

Abstract
Mitochondria are vital and highly dynamic organelles that continuously fuse and divide to maintain mitochondrial quality. Mitochondrial dysfunction impairs cellular integrity and is known to be associated with various human diseases. However, the mechanism by which the quality of mitochondria is maintained remains largely unexplored. Here we show that impaired proteasome function recovers the growth of yeast cells lacking Fzo1, a pivotal protein for mitochondrial fusion. Decreased proteasome activity increased the mitochondrial oxidoreductase protein Mia40 and the ratio of the short isoform of mitochondrial intermembrane protein Mgm1 (s-Mgm1) to the long isoform (l-Mgm1). The increase in Mia40 restored mitochondrial membrane potential, while the increase in the s-Mgm1/l-Mgm1 ratio promoted mitochondrial fusion in an Fzo1-independent manner. Our findings demonstrate a new pathway for mitochondrial quality control that is induced by proteasome impairment.
INTRODUCTION
Mitochondria are highly dynamic organelles that continuously divide and fuse with each other (1, 2). Mitochondrial fusion and fission contribute to quality control of mitochondria. Under moderate-stress conditions, mitochondrial fusion is stimulated to cure damaged mitochondria by fusing to healthy mitochondria. On the other hand, severely damaged mitochondria are eliminated by mitophagy, where uneven distribution of damaged proteins and asymmetrical mitochondrial fission contribute to selective degradation of damaged mitochondria by autophagy (3). Loss of mitochondrial fusion leads to fragmented mitochondria caused by ongoing fission, mitochondrial DNA loss, and decrease in ATP production, manifested by poor growth in yeast or embryonic lethality in mammals.
Previous reports showed that at least two dynamin-related proteins (DRPs), Fzo1/mitofusin and Mgm1/OPA1, are required for mitochondrial fusion, and one DRP, Dnm1/Drp1, regulates mitochondrial fission in yeast/mammals. Mitochondrial outer membrane protein Ugo1 bridges interactions between Fzo1 and Mgm1 (4–8). Although detailed fusion mechanisms remain to be solved, the ubiquitin-proteasome system has been suggested to regulate mitochondrial dynamics. Mutations in Rpn11, which cleaves polyubiquitin chains from substrate proteins, and Blm10, a homolog of proteasome activator PA200, induced mitochondrial fragmentation in a Dnm1-dependent manner (9–11), suggesting that the proteasome plays an important role in mitochondrial fusion-fission balance. In addition, the proteasome positively and negatively regulates mitochondrial fusion: degradation of Fzo1 either downregulates mitochondrial fusion or accomplishes fusion events after transoligomerization of Fzo1, depending on context, in yeast (12–14).
The 26S proteasome is a 2.5-MDa protease complex conserved among eukaryotes. It is made up of 66 subunits and can be divided into two complexes: a catalytic 20S core particle (CP), and a 19S regulatory particle (RP). The proteasome selectively degrades proteins tagged with ubiquitin chains. This selective degradation contributes to a vast array of physiological processes, including stress response, regulation of cell cycle, transcription, apoptosis, metabolism, and protein quality control (15, 16). Hence, dysfunction of the proteasome causes various defects in multiple cellular processes, including mitochondrial function. However, the exact mechanism by which proteasome activity affects mitochondrial function and dynamics remains elusive.
In this study, to discover novel functions of the proteasome, we screened for mutations in budding yeast (Saccharomyces cerevisiae) strains whose phenotypes are affected by proteasome dysfunction and found that proteasome defects restored growth of fzo1Δ cells, suggesting that FZO1 genetically interacts with the proteasome. Proteasome defects increased expression of the mitochondrial oxidoreductase Mia40, and restored mitochondrial membrane potential (ΔΨm). In addition, proteasome defects enhanced mitochondrial fusion in an Fzo1-independent manner, in which the short isoform of Mgm1 (s-Mgm1) played a key role. Thus, proteasome impairment evokes two independent pathways for mitochondrial quality control that have not been recognized to date.
MATERIALS AND METHODS
Strains, media, plasmid construction, and genetic manipulations.
The yeast strains and plasmids used in this study are shown in Tables 1 and 2, respectively. A collection of nonessential gene deletion strains was purchased from Open Biosystems. Complete YPA (1% yeast extract, 2% Bacto peptone, 100 mg/liter adenine sulfate), supplemented with either 2% glucose (YPAD), 0.5% galactose and 2% raffinose (YPAGal), or 3% glycerol (YPAGly), was used. Mitochondrial mutants were maintained and cultivated in YPAGal. To observe their phenotypes by Western blotting, quantitative reverse transcription-PCR (RT-PCR), fluorescence-activated cell sorter (FACS) analysis, and mitochondrial fusion assay, cells were transferred to YPAD and incubated for 2 h. As multicopy vectors, pYO325 and pYO326 were used (pYO326 was previously designated pSQ326) (17). A C-terminal Flag tag was added as previously described (18). Gene disruption and marker switch were performed as previously described (19–21).
TABLE 1.
Yeast strains used in this study
| Strain name | Background | Genotype | Source or reference |
|---|---|---|---|
| BY4741 | BY | MATa | Open Biosystems |
| BY4742 | BY | MATα | Open Biosystems |
| BY4743 | BY | MATa/α | Open Biosystems |
| BY | MATa rpn10Δ::kan | Open Biosystems | |
| BY | MATa rpn13Δ::kan | Open Biosystems | |
| BY | MATa pdr5Δ::kan | Open Biosystems | |
| BY | MATa atg32Δ::kan | Open Biosystems | |
| BY | MATa cox12Δ::kan | Open Biosystems | |
| BY | MATa cox7Δ::kan | Open Biosystems | |
| BY | MATa mrpl35Δ::kan | Open Biosystems | |
| BY | MATa mip1Δ::kan | Open Biosystems | |
| BY | MATa ppa2Δ::kan | Open Biosystems | |
| Y8205 | MATα can1Δ::STE2pr-Sp_his5 lyp1Δ::STE3pr-LEU2 | 20 | |
| YWD228 | BY | MATa pre9Δ::hph | |
| YWD230 | BY | MATa rpn4Δ::hph | |
| YWD242 | BY | MATa sem1Δ::hph | |
| YWD248 | MATα pre9Δ::hph can1Δ::STE2pr-Sp_his5 lyp1Δ::STE3pr-LEU2 | ||
| YWD338 | BY | MATa/α FZO1/fzo1Δ::kan SEM1/sem1Δ::hph | |
| YWD339 | BY | MATa/α MGM1/mgm1Δ::kan | |
| YWD342 | BY | MATa/α UGO1/ugo1Δ::kan SEM1/sem1Δ::hph | |
| YWD344 | W303 | MATa/α FZO1/fzo1Δ::kan | |
| YWD350 | BY | MATa MGM1-FLAG-kan | |
| YWD357 | BY | MATa his3::mt-mCherry-Sp_his5 | 70 |
| YWD364 | BY | MATa/α FZO1/fzo1Δ::nat | |
| YWD368 | BY | MATa fzo1Δ::nat | |
| YWD376 | BY | MATa fzo1Δ::nat his3::mt-mCherry-Sp_his5 | |
| YWD381 | BY | MATα fzo1Δ::nat [p416GPD-mtGFP] | |
| YWD400 | BY | MATα fzo1Δ::nat pdr5Δ::kan | |
| YWD448 | BY | MATa MIA40-FLAG-kan | |
| YWD452 | BY | MATa fzo1Δ::nat MIA40-FLAG-kan pre9Δ::hph | |
| YWD457 | BY | MATa fzo1Δ::nat MIA40-FLAG-kan sem1Δ::hph | |
| YWD460 | BY | MATa fzo1Δ::nat MIA40-FLAG-kan rpn4Δ::hph | |
| YWD467 | BY | MATa fzo1Δ::nat MIA40-FLAG-kan | |
| YWD469 | BY | MATa fzo1Δ::nat [pYO326-MIA40] | |
| YWD477 | BY | MATa fzo1Δ::nat MGM1-FLAG-kan | |
| YWD478 | BY | MATa fzo1Δ::nat MGM1-FLAG-kan pre9Δ::hph | |
| YWD479 | BY | MATa fzo1Δ::nat MGM1-FLAG-kan rpn4Δ::hph | |
| YWD480 | BY | MATa fzo1Δ::nat MGM1-FLAG-kan sem1Δ::hph | |
| YWD481 | BY | MATa fzo1Δ::nat pre9Δ::hph | |
| YWD483 | BY | MATa fzo1Δ::nat sem1Δ::hph | |
| YWD485 | BY | MATa fzo1Δ::nat rpn4Δ::hph | |
| YWD497 | BY | MATa MIA40-FLAG-kan OM45-GFP-HIS3 | |
| YWD507 | BY | MATα dnm1Δ::Sphis5 | |
| YWD508 | BY | MATa dnm1Δ::mt-mCherry-HIS3 | |
| YWD512 | BY | MATa fzo1Δ::nat dnm1Δ::mt-mCherry-HIS3 | |
| YWD513 | BY | MATa fzo1Δ::nat dnm1Δ::mt-mCherry-HIS3 pre9Δ::hph | |
| YWD514 | BY | MATa fzo1Δ::nat dnm1Δ::mt-mCherry-HIS3 rpn4Δ::hph | |
| YWD515 | BY | MATa fzo1Δ::nat dnm1Δ::mt-mCherry-HIS3 sem1Δ::hph | |
| YWD519 | BY | MATα fzo1Δ::nat dnm1Δ::mt-mCherry-HIS3 pre9Δ::hph mgm1Δ::kan | |
| YWD520 | BY | MATα fzo1Δ::nat dnm1Δ::mt-mCherry-HIS3 rpn4Δ::hph mgm1Δ::kan | |
| YWD521 | BY | MATα fzo1Δ::nat dnm1Δ::mt-mCherry-HIS3 sem1Δ::hph mgm1Δ::kan | |
| YWD525 | BY | MATα fzo1Δ::nat dnm1Δ::Sphis5 [p416GPD-mtGFP] | |
| YWD526 | BY | MATα fzo1Δ::nat dnm1Δ::Sphis5 pre9Δ::hph [p416GPD-mtGFP] | |
| YWD527 | BY | MATα fzo1Δ::nat dnm1Δ::Sphis5 rpn4Δ::hph [p416GPD-mtGFP] | |
| YWD528 | BY | MATα fzo1Δ::nat dnm1Δ::Sphis5 sem1Δ::hph [p416GPD-mtGFP] | |
| YWD532 | BY | MATa fzo1Δ::nat dnm1Δ::Sphis5 pre9Δ::hph mgm1Δ::kan [p416GPD-mtGFP] | |
| YWD533 | BY | MATa fzo1Δ::nat dnm1Δ::Sphis5 rpn4Δ::hph mgm1Δ::kan [p416GPD-mtGFP] | |
| YWD534 | BY | MATa fzo1Δ::nat dnm1Δ::Sphis5 sem1Δ::hph mgm1Δ::kan [p416GPD-mtGFP] | |
| YWD538 | BY | MATα fzo1Δ::nat MIA40-FLAG-kan OM45-GFP-HIS3 | |
| YWD539 | BY | MATa fzo1Δ::nat rpn4Δ::hph MIA40-FLAG-kan OM45-GFP-HIS3 | |
| YWD587 | BY | MATa [rho0] | |
| YWD588 | BY | MATa rpn4Δ [rho0] | |
| YWD589 | BY | MATa sem1Δ [rho0] | |
| YWD590 | BY | MATa pre9Δ [rho0] |
TABLE 2.
