

Abstract
The Keap1-Nrf2 system plays a central role in cytoprotection against electrophilic/oxidative stresses. Although Cys151, Cys273, and Cys288 of Keap1 are major sensor cysteine residues for detecting these stresses, it has not been technically feasible to evaluate the functionality of Cys273 or Cys288, since Keap1 mutants that harbor substitutions in these residues and maintain the ability to repress Nrf2 accumulation do not exist. To overcome this problem, we systematically introduced amino acid substitutions into Cys273/Cys288 and finally identified Cys273Trp and Cys288Glu mutations that do not affect Keap1's ability to repress Nrf2 accumulation. Utilizing these Keap1 mutants, we generated stable murine embryonic fibroblast (MEF) cell lines and knock-in mouse lines. Our analyses with the MEFs and peritoneal macrophages from the knock-in mice revealed that three major cysteine residues, Cys151, Cys273, and Cys288, individually and/or redundantly act as sensors. Based on the functional necessity of these three cysteine residues, we categorized chemical inducers of Nrf2 into four classes. Class I and II utilizes Cys151 and Cys288, respectively, while class III requires all three residues (Cys151/Cys273/Cys288), while class IV inducers function independently of all three of these cysteine residues. This study thus demonstrates that Keap1 utilizes multiple cysteine residues specifically and/or collaboratively as sensors for the detection of a wide range of environmental stresses.
INTRODUCTION
Transcription factor Nrf2 (NF-E2-related factor 2) plays a central role in cytoprotection against electrophilic and oxidative insults (1). Under basal unstressed conditions, Nrf2 protein level is maintained at a low level, as Nrf2 is constitutively ubiquitinated by Keap1 (Kelch-like ECH-associated protein 1), an adaptor component of Cul3 (cullin 3)-based ubiquitin E3 ligase complex, resulting in its proteasomal degradation (2, 3). Upon exposure to electrophiles or reactive oxygen species, Nrf2 ubiquitination ceases, leading to the stabilization and nuclear translocation/accumulation of Nrf2, followed by the inducible expression of Nrf2 target genes (4–8).
A variety of Nrf2 inducers have been reported, most of which are electrophilic and readily react with cysteine thiols of Keap1 (9). Keap1 is a cysteine-rich protein possessing 27 and 25 cysteine residues in the human and mouse proteins, respectively. A number of in vitro labeling and mass spectrometry studies have detected covalent modifications of some of the cysteine residues upon exposures of Keap1 to electrophiles (10–17). The functional significance of these cysteine residues has been examined in experimental systems exploiting site-directed mutagenesis of Keap1. The function of mutant Keap1 proteins has been tested by means of reporter cotransfection transactivation experiments with culture cells (in transfecto) (18–20) or by ectopic overexpression experiments in zebrafish embryos (14).
In these analyses, Cys273/Cys288 (10) and Cys151 (18) have emerged as important cysteine residues of Keap1. We also have verified the critical contribution of these three cysteine residues in vivo for the inducible accumulation of Nrf2 through generating transgenic mouse lines expressing cysteine mutant Keap1 proteins and conducting transgenic complementation rescue experiments (21). Of the three cysteine residues, the importance of Cys151 as a sensor for certain specific Nrf2 inducers has been verified (14, 18, 20–22). Utilizing mouse embryonic fibroblasts (MEFs) and peritoneal macrophages from the transgenic mice that complementarily express a Keap1 mutant harboring serine substitution for Cys151, which is referred to as Keap1C151S, the Cys151 residue has been found to be indispensable for the Nrf2 accumulation in response to tert-butyl hydroquinone (tBHQ), diethylmaleate (DEM), sulforaphane (SFN), and dimethylfumarate (DMF) (22). Importantly, we also have observed that the Cys151 mutation does not affect Nrf2 accumulation in response to 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), 9-nitro-octadec-9-enoic acid (9-OA-NO2), and cadmium chloride (CdCl2).
It has been proposed that cysteine residues other than Cys151, especially Cys273 and Cys288, may also contribute to the stress sensor function of Keap1. Indeed, Cys273 and Cys288 residues have been suggested to be reactive to 15d-PGJ2, prostaglandin A2 (PGA2), 9-OA-NO2 and dexamethasone 21-mesylate (Dex-Mes) (10, 14–16). However, there exists a technical difficulty to validate this notion. Since serine or alanine mutations of Cys273 and Cys288 fail to repress Nrf2 activity in reporter cotransfection transactivation assays (18, 19) and transgenic complementation rescue mouse experiments (21), it has not been feasible to evaluate the sensor function of Cys273 and Cys288 residues in Keap1.
To overcome this problem, we systematically introduced amino acid substitutions of the Cys273 and Cys288 residues of Keap1 and successfully identified amino acid residues that do not affect Keap1’s ability to target Nrf2 for degradation in the basal state. We have evaluated these permissive amino acid residues and prepared stable cell lines harboring the substitution mutations in Keap1 which allow it to maintain its basal activity. Since we have identified replacements of Cys273 with tryptophan and Cys288 with glutamate that preserve Keap1 activity, we have established Keap1C273W, Keap1C288E, and Keap1C273W&C288E mutant MEFs utilizing a Keap1-null background. Our experiments utilizing these MEFs demonstrate that Keap1 Cys288 acts as a sensor for 15d-PGJ2. We verified this notion by establishing Keap1C288E knock-in mouse lines. Our results also revealed that the three major cysteine residues, i.e., Cys151, Cys273, and Cys288, act individually and/or redundantly as sensors for various electrophiles. In addition, we present solid lines of evidence that 1-[2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole (CDDO-Im) is a Cys151-preferring inducer. We unequivocally demonstrate here that Keap1 utilizes multiple cysteine residues as sensors to trigger the cytoprotective response governed by the Keap1-Nrf2 system and that the three major cysteine residues of Keap1 indeed act specifically and/or collaboratively as sensors.
MATERIALS AND METHODS
Chemical reagents.
Diethylmaleate (DEM), sodium metaarsenite (NaAsO2), cadmium chloride (CdCl2), zinc chloride (ZnCl2), dexamethasone 21-mesylate (Dex-Mes), and hydrogen peroxide (H2O2) were from Wako Chemicals. 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2), prostaglandin A2 (PGA2), and 9-nitro-octadec-9-enoic acid (9-OA-NO2) were from Cayman Chemical. l-Sulforaphane (SFN) and tert-butyl hydroquinone (tBHQ) were from Sigma-Aldrich. 4-Hydroxy-nonenal (4-HNE) and (±)-S-nitroso-N-acetylpenicillamine (SNAP) were from Santa Cruz and Biomol, respectively. CDDO-Im was kindly provided by Mochida Pharmaceuticals Company, Ltd.
Plasmid construction.
All 19 amino acid substitutions were introduced by PCR into Cys273 and Cys288 sites of human KEAP1 subcloned into simian immunodeficiency virus (SV40) promoter-driven expression vector. The N-terminal domain of human NRF2 was fused to LacZ cDNA (NRF2NT-LacZ) and inserted into an SV40 promoter-driven expression vector (23). Point mutations were also introduced by PCR into hemagglutinin (HA)-tagged mouse Keap1 inserted in EF-1α promoter-driven expression vector as described previously (21). The mouse Keap1 cDNA mutants were inserted into PiggyBac expression vector (PB514B-2; System Biosciences). The Keap1 gene regulatory domain (KRD) and HA-tagged Keap1 cDNA were fused and inserted into pBluescript II vector as described previously (21).
Transfection experiments and measuring β-galactosidase and luciferase activities.
Transfection experiments were performed using Lipofectamine (Invitrogen) as described previously (24). For measuring β-galactosidase activity, HEK293T cells were cotransfected with 15 ng of NRF2-degron LacZ (NRF2NT-LacZ) reporter plasmid and 5, 15, or 45 ng of human KEAP1 mutant expression vector. After culture for 48 h, the relative β-galactosidase activity was measured by using the Beta-Glo assay system (Promega). For measuring luciferase activity, HEK293T cells were transfected with 100 ng of pNqo1-ARE-Luc plasmid, 10 ng of pRL-TK transfection control plasmid, 8 ng of p3×Flag-Nrf2 plasmid, and 8 or 40 ng of wild-type (WT) or mutant Keap1 expression plasmids. The luciferase activity was measured by using a Dual-Luciferase reporter system (Promega).
