been reported, they represent a range of enrichment values from the mRNA-tagging experiment, and DNA constructs containing promoter::GFP reporters have been made available to the C. elegans research community (Dupuy et al., 2004). Pgst-42::GFP and Pelo-6::GFP are expressed strongly in the intestine with some expression in the pharynx. PD2030.5::GFP is expressed in the intestine as well as other tissues. PZK970.2::GFP, PC25E10.8::GFP, and PB0218.8::GFP are expressed at low levels mostly in the midgut. Genes expressed solely in the intestine would be expected to have higher enrichment values from mRNA tagging than genes expressed in the intestine as well as other tissues. gst-42 and B0218.8 have enrichment values of 0.92 and 0.97, respectively, and show intestine-specific expression. By contrast, D2030.5 has a lower enrichment value of 0.68 and is expressed in the intestine as well as most other tissues. A promoter::GFP reporter strain expressing a seventh gene (K11D2.2) in the list of 1938 intestine-expressed genes is also expressed in the intestine (Y.L., unpublished). In summary, all seven GFP reporter genes were expressed in the intestine, indicating that a high fraction of the 1938 genes identified by mRNA tagging are expressed in this tissue.
Fig. 3. Intestinal expression of six genes identified by mRNA tagging.
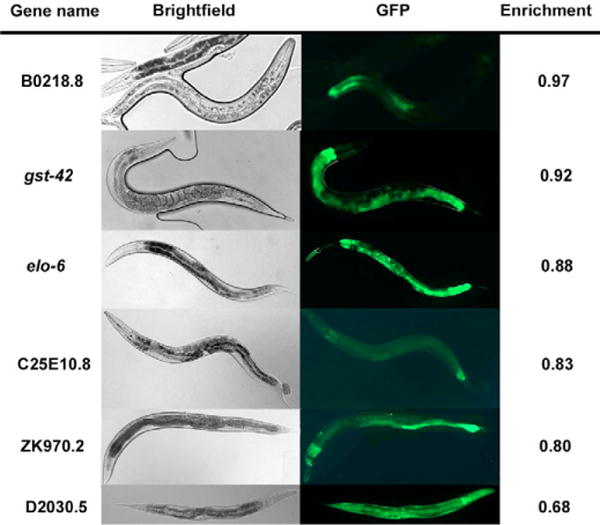
The anatomical expression of six genes chosen from the list of 1938 intestinal genes was visualized by transformation with promoter::GFP transcriptional constructs. B0218.8, C25E10.8, ZK970.2, elo-6 and gst-42 show exclusive intestinal expression and D2030.5 shows expression in the intestine as well as other tissues.
We also generated a list of 22 genes known to be expressed in the intestine, and found 16 in the list of 1938 genes (73%) (see Table S4 in the supplementary material). Finally, we showed that both rare and abundant intestine-expressed genes could be identified by mRNA tagging. The distribution of signal intensities (average normalized hybridization value for each gene in the lysate fraction that was not immunoprecipitated) of the 1938 intestine-expressed genes is nearly the same as the expression levels for the rest of the genes in the genome with only a slight bias toward higher expressed genes being enriched by mRNA tagging (see Fig. S1 in the supplementary material). In summary, these results show that a large fraction of the 1938 genes identified by mRNA tagging are expressed in the intestine (few false positives) and that this list includes a majority of the intestine-expressed genes (low fraction of false negatives).
Comparison of gene expression in the intestine, muscle and germline
Genes specific to the intestine define its unique functions, whereas genes expressed broadly (housekeeping genes) describe the cellular and metabolic functions common to all tissues. Defining the set of genes specifically expressed in the intestine is a necessary first step in order to understand the transcriptional regulatory networks that drive its differentiation. In addition to intestine-enriched genes, genome-wide profiles of muscle-expressed and germ line-enriched genes have been previously defined. A list of 1364 genes expressed in the body-wall muscle was discovered using mRNA tagging (Roy et al., 2002). By comparing gene expression levels in worms with a germline (wild type) with worms without a germline (glp-4 mutants) on DNA microarrays (Reinke et al., 2004), 3144 genes were shown to be enriched in the germline.
We used the lists of intestine, muscle and germline genes to show which are tissue enriched and which are commonly expressed. We defined housekeeping genes as those that were identified in the intestine, muscle and germline with significant enrichment values at P<0.05. This criterion generated a list of 510 genes expressed in these three tissues (see Table S5 in the supplementary material).
In order to characterize the function of the 510 commonly expressed genes, we compared them with other sets of co-expressed genes in the C. elegans gene expression topomap (Kim et al., 2001). The gene expression topomap is an assembly of 553 microarray experiments that can be used to identify genes that are co-expressed across diverse experimental conditions. Groups of co-expressed genes are visualized as gene mountains on a two-dimensional scatter plot such that the distance between two genes indicates the amount of correlation in expression. The 510 commonly expressed genes are enriched on mountains 2, 7, 11,18, 20 and 23 (Fig. 4B). Previous work has shown that all six of these mountains are enriched for genes expressed in the germline, indicating a close association between housekeeping genes and the maternal germline. Housekeeping genes are expressed in the maternal germline and packaged into embryos in order to allow expression of new proteins before the start of transcription at the four-cell stage (Seydoux and Fire, 1994).
Fig. 4. Distribution of intestine-expressed, intestine-, germ line-and muscle-enriched, and commonly-expressed genes on the C. elegans gene expression topomap.