Plasmids used in this study
| Plasmid name | Background vector | Cloned gene | Reference | Function |
|---|---|---|---|---|
| p4339 | pCRII-TOPO | TEFp-natR-TEFt | 20 | Marker switch |
| KOB74 | p416 | mtDHFR-GFP | 21 | |
| KOB154 | pBSII | TEF-mtDHFR-mCherry-CgHIS3 | 70 | |
| pHY68 | pBluescript KS− | 3× FLAG-kanR | 18 | C-terminal Flag tag |
| pFA6a-hph | pFA6a | hph | 71 | Marker switch |
| YEplac195 | 72 | |||
| pRS316 | 73 | |||
| pYO325 | 17 | |||
| pYO326 | 17 | |||
| pT192 | YEplac195 | RPN4 | ||
| pUZ160 | pRS316 | SAL1BY | ||
| pUZ161 | pRS316 | SAL1W303 | ||
| pUZ206 | pYO326 | MIA40 | ||
| pUZ215 | YEplac195 | ERV1 | ||
| PUZ223 | PYO326 | TIM9-TIM10 | ||
| pUZ245 | pYO325 | MIA40 | ||
| pUZ247 | pYO325 | MGM1 | ||
| pUZ248 | pYO325 | s-MGM1 | ||
| pUZ300 | pYO326 | FL-MGM1-FLAG | ||
| pUZ301 | pYO326 | s-MGM1-FLAG |
SGA analysis.
Synthetic genetic array (SGA) analysis was performed as previously described with minor modifications (20). Hygromycin instead of clonNAT was used as a selection marker for the pre9Δ allele. Genetic interaction was confirmed through repetitive tetrad analysis.
Biochemical analysis.
Protein extraction, glycerol density gradient centrifugation, and assays for peptidase activity were performed as described previously (22). Subcellular fractionation was performed as described previously (23).
Western blotting.
Protein was extracted as described previously (24). M2/anti-Flag (F1804; Sigma), antiporin (459500; Invitrogen), and anti-Pgk1 (429250; Invitrogen) antibodies were used. Anti-enhanced green fluorescent protein (anti-EGFP) antiserum was raised in rabbits in this laboratory.
Quantitative RT-PCR.
Yeast cells were disrupted with glass beads, and total RNA was extracted using a High Pure RNA isolation kit (Roche). The RNA was reverse transcribed using a SuperScript VILO cDNA synthesis kit (Invitrogen), and the relative amounts of cDNA for each target gene were quantified using a LightCycler 480 (Roche). The primers and probes are described below. ACT1 and TOM20 were used as the nonmitochondrial and mitochondrial normalization controls, respectively. The primer sequences are as follows: ACT1, probe 25, primer1, CCATCTTCCATGAAGGTCAAG, and primer2, CCACCAATCCAGACGGAGTA; MIA40, probe 143, primer1, GAAGATGGTGAATTGGTTGTTCT, and primer2, TTTGATTCATCAGTATCTTTATCTTCG; and TOM20, probe 38, primer1, TCCATGCAACAAGGTAAGGA, and primer2, GGCTGGCTGAGGGTATACAG.
Measurement of mitochondrial membrane potential by FACS analysis.
Mitochondrial membrane potential was measured as described previously (25). Flow cytometry analysis was performed on a FACSAria II (BD) flow cytometer.
Mitochondrial fusion assay.
The indicated strains were mated on YPAD and incubated at 26°C for 4 h. Fluorescent images were acquired using a Leica sp8 confocal microscope under computer control with a 100× lens and HyD detector. z-stack images were deconvoluted and projected using LAS AF3 software.
Statistical analysis.
Statistical significance was estimated using Welch's t test for mitochondrial membrane potential and Fisher's exact test for the mitochondrial fusion assay.
RESULTS
Proteasome defects rescue the poor growth of fzo1Δ cells.
In search of molecules that potentially have functional implications for proteasome activity, we employed synthetic genetic array (SGA) analysis in Saccharomyces cerevisiae (20), using a strain lacking the α3/Pre9 subunit, which is the only nonessential subunit of the proteasome CP (26). The pre9Δ strain was individually crossed with over 5,000 single-deletion strains that represent the complete set of viable haploid deletion mutants in S. cerevisiae. By comparing the growth of each double mutant strain with that of the corresponding single-deletion strain, we discovered an unexpected genetic interaction in which deletion of the PRE9 gene restored the growth of a strain lacking Fzo1, which has been shown to be essential for mitochondrial fusion (4, 8). Tetrad analysis confirmed that fzo1Δ pre9Δ cells grew better than fzo1Δ cells (Fig. 1A).
FIG 1.

Proteasome impairment restores growth of fzo1Δ cells. (A to E) Spores of heterozygous diploids were dissected on YPAD prior to growth at 26°C for 4 days. Genotypes of each colony are shown. (F) Suc-LLVY-MCA hydrolyzing activities of proteasome mutants used in panels A to E. Fluorescence intensities of cleaved MCA are normalized to that of wild-type cells. (G) Spores of the FZO1/fzo1Δ PDR5/pdr5Δ strain were dissected on YPAD containing dimethyl sulfoxide (DMSO) (vehicle, left panel) or proteasome inhibitor MG132 (right panel) prior to growth at 26°C for 4 days. (H) Spores of FZO1/fzo1Δ cells harboring empty vector or Fzo1-expressing plasmids were dissected on YPAD prior to growth at 26°C for 4 days. Genotypes of each colony are shown. (I) Spores of FZO1/fzo1Δ cells on the BY or W303 strain background were dissected on YPAD prior to growth at 26°C for 4 days. Cells lacking Fzo1 are indicated. (J) Spores of FZO1/fzo1ΔBY cells harboring Sal1BY- or Sal1W303-expressing plasmids were dissected on YPAD prior to growth at 26°C for 4 days. Genotypes of each colony are shown. (K) Spores of FZO1/fzo1ΔBY cells were dissected on YPAD or YPAGal prior to growth at 26°C for 4 days.
To examine whether the ability to restore the growth of the fzo1Δ strain is specific to pre9Δ or is also observed by crossing other proteasome mutants, we investigated the genetic interaction between fzo1Δ and deletion of other nonessential proteasome genes (Fig. 1B to E). Deletion of Rpn4, a transcription factor for proteasome genes, and deletion of Sem1, an RP subunit, rescued fzo1Δ cells. However, deletion of Rpn10 or Rpn13, both of which are RP subunits, did not. Single deletion of Rpn10 or Rpn13 has been shown to affect proteasome activity only modestly (27, 28). Indeed, while a 15 to 40% decrease in proteasome activity was observed in cells lacking PRE9, RPN4, or SEM1, the proteasome activities of rpn10Δ and rpn13Δ were not affected (Fig. 1F). These results suggest that fzo1Δ cells grow better when the proteasome activity is compromised.
To further verify this relationship, we tested whether chemical inhibition of the proteasome by MG132 rescues fzo1Δ cells. To decrease MG132 efflux, we deleted the PDR5 gene, which encodes an ABC transporter (29). Deletion of PDR5 did not affect the growth of fzo1Δ cells (Fig. 1G, left panel). In the pdr5Δ background, while the growth of wild-type (WT) cells was dampened in the presence of MG132, the growth of fzo1Δ cells was markedly restored by MG132 (Fig. 1G, right panel). These results demonstrate that proteasome impairment provides advantages for cell growth of a strain lacking Fzo1 and suggest a functional relationship between mitochondrial fusion and proteasome activity.