Establishment of stable cell lines that express transfected Keap1.
Immortalized Keap1-null MEFs (25, 26) were maintained in Dulbecco modified Eagle medium (Wako Chemical) containing 10% fetal bovine serum (FBS). A PiggyBac transposon vector system (PB514B-2; System Biosciences) was used to establish stable cell lines that express HA-tagged Keap1 cDNA. Cotransfection of PiggyBac expression vector plus transposase plasmid was performed by electroporation with a double 1,100-V pulse for 30 ms. After 2 to 3 days of culture, electroporated cells were selected by puromycin at 2 μg/ml for 7 to 10 days. The expression of red fluorescent protein was verified under a fluorescence microscope, and several single colonies were cloned.
RNA extraction and quantitative real-time PCR.
Total RNAs were prepared from MEFs using a Sepazol-RNA I Super G RNA extraction kit (Nacalai). The cDNAs were synthesized from the total RNA using ReverTra Ace qPCR RT master mix with gDNA Remover (Toyobo). Real-time quantitative PCR was performed using an ABI 7300 system (Applied Biosystems). The primer and probe sequences used for detecting NAD(P)H:quinone oxidoreductase 1 (Nqo1) and glutamate-cysteine ligase catalytic subunit (Gclc) were as described previously (27).
Immunoblotting.
Whole-cell extracts were prepared in a sample buffer (20% glycerol, 4% sodium dodecyl sulfate [SDS], 0.125 M Tris-HCl [pH 6.8], and 0.2 M dithiothreitol) from the MEFs or macrophages treated with Nrf2-inducing chemicals for 3 h. The protein samples were subjected to 8% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred to polyvinylidene difluoride (PVDF) membrane. Specific protein signals were detected by anti-Nrf2 (28), anti-HA (Roche 3F10), anti-Keap1 (29), or anti-α-tubulin (Sigma, DM1A) antibodies.
Generation of Keap1 transgenic mice.
Transgene constructs were injected into fertilized eggs derived from BDF1 parents. Transgenic mice were generated by standard method (21). Transgenic mice expressing wild-type Keap1 line 34 were used as a control (21). Three independent lines of KRD-Keap1C273W&C288E mice were established and mated into a Keap1-null background (25) to obtain compound mutant mice. For hematoxylin and eosin staining, the esophagi of postnatal day 10 (P10) pups were fixed in 3.7% formalin and embedded in paraffin. For immunostaining, the tissues were processed as described earlier (21). Samples were treated with anti-Nrf2 antibody (28), and positive reactivity was visualized with diaminobenzidine (DAB) staining. Hematoxylin was used for nuclear counterstaining.
Generation of Keap1 knock-in mice.
Keap1C151S or Keap1C288E knock-in mice were generated by CRISPR-Cas9 genome editing technology (30). Cas9 mRNA, guide RNA (gRNA), and targeting oligonucleotide were injected into fertilized eggs derived from BDF1 parents. The founder mice were crossed with wild-type mice, and the mutations were transmitted to the germ line. By crossing the heterozygous mutants, homozygotes were obtained. The genotyping were done by TaqMan real-time PCR methods (Applied Biosystems). The sequence information for the guide RNA, targeting oligonucleotide, and genotyping primers is available upon request.
Isolation of thioglycolate-elicited peritoneal macrophages.
Mice were intraperitoneally injected with 2 ml of 4% thioglycolate solution 4 days before collecting peritoneal macrophages (27). Peritoneal lavage fluid was centrifuged and suspended in RPMI 1640 medium containing 10% FBS and penicillin-streptomycin (10 U/0.1 mg/ml) at 37°C and 5% CO2. The adherent macrophages were used for the analyses.
RESULTS
Novel Keap1 mutants that repress Nrf2 activity.
Although several in vitro experiments have shown that Cys151, Cys273, and Cys288 are highly reactive to many electrophiles (10–17), evaluation of the sensor function of Cys273 and Cys288 has not been technically feasible, since replacements of Cys273 and Cys288 with serine or alanine render Keap1 unable to ubiquitinate and repress Nrf2 (18, 19, 21). To overcome this problem, we attempted a systemic analysis by introducing amino acid substitutions into Cys273 and Cys288 sites of human KEAP1 (Fig. 1A). For the series of mutants, we examined KEAP1's ability to repress NRF2 in a reporter cotransfection degron assay (Fig. 1B and C). In order to measure the activity, NRF2 degron-LacZ reporter was used, which enabled us to evaluate the stability of human NRF2 N terminus fused to LacZ (NRF2NT-LacZ) (23). As mentioned above, it is well documented that KEAP1C273S, KEAP1C273A, KEAP1C288S, and KEAP1C288A are unable to repress NRF2 activity (31), and this fact was reproduced in our current experiment (Fig. 1B and C, circled). Expression of KEAP1WT did not further suppress the NRF2NT-LacZ reporter activity, indicating that endogenous KEAP1 sufficiently repress NRF2 activity (WT in Fig. 1B and C). In contrast to the situation with KEAP1WT, the expression of the loss-of-function mutants increased the NRF2NT-LacZ reporter activity in a dose-dependent manner. These results indicate that the loss-of-function mutants of KEAP1 likely bind to NRF2NT-LacZ in a competitive manner and inhibit endogenous KEAP1's ability to repress NRF2 accumulation.
FIG 1.

Novel Keap1 mutants that repress Nrf2 activity. (A) Cysteine residues of Keap1 are shown. Representative reactive cysteine residues against electrophiles are boxed. Keap1 domains: NTR (N-terminal region), BTB (broad complex, tramtrack, and bric-a-brac), IVR (intervening region), Kelch/DGR (double glycine repeat), and CTR (C-terminal region). (B and C) All 19 possible amino acid substitutions were introduced to Cys273 (B) and Cys288 (C) of human KEAP1. HEK293T cells were cotransfected with NRF2-degron LacZ (NRF2NT-LacZ) reporter plasmid (15 ng) and KEAP1 mutant expression vector (5, 15, or 45 ng) and then incubated for 48 h. The relative β-galactosidase activity was measured. Representative results from multiple independent experiments are shown. Circled and boxed terms indicate loss-of-function mutants and mutants that retain activity to repress Nrf2 accumulation, respectively. (D) HEK293T cells were cotransfected with ARE-luciferase reporter vector, Nrf2-overexpressing vector, and vector expressing 8 or 40 ng of Keap1 WT, C273S, C273W, C288S, C288E, or C273W&C288E. At 24 h after transfection, the relative luciferase activity was measured. Boxes indicate mutants that retain activity to repress Nrf2 accumulation.
Consistent with our expectation, we found that overexpression of two KEAP1C273 mutants, i.e., KEAP1C273M and KEAP1C273W, did repress the activity of the NRF2NT-LacZ reporter (Fig. 1B, boxed), indicating that these two KEAP1 mutants still retain the ability to repress NRF2 accumulation. In the case for Cys288, KEAP1C288E, KEAP1C288N, and KEAP1C288R repressed the LacZ reporter activity (Fig. 1C, boxed), indicating these three mutants of Cys288 still retain the ability to repress NRF2 accumulation. These results for the first time indicate that the Cys273 and Cys288 residues of Keap1 can be replaced by two and three amino acids, respectively, and still retain the ability to ubiquitinate NRF2. Of note, the substitution profiles of amino acid residues that repress the LacZ reporter activity are distinct between Cys273 and Cys288, clearly demonstrating that Cys273 and Cys288 are not equivalent in supporting KEAP1 activity.