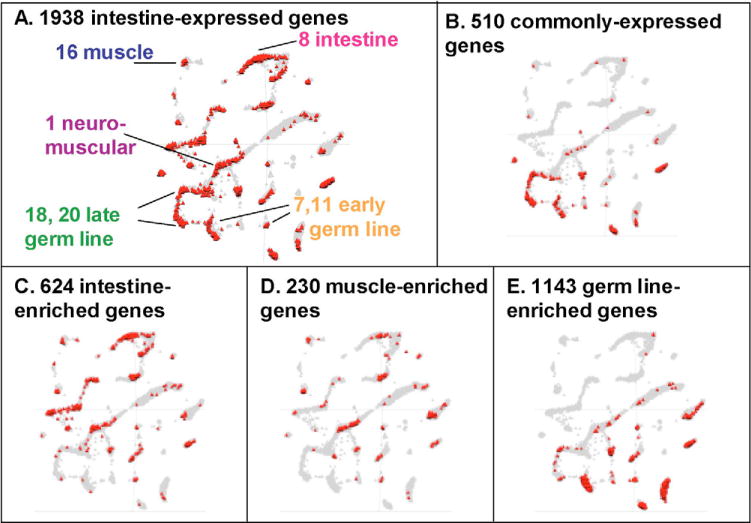
The gene expression topomap is an assembly of data from 553 microarray experiments in which genes that are strongly co-expressed are plotted in close proximity to each other on the map (Kim et al., 2001). The gray triangles represent all the genes in the topomap (17,817) and the red triangles represent the genes in each list.
We identified tissue-enriched genes by counting genes that have a P value of less than 0.01 for one tissue, but greater than 0.5 for the remaining two tissues. Using these criteria, we identified 624 intestine-enriched, 230 muscle-enriched and 1135 germ line-enriched genes (see Tables S6, S7 and S8 in the supplementary material). We plotted the tissue-enriched lists on the gene expression topomap and found that the 624 intestine-expressed genes are highly enriched on mountain 8, which was previously found to be enriched for intestine genes (151 genes, representation factor 5.8, P<4.3 10−74) (Kim et al., 2001) (Fig. 4C). Intestine-enriched genes are also enriched on mountains 19 and 21, which contain lipid metabolism genes. This observation is consistent with the role of the intestine as the fat storage and lipid metabolism organ in C. elegans. The 1135 germ line-enriched genes overlap with mountains 7 and 11 (enriched for early germline genes) and the 230 muscle-enriched genes overlap mountain 16 (enriched for muscle genes) and mountain 1 (enriched for neuromuscular genes) (Fig. 4D,E). Genes expressed in the muscle and germ line have been discussed in previous work, and are not discussed further for the sake of brevity (Reinke et al., 2004; Roy et al., 2002).
The 624 intestine-enriched genes include 329 that encode proteins with motifs suggesting their biochemical functions and serve to identify genetic pathways and processes that are specialized for the intestine and the functions it performs. For example, 33 genes encode proteases and lipases that may be involved in digestion, such as Y75B8A.4, a Lon protease. Eight genes encode extracellular molecules, such as B0218.6, a C-type lectin. These extracellular proteins might either be secreted into the lumen or expressed on the apical membrane domain of the intestine to form a barrier between the bacterial food and the intestinal cells. There are a large number of regulatory molecules expressed in the intestine (20 transcription factors and 23 signaling molecules). It is unlikely that these regulatory genes are involved primarily in intestinal differentiation, as this process is finished by the fourth larval stage, which was the stage used for mRNA tagging. The intestine is a major organ used by nematodes to interact with the environment, based on what they ingest. The intestine is the first line of defense against pathogenic bacteria, alerts the worm to noxious chemicals in the environment, and signals when food is abundant or scarce. Transcription factors and signaling molecules expressed in the mature intestine may regulate how this organ responds to these environmental cues.
There are 295 intestine-enriched genes that do not show sequence similarity to genes that have been studied previously. We made use of the gene expression topomap to identify possible functions for these novel genes. Sixty-six genes are in mountain 8 (enriched for intestine genes), further reinforcing their role in the intestine. Intestine-enriched genes are also overrepresented on mountain 19 (26 genes, representation factor 7.4, P<1.38×10−15) and mountain 27 (5 genes, representation factor 3.1, P<0.023). These two gene expression mountains are each enriched for genes known to function in amino acid metabolism, lipid metabolism and energy generation. The 31 intestine-enriched genes in these mountains may also function in these metabolic and energy pathways.
A bias in chromosomal clustering of commonly-expressed versus tissue-enriched genes
Previous work has shown that co-expressed genes cluster on the chromosomes of yeast, worms, fruit flies, humans, mice and rats (Boutanaev et al., 2002; Cohen et al., 2000; Kruglyak and Tang, 2000; Lercher et al., 2003; Lercher et al., 2002; Roy et al., 2002; Spellman and Rubin, 2002). One possibility is that clustering of co-expressed genes could be due to the influence of chromatin domains on gene expression. This explanation is compatible with the longstanding hypothesis that open areas of chromatin are accessible to transcription factors and are therefore areas of active gene expression. Another possibility is that a single locus control region could activate a cluster of closely spaced genes.