The Sal1 polymorphism affects growth of fzo1Δ cells.
The fzo1Δ cells we used showed severe growth defects on YPAD, where respiration is dispensable for yeast cell growth. We confirmed that expression of Fzo1 protein completely reversed the growth defect of fzo1Δ cells (Fig. 1H), indicating that this severe growth defect of the fzo1Δ strain we used in this study indeed resulted from the deletion of the FZO1 gene and not from an unidentified mutation in this strain. However, this result is inconsistent with a previous finding that fzo1Δ cells show only a slight growth defect on YPD, which is essentially the same as YPAD, except that extra adenine is not added (8). In contrast, another paper reported a severe growth defect of fzo1Δ cells on YPD (4). The former study used the W303 strain, while the latter study and we used the BY strain. We speculated that the differences in strain backgrounds caused this discrepancy. To test this possibility, we made an fzo1Δ strain in the W303 background (fzo1ΔW303) and examined the growth phenotype. As expected, fzo1ΔW303 cells grew better than an fzo1Δ strain in the BY background (fzo1ΔBY) cells (Fig. 1I). This result indicates that the growth difference in fzo1Δ cells reported in the previous studies resulted from different strain backgrounds.
Sal1, a mitochondrial ATP/ADP transporter that localizes at the inner membrane, is known to be polymorphic between the W303 and BY strains; Sal1 is functional in the W303 strain, while it is truncated and defective in the BY strain (30). We cloned SAL1 from both the W303 and BY strains (referred to as the SAL1W303 and SAL1BY strains, respectively) and introduced each type of SAL1 into the fzo1ΔBY strain. Expression of Sal1W303 recovered growth of the fzo1ΔBY strain, but Sal1BY did not (Fig. 1J, compare colonies indicated with dashed triangles). This result indicates that the lack of functional Sal1 is the cause of the severe growth defect of the fzo1ΔBY strain on YPAD.
We noticed that the fzo1ΔBY strain grew better on galactose-containing medium (YPAGal) than on glucose-containing medium (YPAD) (Fig. 1K). This result suggests that the type of carbon source regulates mitochondrial functions, although the precise reason for this phenotype remains to be clarified. Taking advantage of this phenotype, we used galactose-containing medium for maintenance and preculture of the fzo1ΔBY strain in the following studies.
Overexpression of Mia40 restores growth of fzo1Δ cells.
The observation that proteasome inhibition improved the growth of fzo1Δ cells raises the possibility that some proteins that are stabilized under proteasome impairment assist the growth of fzo1Δ cells. Therefore, we screened for a multicopy suppressor of fzo1Δ using a yeast genomic library to understand the molecular mechanism of the rescue. This screening identified the mitochondrial thiol oxidoreductase Mia40, which is one of the main components of the mitochondrial intermembrane space import and assembly (MIA) pathway and resides in the intermembrane space of mitochondria as a multicopy suppressor of the fzo1Δ strain (Fig. 2A, left panel). The sulfhydryl oxidase Erv1 plays an important role in the MIA pathway by oxidizing Mia40 (31, 32). We tested whether overexpression of Erv1 also suppresses the growth defect of the fzo1Δ strain. However, Erv1 overexpression did not restore the poor growth of fzo1Δ cells (Fig. 2A, right panel). This result implies that an increase in Mia40, rather than activation of the MIA pathway, is specifically required for the rescue mechanism.
FIG 2.

Proteasome defects increase Mia40. (A) Spores of FZO1/fzo1Δ cells harboring the Mia40 or Erv1 expression plasmid were dissected on YPAD prior to growth at 26°C for 4 days. Triangles and pentagons indicate fzo1Δ cells with or without the Mia40 or Erv1 expression plasmid, respectively. (B) Lysates of cells expressing chromosomally encoded Mia40-Flag were subjected to Western blotting, and proteins were detected using the indicated antibodies. Pgk1 and porin were used as cytosolic and mitochondrial loading controls, respectively. (C) Total RNA of the indicated strains was extracted. The amount of MIA40 mRNA was measured and standardized to TOM20 or ACT1 mRNA. TOM20 and ACT1 were used as mitochondrial and nonmitochondrial standards, respectively. (D) The indicated yeast cells were treated with Zymolyase 100T to digest the cell wall. Spheroplasts were homogenized, and the resultant homogenates were fractionated by differential centrifugation. Total, total homogenate; PMS, postmitochondrial supernatant; Mito, rough mitochondria. The total and rough mitochondrial fractions were diluted by 5-fold and 8-fold, respectively, so that Mia40-Flag in the PMS fraction could be detected. Pgk1 was used as a cytosolic loading control, and Om45-GFP was used as a mitochondrial loading control.
Next, we tested whether proteasome defects increased the protein level of endogenous Mia40 in the fzo1Δ mutant. All of the proteasome gene deletions that restored the growth of fzo1Δ cells, rpn4Δ, sem1Δ, and pre9Δ, considerably increased the amount of endogenous Mia40 on the fzo1Δ background (Fig. 2B). To see whether the increase in Mia40 protein was a result of an increase in mRNA expression of MIA40, we performed quantitative RT-PCR analysis using the expression levels of TOM20 and ACT1 as mitochondrial and cytoplasmic normalization controls, respectively. The MIA40 mRNA level in fzo1Δ rpn4Δ cells was comparable to that in fzo1Δ cells (Fig. 2C). In fzo1Δ sem1Δ and fzo1Δ pre9Δ cells, the MIA40 mRNA levels exhibited a 1.2- to 1.5-fold increase, but the extent was much smaller than that of the increase in Mia40 protein levels. These results suggest that proteasome defects restored growth of fzo1Δ cells by increasing Mia40 protein, presumably due to decreased degradation of Mia40 and in part due to upregulation of MIA40 transcription, which might be dependent on Rpn4.
Mia40 is synthesized as a precursor protein and processed after import into the intermembrane space (33). To determine whether Mia40 was increased as a mature protein in mitochondria, we fractionated lysates of fzo1Δ rpn4Δ cells to eliminate the effect of transcriptional upregulation of MIA40 mRNA (Fig. 2C). The mature form of Mia40 was increased in the mitochondrial fraction in fzo1Δ rpn4Δ cells compared to fzo1Δ cells (Fig. 2D). The precursor form of Mia40 was also increased in the postmitochondrial supernatant (PMS) of fzo1Δ rpn4Δ cells, although MIA40 mRNA was not increased in this strain (Fig. 2C and D). Considering that the proteasome, which does not localize inside mitochondria, can encounter Mia40 only in the cytosol, these results suggest that the proteasome regulates the amount of Mia40 in the cytosol and that the defect in proteasome activity leads to an increase in mitochondrial mature Mia40.
Mitochondrial DNA is not restored by proteasome defects or Mia40.
Cells lacking Fzo1 lose mitochondrial genomes (mitochondrial DNA [mtDNA]) (4, 8). Loss of mtDNA leads to a respiratory defect, which is manifested by an inability to grow on nonfermentable carbon sources such as glycerol. We next examined whether the proteasome defect and Mia40 overexpression restored mtDNA of fzo1Δ cells. DAPI (4′,6-diamidino-2-phenylindole) staining detected mtDNA as small dots around the nuclear DNA in wild-type cells but not in fzo1Δ cells (Fig. 3A). The lack of mtDNA in fzo1Δ cells was not suppressed by loss of α3/Pre9, Rpn4, or Sem1 (Fig. 3A). Additionally, overexpression of Mia40, which rescued growth of fzo1Δ cells (Fig. 2A), did not restore mtDNA, suggesting that neither proteasome defects nor Mia40 overexpression prevents mtDNA loss induced by fzo1Δ (Fig. 3A). Consistent with this observation, growth on glycerol-containing medium (YPAGly) was not restored by either proteasome defects or Mia40 overexpression, while growth on glucose medium (YPAD) was restored (Fig. 3B). These results suggest that proteasome defects or Mia40 overexpression does not contribute to maintenance of mtDNA and that mtDNA maintenance is not involved in growth restoration of fzo1Δ cells by proteasome impairment or Mia40 overexpression.
FIG 3.

Proteasome defects or Mia40 overexpression do not prevent mitochondrial DNA loss in fzo1Δ cells. (A) The indicated yeast cells were fixed with ethanol and stained with DAPI. Images merged by differential interference contrast (DIC) are also shown. Scale bars, 5 μm. (B) Spot dilution assay. The indicated strains were sequentially diluted by 5-fold and spotted onto YPAD or YPAGly. Plates were incubated at 26°C for 2 days.
Proteasome defects are beneficial for growth of [rho0] cells.
We next examined whether proteasome defects rescue not only growth of fzo1Δ cells but also other mitochondrial mutants. We found that the growth defects of cells lacking MRPL35, MIP1, or PPA2, which encode a mitochondrial ribosome subunit, mitochondrial DNA polymerase, or mitochondrial pyrophosphatase, respectively, were suppressed by proteasome defects (Fig. 4A). Overexpression of Mia40 also restored growth of mrpl35Δ cells, similar to the observation in fzo1Δ cells (Fig. 4B). Not all of the mitochondrial mutations are suppressed by proteasome defects, since sem1Δ did not rescue growth of cells lacking Ugo1, which is a mitochondrial outer membrane protein required for mitochondrial fusion (Fig. 4C).
FIG 4.