While we exploited the human KEAP1-NRF2 system for the above series of experiments, we decided to switch the experimental system to mouse Keap1-Nrf2 for our extended analyses to examine how these novel substitutions affect Keap1's ability to sense oxidative and electrophilic stresses. To this end, we first introduced identical mutations into the mouse Keap1 protein and examined the reproducibility of our original result. We determined whether the mouse Keap1 mutants exhibited ubiquitin ligase activity and repressed Nrf2 accumulation by conducting similar cotransfection assay using the pNqo1-ARE luciferase reporter (Fig. 1D). As was the case for the NRF2NT-LacZ reporter assay, Keap1C273W and Keap1C288E reproducibly repressed the reporter activity like Keap1WT (Fig. 1D, boxed). In contrast, expression of Keap1C273S or Keap1C288S did not efficiently repress the luciferase activity induced by Nrf2. In addition, we generated a double mutant, Keap1C273W&C288E, that was able to repress Nrf2 activity (Fig. 1D). The Keap1C273W&C288E mutant was also able to repress the Luc reporter indicating that the double mutation does not affect the ubiquitin ligase activity of Keap1.
Comparisons of Cys273 and Cys288 among KLHL family.
Keap1 is a member of the Kelch-like (KLHL) family that generally possesses BTB and Kelch domains. KLHL family members have a potential to form a Cul3-based ubiquitin ligase complex (32). Alignment of human KLHL family revealed that the cysteine residues at the positions of Cys273 and Cys288 are conserved in various mammalians but not in the other KLHL family proteins (Fig. 2). This observation suggests that these cysteine residues harbor special characteristics for Keap1 to gain stress-sensing function. Since leucine residue is frequently found at position corresponding to Cys273 in the other KLHL proteins, we initially hypothesized that replacement of Cys273 with leucine (C273L) may maintain Keap1 activity. However, Keap1C273L failed to fully repress the reporter activities in the NRF2NT-LacZ assay (Fig. 1B) and in the pNqo1-ARE luciferase assay (data not shown).
FIG 2.

Comparison of Cys273 and Cys288 between Keap1 and human KLHL (Kelch-like) family members. Cys273 and 288 are highly conserved among mammal Keap1 but not in other KLHL family members. Note that whereas Cys273 of Keap1 corresponds to leucine (L) in other KLHL family members, Cys288 of Keap1 corresponds to glutamate (E) or glutamine (N) in other KLHL family members.
In contrast, glutamic acid and asparagine residues are frequently found at the position corresponding to Cys288 of Keap1 (Fig. 2), and Keap1C288E and Keap1C288N demonstrated the ability to repress the reporter activity. These results imply that while the structure surrounding Cys273 has been diversified among KLHL proteins, KLHL proteins, including Keap1, possess a similar structure surrounding Cys288 to act as Cul3-based ubiquitin ligase (33). Based on these observations, we decided to utilize the novel cysteine mutant Keap1C273W&C288E for further investigation.
Keap1C273W&C288E mutant represses Nrf2 activity in vivo.
To verify the ability of the Keap1C273W&C288E mutant to repress Nrf2 accumulation in vivo, we conducted transgenic complementation rescue assays (21). For this purpose, we exploited a 5.7-kb genomic region of Keap1 harboring regulatory domain (KRD) and generated transgenic mice expressing Keap1C273W&C288E under the regulation of KRD (Fig. 3A). We established three lines of transgenic mice, which were crossed with Keap1-null mice (25) and Keap1−/−::Tg-Keap1C273W&C288E mice were obtained. Of the three lines of mice expressing Keap1C273W&C288E, two lines (lines 30 and 18) were able to rescue Keap1-null mice from juvenile lethality. The rescued Keap1−/−::Tg-Keap1C273W&C288E mice had a normal appearance and growth curve (Fig. 3B) and were indistinguishable from the Keap1−/−::Tg-Keap1WT mice (21).
FIG 3.
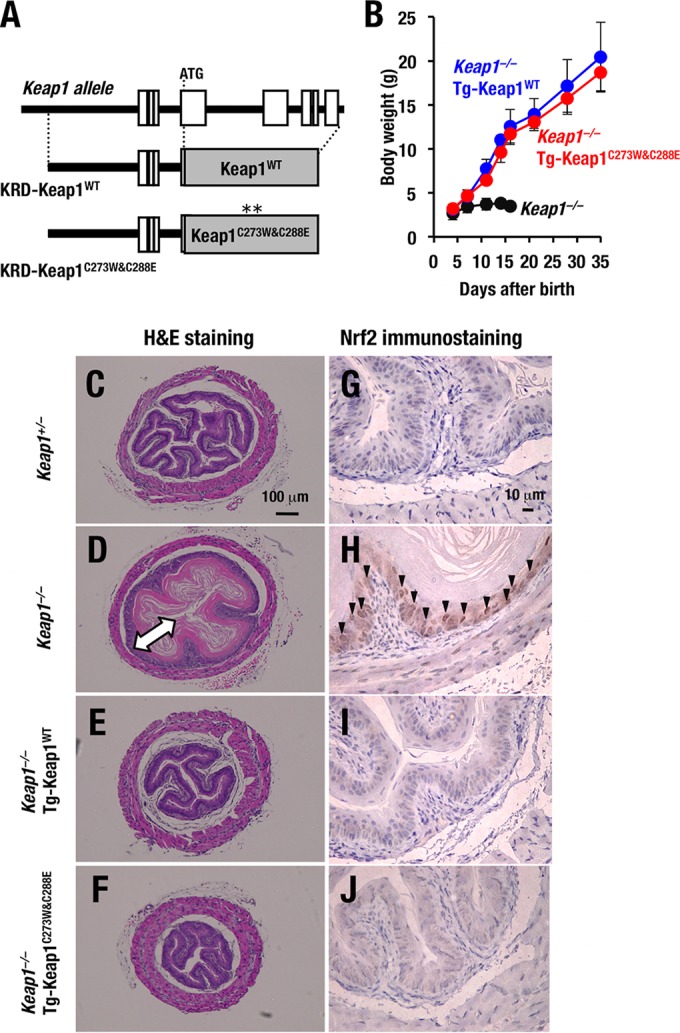
Keap1C273W&C288E represses Nrf2 activity in vivo. (A) A schematic presentation of KRD-Keap1C273W&C288E transgene that expresses Keap1 under the regulation of KRD is shown. (B) Growth curves for Keap1−/−, Keap1−/−::Tg-Keap1WT mice (line 34) and Keap1−/−::Tg-Keap1C273W&C288E mice (line 30). Note that mice of the last two genotypes grew normally. (C to F) Hematoxylin-eosin staining of esophagus transverse sections of Keap1+/− (C), Keap1−/− (D), Keap1−/−::Tg-Keap1WT (E), and Keap1−/−::Tg-Keap1C273W&C288E (F) mice at P10. The arrow in panel D indicates the thickened cornified layer. (G to J) Nrf2 immunostaining of esophagus transverse sections of Keap1+/− (G), Keap1−/− (H), Keap1−/−::Tg-Keap1WT (I), and Keap1−/−::Tg-Keap1C273W&C288E (J) mice at P10. Arrowheads indicate Nrf2 accumulation in basal layer cells of esophagi.
It has been shown that Keap1-null mutant mice suffer from severe hyperkeratosis of the esophagi. To examine whether Keap1C273W&C288E expression driven by KRD improve the upper digestive tract abnormalities that are usually found in Keap1-null mutant mice, we performed histological analyses of 10-day-old (P10) mice (25). Consistent with our previous findings, Keap1-null mutant mice showed severe hyperkeratosis of the esophagi, while Keap1+/− mice did not show such a phenotype (Fig. 3C and D). Of note, the esophagi of Keap1−/−::Tg-Keap1C273W&C288E mice were histologically indistinguishable from those of Keap1−/−::Tg-Keap1WT mice (21) (Fig. 3E and F).