To determine if the 1938 intestine-expressed genes are physically clustered, we plotted their chromosomal position and counted the number of times there were two or more genes with translation start sites within 10 kilobases (kb) of each other (Table 1). We excluded genes that are in operons or are a result of recent gene duplications because these genes have similar regulatory elements and would be expected to be co-regulated. Fig. 5 shows an example of an intestinal gene cluster composed of eight genes: two that are highly enriched in the intestine (P<0.001), four that are moderately enriched (P<0.01) and two that are not enriched in the intestine. Out of 1746 intestine-expressed genes, 684 have chromosomal positions within 10 kb of each other, which is significantly more than the number that we would expect to see by chance (519 genes) when 1746 genes are sampled randomly from the genome 10,000 times (P<1 10−15). The gene clusters include 291 genes that were not selected in the mRNA-tagging experiment, of which 24 have P-values less than 0.05 and could therefore also be expressed in the intestine. The observation that intestine-expressed genes are clustered in close proximity to each other on the chromosomes confirms and extends previous results showing clustering of genes that are similarly expressed.
Table 1.
Results of chromosomal clustering analysis
| Gene list | Number of genes | Genes expected to cluster | Genes observed to cluster | P |
|---|---|---|---|---|
| Intestine expressed | 1746 | 519 | 684 | 1.0×10−15 |
| Intestine enriched | 613 | 74 | 96 | 0.037 |
| Muscle enriched | 219 | 10 | 13 | 0.16 |
| Common genes | 510 | 45 | 116 | 1.0×10−15 |
Fig. 5. A chromosomal cluster of intestine-expressed genes.
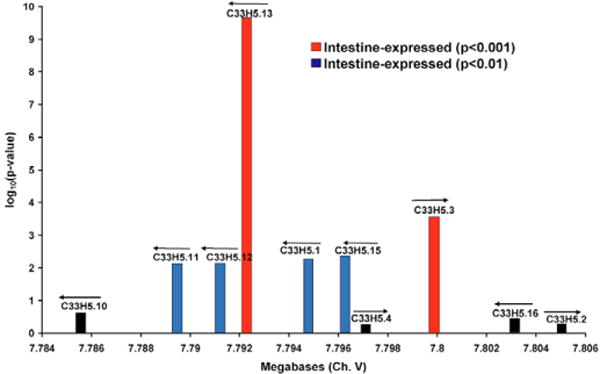
Genes are represented by vertical bars. The distance between genes is indicated on the x-axis and the log10(p-value) of enrichment in the intestine by mRNA tagging is indicated on the y-axis. Arrows indicate the direction of transcription for each gene.
The two main models for chromosomal clustering (chromatin domains and locus controllers) make different predictions about the relative amounts of clustering in tissue-enriched versus housekeeping genes. The chromatin domain model predicts that housekeeping genes are constrained to sections of chromatin that are open in all tissues, whereas tissue-enriched genes could occur in any open chromatin domain in the corresponding tissue. Thus, there are fewer chromatin domains available for housekeeping genes than for tissue-specific genes, predicting that housekeeping genes should show a higher degree of chromosomal clustering than tissue-enriched genes. By contrast, the locus control model predicts that locus control regions could drive expression of either housekeeping or tissue specific genes, such that both classes of genes would show chromosomal clustering. To distinguish between these two models, we determined whether tissue-enriched and housekeeping genes show equivalent degrees of chromosomal clustering.
We carried out the chromosomal clustering analysis separately on the lists of tissue-enriched and commonly expressed genes, as described above (Table 1). We did not analyze clustering in germline-enriched genes because many of these are housekeeping genes supplied by the maternal germline to the developing embryo. We found that muscle- and intestine-enriched genes are not significantly clustered, but commonly expressed genes show strong chromosomal clustering (P<1.0×10−15). These results indicate that genes that are commonly expressed are more significantly clustered on the chromosomes than are tissue-enriched genes, as predicted by the chromatin domain model.
Regulation of intestine gene expression by GATA transcription factors
To uncover transcriptional and regulatory networks that drive gene expression in the intestine, we looked for genes that encode putative transcription factors in the list of 1938 intestine-expressed genes. There are roughly 473 genes that encode putative transcription factors in the genome, of which 29 are present in our intestine gene list. Sixteen of these genes are from the nuclear hormone receptor/zinc finger protein family, three genes have BZIP domains, four have homeodomains, two are GATA transcription factors, two have DM (dsx and mab-3)-DNA binding domains, one has a helix-turn-helix DNA binding domain, and one has a domain similar to the vertebrate transcription factor enhancer protein TEF-1.
Next, we wanted to identify cis-acting regulatory elements that could control expression of intestine genes. We used CompareProspector (Liu et al., 2004) (available at http://CompareProspector.stanford.edu) to search for DNA sequence motifs that are over-represented in the promoter regions of the 1938 intestine-expressed genes. In this search, CompareProspector started with the 1000 base pairs upstream of the ATG translation start site and narrowed the search region by selecting sequences that are conserved between C. elegans and C. briggsae. A Gibbs sampling algorithm was employed to search for sequences that are overrepresented compared with random DNA sequence.
The top-ranking DNA motif found by CompareProspector was the consensus sequence T/AGATAA/T, which is the binding site for GATA transcription factors (Fig. 6A). The GATA motif is found in 820 out of 1750 intestine-expressed genes, representing a twofold enrichment over the rest of the genes in the genome (see Table S9 in the supplementary material). GATA transcription factors direct the development of the intestine in a regulatory cascade, as detailed in the Introduction of this paper.
Fig. 6. The GATA sequence motif is important for intestinal gene expression.