Proteasome defects rescue [rho0] cells and mitochondrial mutants other than fzo1Δ cells. (A and C) Spores of heterozygous diploids were dissected on YPAD prior to growth at 26°C for 4 days. Genotypes of each colony are shown. (B) Spores of MRPL35/mrpl35Δ cells harboring empty vector or Mia40-expressing plasmids were dissected on YPAD prior to growth at 26°C for 4 days. Genotypes of each colony are shown. (D) Wild-type and proteasome mutant cells were cultured on YPAGal containing 20 μg/ml ethidium bromide (EtBr) to deplete mitochondrial DNA. The resultant [rho0] cells and control (WT [rho+]) cells were sequentially diluted by 5-fold and spotted onto YPAD (left panels) or YPAGly (right panels). Plates were incubated at 26°C for 2 days.
Mutants lacking mitochondrial genes often lose mtDNA. We examined whether proteasome defects restore growth of cells lacking mtDNA ([rho0]) without any specific deletion of mitochondrial genes. [rho0] cells were generated by treating cells with ethidium bromide (EtBr). EtBr-treated wild-type cells (WT [rho0]) grew slower than wild-type cells containing mtDNA (WT [rho+]) on YPAD and did not grow on YPAGly, confirming respiratory defects (Fig. 4D, upper panels). However, rpn4Δ, sem1Δ, and pre9Δ cells depleted of mtDNA grew better than WT [rho0] cells on YPAD (Fig. 4D, upper panels). Overexpression of Mia40 also restored growth of [rho0] cells, similar to the observation in fzo1Δ and mrpl35Δ cells (Fig. 2A and Fig. 4B and D, lower panels). These results indicate that proteasome defects are also beneficial for chemically induced [rho0] cells and that rescue by proteasome impairment is not specific to fzo1Δ.
Deletion of Mia40 substrates does not compromise growth rescue of mitochondrial mutants by proteasome defects.
A previous report showed that Mia40 substrate proteins are increased under proteasome impairment (34). Therefore, we assumed that an increase in a Mia40 substrate was specifically required for the growth rescue of mitochondrial mutant cells. If a specific Mia40 substrate is required for the rescue mechanism, deletion of the protein would impair growth of mitochondrial mutants rescued by proteasome defects. Alternatively, overexpression of such proteins would rescue the mitochondrial mutants. However, deletion of nonessential Mia40 substrates such as Cmc1, Cmc3/Coa4, Cmc4, Cox17, Cox23, Emi1, Mix14, Mrp10, Pet191, and Som1 did not impair the growth rescue (Fig. 5A). In this experiment, we used mrpl35Δ sem1Δ cells as a strain that has both mitochondrial and proteasome defects, because the MRPL35 and SEM1 genes are located adjacently on the same chromosome, which is convenient for obtaining triple mutants by tetrad analysis. These results indicate that the 10 Mia40 substrates we tested are not specifically required for the rescue mechanism. We have not tested multiple deletions of the Mia40 substrates and therefore cannot exclude the possibility that these proteins function redundantly in the rescue mechanism.
FIG 5.

Mia40 substrates are not specifically required for growth rescue of mitochondrial mutants by proteasome defects. (A) Spores of heterozygous diploids were dissected on YPAD prior to growth at 26°C for 4 days. Genotypes of each colony are shown. (B) Spores of heterozygous diploids harboring empty vector or Tim9-Tim10 expression plasmid were dissected on YPAD prior to growth at 26°C for 4 days. Genotypes of each colony are shown.
We also tested overexpression of essential Mia40 substrates, Erv1 and the Tim9-Tim10 complex. However, we did not observe growth recovery of mitochondrial mutant cells by these proteins (Fig. 2A and 5B). From these results, we are unable to conclude that an increase in a specific Mia40 substrate is involved in the mechanism by which an increase in Mia40 rescues the growth of mitochondrial mutants.
Proteasome defects and Mia40 overexpression restore mitochondrial membrane potential.
It has been shown that mitochondria in fzo1Δ cells are fragmented due to impairment of fusion and have decreased mitochondrial membrane potential (ΔΨm) (35–37). Since ΔΨm is essential for mitochondrial functions such as ATP production, protein import, and protein synthesis (38, 39), we assumed that proteasome defects and Mia40 overexpression would recover ΔΨm in fzo1Δ cells. Accordingly, we measured ΔΨm using rhodamine 123, a ΔΨm-dependent mitochondrion-specific fluorescent dye (25). Consistent with a previous report using Candida albicans (37), fzo1Δ cells displayed a lower ΔΨm than wild-type cells (Fig. 6A). On the other hand, deletion of RPN4, SEM1, or PRE9 ameliorated ΔΨm of fzo1Δ cells (Fig. 6A). Overexpression of Mia40 also increased ΔΨm in fzo1Δ cells (Fig. 6B). Although we were unable to examine whether the increase in Mia40 is essential for recovery of ΔΨm of fzo1Δ cells by deleting MIA40, which is essential for cell viability, these results, together with the results in Fig. 2A and B, suggest that proteasome impairment ameliorates mitochondrial membrane potential by increasing Mia40 protein and that the recovery from low ΔΨm might be one of the causes of growth recovery of fzo1Δ cells mediated by proteasome impairment.
FIG 6.

Proteasome defects and Mia40 overexpression enhance mitochondrial membrane potential. (A to C) The indicated strains were stained with rhodamine 123, and fluorescence intensity was measured by FACS analysis. Mean fluorescence intensities ± standard deviations (SD) are shown. Asterisks indicate P < 0.01. (D) Proteasome activity of cells treated with antimycin A relative to control cells (100%). Proteasome activity was measured as in Fig. 1F. (E) Lysates of antimycin A-treated cells expressing chromosomally encoded Mia40-Flag and Om45-GFP were subjected to Western blotting using the indicated antibodies. Pgk1 was used as a loading control. (F) Mitochondrial membrane potential (ΔΨm) of normal and Rpn4-overexpressing cells treated with or without antimycin A. ΔΨm was measured as in panel A. (G) Spores of an FZO1/fzo1Δ ATG32/atg32Δ strain were dissected on YPAD prior to growth at 26°C for 4 days. Genotypes of each colony are shown. (H) Spores of FZO1/fzo1Δ SEM1/sem1Δ ATG32/atg32Δ strain were dissected on YPAD prior to growth at 26°C for 4 days. Genotypes of each colony are shown.
Several reports showed that Mia40 is functionally related to the respiratory chain, which generates ΔΨm (40–42). Mia40 transfers electrons from substrate proteins to cytochrome c and complex IV of the electron transport chain, which then pumps protons and generates ΔΨm, via a disulfide relay system. Thus, we hypothesized that Mia40 overexpression enhanced membrane potential by increasing electron flux into complex IV. To verify this hypothesis, we tested whether Mia40 overexpression is able to increase membrane potential in cells that lack subunits of complex IV, Cox12 or Cox7. Mia40 expression enhanced membrane potential only in wild-type and fzo1Δ cells but not in cox12Δ and cox7Δ cells (Fig. 6C). This result suggests that respiratory complex IV is required for membrane potential recovery by Mia40 overexpression.
Mitochondrial damage induces proteasome impairment followed by Mia40 increase.
We have shown that proteasome defects restored growth of fzo1Δ cells by increasing Mia40 and ΔΨm. To examine whether cells employ this mechanism upon mitochondrial damage other than by gene disruption, cells were treated with antimycin A, an inhibitor of electron transport chain complex III. Previous studies showed that mitochondrial impairment increases production of reactive oxygen species (ROS) (3, 43, 44), which then causes dissociation of the 26S proteasome and a decrease in proteasome activity (44–46). Consistent with these reports, treatment with antimycin A decreased proteasome activity in cells (Fig. 6D).
We next examined whether Mia40 was increased by antimycin A treatment. Green fluorescent protein (GFP)-fused mitochondrial outer membrane protein Om45-GFP was drastically decreased by antimycin A (Fig. 6E), indicating that the amount of mitochondria was decreased by mitochondrial damage. Nevertheless, the total amount of Mia40 was not decreased, which means that Mia40 per mitochondrion was increased by antimycin A treatment (Fig. 6E). This relative increase in Mia40 presumably contributed to enhanced ΔΨm in the antimycin A-treated cells compared to the nontreated cells (Fig. 6F). The increase in ΔΨm was cancelled by Rpn4 overexpression, which increases the proteasome amount and activity (47, 48) (Fig. 6F). These results suggest that proteasome impairment is essential for ΔΨm increase upon antimycin A treatment and that yeast cells are equipped with a protective mechanism by which cells decrease proteasome activity to increase Mia40 protein and enhance ΔΨm in response to mitochondrial damage.
Mitophagy is not involved in the rescue mechanism.
Decrease in ΔΨm has been shown to evoke mitophagy, the specific autophagic elimination of mitochondria, in mammalian cells (3, 49). In yeast cells, several reports showed that mitophagy is induced under mitochondrial impairment, such as temperature-sensitive mutation of Fmc1, which is required for assembly or stability of the F1 sector of F1Fo ATP synthase, and depletion of mitochondrial translational activator Mdm38 (50, 51).
We first predicted that deletion of FZO1 induced excess mitophagy and that restoration of ΔΨm by either proteasome impairment or Mia40 overexpression attenuated mitophagy, leading to recovered growth of fzo1Δ cells. Therefore, we tested whether inhibition of mitophagy by deleting ATG32, an essential gene for mitophagy in budding yeast (21, 52), also rescues the poor growth of fzo1Δ cells. However, fzo1Δ atg32Δ double mutant cells grew as poorly as fzo1Δ single mutant cells (Fig. 6G), suggesting that excess mitophagy is not the cause of poor growth of fzo1Δ cells.