In good agreement with the histology of the esophagi, Nrf2 accumulation in basal layer cells of esophagi was clearly observed in Keap1−/− mice, while the Nrf2 accumulation disappeared in the Keap1−/−::Tg-Keap1C273W&C288E mice in immunostaining analyses with anti-Nrf2 antibody (Fig. 3G to J). These results indicate that the Keap1C273W&C288E mutant indeed retains the ability to repress Nrf2 accumulation in vivo.
Stable cell lines that complementarily express transfected Keap1.
Capitalizing on the novel mutants of Cys273 and Cys288 residues described above, we addressed the importance of these cysteine residues as stress sensors. In this regard, it should be noted that upon the evaluation of sensor function, overexpression experiments in transfecto may be misleading, since the expression levels of Keap1 mutant proteins fluctuate significantly, and endogenous Keap1 seems to interfere with the activity of the transfected Keap1 (22). Therefore, in order to develop a stable system for the fine evaluation of Keap1 sensor function, we decided to prepare stable cell lines by introducing an HA-Keap1 expression vector ligated to the PiggyBac transposon system. Expression plasmids for HA-Keap1 were transfected into Keap1-null MEF cells, and several HA-Keap1 expression lines were established by cloning from single colonies that survived after culture with puromycin (Fig. 4A). The expression levels of Keap1 protein within the cell lines were verified by means of Western blotting analyses. We found that the ability of Keap1 to repress Nrf2 accumulation in the basal state (i.e., without DEM) was dependent on the expression levels of transduced Keap1 (Fig. 4B). Accumulation of Nrf2 in response to DEM, a representative electrophilic inducer, was observed in the middle and high expresser of Keap1WT (Fig. 4B). Therefore, in this study the middle expresser cell lines were used for further analyses.
FIG 4.

Stable cell lines of Keap1-null MEFs with Keap1 complementation. (A) Scheme for complementation of Keap1 in Keap1−/− MEFs. PiggyBac vector expressing HA-tagged Keap1WT cDNA and transposase expression vector were cotransfected to Keap1−/− MEFs. Subsequently several lines of Keap1−/−::HA-Keap1WT MEFs (Keap1WT) were established by cloning from a single colony survived after culture with puromycin. (B) Whole-cell extracts of Keap1WT or Keap1−/− mock cells after incubation with 0, 30, or 100 μM DEM for 3 h were examined by Western blotting. Low-, middle-, and high-level expressers of Keap1WT MEFs are shown. (C) Schematic structure of Keap1C273W&C288E. (D) Western blot analysis of Keap1 expression in Keap1WT and Keap1C273W&C288E MEFs. (E) Keap1WT and Keap1C273W&C288E MEFs treated with 0, 3, or 10 μM 15d-PGJ2 for 3 h were examined by Western blotting. (F) Graphical representation of the results shown in panel E (n = 3). Asterisks indicate statistically significant differences (*, P < 0.05; **, P < 0.01). (G and H) Keap1WT and Keap1C273W&C288E MEFs treated with 0, 3, or 10 μM 15d-PGJ2 for 12 h. The expression levels of Nqo1 (G) and Gclc (H) were examined by RT-qPCR with the 18S ribosomal subunit as an internal control. Representative results are shown from multiple independent experiments.
We then tested the importance of Keap1 Cys273 and Cys288 residues in the stress response. For this purpose, we first prepared Keap1-deficient MEFs complemented with the PiggyBac expression system harboring Keap1C273W&C288E double cysteine mutant (Fig. 4C). Of the multiple stable clones, we selected one clone that expresses a comparable level of the Keap1C273W&C288E protein with control cells expressing Keap1WT at the middle level (Fig. 4D). Showing very good agreement with the transient-transfection experiment, Keap1C273W&C288E reproducibly repressed basal Nrf2 accumulation in the complemented MEFs (Fig. 4E and F).
We then challenged the Keap1C273W&C288E MEFs with 15d-PGJ2, an Nrf2-inducing chemical. As expected, Nrf2 accumulation by 15d-PGJ2 was markedly decreased in MEFs expressing Keap1C273W&C288E compared to MEFs expressing Keap1WT (Fig. 4E and F), indicating that the Keap1C273W&C288E mutant could not respond to 15d-PGJ2. Consistent with this observation, Nqo1, a prototype Nrf2 target gene, was induced at a level 5.5-fold higher than that under the basal state in Keap1WT MEFs by 10 μM 15d-PGJ2 treatment, whereas only a marginal increase was observed in Keap1C273W&C288E MEFs (Fig. 4G). Another Nrf2 target gene, Gclc, was induced at a level 7.2-fold higher than that under the basal state in Keap1WT MEFs by 10 μM 15d-PGJ2, whereas only a marginal increase was observed in Keap1C273W&C288E MEFs (Fig. 4H). These results thus indicate that both Cys273 and Cys288, or either one of these residues, are essential for 15d-PGJ2-mediated activation of Nrf2 signaling.
15d-PGJ2 is a Cys288-preferring inducer of Nrf2.
We next sought to determine whether either Cys273 or Cys288 residues (or both) are required for 15d-PGJ2-mediated activation of Nrf2 signaling. To address this question, we generated MEFs expressing either Keap1C273W or Keap1C288E (Fig. 5A). The expression levels of the Keap1C273W and Keap1C288E proteins were comparable to that of control Keap1WT (Fig. 5B). We found that Nrf2 accumulation by 15d-PGJ2 was significantly reduced in MEFs expressing Keap1C288E but not in MEFs expressing Keap1C273W (Fig. 5C and D). This was somewhat of a surprise since we found that 15d-PGJ2 induces Nrf2 accumulation in a Cys273-preferring manner in an overexpression experiment using zebra fish embryos (14). However, our present results indicate that Cys288 of Keap1, but not Cys273, is essential for 15d-PGJ2-mediated accumulation of Nrf2. The reason for this difference between mouse and zebra fish is unclear at present.
FIG 5.
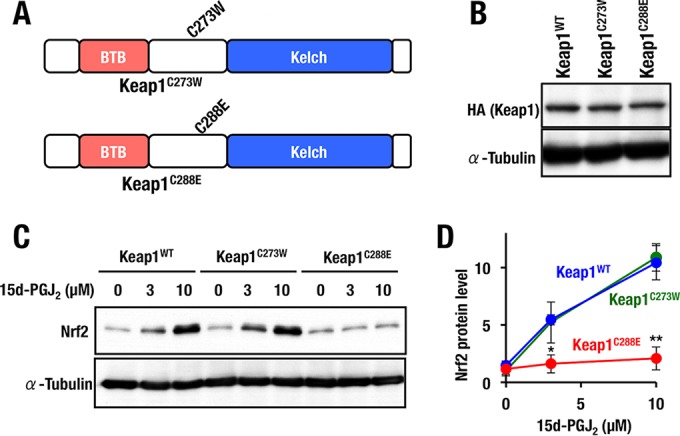
Keap1 Cys288 is a functional sensor for 15d-PGJ2. (A) Schematic structures of Keap1C273W and Keap1C288E. (B) Western blot analysis of Keap1 expression in Keap1WT, Keap1C273W, and Keap1C288E MEFs. (C) Keap1WT, Keap1C273W, and Keap1C288E MEFs treated with 0, 3, or 10 μM 15d-PGJ2 for 3 h were examined by Western blotting. (D) Graphical representation of the results shown in panel C (n = 3). Asterisks indicate statistically significant differences (*, P < 0.05; **, P < 0.01).
Generation and analyses of Keap1C288E knock-in mice.
To further validate the importance of Keap1 Cys288 residue in sensing 15d-PGJ2 in vivo, we generated Keap1C288E knock-in lines of mice using a genome-editing technology (30). Cas9 mRNA, guide RNA (gRNA), and targeting oligonucleotide were injected into fertilized eggs derived from BDF1 parents. Point mutation of Keap1C288E was identified through sequencing of genomic DNA from injected mice. The founder mice were crossed with wild-type mice, and the mutation was successfully transmitted to the germ line (Fig. 6A). Genotyping of Keap1C288E/C288E mice was performed by genomic sequencing. Thioglycolate-elicited peritoneal macrophages were obtained from homozygous Keap1C288E knock-in (Keap1C288E/C288E) mice, and the macrophages were used for further analyses.