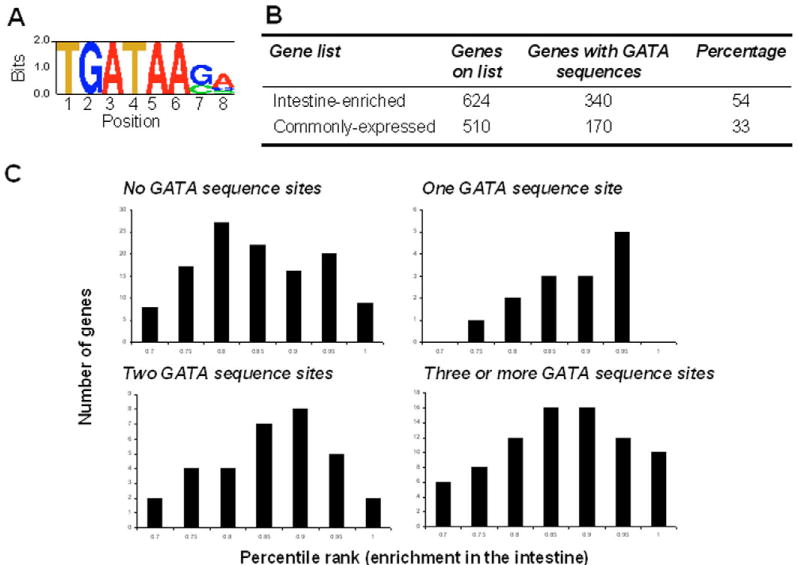
(A) Motif logo for the consensus GATA sequence sites identified in 820 out of 1750 intestine-expressed genes by CompareProspector. The y-axis represents the information content for each position. (B) Percentage of genes with GATA sites in the intestine and commonly-expressed gene lists. (C) Histogram of the average percentile rank for intestine enrichment of intestine-expressed genes with 0, 1, 2 or 3+ GATA sequence sites.
If the GATA motif were functional in the intestine of the L4/young adult worm, then genes with more GATA motifs should be more tightly co-regulated. To see if this was true, we calculated the average pairwise Pearson correlation to measure the co-regulation of these genes across 979 C. elegans microarray experiments (Stuart et al., 2003). The list of 1938 intestine-expressed genes contains 554 genes with one GATA site, 193 with two GATA sites and 73 with three or more GATA sites. The average pairwise Pearson correlation increases from 0.11 to 0.13 to 0.17 for genes with one, two and three GATA motifs, respectively, further indicating that the GATA motif is functional in intestinal gene expression.
We determined that there is a higher enrichment of genes with GATA sequence sites in the list of 624 intestine-enriched genes (54%) compared with 510 commonly expressed genes (33%) (Fig. 6B). Furthermore, we showed that a higher number of GATA sequence sites per promoter region generally indicate a higher enrichment value in the intestine. As the number of GATA sites increases, the distribution of enrichment in the intestine shifts to higher values, which we previously found was correlated with intestine-specific expression (Fig. 6C). These results suggest that GATA sequence sites are preferentially associated with genes that are expressed specifically in the intestine rather than generally in all cells.
We experimentally confirmed the function of GATA DNA sequence sites in the upstream regions of three intestine-expressed genes in vivo. One of the GATA sites in each of the promoter regions of the gst-42, elo-6 and D2030.5 GFP reporter constructs was changed with site-directed mutagenesis. The mutagenized constructs were used to generate transgenic GFP reporters. We generated three wild-type GFP reporter lines for gst-42, two for D2030.5 and one for elo-6, as well as two or three GFP reporter lines for each GATA mutant construct. The amount of GFP expression in 20 individual worms from each of the wild-type and GATA-site mutant GFP reporter lines was quantified. Mutation of the GATA DNA sequence consistently resulted in a reduction in GFP expression for all three genes (Fig. 7), indicating that the GATA site is necessary for wild-type intestinal gene expression in cis.
Fig. 7. The GATA sequence is necessary for intestinal gene expression.
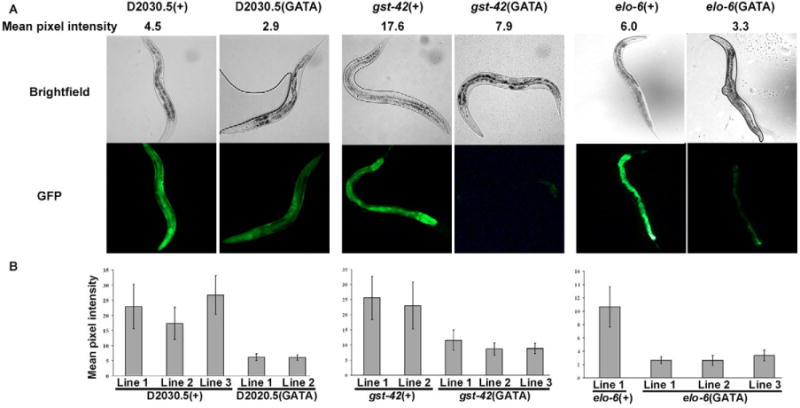
The GATA sequence in the promoters of elo-6, gst-42 and D2030.5 was mutated and used to generate transgenic GFP reporter strains. (+) indicates the wild-type promoter for each gene and (GATA) indicates that one copy of the GATA motif in the promoter was mutated. Mean pixel intensity was calculated by averaging the image pixel intensity of 20 young adult hermaphrodite animals. (A) Bright-field and GFP images of wild-type and mutant strains. (B) Quantification of GFP expression indicates that the variation between lines is less than the variation between wild-type and mutant strains (P<0.001), indicating that the difference between wild-type and GATA mutant expression levels is not due to variability in the transformation procedure. There is little difference in expression between different lines formed from the same DNA construct, not only in the five lines shown here, but also for three other lines used in a different study (Y.L., unpublished).