We then speculated that mitophagy-mediated quality control of mitochondria, which might be promoted by proteasome impairment, is a cause of growth recovery of fzo1Δ cells. If so, mitophagy impairment by ATG32 deletion would inhibit growth restoration of fzo1Δ cells under proteasome impairment. However, restoration of growth of fzo1Δ by sem1Δ was not affected by atg32Δ (Fig. 6H). These results suggest that Atg32-dependent mitophagy is not involved in growth restoration of fzo1Δ cells by proteasome impairment.
Proteasome impairment promotes Fzo1-independent mitochondrial fusion.
ΔΨm, especially the proton gradient across the inner membrane, is essential for fusion of the mitochondrial outer membrane, where Fzo1 is required (53). Since proteasome impairment restored ΔΨm in fzo1Δ cells, we surmised that proteasome impairment enhances mitochondrial fusion even in the absence of Fzo1. To investigate mitochondrial fusion, cells expressing mitochondrially targeted GFP (mt-GFP) were mated to isogenic cells of opposite mating type that express mitochondrially targeted mCherry (mt-mCherry) (4, 21). Because the contents of fused mitochondria are mixed with each other, colocalization of mt-GFP and mt-mCherry at the same mitochondria assessed by overlay and morphology indicates fusion of mitochondria (Fig. 7A, top). To prevent excess fragmentation of mitochondria, Dnm1, a DRP for mitochondrial fission, was deleted.
FIG 7.
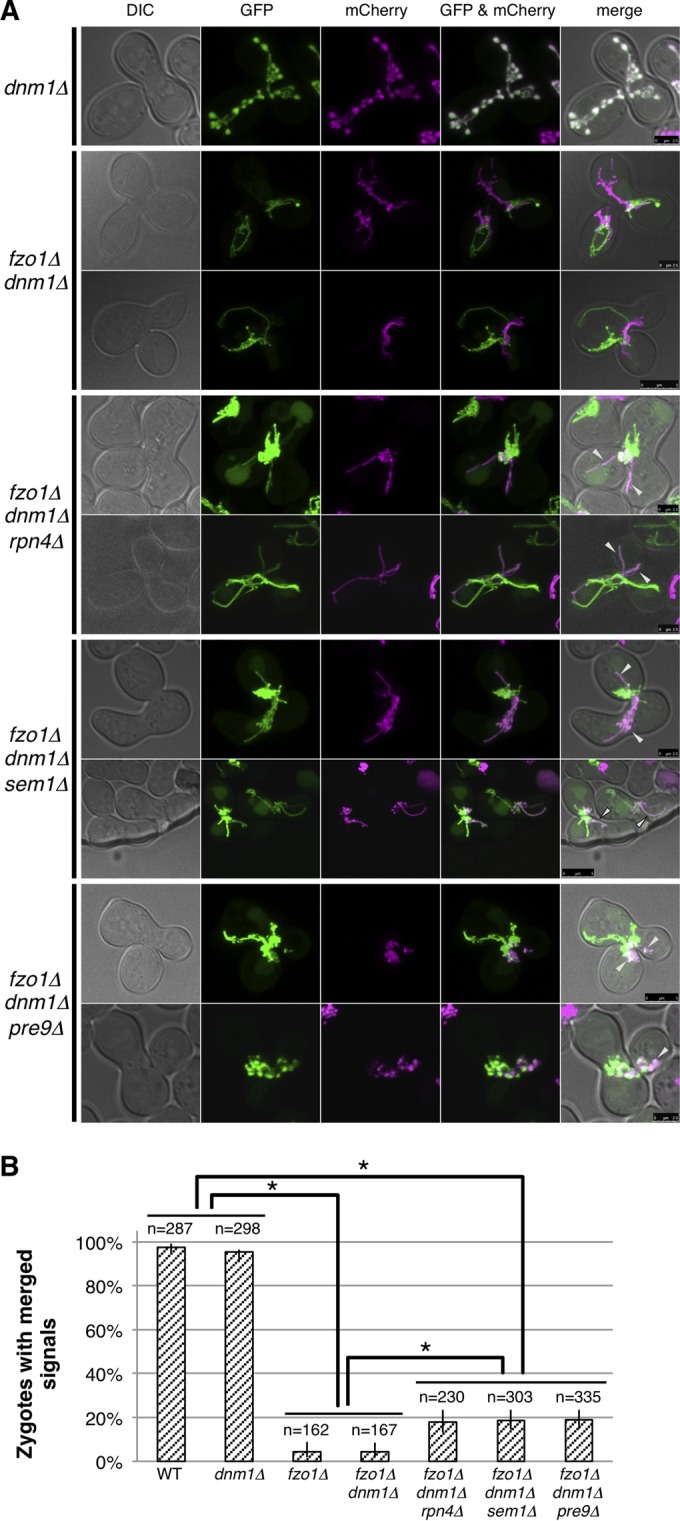
Proteasome defects enhance Fzo1-independent mitochondrial fusion. (A) Cells expressing mitochondrially targeted GFP were mated to cells with mitochondrially targeted mCherry on YPAD and observed by confocal microscopy after 4 h. Images of DIC, GFP, mCherry, GFP/mCherry were merged as shown in the panels from left to right. (B) Percentage of cells with merged signals in mitochondrial fusion assay and 95% confidence intervals are shown. Counted cell numbers are indicated. Fisher's exact test was performed. Asterisks indicate P < 0.01.
As reported previously (4, 54), mitochondrial fusion was compromised in fzo1Δ dnm1Δ cells (Fig. 7A, second and third rows). However, we found that proteasome defects (rpn4Δ, sem1Δ, and pre9Δ) increased colocalization of mt-GFP and mt-mCherry (Fig. 7A, arrowheads). We calculated the percentage of zygotes in which mt-GFP and mt-mCherry showed colocalization. The result revealed a slight but significant increase in mitochondrial fusion in rpn4Δ, sem1Δ, and pre9Δ cells on the fzo1Δ background (Fig. 7B). These results suggest that proteasome impairment enhances Fzo1-independent mitochondrial fusion.
The short isoform of Mgm1 triggers Fzo1-independent mitochondrial fusion.
Next, we tried to uncover the molecular mechanism of Fzo1-independent mitochondrial fusion. It was reported that Mgm1, the other DRP for mitochondrial fusion, is required for fusion of the mitochondrial outer membrane (55). Another paper reported that overexpression of a Mgm1 GTPase mutant triggers mitochondrial fragmentation and aggregation (5). Therefore, we assumed that Mgm1 might be involved in Fzo1-independent mitochondrial fusion. Indeed, we found that loss of Mgm1 interfered with Fzo1-independent mitochondrial fusion caused by proteasome defects on the fzo1Δ background (Fig. 8A).
FIG 8.

Overexpression of s-Mgm1 enhances Fzo1-independent mitochondrial fusion. (A) Percentage of cells with merged signals in mitochondrial fusion assay and 95% confidence intervals are shown. Counted cell numbers are indicated. Fisher's exact test was performed. Asterisks indicate P < 0.01. (B) Lysates of cells expressing chromosomally encoded Mgm1-Flag were subjected to Western blotting, and proteins were detected using the indicated antibodies. Pgk1 was used as a loading control. (C) Lysates of Mgm1-Flag-expressing cells harboring empty vector, the short isoform of Mgm1 (s-Mgm1)-Flag-, and full-length Mgm1 (FL-Mgm1)-Flag expression plasmids were subjected to Western blotting, and proteins were detected using the indicated antibodies. Pgk1 was used as loading control. The ratios of s-Mgm1 to l-Mgm1 were calculated. (D) The indicated yeast cells were treated with Zymolyase 100T to digest cell wall. Spheroplasts were homogenized, and the resultant homogenates were fractionated by differential centrifugation. Total, total homogenate; PMS, postmitochondrial supernatant; Mito, rough mitochondria. Pgk1 was used as a cytosolic loading control, and Om45-GFP was used as a mitochondrial loading control. (E) The mitochondrial fusion assay was performed as in Fig. 7A. FL, full length. (F) Percentages of cells with merged signals in the mitochondrial fusion assay and 95% confidence intervals are shown. Counted cell numbers are also indicated. Fisher's exact test was performed. Asterisks indicate P < 0.01. n.s., not significant.
Mgm1 protein has two isoforms. Following import into the mitochondria, the presequence of Mgm1 is cleaved to generate the long isoform of Mgm1 (l-Mgm1), which is inserted into the inner membrane. The transmembrane domain of l-Mgm1 is further processed by the rhomboid protease Pcp1, resulting in the short isoform of Mgm1 (s-Mgm1), which is released to the intermembrane space (56–58). Thus, we examined whether protein expression and processing of Mgm1 are altered by proteasome impairment. Western blot analysis detected two bands corresponding to l-Mgm1 and s-Mgm1, where the s-Mgm1/l-Mgm1 ratio was consistently increased in the absence of RPN4, SEM1, or PRE9 on the fzo1Δ background (Fig. 8B). Taken together, these results indicate that Mgm1 is essential for Fzo1-independent mitochondrial fusion and suggest that s-Mgm1, not l-Mgm1, is a key molecule in this process.