FIG 6.

Generation and analyses of Keap1C288E knock-in mice. (A) Experimental scheme for isolation of thioglycolate-elicited peritoneal macrophages from wild-type and Keap1C288E/C288E mice. Representative sequencing data show the successful replacement of a cysteine (Cys) with glutamic acid (Glu) at position 288 (C288E) of Keap1C288E/C288E mice. (B to F) Peritoneal macrophages from WT and Keap1C288E/C288E mice treated with 0, 3, or 10 μM 15d-PGJ2 (B), 0, 30, or 100 μM DEM (C), 0, 3, or 10 μM 9-OA-NO2 (D), 0, 5, or 15 μM 4-HNE (E), or 0, 10, or 30 μM PGA2 (F) for 3 h were examined by Western blotting.
The Keap1C288E/C288E mice exhibited a normal appearance and a normal growth curve, indicating that the Keap1C288E maintains the ability to repress Nrf2 accumulation in vivo. In the peritoneal macrophages derived from Keap1C288E/C288E mice, Nrf2 accumulation in response to 15d-PGJ2 was decreased significantly compared to wild-type macrophages (Fig. 6B), showing very good agreement with the result in the Keap1C288E-complemented MEFs. This result provides further evidence that Cys288 of Keap1 is essential for Nrf2 accumulation in response to 15d-PGJ2 in vivo.
We also examined how Keap1 activity responds to the other Nrf2 inducers utilizing the Keap1C288E/C288E macrophages. Upon treatment with DEM, the accumulation level of Nrf2 in the Keap1C288E/C288E macrophages was comparable to that in wild-type macrophages (Fig. 6C), a finding consistent with the notion that DEM is a Cys151-preferring inducer (22). We also found that treatment with 9-OA-NO2 (Fig. 6D) and 4-HNE (Fig. 6E), both of which were reported to exploit Cys273 and Cys288 to modify Keap1 activity (15, 16, 34), resulted in the accumulation of Nrf2 in both wild-type and Keap1C288E/C288E macrophages.
To our surprise, PGA2 induced Nrf2 accumulation in Keap1C288E/C288E macrophages (Fig. 6F), demonstrating that PGA2 apparently utilizes cysteine residues other than Cys288 to stabilize Nrf2. The observation that PGA2 utilizes distinct cysteine residues of Keap1 from that utilized by 15d-PGJ2, i.e., Cys288, for the induction of Nrf2 is particularly intriguing, since PGA2 is a member of the prostaglandin family and shares similar structural features with 15d-PGJ2. These results thus demonstrate that 9-OA-NO2, 4-HNE, and PGA2 utilize cysteine residues other than Cys288.
Simultaneous substitution of three major cysteine residues.
As for the specificity of the three major reactive cysteine residues, i.e., Cys151, Cys273, and Cys288, against various Nrf2-inducing chemicals, there is some confusion. We surmise two plausible reasons for this. One is that these cysteine residues may act redundantly, and the other is that some of the inducers may bind and utilize other cysteine residues of Keap1. To address this issue, we generated Keap1C151S&C273W&C288E MEFs that expressed Keap1 harboring all three major cysteine mutations (Fig. 7A). We selected MEF clones expressing levels of Keap1C151S&C273W&C288E protein comparable to those of the control Keap1WT protein (Fig. 7B). Importantly, simultaneous substitutions of these three major cysteine residues could repress Nrf2 accumulation in the basal unstressed conditions, indicating that the substitution did not affect Keap1's ability to promote the proteasomal degradation of Nrf2 (see Fig. 7C and subsequent panels).
FIG 7.

Keap1 mutant with triple sensor cysteine mutations. (A) Schematic structure of Keap1C151S&C273W&C288E. (B) Keap1C151S&C273W&C288E protein levels of stable complemented MEFs examined by Western blotting. (C to L) Keap1WT and Keap1C151S&C273W&C288E MEFs treated with 0, 3, or 10 μM 9-OA-NO2 (C), 0, 5, or 15 μM 4-HNE (D), 0, 3, or 10 μM NaAsO2 (E), 0, 100, or 300 μM SNAP (F), 0, 10, or 30 nM CDDO-Im (G), 0, 10, or 30 μM PGA2 (H), 0, 30, or 90 μM ZnCl2 (I), 0, 20, or 60 μM CdCl2 (J), 0, 3, or 10 μM Dex-Mes (K), or 0, 130, or 400 μM H2O2 (L) for 3 h were examined by Western blotting.
Exploiting the triple substitution mutant, we first examined whether the Nrf2-inducing activity of various chemical Nrf2 inducers was affected in the Keap1C151S&C273W&C288E MEFs. For this purpose, we examined Nrf2 accumulation in the Keap1C151S&C273W&C288E MEFs upon treatment with 9-OA-NO2, 4-HNE, NaAsO2, SNAP, CDDO-Im, PGA2, ZnCl2, CdCl2, Dex-Mes, and H2O2 (Fig. 7C to L). We found that these Nrf2 inducers are classified into two groups; one resulted in weakened Nrf2 accumulation in Keap1C151S&C273W&C288E MEFs compared to that observed in Keap1WT MEFs, while the other resulted in a level of Nrf2 accumulation in Keap1C151S&C273W&C288E MEFs similar to that in Keap1WT MEFs.
Nrf2 accumulation in Keap1C151S&C273W&C288E MEFs was significantly decreased in response to 9-OA-NO2, 4-HNE, NaAsO2, SNAP, and CDDO-Im (Fig. 7C to G), indicating that part or all the three cysteine residues are important for Nrf2 accumulation in response to these inducers. In contrast, Nrf2 accumulation by PGA2, ZnCl2, CdCl2, Dex-Mes, and H2O2 was not affected in Keap1C151S&C273W&C288E MEFs (Fig. 7H to L), indicating that these three cysteine residues are dispensable for Nrf2 activation in response to this group of inducers.
Fine dissection of three major cysteine residues.
We then examined the Nrf2 inducers that failed to accumulate Nrf2 in Keap1C151S&C273W&C288E MEFs in detail. Using the newly generated Keap1C151S and Keap1C273W&C288E MEFs, we sought to determine whether one residue or all three—Cys151, Cys273 and Cys288—are required for the response to these inducers (Fig. 8A). Again, we selected stable clones that expressed Keap1C151S and Keap1C273W&C288E proteins at a level comparable to that of control Keap1WT (Fig. 8B). The accumulation of Nrf2 in response to DEM, SFN, and tBHQ, which are Cys151-preferring inducers, was indeed decreased in Keap1C151S MEFs (Fig. 8C to E). These results are consistent with our previous studies (14, 22), confirming the credibility of our Keap1C151S systems.
FIG 8.

Dissection of three major cysteine residues. (A) Schematic presentations of Keap1C151S and Keap1C273W&C288E structures. (B) Keap1C151S and Keap1C273W&C288E protein levels of stable complemented MEFs examined by Western blotting. (C to E) Keap1WT and Keap1C151S MEFs were treated with 0, 30, or 100 μM DEM (C), 0, 1, or 3 μM SFN (D), or 0, 10, or 30 μM tBHQ (E) for 3 h were examined by Western blotting. (F to J) Keap1WT, Keap1C151S, and Keap1C273W&C288E MEFs were treated with 0, 3, or 10 μM 9-OA-NO2 (F), 0, 5, or 15 μM 4-HNE (G), 0, 3, or 10 μM NaAsO2 (H), 0, 100, or 300 μM SNAP (I), or 0, 10, or 30 nM CDDO-Im (J) for 3 h were examined by Western blotting.