We also wanted to know which GATA transcription factors are important for the regulation of intestine expressed genes in trans. There are 11 putative GATA transcription factors in the C. elegans genome. For seven of these, we used RNAi to determine whether reducing their activity would affect expression of six of the GFP reporters with GATA sites. The seven GATA transcription factor genes include elt-2 and elt-3, which are in the list of 1938 intestine genes, and were previously known to have intestinal expression in the adult. end-1 and end-3 are expressed in the intestinal E lineage in the embryo (Maduro and Rothman, 2002; Zhu et al., 1997). egl-18, elt-1 and elt-6 are not reported to be expressed in the intestine (Koh and Rothman, 2001; Page et al., 1997).
Table 2 shows the results of using RNAi on the GATA transcription factor genes for the six GFP intestinal markers. We quantitatively determined the average level of GFP expression of animals growing on bacteria expressing double-stranded RNA for one of the GATA transcription factors and animals growing on bacteria expressing an empty vector. Student’s t-test was used to determine if there was a significant reduction in GFP expression (P<0.001). The reduction of elt-2 function by RNAi decreased GFP expression for four intestinal markers. Reducing the function of end-1, end-3 and elt-3 each reduced GFP expression of 1 or 2 genes. Although egl-18, elt-1 and elt-6 do not have reported intestinal expression, RNAi treatment of these genes decreased expression of two to five intestinal markers. This observation could be due to previously undetected expression of these genes in the intestine or to effects on intestinal expression in response to RNAi treatment in other tissues. As a negative control, we showed that RNAi of unc-22 (a gene expressed in the body wall muscle) did not significantly reduce expression of four out of four GFP markers tested (data not shown).
Table 2.
Effects of RNAi of GATA transcription factors on intestinal GFP markers
| GATA TFs | p::GFP reporters
|
|||||
|---|---|---|---|---|---|---|
| gst-42 | elo-6 | D2030.5 | ZK970.2 | C25E10.8 | B0218.8 | |
| elt-2 | Reduced (P<0.001) | No change | No change | Reduced (P<0.001) | Reduced (P<0.001) | Reduced (P<0.001) |
| end-3 | Reduced (P<0.001) | No change | No change | Reduced (P<0.001) | Reduced (P<0.001) | No change |
| end-1 | No change | No change | No change | Reduced (P<0.001) | Reduced (P<0.001) | No change |
| elt-3 | No change | No change | No change | No change | Reduced (P<0.001) | No change |
| elt-1* | No change | No change | No change | Reduced (P<0.001) | Reduced (P<0.001) | No change |
| egl-18* | Reduced (P<0.001) | No change | Reduced (P<0.001) | Reduced (P<0.001) | Reduced (P<0.001) | Reduced (P<0.001) |
| elt-6* | No change | No change | No change | Reduced (P<0.001) | Reduced (P<0.001) | No change |
elt-6, elt-1 and egl-18 GFP reporters are not expressed in the intestine, and yet RNAi treatment of these genes appears to regulate intestinal GFP reporters. Either these genes are indeed expressed in the intestine (GFP expression may not reflect true in vivo patterns) or the regulation is indirect.
Taken together, the results from the GATA transcription factor RNAi and GATA DNA motif mutagenesis experiments provide verification for a GATA transcription factor regulatory circuit driving intestinal gene expression. The GATA transcription factors may drive the expression of 820 intestine-expressed genes with GATA sequence motifs identified in our study. As the vast majority of these genes were not previously known to be targets of GATA transcription factors in the intestine, their identification helps illuminate the molecular pathways used by GATA transcription factors to specify the intestinal cell fate.
DISCUSSION
One goal of a systems level analysis of development is to use DNA microarrays to examine the tissue specificity of nearly every gene in the genome. In this study we have identified 1938 genes expressed in the intestine of C. elegans, adding to previous work showing that 1354 genes are expressed in the muscle and 3144 genes are expressed in the germline (Reinke et al., 2004; Roy et al., 2002). Gene expression profiles of the hypodermis and pan-neuronal tissues would complete the transcriptional identities of the five main tissue types in the adult hermaphrodite.
However, purifying RNA from specific tissues for gene expression analysis in C. elegans is not trivial because of its microscopic size. For this reason, alternate methods have been described to profile tissue-specific gene expression in the worm. One such method uses FACS sorting to isolate cells from primary embryonic cell culture that express GFP from a tissue-specific promoter (Christensen et al., 2002; Zhang et al., 2002). mRNA purified from these cells is then profiled on microarrays (Zhang et al., 2002). However, this method captures mRNA from tissue culture, which may differ in expression from the intact organism. A second approach compares gene expression in wild-type animals with mutants that lack specific tissues. This method was employed by Reinke et al. (Reinke et al., 2000; Reinke et al., 2004) to compare animals with and without a germline to identify germline-expressed genes and by Gaudet and Mango (Gaudet and Mango, 2002) to compare mutant embryos that produced either excess or no pharyngeal cells to identify candidate pharyngeal genes. However, many tissues are necessary for the development and survival of the animal (such as muscle, intestine, and pharynx). Mutants lacking these tissues die before hatching and thus RNA must be prepared from embryos before development has been completed.