To test whether the increase in the s-Mgm1/l-Mgm1 ratio mediates Fzo1-independent mitochondrial fusion, we constructed full-length Mgm1 (FL-Mgm1) and s-Mgm1 overexpression plasmids and observed mitochondrial fusion of cells expressing FL-Mgm1 and s-Mgm1. The amount of plasmid-derived FL-Mgm1 and s-Mgm1 was much higher than endogenous levels (Fig. 8C). Ectopic expression of s-Mgm1 led to the increase in the s-Mgm1/l-Mgm1 ratio up to 319%, whereas FL-Mgm1 expression did not. We confirmed that ectopically expressed FL-Mgm1 and s-Mgm1 were correctly targeted to mitochondria by fractionation analysis (Fig. 8D). While there was no significant increase of mitochondrial fusion by expression of FL-Mgm1, s-Mgm1 expression promoted mitochondrial fusion (Fig. 8E, arrowheads). Counting of zygotes showing colocalization of mt-GFP and mt-mCherry confirmed that expression of s-Mgm1 significantly increased mitochondrial fusion in the absence of Fzo1 (Fig. 8F). We also tested whether overexpression of Mia40, which recovered ΔΨm and growth of fzo1Δ cells (Fig. 2A and 6B), promotes Fzo1-independent mitochondrial fusion. However, we could not detect a significant increase in the fusion by Mia40 expression (Fig. 8F). This result suggests that Mia40 contributes to the growth recovery of fzo1Δ cells independently of mitochondrial fusion.
We next tested whether the increase in the s-Mgm1/l-Mgm1 ratio is sufficient for the growth recovery of fzo1Δ cells. However, overexpression of s-Mgm1 did not restore cellular growth (Fig. 9A), the ability to utilize nonfermentable carbon source glycerol irrespective of the presence of Mia40 (Fig. 9B), and membrane potential in fzo1Δ cells (Fig. 9C), suggesting that s-Mgm1-mediated, Fzo1-independent mitochondrial fusion is not sufficient to support growth of fzo1Δ cells and that Mia40-dependent recovery of ΔΨm is a key event, at least with respect to growth recovery of fzo1Δ cells.
FIG 9.

Overexpression of s-Mgm1 does not restore fzo1Δ growth phenotypes. (A) Spores of FZO1/fzo1Δ cells harboring an s-Mgm1 expression plasmid were dissected on YPAD prior to growth at 26°C for 4 days. Genotypes of each colony are shown. The triangle and pentagon indicate fzo1Δ cells with or without the s-Mgm1 plasmid, respectively. (B) The indicated strains were streaked on YPAD and YPAGly prior to growth at 26°C for 3 days. (C) The indicated strains were stained with rhodamine 123, and fluorescence intensities were measured by FACS analysis. Mean fluorescence intensities ± SD are shown. Asterisks indicate P < 0.01. n.s., not significant.
DISCUSSION
In this study, we revealed that proteasome impairment ameliorates the poor growth of fzo1Δ cells. Through analysis of the molecular mechanism, we identified an increase in Mia40 upon proteasome impairment as a key process of this growth recovery. We also identified, for the first time, an Fzo1-independent mitochondrial fusion pathway mediated by s-Mgm1. Figure 10 summarizes our findings. Cells lacking Fzo1 show a severe growth defect accompanied by low ΔΨm and a mitochondrial fusion defect, each of which is restored by proteasome defects via distinct pathways.
FIG 10.
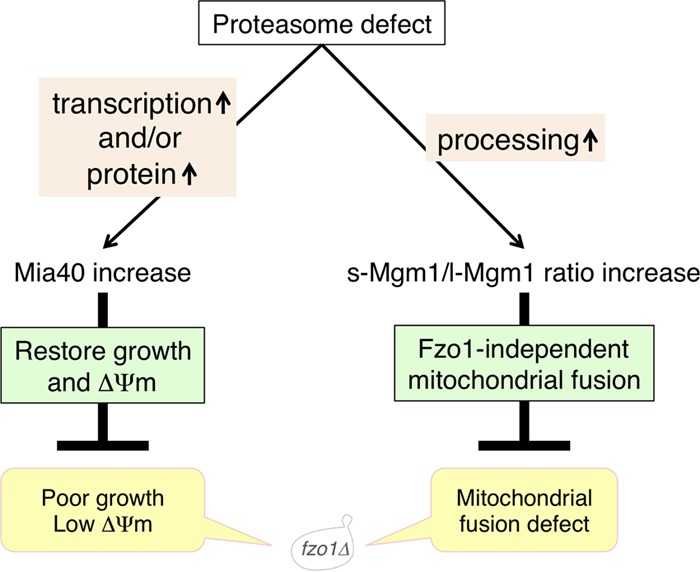
Novel two independent pathways for mitochondrial quality control induced by proteasome defects. Cells lacking Fzo1 show poor growth, low mitochondrial membrane potential (ΔΨm), and defects in mitochondrial fusion (bottom). Proteasome defects increase Mia40 (left pathway) and the s-Mgm1/l-Mgm1 ratio (right pathway) in fzo1Δ cells. Increased Mia40 restores poor growth and mitochondrial membrane potential. Increase of the s-Mgm1/l-Mgm1 ratio induces Fzo1-independent mitochondrial fusion.
Proteasome defects restore ΔΨm and cell growth by increasing Mia40 expression (Fig. 10, left pathway), although it remains unclear whether ΔΨm recovery is the cause of the growth rescue. This pathway may function as a protective mechanism for cells in response to mitochondrial damage, as shown in Fig. 6D to F. Dunn et al. previously reported that rpn4Δ and pre9Δ strains grew better than the wild-type strain after mitochondrial genome loss induced by EtBr (59). However, the reason why these proteasome mutants were highly resistant to EtBr remained an unanswered question. We confirmed that proteasome defects are beneficial for EtBr-induced [rho0] cells (Fig. 4D). Our finding that proteasome defects enhance ΔΨm via Mia40 increase may provide a mechanism by which proteasome mutants show resistance to EtBr. At the same time, Fzo1-independent mitochondrial fusion is manifested by proteasome defects, which is mediated by an increase in the s-Mgm1/l-Mgm1 ratio (Fig. 10, right pathway). These two pathways seem to be separable because Fzo1-independent mitochondrial fusion is not sufficient to recover growth and ΔΨm of fzo1Δ cells (Fig. 9) and because expression of Mia40 did not significantly enhance Fzo1-independent mitochondrial fusion (Fig. 8F). We showed that an elevated level of Mia40 increased ΔΨm in fzo1Δ cells and provided a benefit in growth (Fig. 2A and 6B), but overexpression of Erv1, which is required for Mia40 function, did not rescue fzo1Δ cells (Fig. 2A). There are two possibilities. (i) There is an alternative, Erv1-independent pathway in which Mia40 transport electrons to the respiratory complex, although it is difficult to verify this model because Erv1 is essential for cell viability, making it impossible to obtain and analyze erv1Δ cells. (ii) Erv1 is not a rate-limiting factor of the disulfide relay system that transfers electrons from Mia40 to respiratory complex IV. Erv1 oxidizes the reduced form of Mia40 and receives electrons. However, Mia40 seems to be highly oxidized, and the reduced form of Mia40 was not always detected in a previous study (42), suggesting that Erv1 overexpression might not increase electron flux from Mia40 to respiratory complex IV.
Our results also suggest that Rpn4, which is a transcription factor stabilized under proteasome impairment that activates transcription of multiple genes, including the proteasome (47, 48), promotes transcription of MIA40 (Fig. 2C). Indeed, the promoter region of MIA40 contains a reported Rpn4-binding sequence, 5′-CGCCACC-3′ (60–62). A previous report showed that Mia40 substrates were increased under proteasome impairment, while an increase in Mia40 itself under proteasome impairment was not clearly shown (34). Our results showed that proteasome defects increased Mia40 in both transcriptional and protein levels (Fig. 2B and C). Considering that damaged proteins are accumulated in mitochondria when the proteasome is inhibited (63, 64), induction of Mia40 is a rational response to maintain the quality of mitochondria under proteasome impairment.
Fzo1 has been believed to be essential for mitochondrial fusion (4, 65, 66). To our surprise, however, proteasome defects increased mitochondrial fusion in cells lacking Fzo1, suggesting that Fzo1 is not absolutely necessary for mitochondrial fusion. This Fzo1-independent fusion is the first report of a specific function of the Mgm1 short isoform. A previous study suggested that the two isoforms of Mgm1, l-Mgm1 and s-Mgm1, have distinct functions, although the details remained unclear (67). s-Mgm1 was enriched in the outer membrane and inner boundary membrane, while l-Mgm1 mainly localized to the crista membrane (67), suggesting a specific function of s-Mgm1 associated with the outer membrane. Since s-Mgm1 and l-Mgm1 bind to each other (67), l-Mgm1 potentially inhibits s-Mgm1. Consistent with this view, a high s-Mgm1/l-Mgm1 ratio was associated with mitochondrial fusion: “free” s-Mgm1 that is not bound to l-Mgm1 and liberated from inner membrane promoted Fzo1-independent mitochondrial fusion (Fig. 8C to F). Supporting this idea, the s-Mgm1/l-Mgm1 ratio was higher than 300% in s-Mgm1-expressing or proteasome mutant fzo1Δ cells (Fig. 8B and C). In contrast, the ratio was lower than 300% in cells where Fzo1-independent fusion was not observed (Fig. 8B to F). This result demonstrated a correlation between Mgm1 processing and Fzo1-independent fusion. However, the biological significance of Fzo1-independent mitochondrial fusion is still unclear, because Fzo1-independent fusion cannot support growth and respiration of fzo1Δ cells (Fig. 9A and B). Moreover, how s-Mgm1, which localizes to the mitochondrial intermembrane space, affects the fusion of the mitochondrial outer membrane remains elusive.