Importantly, upon treatment with 9-OA-NO2, 4-HNE, and NaAsO2, Nrf2 accumulated significantly both in Keap1C151S and in Keap1C273W&C288E MEFs (Fig. 8F to H). Considering the results derived from Keap1C151S&C273W&C288E MEFs that Nrf2 accumulation is markedly decreased in response to 9-OA-NO2, 4-HNE, and NaAsO2 (Fig. 7C to E), these results indicate that all three cysteine-residues—Cys151, Cys273, and Cys288—are indispensable for Keap1’s ability to sense 9-OA-NO2, 4-HNE, and NaAsO2.
In contrast to the former type of inducers, we found that Nrf2 accumulation upon exposure to SNAP and CDDO-Im was notably reduced in Keap1C151S MEFs but not in Keap1C273W&C288E MEFs (Fig. 8I and J). This observation was in contrast to our previous observations (22). We repeated the analysis three times and also repeated similar experiments four times utilizing other lines of Keap1C151S and Keap1C273W&C288E MEFs, and the results were reproducible. Therefore, we conclude that Cys151 of Keap1 is a critical sensor for SNAP and CDDO-Im. To further validate this conclusion, we decided to challenge the generation of the germ line mutant mice, as described below.
Generation and analyses of Keap1C151S knock-in mice.
We generated Keap1C151S knock-in mice by exploiting the CRISPR/Cas9 genome editing technology (Fig. 9A), as was the case for the generation of Keap1C288E knock-in mice (Fig. 6). Details of the knock-in mouse preparation were as described in Materials and Methods. Genomic substitution with Keap1C151S was confirmed by genome sequencing.
FIG 9.

Generation and analyses of Keap1C151S knock-in mice. (A) Experimental schema for isolation of thioglycolate-elicited peritoneal macrophages from wild-type (WT) and Keap1C151S/C151S mice. Representative sequencing data showing replacement of a cysteine (Cys) with serine (Ser) at position 151 (C151S) of WT and Keap1C151S/C151S mice. (B to F) Peritoneal macrophages from WT or Keap1C151S/C151S mice were treated with 0, 30, or 100 μM DEM (B), 0, 3, or 10 μM 15d-PGJ2 (C), 0, 10, or 30 μM PGA2 (D), 0, 100, or 300 μM SNAP (E), or 0, 10, 30, or 100 nM CDDO-Im (F) for 3 h were examined by Western blotting.
We obtained thioglycolate-elicited peritoneal macrophages from Keap1C151S knock-in (Keap1C151S/C151S) and wild-type mice (Fig. 9A) and first challenged them with DEM, as DEM is a well-known Cys151-dependent inducer of Nrf2 accumulation. Showing very good agreement with previous (14, 22) and current (Fig. 8C) results, DEM-mediated accumulation of Nrf2 was abrogated substantially (Fig. 9B). We also challenged the cells with 15d-PGJ2 and PGA2, since our study with mutant MEFs proved that 15d-PGJ2 was a Cys288-preferring inducer (Fig. 5C) and that PGA2 was a Cys151/Cys273/Cys288-independent inducer (Fig. 7H), respectively. Supporting our conclusion from the mutant MEFs, Nrf2 was accumulated in Keap1C151S/C151S macrophages to a similar extent as in wild-type macrophages upon treatment with 15d-PGJ2 (Fig. 9C) and PGA2 (Fig. 9D). These results clearly indicate that the Keap1C151S knock-in mouse macrophages provide a stable and reliable ex vivo system for evaluating the necessity of Cys151 for the stress sensing function of the Keap1-Nrf2 pathway.
Finally, we challenged Keap1C151S/C151S macrophages with SNAP and CDDO-Im. Consistent with the results derived from Keap1C151S MEFs, we found that Nrf2 accumulation by the SNAP treatment in the Keap1C151S/C151S macrophages was markedly decreased compared to that in wild-type macrophages (Fig. 9E), conclusively demonstrating that SNAP is a Cys151-preferring inducer. Similarly, the CDDO-Im treatment resulted in a markedly decreased accumulation of Nrf2 in the Keap1C151S/C151S macrophages compared to that in wild-type macrophages (Fig. 9F), but in this case marginal accumulation of Nrf2 was observed, indicating that CDDO-Im is a Cys151-preferring inducer but may also interact weakly with certain other cysteine residues.
DISCUSSION
To date, it has not been technically feasible to test the requirements of Cys273 and Cys288 for Nrf2 induction in response to chemical inducers. In order to conduct a functional evaluation of the Keap1 cysteine activity, it is a mandatory prerequisite for Keap1 mutants to retain the Nrf2 ubiquitination activity that represses Nrf2 accumulation; otherwise, the loss of Keap1 activity automatically promotes Nrf2 accumulation, and the activity of chemical Nrf2 inducers is easily masked. In the present study, we conducted systematic substitution studies of Keap1 Cys273 and Cys288 and successfully identified the amino acid substitutions that preserve Keap1’s ability to ubiquitinate and degrade Nrf2. The C273W and C288E substitution mutants have enabled us to examine the necessity of Cys273 and Cys288 in response to various chemical Nrf2 inducers. Our analyses with the stable Keap1C288E MEF lines and peritoneal macrophages from Keap1C288E knock-in mice revealed that Keap1 Cys288 acts as a functional sensor for 15d-PGJ2, an important mediator in the resolution of inflammation. Our results also revealed that three major cysteine residues, i.e., Cys151, Cys273, and Cys288, function collaboratively for sensing of 9-OA-NO2, 4-HNE, and NaAsO2. Our analyses also provide convincing lines of evidence to suggest that SNAP and CDDO-Im are Cys151-preferring inducers. In contrast, PGA2, CdCl2, ZnCl2, Dex-Mes, and H2O2 appear to be Cys151/Cys273/Cys288 independent. Thus, as shown in Fig. 10, we propose, based on this study, that the chemical inducers of Nrf2 be categorized into at least four classes:class I (Cys151 preferring), class II (Cys288 preferring), class III (Cys151/Cys273/Cys288 collaboration preferring), and class IV (Cys151/Cys273/Cys288 independent).
FIG 10.

Model for multiple stress-sensing mechanisms by Keap1. We examined the reactivity of three cysteine mutants against various chemical Nrf2 inducers. Based on the results, we propose a classification of Nrf2 inducers into four classes, namely, class I (Cys151 preferring), class II (Cys288 preferring), class III (Cys151/Cys273/Cys288 collaboration preferring), and class IV (Cys151/Cys273/Cys288 independent). Representative chemicals for each class are also shown in the figure. The molecular basis for the classification is of interest.
Our comparison of Cys273 and Cys288 among KLHL family members (shown in Fig. 2) suggests that KLHL proteins, including Keap1, likely possess a similar structure surrounding Cys288, which may be required in order to function as an ubiquitin ligase (33). In light of this information, we conclude that electrophilic modification of Cys273 and Cys288 disrupts the specific conformation of Keap1, which is critical for its ubiquitin ligase activity. In this regard, we first generated cysteine substitution mutants of Keap1 utilizing serine and alanine residues (18, 19, 21). However, in the case of Cys273/Cys288, replacements of the cysteine residues with serine or alanine result in the loss of Keap1’s ability to ubiquitinate and degrade Nrf2. In contrast, we found here that Keap1 mutants retain the ability to repress Nrf2 accumulation if Cys273 of Keap1 is replaced with either methionine or tryptophan and Cys288 is changed to either glutamate, asparagine, or arginine. Since all of these substituted amino acid residues exhibit bulky characteristics, electrophilic modification of Cys273 and Cys288 which results in Keap1 inactivation may be independent from the simple space-filling thiol modification model that has been proposed for Cys151 (20). In addition, methionine and tryptophan are hydrophobic residues, whereas glutamate, asparagine, and arginine are hydrophilic, suggesting that in unstressed conditions Cys273 and Cys288 are found in hydrophobic and hydrophilic states, respectively. We surmise that these characteristics of Cys273 and Cys288 are critical for the overall structure of the Keap1 protein, allowing it to maintain its function as a ubiquitin ligase. However, but conclusive insight into these structural requirements awaits the elucidation of the structure surrounding the intervening region (IVR) that includes Cys273 and Cys288.