A third method is mRNA tagging, which was devised by Roy et al. (Roy et al., 2002) to identify genes expressed in body wall muscles and used by Kunitomo et al. (Kunitomo et al., 2005) to identify genes expressed in ciliated sensory neurons. We used mRNA tagging to identify genes expressed in the intestine because it allowed us to look at gene expression in intact organisms. Once we had identified intestine-expressed genes, we compared them with previously identified muscle-expressed and germ line-enriched genes. We identified genes that are commonly expressed between tissues or enriched in one tissue, thus implicating them in housekeeping versus tissue-specific pathways.
Previous work has shown that genes in close proximity show correlated expression in yeast, worms, fruit flies, humans, mice and rats (Boutanaev et al., 2002; Cohen et al., 2000; Kruglyak and Tang, 2000; Lercher et al., 2003; Lercher et al., 2002; Roy et al., 2002; Spellman and Rubin, 2002). What mechanisms could cause co-expressed genes to be positionally clustered on chromosomes? One possibility is that chromatin domains cause chromosomal clustering (Weintraub, 1984). In any particular tissue or cell, the genome is divided into regions of open and closed chromatin, corresponding to regions of active or inactive gene expression. Genes that are expressed in that tissue would be clustered in open chromatin regions. Another possibility (not exclusive of the first) is that a single DNA site simultaneously induces the expression of several genes in close proximity. For example, the globin genes in mammals are located in a gene cluster, and high levels of expression of these globin genes requires a single locus controller that affects expression of each gene in the cluster (Hebbes et al., 1994; Stalder et al., 1980). In C. elegans, DAF-12 DNA response elements have been shown to reside within clusters of DAF-12-regulated genes (Shostak et al., 2004). Enhancer elements can act over very large distances and many are located in regions 3′ to a gene (Valarche et al., 1997). It is possible that many enhancers have effects on nearby genes in a manner similar to the globin locus control region or DAF-12-regulated gene clusters.
These mechanisms for chromosomal clustering make distinct predictions about the relative amounts of chromosomal clustering in housekeeping versus tissue-specific genes. The chromatin domain mechanism predicts that housekeeping genes should show a higher level of chromosomal clustering than tissue-specific genes because housekeeping genes are constrained to be in chromatin domains that are open in all tissues (which are just a subset of the open chromatin domains in a particular tissue). The locus controller/enhancer mechanism does not necessarily predict that there would be a difference in chromosomal clustering between housekeeping and tissue-specific genes because enhancers could act over a distance for both sets of genes.
The data in this paper help distinguish between the two mechanisms for chromosomal clustering. We have shown that genes that are commonly expressed show more significant clustering than genes that are specific to intestine and muscle. Similarly, Lercher et al. (Lercher et al., 2002) found that genes commonly expressed by 14 human tissues were chromosomally clustered. By contrast, genes that were specific to those tissues were not clustered. These results support the chromatin domain mechanism for chromosomal clustering.
To gain further insight into the regulation of intestine-expressed genes, we searched for over-represented DNA sequence motifs in the promoters of these genes and found an enrichment of GATA sequence sites. Several GATA transcription factors are necessary for the development of the intestine. Specifically, two redundant genes elt-2 and elt-7 are responsible for maintaining intestinal cell identity (Fukushige et al., 1998). There are only a few targets known to be regulated by elt-2, including a cysteine protease gene (gcp-1) (Ray and McKerrow, 1992), two metallothionein genes (mtl-1 and mtl-2) (Moilanen et al., 1999), and several vitellogenin genes (vit-2, vit-5 and vit-6) (MacMorris et al., 1992; MacMorris et al., 1994; Spieth et al., 1985; Spieth et al., 1991; Zucker-Aprison and Blumenthal, 1989). By generating a molecular profile of the intestine, we have identified 820 intestine-expressed genes that have GATA sequence sites in their promoters and may be targets of GATA transcription factors, such as elt-2 and elt-7. These target genes may maintain intestinal cell identity in the adult worm and may be involved in intestinal processes such as cell polarity, secretion, digestion, nourishment of embryos and defense against pathogens.