Although Fzo1-independent fusion occurs at most in 20% of the population of proteasome-defective cells, this unconventional fusion event is worthy of further investigation. Impairment in mitochondrial fusion has been suggested to be involved in the mechanisms of human diseases (68, 69). The Fzo1-independent fusion mechanism identified in this study may therefore provide a new understanding of diseases associated with mitochondrial dynamics.
ACKNOWLEDGMENTS
We thank C. Boone (University of Toronto), K. Okamoto (Osaka University), and Y. Ohya (University of Tokyo) for providing the p4339 plasmid and Y8205 yeast strain, the mt-GFP and mt-mCherry constructs, and the pYO325 and pYO326 plasmids, respectively, and Y. Sakurai for critical reading.
This work was supported by grants from the Ministry of Education, Science and Culture of Japan to S.M.
REFERENCES
- 1.Hermann GJ, Shaw JM. 1998. Mitochondrial dynamics in yeast. Annu Rev Cell Dev Biol 14:265–303. doi: 10.1146/annurev.cellbio.14.1.265. [DOI] [PubMed] [Google Scholar]
- 2.Okamoto K, Shaw JM. 2005. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet 39:503–536. doi: 10.1146/annurev.genet.38.072902.093019. [DOI] [PubMed] [Google Scholar]
- 3.Youle RJ, van der Bliek AM. 2012. Mitochondrial fission, fusion, and stress. Science 337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hermann GJ, Thatcher JW, Mills JP, Hales KG, Fuller MT, Nunnari J, Shaw JM. 1998. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J Cell Biol 143:359–373. doi: 10.1083/jcb.143.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong ED, Wagner JA, Scott SV, Okreglak V, Holewinske TJ, Cassidy-Stone A, Nunnari J. 2003. The intramitochondrial dynamin-related GTPase, Mgm1p, is a component of a protein complex that mediates mitochondrial fusion. J Cell Biol 160:303–311. doi: 10.1083/jcb.200209015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sesaki H, Jensen RE. 2004. Ugo1p links the Fzo1p and Mgm1p GTPases for mitochondrial fusion. J Biol Chem 279:28298–28303. doi: 10.1074/jbc.M401363200. [DOI] [PubMed] [Google Scholar]
- 7.Escobar-Henriques M, Anton F. 2013. Mechanistic perspective of mitochondrial fusion: tubulation vs. fragmentation. Biochim Biophys Acta 1833:162–175. doi: 10.1016/j.bbamcr.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 8.Rapaport D, Brunner M, Neupert W, Westermann B. 1998. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae. J Biol Chem 273:20150–20155. doi: 10.1074/jbc.273.32.20150. [DOI] [PubMed] [Google Scholar]
- 9.Rinaldi T, Pick E, Gambadoro A, Zilli S, Maytal-Kivity V, Frontali L, Glickman MH. 2004. Participation of the proteasomal lid subunit Rpn11 in mitochondrial morphology and function is mapped to a distinct C-terminal domain. Biochem J 381:275–285. doi: 10.1042/BJ20040008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rinaldi T, Hofmann L, Gambadoro A, Cossard R, Livnat-Levanon N, Glickman MH, Frontali L, Cedex O. 2008. Dissection of the carboxyl-terminal domain of the proteasomal subunit Rpn11 in maintenance of mitochondrial structure and function. Mol Biol Cell 19:1022–1031. doi: 10.1091/mbc.E07-07-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tar K, Dange T, Yang C, Yao Y, Bulteau A, Salcedo F, Braigen S, Finley D, Schmidt M, Fernandez Salcedo E, Bouillaud F. 2014. Proteasomes associated with the Blm10 activator protein antagonize mitochondrial fission through degradation of the fission protein Dnm1. J Biol Chem 289:12145–12156. doi: 10.1074/jbc.M114.554105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen MM, Amiott EA, Day AR, Leboucher GP, Pryce EN, Glickman MH, McCaffery JM, Shaw JM, Weissman AM. 2011. Sequential requirements for the GTPase domain of the mitofusin Fzo1 and the ubiquitin ligase SCFMdm30 in mitochondrial outer membrane fusion. J Cell Sci 124:1403–1410. doi: 10.1242/jcs.079293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anton F, Fres JM, Schauss A, Pinson B, Praefcke GJK, Langer T, Escobar-Henriques M. 2011. Ugo1 and Mdm30 act sequentially during Fzo1-mediated mitochondrial outer membrane fusion. J Cell Sci 124:1126–1135. doi: 10.1242/jcs.073080. [DOI] [PubMed] [Google Scholar]
- 14.Anton F, Dittmar G, Langer T, Escobar-Henriques M. 2013. Two deubiquitylases act on mitofusin and regulate mitochondrial fusion along independent pathways. Mol Cell 49:487–498. doi: 10.1016/j.molcel.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 15.Hershko A, Ciechanover A. 1998. The ubiquitin system. Annu Rev Biochem 67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 16.Baumeister W, Walz J, Zühl F, Seemüller E. 1998. The proteasome: paradigm of a self-compartmentalizing protease. Cell 92:367–380. doi: 10.1016/S0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 17.Qadota H, Ishii I, Fujiyama A, Ohya Y, Anraku Y. 1992. RHO gene products, putative small GTP-binding proteins, are important for activation of the CAL1/CDC43 gene product, a protein geranylgeranyltransferase in Saccharomyces cerevisiae. Yeast 8:735–741. doi: 10.1002/yea.320080906. [DOI] [PubMed] [Google Scholar]
- 18.Yashiroda H, Tanaka K. 2004. Hub1 is an essential ubiquitin-like protein without functioning as a typical modifier in fission yeast. Genes Cells 9:1189–1197. doi: 10.1111/j.1365-2443.2004.00807.x. [DOI] [PubMed] [Google Scholar]
- 19.Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953–961. [DOI] [PubMed] [Google Scholar]
- 20.Tong AHY, Boone C. 2007. High-throughput strain construction and systematic synthetic lethal screening in Saccharomyces cerevisiae. Methods Microbiol 36:369–386. doi: 10.1016/S0580-9517(06)36016-3. [DOI] [Google Scholar]
- 21.Okamoto K, Kondo-Okamoto N, Ohsumi Y. 2009. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 22.Yashiroda H, Mizushima T, Okamoto K, Kameyama T, Hayashi H, Kishimoto T, Niwa S, Kasahara M, Kurimoto E, Sakata E, Takagi K, Suzuki A, Hirano Y, Murata S, Kato K, Yamane T, Tanaka K. 2008. Crystal structure of a chaperone complex that contributes to the assembly of yeast 20S proteasomes. Nat Struct Mol Biol 15:228–236. doi: 10.1038/nsmb.1386. [DOI] [PubMed] [Google Scholar]
- 23.Gregg C, Kyryakov P, Titorenko VI. 24 August 2009. Purification of mitochondria from yeast cells. J Vis Exp doi: 10.3791/1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kimura Y, Koitabashi S, Kakizuka A, Fujita T. 2001. Initial process of polyglutamine aggregate formation in vivo. Genes Cells 6:887–897. doi: 10.1046/j.1365-2443.2001.00472.x. [DOI] [PubMed] [Google Scholar]
- 25.Ludovico P, Sansonetty F, Co M, Côrte-Real M. 2001. Assessment of mitochondrial membrane potential in yeast cell populations by flow cytometry. Microbiology 147:3335–3343. doi: 10.1099/00221287-147-12-3335. [DOI] [PubMed] [Google Scholar]
- 26.Velichutina I, Connerly PL, Arendt CS, Li X, Hochstrasser M. 2004. Plasticity in eucaryotic 20S proteasome ring assembly revealed by a subunit deletion in yeast. EMBO J 23:500–510. doi: 10.1038/sj.emboj.7600059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glickman MH, Rubin DM, Coux O, Wefes I, Pfeifer G, Cjeka Z, Baumeister W, Fried VA, Finley D. 1998. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell 94:615–623. doi: 10.1016/S0092-8674(00)81603-7. [DOI] [PubMed] [Google Scholar]
- 28.Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, Dikic I. 2008. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453:481–488. doi: 10.1038/nature06926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haarer B, Aggeli D, Viggiano S, Burke DJ, Amberg DC. 2011. Novel interactions between actin and the proteasome revealed by complex haploinsufficiency. PLoS Genet 7:e1002288. doi: 10.1371/journal.pgen.1002288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen XJ. 2004. Sal1p, a calcium-dependent carrier protein that suppresses an essential cellular function associated with the Aac2 isoform of ADP/ATP translocase in Saccharomyces cerevisiae. Genetics 167:607–617. doi: 10.1534/genetics.103.023655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mokranjac D, Neupert W. 2009. Thirty years of protein translocation into mitochondria: unexpectedly complex and still puzzling. Biochim Biophys Acta 1793:33–41. doi: 10.1016/j.bbamcr.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 32.Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. 2009. Importing mitochondrial proteins: machineries and mechanisms. Cell 138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Terziyska N, Lutz T, Kozany C, Mokranjac D, Mesecke N, Neupert W, Herrmann JM, Hell K. 2005. Mia40, a novel factor for protein import into the intermembrane space of mitochondria is able to bind metal ions. FEBS Lett 579:179–184. doi: 10.1016/j.febslet.2004.11.072. [DOI] [PubMed] [Google Scholar]
- 34.Bragoszewski P, Gornicka AA, Sztolsztener ME, Chacinska A. 2013. The ubiquitin-proteasome system regulates mitochondrial intermembrane space proteins. Mol Cell Biol 33:2136–2148. doi: 10.1128/MCB.01579-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sugioka R, Shimizu S, Tsujimoto Y. 2004. Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem 279:52726–52734. doi: 10.1074/jbc.M408910200. [DOI] [PubMed] [Google Scholar]