One of the most important findings that emerged from this study is how distinct cysteine residues recognize each Nrf2-inducing chemical. We verified that NO and CDDO-Im are Cys151-preferring inducers (class I above), whereas 15d-PGJ2 is a Cys288-preferring inducer (class II). These two groups of Nrf2-inducing chemicals are apparently recognized by the specific cysteine residue of Keap1. On the other hand, recognition of 9-OA-NO2, 4-HNE, and NaAsO2 requires all three major cysteine residues, i.e., Cys151, Cys273, and Cys288 (class III), indicating that the preference of these inducers for Keap1 cysteine residues is relatively ambiguous. Of note, although both 15d-PGJ2 and PGA2 belong to the prostaglandin family and share similar structural characteristics in relation to their interaction with Keap1, 15d-PGJ2 and PGA2 display class II and class IV specificities, respectively. These observations support our contention that the simple chemical structure of Nrf2 inducers may not be the critical factor in determining which Keap1 sensor cysteine residues they react with. We surmise that combinations of size, shape, and electrophilicity en bloc contribute to the determination of which cysteine residue(s) the Nrf2-inducing chemicals recognize.
Mass spectrometry of the Keap1 protein treated with electrophilic reagents in vitro demonstrates that different electrophilic reagents give rise to different patterns of cysteine residue modification of Keap1 (10–12). Based on these observations, we have proposed the cysteine code hypothesis, wherein each electrophile prefers a specific set of cysteine residues of Keap1 (21). However, this hypothesis has been obscure, because studies on the structure-function relationships have not been technically feasible, as described here. The present study provides for the first time reliable and solid evidence that Keap1 functionally utilizes a distinct set of cysteine residues for the sensing of Nrf2-inducing chemicals. It should be noted that, even when all three cysteine residues (Cys151, Cys273, and Cys288) were replaced, certain Nrf2 inducers (i.e., CdCl2, ZnCl2, Dex-Mes, and H2O2) still induced Nrf2 accumulation. This suggests that there are cysteine residues other than the three residues discussed here that are critical for Keap1 sensor activity. Indeed, Cys226, Cys434, and Cys613 have been suggested to be important for Nrf2 activation (35, 36), although further biological investigations utilizing an evaluation system similar to the one used here are necessary to confirm this.
An important but remaining issue is how these Keap1 cysteine modifications provoked by the Nrf2-inducing chemicals actually cause the stabilization and accumulation of Nrf2. An important finding for the Cys273 and Cys288 case is that these cysteine residues are in the IVR domain, which is located near the Kelch-DGR domain, so that the modifications of these residues by class II chemicals likely affects Keap1 binding to Nrf2 and ceases the ubiquitination of Nrf2. This model has been referred to as the “hinge and latch” mechanism and has been firmly supported by wide-ranging observations (37–40). For instance, the IVR domain was found to surround the Kelch domain in our electron microscope analysis (37), and the proximity between the IVR and Kelch-DGR domains supports the notion that the modification of Cys273 and Cys288 is conveyed as a conformational distortion to the Kelch domain where Nrf2 binds (38, 39). Further structural, kinetic, and thermodynamic analyses of the Keap1-Nrf2 interaction (39), as well as somatic mutations analyses of Keap1 and Nrf2 in human cancers (40), also provide convincing lines of evidence that support this model. Thus, our conclusion for the consequence of Cys273 and Cys288 modification is that the conformational change in Keap1 elicited by the cysteine modification disrupts or distorts the Keap1-Nrf2 interaction and results in Nrf2 stabilization.
As for the question of how Cys151-preferring class I inducers induce Nrf2 accumulation, a model based on the disruption of Keap1-Cul3 interaction has been proposed (41–43). We found in the present study that Cys151 in the Keap1 BTB domain is important for Nrf2 activation in response to CDDO-Im, which supports the idea based on cocrystal analysis of CDDO and BTB domain of Keap1 that CDDO interferes with the Keap1-Cul3 interaction (44). In light of the cocrystal analysis, the class I inducers need to be relatively large for the inhibition of Keap1-Cul3 interaction. However, it seems unlikely that relatively small Cys151-preferring inducers, such as NO and DEM, dissociate the Keap1-Cul3 interaction. Indeed, conflicting results against the Keap1-Cul3 dissociation hypothesis have been proposed, showing that Keap1 and Cul3 do not dissociate upon exposure to the chemicals (45, 46). Thus, a full description of the mechanisms by which Cys151-preferring class I inducers stabilize Nrf2 must await further studies. Similarly, the molecular mechanisms underlying the activities of 9-OA-NO2, 4-HNE, and NaAsO2, which attack redundantly these three cysteine residues, remain to be clarified.
In conclusion, we have approached a long-lasting question as to how the body senses oxidative and electrophilic stresses, and we have uncovered important lines of evidence that enable us to categorize Nrf2-inducing chemicals into four classes based on the necessity of Cys151, Cys273, and Cys288. Our findings also provide insights into the multiple sensing mechanisms for the detection of various environmental stimuli.
ACKNOWLEDGMENTS
We thank Tadayuki Tsujita, Keiko Taguchi, Eri H. Kobayashi, and Hozumi Motohashi for discussion and advice.
REFERENCES
- 1.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. 1997. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 2.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. 1999. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, Yamamoto M. 2004. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 24:7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki T, Shibata T, Takaya K, Shiraishi K, Kohno T, Kunitoh H, Tsuta K, Furuta K, Goto K, Hosoda F, Sakamoto H, Motohashi H, Yamamoto M. 2013. Regulatory nexus of synthesis and degradation deciphers cellular Nrf2 expression levels. Mol Cell Biol 33:2402–2412. doi: 10.1128/MCB.00065-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taguchi K, Hirano I, Itoh T, Tanaka M, Miyajima A, Suzuki A, Motohashi H, Yamamoto M. 2014. Nrf2 enhances cholangiocyte expansion in Pten-deficient livers. Mol Cell Biol 34:900–913. doi: 10.1128/MCB.01384-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uruno A, Furusawa Y, Yagishita Y, Fukutomi T, Muramatsu H, Negishi T, Sugawara A, Kensler TW, Yamamoto M. 2013. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol Cell Biol 33:2996–3010. doi: 10.1128/MCB.00225-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki T, Yamamoto M. Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med 88:93–100. doi: 10.1016/j.freeradbiomed.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki T, Motohashi H, Yamamoto M. 2013. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol Sci 34:340–346. doi: 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Dinkova-Kostova AT, Talalay P. 2008. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res 52(Suppl 1):S128–S138. doi: 10.1002/mnfr.200700195. [DOI] [PubMed] [Google Scholar]
- 10.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. 2002. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A 99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hong F, Freeman ML, Liebler DC. 2005. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chem Res Toxicol 18:1917–1926. doi: 10.1021/tx0502138. [DOI] [PubMed] [Google Scholar]