Supplementary Material
{kind=link}
Acknowledgments
We thank the members of the Kim laboratory for thoughtful discussions and revisions of the manuscript; D. Dupuy, M. Vidal, A. Fire and P. Roy for providing DNA constructs; the programmers at the Stanford Microarray Database for microarray analysis and database management; and Wormbase for annotation of C. elegans genes. This work was supported by grants from the National Institutes of Health and F.P. was supported by the Stanford Genome Training Program (Grant Number T32 HG00044 from the National Human Genome Research Institute). The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/cgi/content/full/133/2/287/DC1
References
- Boutanaev AM, Kalmykova AI, Shevelyov YY, Nurminsky DI. Large clusters of co-expressed genes in the Drosophila genome. Nature. 2002;420:666–669. doi: 10.1038/nature01216. [DOI] [PubMed] [Google Scholar]
- Christensen M, Estevez A, Yin X, Fox R, Morrison R, McDonnell M, Gleason C, Miller DM, 3rd, Strange K. A primary culture system for functional analysis of C. elegans neurons and muscle cells. Neuron. 2002;33:503–514. doi: 10.1016/s0896-6273(02)00591-3. [DOI] [PubMed] [Google Scholar]
- Cohen BA, Mitra RD, Hughes JD, Church GM. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat Genet. 2000;26:183–186. doi: 10.1038/79896. [DOI] [PubMed] [Google Scholar]
- Dupuy D, Li QR, Deplancke B, Boxem M, Hao T, Lamesch P, Sequerra R, Bosak S, Doucette-Stamm L, et al. A first version of the Caenorhabditis elegans Promoterome. Genome Res. 2004;14:2169–2175. doi: 10.1101/gr.2497604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushige T, Hawkins MG, McGhee JD. The GATA-factor elt-2 is essential for formation of the Caenorhabditis elegans intestine. Dev Biol. 1998;198:286–302. [PubMed] [Google Scholar]
- Gaudet J, Mango SE. Regulation of organogenesis by the Caenorhabditis elegans FoxA protein PHA-4. Science. 2002;295:821–825. doi: 10.1126/science.1065175. [DOI] [PubMed] [Google Scholar]
- Hebbes TR, Clayton AL, Thorne AW, Crane-Robinson C. Core histone hyperacetylation co-maps with generalized DNase I sensitivity in the chicken beta-globin chromosomal domain. EMBO J. 1994;13:1823–1830. doi: 10.1002/j.1460-2075.1994.tb06451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedgecock EM, White JG. Polyploid tissues in the nematode Caenorhabditis elegans. Dev Biol. 1985;107:128–133. doi: 10.1016/0012-1606(85)90381-1. [DOI] [PubMed] [Google Scholar]
- Jiang M, Ryu J, Kiraly M, Duke K, Reinke V, Kim SK. Genome-wide analysis of developmental and sex-regulated gene expression profiles in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2001;98:218–223. doi: 10.1073/pnas.011520898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/s1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- Kim SK, Lund J, Kiraly M, Duke K, Jiang M, Stuart JM, Eizinger A, Wylie BN, Davidson GS. A gene expression map for Caenorhabditis elegans. Science. 2001;293:2087–2092. doi: 10.1126/science.1061603. [DOI] [PubMed] [Google Scholar]
- Kimble J, Sharrock WJ. Tissue-specific synthesis of yolk proteins in Caenorhabditis elegans. Dev Biol. 1983;96:189–196. doi: 10.1016/0012-1606(83)90322-6. [DOI] [PubMed] [Google Scholar]
- Knust E, Bossinger O. Composition and formation of intercellular junctions in epithelial cells. Science. 2002;298:1955–1959. doi: 10.1126/science.1072161. [DOI] [PubMed] [Google Scholar]
- Koh K, Rothman JH. ELT-5 and ELT-6 are required continuously to regulate epidermal seam cell differentiation and cell fusion in C. elegans. Development. 2001;128:2867–2880. doi: 10.1242/dev.128.15.2867. [DOI] [PubMed] [Google Scholar]
- Kruglyak S, Tang H. Regulation of adjacent yeast genes. Trends Genet. 2000;16:109–111. doi: 10.1016/s0168-9525(99)01941-1. [DOI] [PubMed] [Google Scholar]
- Kunitomo H, Uesugi H, Kohara Y, Iino Y. Identification of ciliated sensory neuron-expressed genes in Caenorhabditis elegans using targeted pull-down of poly(A) tails. Genome Biol. 2005;6:R17. doi: 10.1186/gb-2005-6-2-r17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lercher MJ, Urrutia AO, Hurst LD. Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nat Genet. 2002;31:180–183. doi: 10.1038/ng887. [DOI] [PubMed] [Google Scholar]
- Lercher MJ, Blumenthal T, Hurst LD. Coexpression of neighboring genes in Caenorhabditis elegans is mostly due to operons and duplicate genes. Genome Res. 2003;13:238–243. doi: 10.1101/gr.553803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung B, Hermann GJ, Priess JR. Organogenesis of the Caenorhabditis elegans intestine. Dev Biol. 1999;216:114–134. doi: 10.1006/dbio.1999.9471. [DOI] [PubMed] [Google Scholar]
- Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain PO, Han JD, Chesneau A, Hao T, et al. A map of the interactome network of the metazoan C. elegans. Science. 2004;303:540–543. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu XS, Wei L, Altman RB, Batzoglou S. Eukaryotic regulatory element conservation analysis and identification using comparative genomics. Genome Res. 2004;14:451–458. doi: 10.1101/gr.1327604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMorris M, Broverman S, Greenspoon S, Lea K, Madej C, Blumenthal T, Spieth J. Regulation of vitellogenin gene expression in transgenic Caenorhabditis elegans: short sequences required for activation of the vit-2 promoter. Mol Cell Biol. 1992;12:1652–1662. doi: 10.1128/mcb.12.4.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacMorris M, Spieth J, Madej C, Lea K, Blumenthal T. Analysis of the VPE sequences in the Caenorhabditis elegans vit-2 promoter with extrachromosomal tandem array-containing transgenic strains. Mol Cell Biol. 1994;14:484–491. doi: 10.1128/mcb.14.1.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro MF, Rothman JH. Making worm guts: the gene regulatory network of the Caenorhabditis elegans endoderm. Dev Biol. 2002;246:68–85. doi: 10.1006/dbio.2002.0655. [DOI] [PubMed] [Google Scholar]