- 36.Gerstenberger JP, Occhipinti P, Amy S, Gladfelter AS. 2012. Heterogeneity in mitochondrial morphology and membrane potential is independent of the nuclear division cycle in multinucleate fungal cells. Eukaryot Cell 11:353–367. doi: 10.1128/EC.05257-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas E, Roman E, Claypool S, Manzoor N, Pla J, Panwar SL. 2013. Mitochondria influence CDR1 efflux pump activity, Hog1-mediated oxidative stress pathway, iron homeostasis, and ergosterol levels in Candida albicans. Antimicrob Agents Chemother 57:5580–5599. doi: 10.1128/AAC.00889-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen XJ, Clark-Walker GD. 2000. The petite mutation in yeasts: 50 years on. Int Rev Cytol 194:197–238. [DOI] [PubMed] [Google Scholar]
- 39.Chen L. 1988. Mitochondrial membrane potential in living cells. Annu Rev Cell Biol 4:155–181. doi: 10.1146/annurev.cb.04.110188.001103. [DOI] [PubMed] [Google Scholar]
- 40.Allen S, Balabanidou V, Sideris DP, Lisowsky T, Tokatlidis K. 2005. Erv1 mediates the Mia40-dependent protein import pathway and provides a functional link to the respiratory chain by shuttling electrons to cytochrome c. J Mol Biol 353:937–944. doi: 10.1016/j.jmb.2005.08.049. [DOI] [PubMed] [Google Scholar]
- 41.Dabir DV, Leverich EP, Kim S, Tsai FD, Hirasawa M, Knaff DB, Koehler CM. 2007. A role for cytochrome c and cytochrome c peroxidase in electron shuttling from Erv1. EMBO J 26:4801–4811. doi: 10.1038/sj.emboj.7601909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bihlmaier K, Mesecke N, Terziyska N, Bien M, Hell K, Herrmann JM. 2007. The disulfide relay system of mitochondria is connected to the respiratory chain. J Cell Biol 179:389–395. doi: 10.1083/jcb.200707123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Balaban RS, Nemoto S, Finkel T. 2005. Mitochondria, oxidants, and aging. Cell 120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 44.Livnat-Levanon N, Kevei E, Kleifeld O, Krutauz D, Segref A, Rinaldi T, Erpapazoglou Z, Cohen M, Reis N, Hoppe T, Glickman MH. 2014. Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep 7:1371–1380. doi: 10.1016/j.celrep.2014.04.030. [DOI] [PubMed] [Google Scholar]
- 45.Brohem C, Massaro R, Tiago M, Marinho C, Jasiulionis M, de Almeida R, Rivelli D, Albuquerque R, de Oliveira T, de Melo Loureiro A, Okada S, Soengas M, de Moraes Barros S, Maria-Engler S. 2012. Proteasome inhibition and ROS generation by 4-nerolidylcatechol induces melanoma cell death. Pigment Cell Melanoma Res 25:354–369. doi: 10.1111/j.1755-148X.2012.00992.x. [DOI] [PubMed] [Google Scholar]
- 46.Wang X, Yen J, Kaiser P, Huang L. 2010. Regulation of the 26S proteasome complex during oxidative stress. Sci Signal 3:ra88. doi: 10.1126/scisignal.2001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mannhaupt G, Schnall R, Karpov V, Vetter I, Feldmann H. 1999. Rpn4p acts as a transcription factor by binding to PACE, a nonamer box found upstream of 26S proteasomal and other genes in yeast. FEBS Lett 450:27–34. doi: 10.1016/S0014-5793(99)00467-6. [DOI] [PubMed] [Google Scholar]
- 48.Xie Y, Varshavsky A. 2001. RPN4 is a ligand, substrate, and transcriptional regulator of the 26S proteasome: a negative feedback circuit. Proc Natl Acad Sci USA 98:3056–3061. doi: 10.1073/pnas.071022298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanki T, Klionsky DJ, Okamoto K. 2011. Mitochondria autophagy in yeast. Antioxid Redox Signal 14:1989–2001. doi: 10.1089/ars.2010.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Priault M, Salin B, Schaeffer J, Vallette FM, di Rago J-P, Martinou J-C. 2005. Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ 12:1613–1621. doi: 10.1038/sj.cdd.4401697. [DOI] [PubMed] [Google Scholar]
- 51.Nowikovsky K, Reipert S, Devenish RJ, Schweyen RJ. 2007. Mdm38 protein depletion causes loss of mitochondrial K+/H+ exchange activity, osmotic swelling and mitophagy. Cell Death Differ 14:1647–1656. doi: 10.1038/sj.cdd.4402167. [DOI] [PubMed] [Google Scholar]
- 52.Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. 2009. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 17:98–109. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meeusen S, McCaffery JM, Nunnari J. 2004. Mitochondrial fusion intermediates revealed in vitro. Science 305:1747–1752. doi: 10.1126/science.1100612. [DOI] [PubMed] [Google Scholar]
- 54.Wong ED, Wagner JA, Gorsich SW, McCaffery JM, Shaw JM, Nunnari J. 2000. The dynamin-related GTPase, Mgm1p, is an intermembrane space protein required for maintenance of fusion competent mitochondria. J Cell Biol 151:341–352. doi: 10.1083/jcb.151.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sesaki H, Southard SM, Yaffe MP, Jensen RE. 2003. Mgm1p, a dynamin-related GTPase, is essential for fusion of the mitochondrial outer membrane. Mol Biol Cell 14:2342–2356. doi: 10.1091/mbc.E02-12-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herlan M, Bornhövd C, Hell K, Neupert W, Reichert AS. 2004. Alternative topogenesis of Mgm1 and mitochondrial morphology depend on ATP and a functional import motor. J Cell Biol 165:167–173. doi: 10.1083/jcb.200403022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McQuibban GA, Saurya S, Freeman M. 2003. Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature 423:537–541. doi: 10.1038/nature01633. [DOI] [PubMed] [Google Scholar]
- 58.Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, Meisinger C. 2014. The protein import machinery of mitochondria—a regulatory hub in metabolism, stress, and disease. Cell Metab 19:357–372. doi: 10.1016/j.cmet.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 59.Dunn CD, Lee MS, Spencer FA, Jensen RE. 2006. A genomewide screen for petite-negative yeast strains yields a new subunit of the i-AAA protease complex. Mol Biol Cell 17:213–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Badis G, Chan ET, van Bakel H, Pena-Castillo L, Tillo D, Tsui K, Carlson CD, Gossett AJ, Hasinoff MJ, Warren CL, Gebbia M, Talukder S, Yang A, Mnaimneh S, Terterov D, Coburn D, Li Yeo A, Yeo ZX, Clarke ND, Lieb JD, Ansari AZ, Nislow C, Hughes TR. 2008. A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol Cell 32:878–887. doi: 10.1016/j.molcel.2008.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu C, Byers KJ, McCord RP, Shi Z, Berger MF, Newburger DE, Saulrieta K, Smith Z, Shah MV, Radhakrishnan M, Philippakis AA, Hu Y, De Masi F, Pacek M, Rolfs A, Murthy T, Labaer J, Bulyk ML. 2009. High-resolution DNA-binding specificity analysis of yeast transcription factors. Genome Res 19:556–566. doi: 10.1101/gr.090233.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shirozu R, Yashiroda H, Murata S. 2015. Identification of minimum Rpn4-responsive elements in genes related to proteasome functions. FEBS Lett 589:933–940. doi: 10.1016/j.febslet.2015.02.025. [DOI] [PubMed] [Google Scholar]
- 63.Livnat-Levanon N, Glickman MH. 2011. Ubiquitin-proteasome system and mitochondria-reciprocity. Biochim Biophys Acta 1809:80–87. doi: 10.1016/j.bbagrm.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 64.Tatsuta T, Langer T. 2008. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J 27:306–314. doi: 10.1038/sj.emboj.7601972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoppins S, Nunnari J. 2009. The molecular mechanism of mitochondrial fusion. Biochim Biophys Acta 1793:20–26. doi: 10.1016/j.bbamcr.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 66.Chan DC. 2006. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol 22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 67.Zick M, Duvezin-Caubet S, Schäfer A, Vogel F, Neupert W, Reichert AS. 2009. Distinct roles of the two isoforms of the dynamin-like GTPase Mgm1 in mitochondrial fusion. FEBS Lett 583:2237–2243. doi: 10.1016/j.febslet.2009.05.053. [DOI] [PubMed] [Google Scholar]
- 68.Santel A. 2006. Get the balance right: mitofusins roles in health and disease. Biochim Biophys Acta 1763:490–499. doi: 10.1016/j.bbamcr.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 69.Archer SL. 2013. Mitochondrial dynamics—mitochondrial fission and fusion in human diseases. N Engl J Med 369:2236–2251. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 70.Kondo-Okamoto N, Noda NN, Suzuki SW, Nakatogawa H, Takahashi I, Matsunami M, Hashimoto A, Inagaki F, Ohsumi Y, Okamoto K. 2012. Autophagy-related protein 32 acts as an autophagic degron and directly initiates mitophagy. J Biol Chem 287:10631–10638. doi: 10.1074/jbc.M111.299917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goldstein AL, McCusker JH. 1999. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 15:1541–1553. [DOI] [PubMed] [Google Scholar]
- 72.Gietz R, Sugino A. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- 73.Sikorski R, Hieter P. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]