- 12.Eggler AL, Liu G, Pezzuto JM, van Breemen RB, Mesecar AD. 2005. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc Natl Acad Sci U S A 102:10070–10075. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eggler AL, Luo Y, van Breemen RB, Mesecar AD. 2007. Identification of the highly reactive cysteine 151 in the chemopreventive agent-sensor Keap1 protein is method dependent. Chem Res Toxicol 20:1878–1884. doi: 10.1021/tx700217c. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi M, Li L, Iwamoto N, Nakajima-Takagi Y, Kaneko H, Nakayama Y, Eguchi M, Wada Y, Kumagai Y, Yamamoto M. 2009. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol Cell Biol 29:493–502. doi: 10.1128/MCB.01080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsujita T, Li L, Nakajima H, Iwamoto N, Nakajima-Takagi Y, Ohashi K, Kawakami K, Kumagai Y, Freeman BA, Yamamoto M, Kobayashi M. 2011. Nitro-fatty acids and cyclopentenone prostaglandins share strategies to activate the Keap1-Nrf2 system: a study using green fluorescent protein transgenic zebrafish. Genes Cells 16:46–57. doi: 10.1111/j.1365-2443.2010.01466.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kansanen E, Bonacci G, Schopfer FJ, Kuosmanen SM, Tong KI, Leinonen H, Woodcock SR, Yamamoto M, Carlberg C, Ylä-Herttuala S, Freeman BA, Levonen AL. 2011. Electrophilic nitro-fatty acids activate NRF2 by a KEAP1 cysteine 151-independent mechanism. J Biol Chem 286:14019–14027. doi: 10.1074/jbc.M110.190710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu C, Eggler AL, Mesecar AD, van Breemen RB. 2011. Modification of keap1 cysteine residues by sulforaphane. Chem Res Toxicol 24:515–521. doi: 10.1021/tx100389r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang DD, Hannink M. 2003. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol 23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW, Talalay P. 2004. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci U S A 101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eggler AL, Small E, Hannink M, Mesecar AD. 2009. Cul3-mediated Nrf2 ubiquitination and antioxidant response element (ARE) activation are dependent on the partial molar volume at position 151 of Keap1. Biochem J 422:171–180. doi: 10.1042/BJ20090471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H, Yamamoto M. 2008. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol 28:2758–2770. doi: 10.1128/MCB.01704-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takaya K, Suzuki T, Motohashi H, Onodera K, Satomi S, Kensler TW, Yamamoto M. 2012. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic Biol Med 53:817–827. doi: 10.1016/j.freeradbiomed.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirotsu Y, Katsuoka F, Itoh K, Yamamoto M. 2011. Nrf2 degron-fused reporter system: a new tool for specific evaluation of Nrf2 inducers. Genes Cells 16:406–415. doi: 10.1111/j.1365-2443.2011.01496.x. [DOI] [PubMed] [Google Scholar]
- 24.Kang MI, Kobayashi A, Wakabayashi N, Kim SG, Yamamoto M. 2004. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc Natl Acad Sci U S A 101:2046–2051. doi: 10.1073/pnas.0308347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, Imakado S, Kotsuji T, Otsuka F, Roop DR, Harada T, Engel JD, Yamamoto M. 2003. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet 35:238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 26.Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, Suzuki T, Kobayashi A, Yokota J, Sakiyama T, Shibata T, Yamamoto M, Hirohashi S. 2008. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res 68:1303–1309. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki T, Kelly VP, Motohashi H, Nakajima O, Takahashi S, Nishimura S, Yamamoto M. 2008. Deletion of the selenocysteine tRNA gene in macrophages and liver results in compensatory gene induction of cytoprotective enzymes by Nrf2. J Biol Chem 283:2021–2030. doi: 10.1074/jbc.M708352200. [DOI] [PubMed] [Google Scholar]
- 28.Maruyama A, Tsukamoto S, Nishikawa K, Yoshida A, Harada N, Motojima K, Ishii T, Nakane A, Yamamoto M, Itoh K. 2008. Nrf2 regulates the alternative first exons of CD36 in macrophages through specific antioxidant response elements. Arch Biochem Biophys 477:139–145. doi: 10.1016/j.abb.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Watai Y, Kobayashi A, Nagase H, Mizukami M, McEvoy J, Singer JD, Itoh K, Yamamoto M. 2007. Subcellular localization and cytoplasmic complex status of endogenous Keap1. Genes Cells 12:1163–1178. doi: 10.1111/j.1365-2443.2007.01118.x. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suzuki T, Maher J, Yamamoto M. 2011. Select heterozygous Keap1 mutations have a dominant-negative effect on wild-type Keap1 in vivo. Cancer Res 71:1700–1709. doi: 10.1158/1538-7445.AM2011-1700. [DOI] [PubMed] [Google Scholar]
- 32.Dhanoa BS, Cogliati T, Satish AG, Bruford EA, Friedman JS. 2013. Update on the Kelch-like (KLHL) gene family. Hum Genomics 7:13. doi: 10.1186/1479-7364-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Canning P, Cooper CD, Krojer T, Murray JW, Pike AC, Chaikuad A, Keates T, Thangaratnarajah C, Hojzan V, Ayinampudi V, Marsden BD, Gileadi O, Knapp S, von Delft F, Bullock AN. 2013. Structural basis for Cul3 protein assembly with the BTB-Kelch family of E3 ubiquitin ligases. J Biol Chem 288:7803–7814. doi: 10.1074/jbc.M112.437996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McMahon M, Lamont DJ, Beattie KA, Hayes JD. 2010. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc Natl Acad Sci U S A 107:18838–18843. doi: 10.1073/pnas.1007387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hourihan JM, Kenna JG, Hayes JD. 2013. The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid Redox Signal 19:465–481. doi: 10.1089/ars.2012.4944. [DOI] [PubMed] [Google Scholar]
- 36.Fujii S, Sawa T, Ihara H, Tong KI, Ida T, Okamoto T, Ahtesham AK, Ishima Y, Motohashi H, Yamamoto M, Akaike T. 2010. The critical role of nitric oxide signaling, via protein S-guanylation and nitrated cyclic GMP, in the antioxidant adaptive response. J Biol Chem 285:23970–23984. doi: 10.1074/jbc.M110.145441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogura T, Tong KI, Mio K, Maruyama Y, Kurokawa H, Sato C, Yamamoto M. 2010. Keap1 is a forked-stem dimer structure with two large spheres enclosing the intervening, double glycine repeat, and C-terminal domains. Proc Natl Acad Sci U S A 107:2842–2847. doi: 10.1073/pnas.0914036107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tong KI, Padmanabhan B, Kobayashi A, Shang C, Hirotsu Y, Yokoyama S, Yamamoto M. 2007. Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol Cell Biol 27:7511–7521. doi: 10.1128/MCB.00753-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fukutomi T, Takagi K, Mizushima T, Ohuchi N, Yamamoto M. 2014. Kinetic, thermodynamic, and structural characterizations of the association between Nrf2-DLGex degron and Keap1. Mol Cell Biol 34:832–846. doi: 10.1128/MCB.01191-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M, Hirohashi S. 2008. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A 105:13568–13573. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang DD, Lo SC, Cross JV, Templeton DJ, Hannink M. 2004. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol 24:10941–10953. doi: 10.1128/MCB.24.24.10941-10953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao L, Wang J, Sekhar KR, Yin H, Yared NF, Schneider SN, Sasi S, Dalton TP, Anderson ME, Chan JY, Morrow JD, Freeman ML. 2007. Novel n-3 fatty acid oxidation products activate Nrf2 by destabilizing the association between Keap1 and Cullin3. J Biol Chem 282:2529–2537. doi: 10.1074/jbc.M607622200. [DOI] [PubMed] [Google Scholar]
- 43.Rachakonda G, Xiong Y, Sekhar KR, Stamer SL, Liebler DC, Freeman ML. 2008. Covalent modification at Cys151 dissociates the electrophile sensor Keap1 from the ubiquitin ligase CUL3. Chem Res Toxicol 21:705–710. doi: 10.1021/tx700302s. [DOI] [PubMed] [Google Scholar]
- 44.Cleasby A, Yon J, Day PJ, Richardson C, Tickle IJ, Williams PA, Callahan JF, Carr R, Concha N, Kerns JK, Qi H, Sweitzer T, Ward P, Davies TG. 2014. Structure of the BTB domain of Keap1 and its interaction with the triterpenoid antagonist CDDO. PLoS One 9:e98896. doi: 10.1371/journal.pone.0098896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Y, Paonessa JD, Zhang Y. 2012. Mechanism of chemical activation of Nrf2. PLoS One 7:e35122. doi: 10.1371/journal.pone.0035122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baird L, Dinkova-Kostova AT. 2013. Diffusion dynamics of the Keap1-Cullin3 interaction in single live cells. Biochem Biophys Res Commun 433:58–65. doi: 10.1016/j.bbrc.2013.02.065. [DOI] [PubMed] [Google Scholar]