- Maduro MF, Meneghini MD, Bowerman B, Broitman-Maduro G, Rothman JH. Restriction of mesendoderm to a single blastomere by the combined action of SKN-1 and a GSK-3beta homolog is mediated by MED-1 and -2 in C. elegans. Mol Cell. 2001;7:475–485. doi: 10.1016/s1097-2765(01)00195-2. [DOI] [PubMed] [Google Scholar]
- Mello C, Fire A. DNA transformation. Methods Cell Biol. 1995;48:451–482. [PubMed] [Google Scholar]
- Moilanen LH, Fukushige T, Freedman JH. Regulation of metallothionein gene transcription. Identification of upstream regulatory elements and transcription factors responsible for cell-specific expression of the metallothionein genes from Caenorhabditis elegans. J Biol Chem. 1999;274:29655–29665. doi: 10.1074/jbc.274.42.29655. [DOI] [PubMed] [Google Scholar]
- Page BD, Zhang W, Steward K, Blumenthal T, Priess JR. ELT-1, a GATA-like transcription factor, is required for epidermal cell fates in Caenorhabditis elegans embryos. Genes Dev. 1997;11:1651–1661. doi: 10.1101/gad.11.13.1651. [DOI] [PubMed] [Google Scholar]
- Rasband WS. ImageJ. Bethesda (MD): National Institues of Health; 2004. [Google Scholar]
- Ray C, McKerrow JH. Gut-specific and developmental expression of a Caenorhabditis elegans cysteine protease gene. Mol Biochemical Parasitol. 1992;51:239–249. doi: 10.1016/0166-6851(92)90074-t. [DOI] [PubMed] [Google Scholar]
- Reinke V, Smith HE, Nance J, Wang J, Van Doren C, Begley R, Jones SJ, Davis EB, Scherer S, Ward S, et al. A global profile of germline gene expression in C. elegans. Mol Cell. 2000;6:605–616. doi: 10.1016/s1097-2765(00)00059-9. [DOI] [PubMed] [Google Scholar]
- Reinke V, Gil IS, Ward S, Kazmer K. Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development. 2004;131:311–323. doi: 10.1242/dev.00914. [DOI] [PubMed] [Google Scholar]
- Roy PJ, Stuart JM, Lund J, Kim SK. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature. 2002;418:975–979. doi: 10.1038/nature01012. [DOI] [PubMed] [Google Scholar]
- Seydoux G, Fire A. Soma-germline asymmetry in the distributions of embryonic RNAs in Caenorhabditis elegans. Development. 1994;120:2823–2834. doi: 10.1242/dev.120.10.2823. [DOI] [PubMed] [Google Scholar]
- Shostak Y, Van Gilst MR, Antebi A, Yamamoto KR. Identification of C. elegans DAF-12-binding sites, response elements and target genes. Genes Dev. 2004;18:2529–2544. doi: 10.1101/gad.1218504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellman PT, Rubin GM. Evidence for large domains of similarly expressed genes in the Drosophila genome. J Biol. 2002;1:5. doi: 10.1186/1475-4924-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spieth J, Denison K, Kirtland S, Cane J, Blumenthal T. The C. elegans vitellogenin genes: short sequence repeats in the promoter regions and homology to the vertebrate genes. Nucleic Acids Res. 1985;13:5283–5295. doi: 10.1093/nar/13.14.5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spieth J, Nettleton M, Zucker-Aprison E, Lea K, Blumenthal T. Vitellogenin motifs conserved in nematodes and vertebrates. J Mol Evol. 1991;32:429–438. doi: 10.1007/BF02101283. [DOI] [PubMed] [Google Scholar]
- Stalder J, Larsen A, Engel JD, Dolan M, Groudine M, Weintraub H. Tissue-specific DNA cleavages in the globin chromatin domain introduced by DNAase I. Cell. 1980;20:451–460. doi: 10.1016/0092-8674(80)90631-5. [DOI] [PubMed] [Google Scholar]
- Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. [see comment] Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977;56:110–156. doi: 10.1016/0012-1606(77)90158-0. [DOI] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983;100:64–119. doi: 10.1016/0012-1606(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Valarche I, de Graaff W, Deschamps J. A 3′ remote control region is a candidate to modulate Hoxb-8 expression boundaries. Int J Dev Biol. 1997;41:705–714. [PubMed] [Google Scholar]
- Wang E, Miller LD, Ohnmacht GA, Liu ET, Marincola FM. High-fidelity mRNA amplification for gene profiling. Nat Biotechnol. 2000;18:457–459. doi: 10.1038/74546. [DOI] [PubMed] [Google Scholar]
- Weintraub H. Histone-H1-dependent chromatin superstructures and the suppression of gene activity. Cell. 1984;38:17–27. doi: 10.1016/0092-8674(84)90522-1. [DOI] [PubMed] [Google Scholar]
- White J. The Anatomy. In: Wood WB, editor. The Nematode Caenorhabditis elegans. New York: Cold Spring Harbor Laboratory Press; 1988. pp. 103–105. [Google Scholar]
- Zhang Y, Ma C, Delohery T, Nasipak B, Foat BC, Bounoutas A, Bussemaker HJ, Kim SK, Chalfie M. Identification of genes expressed in C. elegans touch receptor neurons. Nature. 2002;418:331–335. doi: 10.1038/nature00891. [DOI] [PubMed] [Google Scholar]
- Zhu J, Hill RJ, Heid PJ, Fukuyama M, Sugimoto A, Priess JR, Rothman JH. end-1 encodes an apparent GATA factor that specifies the endoderm precursor in Caenorhabditis elegans embryos. Genes Dev. 1997;11:2883–2896. doi: 10.1101/gad.11.21.2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker-Aprison E, Blumenthal T. Potential regulatory elements of nematode vitellogenin genes revealed by interspecies sequence comparison. J Mol Evol. 1989;28:487–496. doi: 10.1007/BF02602929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.